Human Immunodeficiency Virus Type 1 Assembly, Budding, and Cell-Cell Spread in T Cells Take Place in Tetraspanin-Enriched Plasma Membrane Domains (original) (raw)
Abstract
Human immunodeficiency virus type-1 (HIV-1) egress from infected CD4+ T cells is thought to be via assembly and budding at the plasma membrane and may involve components of the T-cell secretory apparatus, including tetraspanins. However, many studies on HIV-1 assembly have examined the trafficking of viral proteins in isolation, and most have used immortalized epithelial, fibroblastic, or hematopoietic cell lines that may not necessarily reflect natural infection of susceptible T cells. Here we have used immunofluorescence and cryoimmunoelectron microscopy (CEM) to examine protein transport during HIV-1 assembly in productively infected Jurkat CD4+ T cells and primary CD4+ T cells. The HIV-1 envelope glycoprotein (Env) and the core protein (Gag) colocalize strongly with CD63 and CD81 and less strongly with CD9, whereas no colocalization was seen between Env or Gag and the late endosome/lysosomal marker Lamp2. CEM revealed incorporation of CD63 and CD81 but not Lamp2 into virions budding at the plasma membrane, and this was supported by immunoprecipitation studies, confirming that HIV-1 egress in T cells is trafficked via tetraspanin-enriched membrane domains (TEMs) that are distinct from lysosomal compartments. CD63, CD81, and, to a lesser extent, CD9 were recruited to the virological synapse (VS), and antibodies against these tetraspanins reduced VS formation. We propose that HIV-1 promotes virus assembly and cell-cell transfer in T cells by targeting plasma membrane TEMs.
The major targets of human immunodeficiency virus type 1 (HIV-1) infection in vivo are CD4+ T cells and macrophages. In both cell types, infection culminates in release of progeny virions by budding and release; however, the sites at which HIV-1 assembles and buds differ between these two cell types. In CD4+ T cells, it is generally accepted that HIV-1 buds from the plasma membrane (13, 15, 16, 23, 27, 38, 39, 41, 43, 49). In contrast, the principal site of viral assembly in macrophages has been proposed to be intracellular vesicles potentially corresponding to multivesicular bodies (MVBs), where virus may remain infectious for subsequent exocytosis (31, 36, 42, 44, 51, 54), although this has been challenged (30, 63).
The molecular pathways regulating the trafficking of HIV-1 proteins have been intensely studied over recent years and have provided many important insights into virus assembly. In infected T cells, both HIV-1 Env and Gag are targeted to GM-1-rich, lipid raft-like plasma membrane, in part by palmitoylation and myristoylation signals (3, 7, 55, 56). At the plasma membrane, HIV-1 Env associates with the outer leaflet, whereas Gag is transported independently of Env and associates with the inner leaflet. The integrity of lipid raft-like plasma membrane domains is crucial for virus assembly and budding of infectious virions (6, 10, 18, 24, 27, 32, 43, 48). Self-multimerization of Gag at the appropriate cellular membrane appears to drive viral core formation, and capsids acquire Env during the process of budding and envelopment to produce infectious virions. Cellular components are also involved in the trafficking of Env and Gag, including the clathrin adaptor complex proteins AP1, AP2, and AP3 (1, 4, 9, 11, 64) and the endosomal sorting complex required for transport (ESCRT) (41). Moreover, the lipid second messenger phosphatidylinositol 4,5 bisphosphate (PIP2) has been implicated in HIV-1 Gag trafficking and influences the site of virus budding in different cell types (46). Finally, elements of the cellular cytoskeleton and the secretory pathway are also likely to be major factors that spatially and temporally regulate HIV-1 egress (5, 9, 44, 45, 50, 52).
Although much has been done to elucidate the molecular cell biology of HIV-1 morphogenesis pathways, most of the work has been performed using viral gene expression systems in immortalized cell lines of epithelial or fibroblastic origin, not least because of their facility for gene transfer and ease of imaging. The weight of evidence suggests that most nonlymphoid immortalized cell lines demonstrate a mixed phenotype, with viral proteins trafficking both to the plasma membrane and into vesicular compartments resembling MVBs (19, 44, 45, 52). The association of tetraspanins such as CD63, CD81, and CD82, the lysosomal marker Lamp1, and major histocompatibility complex II (MHC class II) molecules with the budding virion compartment and their presence within the virion have been taken as support for the late endosome (LE)/MVB classification of this compartment in nonlymphoid cell types (44, 47, 51). Moreover, many of these studies have followed Gag expressed in isolation (5, 47, 57), whereas the trafficking of Gag is influenced by other viral gene products such as Env (9, 33, 34). Thus, relatively little work has been done in the context of productive infection in T cells, particularly primary T cells. Although there is evidence that recombinant Gag expressed in Jurkat T cells colocalizes with CD81 (5), there are conflicting reports as to whether Gag and CD63 colocalize in T cells (5, 44, 47). Even less well studied is the association between Env and tetraspanins, although recent data suggest Env may colocalize with CD9 and CD63 at the surface of the Jurkat T-cell line (45).
To address this, we have analyzed viral trafficking and assembly in Jurkat T cells and primary CD4+ T cells infected with replication-competent HIV-1. We find that Env and Gag colocalize with the tetraspanin proteins CD63 and CD81 both intracellularly and at the plasma membrane, with the majority of viral antigen located at the plasma membrane. HIV-1 assembles in actin-dependent tetraspanin-enriched membrane (TEM)-enriched plasma membrane caps and patches, and both CD63 and CD81 are incorporated into nascent virions. In conjugates formed between HIV-1-infected and uninfected CD4+ T cells, viral antigens and receptors are trafficked towards the virological synapse (VS) (26, 28) and TEMs are recruited along with Gag and Env to the VS. Based upon these data, we propose that HIV-1 infection intersects the secretory pathway in T cells, spatially and temporally organizing viral assembly to promote viral budding and cell-cell spread.
MATERIALS AND METHODS
Cells and tissue culture.
The CD4+/CXCR4+ T-cell line Jurkat CE6.1 (from the American Type Culture Collection [ATCC]) was maintained in RPMI 1640 supplemented with streptomycin (100 μg/ml), penicillin (100 U/ml), and 10% fetal calf serum (FCS). Primary cells, purified from the peripheral blood of healthy donors, were stimulated every 21 to 28 days with irradiated allogeneic peripheral blood mononuclear cells and 1 μg/ml phytohemagglutinin and maintained in RPMI 1640 (Gibco BRL) containing 5% human serum, 10 mM HEPES, 50 μM mercaptoethanol, and 10 to 100 U interleukin-2. To prepare cells infected with the T-cell-line-adapted HIV-1 strain LAI (termed JurkatLAI), 5 × 106 Jurkat cells were infected at a multiplicity of infection of approximately 0.01 and cultured for up to 10 days. Alternatively, 2 × 106 to 4 × 106 primary cells were infected (primaryLAI) at a multiplicity of infection of approximately 0.01 and cultured for up to 7 days. Cells were phenotyped for surface Env expression using pooled human anti-HIV immunoglobulin (HIVIg; from the NIH AIDS Research and Reference Reagent Program, Rockville, MD) at 50 μg/ml, detected by anti-human IgG-phycoerythrin (Jackson Immunoresearch) and for CD4 expression using monoclonal antibody (MAb) 13B.8.2 (Center for AIDS Research [CFAR]). Jurkat cells were used between days 7 and 14 postinfection, when Env expression was readily detectable and CD4 expression was negative. Primary cells do not down regulate CD4 detectably but do express viral Env and were used 5 to 7 days postinfection. For conjugate experiments, peripheral blood mononuclear cells were separated from fresh blood of a healthy HIV-1-seronegative donor using a Ficoll-Hypaque gradient and negatively enriched for CD4+ T cells (hereafter termed “target cells”) by magnetic cell sorting according to the manufacturer's instructions (Miltenyi Biotec), and this routinely gave >90% pure CD4+ T cells. Cells were diluted in RPMI 1640-1% FCS (wash buffer [WB]) and used immediately.
Immunofluorescence and confocal microscopy.
CD4+ T cells (2 × 105 to 5 × 105) were washed in RPMI 1640-1% FCS and incubated on poly-l-lysine (Sigma)-treated coverslips at 37°C for up to 60 min. Cells were then fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS)-1% bovine serum albumin (BSA) for 15 min followed by quenching in ammonium chloride for 10 min at room temperature. For intracellular staining of tetraspanins and HIV Gag, the cells were permeabilized in 0.1%Triton X-100-5% FCS for 20 min at room temperature and washed extensively in PBS-1% BSA. For surface staining of tetraspanins, the cells were first adhered at 37°C and then incubated with antibodies for 30 min at 4°C before being fixed. Immunostaining was done with the following antibodies. CD63 was detected with an anti-CD63 hybridoma supernatant (Developmental Studies Hybridoma Cell Bank, Iowa City, IA) or an IgG2b anti-CD63 MAb (a generous gift from Mark Marsh, University College, London, United Kingdom). Mouse MAbs to CD81 (clone JS-81) and CD9 (clone M-L13) were purchased from BD Pharmingen. Rabbit antiserum against Lamp2 was from the Developmental Studies Hybridoma Cell Bank. HIV-1 Env was detected with the gp41-specific MAb 50-69 (CFAR) or the carbohydrate-specific anti-Env MAb 2G12 (Polymun Scientific), and Gag was stained with rabbit antisera against p17 and p24 or a mouse MAb specific for p24 (CFAR). Primary antibodies were detected with fluorescein isothiocyanate (FITC)-, tetramethyl rhodamine isocyanate (TRITC)-, or Cy5-conjugated donkey anti-mouse, human or rabbit secondary antibodies that were tested for an absence of interspecies reactivity (Jackson Immunoresearch). Alternatively, isotype-specific (anti-IgG1 and anti-IgG2b) Alexa-Fluor 568 and Alexa-Fluor 488 secondary antibodies were used (Molecular Probes). Coverslips were mounted with ProLong antifade mounting solution (Molecular Probes), and confocal analysis was performed using a Bio-Rad Radiance 2000 MP laser-scanning confocal microscope. Image processing and three-dimensional reconstruction of sequential z series was performed using MetaMorph version 6.1 and Adobe Photoshop version 7.
Quantification of copolarization.
JurkatLAI cells alone were stained for Env, Gag, and either CD63 or CD81 as described above, and multiple random sections of low-power fields were acquired using confocal microscopy. The total number of effector cells was counted, and each effector cell was analyzed for cocapping of Env or Gag with CD63 or CD81. Statistical analysis was performed using a one-way analysis of variance (ANOVA), and statistical significance was assumed when P was <0.05.
Inhibition of the actin cytoskeleton.
Actin remodeling was blocked by treating JurkatLAI cells with 1 μM latrunculin A (Molecular Probes) in WB at 37°C for 60 min prior to fixing and staining for HIV-1 Env, Gag, and tetraspanins. Quantification of the disruption of Env-Gag-tetraspanin colocalization in untreated or inhibitor-treated cells was performed. Multiple random sections of low-power fields were acquired using confocal microscopy, the total number of effector cells was counted, and each effector cell was analyzed for colocalization of Env and Gag with CD63 or CD81. Statistical analysis was performed using an unpaired, two-tailed Student's t test with Bonferroni correction for multiple comparisons, and statistical significance was assumed when P was <0.05.
Flow cytometry of surface tetraspanin expression.
Uninfected CD4+ cells or HIV-1-infected cells (primaryLAI and JurkatLAI) were washed in cold fluorescence-activated cell sorter (FACS) WB (FWB: PBS with 1% FCS and 0.01% sodium azide) and cells were incubated on ice for 1 h with saturating concentrations of antibodies specific for CD63, CD81, or CD9. An irrelevant antibody was also included as a control. Cells were subsequently washed in cold FWB, incubated with anti-mouse IgG-phycoerythrin for 30 min on ice, and fixed in FWB with 1% formaldehyde. Acquisition and analysis were performed using a Becton Dickinson FACS Calibur and CellQuest software.
CEM.
For cryoimmunoelectron microscopy (CEM), JurkatLAI cells (2 × 106) were washed, pelleted by low-speed centrifugation, and fixed in 250 mM HEPES containing 4% paraformaldehyde for 30 min. This was replaced with 8% paraformaldehyde, and cells were incubated for 2 h at 4°C. Fixed cells were washed in 250 mM HEPES, embedded in 2% gelatin, and infused with 2.3 M sucrose and frozen in liquid nitrogen. Ultrathin cryosections (50 nm) were quenched in 20 mM glycine and blocked in PBS-1% BSA. For double-labeling experiments, staining was performed first with either mouse anti-CD63 (Caltag), mouse anti-CD81 (kindly provided by Fedor Berditchevski, University of Birmingham, United Kingdom), or rabbit anti-Lamp2 followed by 5-nm mouse- or 10-nm rabbit-specific gold colloids (Agar). Sections were then washed and labeled with rabbit antiserum raised against HIV Gag (CFAR) or a mouse MAb specific for Gag p55/p17 (CFAR). This was followed by either 10-nm rabbit- or 5-nm mouse-specific gold colloids. Ultrathin sections were examined using a Phillips FEI Technai 12 transmission electron microscope, and digital images were captured using Soft Imaging software and processed using Photoshop. The scale bar represents 100 nm.
Virus precipitation assay.
Immunoprecipitations were based on methods described by others (43, 51). Briefly, cell-free virus containing supernatants prepared from infected T cells was diluted in PBS-3% BSA and incubated overnight at 4°C with 10 μg/ml of either anti-Env antibodies (2G12 and B12; Polymun Scientific and CFAR, respectively), pooled human serum from infected individuals (HIVIg), CD63, CD81, CD9, or a dengue virus-specific control antibody (a gift from Joanna Miller, University of Oxford, United Kingdom). Pansorbin cells (formalin-fixed Staphylococcus aureus; Calbiochem) were blocked in PBS-3% BSA and added to the virus-antibody mixture at a final concentration of 2%. After 1 h at room temperature, captured virus was pelleted by centrifugation at 4,000 rpm for 15 min. Precipitated material was analyzed for p24 Gag content by enzyme-linked immunosorbent assay (ELISA). To measure the effect of actin depolymerization on the incorporation of tetraspanins into virions, 1 × 107 JurkatLAI cells were treated with 1 μM latrunculin A for 3 h at 37°C, washed, and incubated in RPMI 1640-10% FCS without latrunculin A for 3 h. Viral supernatants were collected, pelleted, and resuspended in PBS-3% BSA, and immunoprecipitations were performed as described above.
p24 Gag ELISA.
Antibody-precipitated pellets were resuspended in PBS-3% BSA and inactivated at 56°C for 30 min with 1% Empigen (Calbiochem). Samples were diluted in Tris-buffered saline (pH 7.4) containing 10% FCS and 1% Empigen, and viral p24 was captured with the sheep anti-p24 antibody D7320 (CFAR). Plates were washed, and bound p24 was detected with biotinylated mouse anti-p24 (Aalto Scientific) followed by streptavidin-horseradish peroxidase (Serotec). Plates were developed with the 1-Step Ultra TMB-ELISA (Pierce), and optical density was measured at 450 nm. The p24 content for each precipitation was calculated after subtracting the nonspecific background signal determined by incubating viral supernatants with preblocked Pansorbin cells in the absence of antibody.
VS formation, analysis, and inhibition.
JurkatLAI cells (5 × 105) were mixed with an equal number of primary CD4+ target T cells, and conjugates were allowed to evolve by incubating on poly-l-lysine-treated coverslips for 60 min at 37°C. Surface staining of HIV Env was performed using 10 μg/ml of MAb 50-69 (CFAR) that was included during conjugate formation as this MAb has previously been shown not to be inhibitory in this system (26). Conjugate evolution was arrested by fixation with cold 4% paraformaldehyde, and cells were permeabilized and stained for immunofluorescence analysis as described above. Inhibition of synapse formation with antitetraspanin antibodies was performed as follows. JurkatLAI cells (5 × 105) were mixed with primary CD4+ target T cells (prelabeled with Cell Tracker dye from Invitrogen) in the presence of anti-CD63 (diluted 1/250), anti-CD81 (20 μg/ml), or anti-CD9 (20 μg/ml) antibodies that had been dialyzed to remove sodium azide. Conjugates were allowed to evolve for 60 min at 37°C, and the cells were then fixed, permeabilized, and stained for HIV-1 Gag and Env as described above. Coverslips were analyzed by immunofluorescence, and multiple random sections of low-power fields were acquired. The total number of effector cells was counted, and the percentage of effectors within conjugates was quantified. Conjugates were defined as closely apposed pairs of cells containing at least one target T cell and one Env+ or Gag+ cell. Each effector cell was analyzed for polarization to the cell-cell interface of Env and Gag. Statistical analysis was performed using a one-way ANOVA with Bonferroni correction for multiple comparisons, and statistical significance was assumed when P was <0.05.
RESULTS
Expression of tetraspanins CD63, CD81, and CD9 in Jurkat T cells.
To examine the pathways that regulate the assembly, budding, and cell-cell spread of HIV-1 in T cells, we used both primary CD4+ T cells and the CD4+ T-cell line Jurkat CE6.1, as well as infectious, replication-competent virus containing the full complement of viral proteins. Although there are many studies detailing the subcellular localization of tetraspanins in MHC class-II-expressing antigen-presenting cells (12, 35, 54) and colocalization of HIV-1 proteins with tetraspanins in dendritic cells, macrophages, and HeLa cells (14, 45, 51, 54), the distribution of CD63, CD81, and CD9 is not well characterized in T cells. We therefore first sought to determine their expression levels and cellular distribution in uninfected Jurkat cells. Jurkat cells were adhered to poly-l-lysine-coated coverslips; fixed, permeabilized, and stained with MAbs for CD63, CD81, and CD9; and examined by laser-scanning confocal microscopy (LSCM). Figure 1A shows single optical sections taken through the middle of Jurkat cells labeled for the different tetraspanins. Staining of CD63 was relatively weak and, consistent with that reported for other cell types, was principally in the cytoplasm in distinct structures resembling vesicles, with little staining at the cell periphery. In contrast, most CD81 and CD9 staining appeared to be located proximal to or at the plasma membrane, with minimal labeling of cytoplasmic structures. Distribution of tetraspanins at the surface of Jurkat cells was confirmed by staining live cells for CD63, CD81, and CD9 followed by flow cytometric analysis (Fig. 1B). There was strong surface expression of CD81 (approximately 20-fold higher than CD63), consistent with the fluorescence images indicating predominantly plasma membrane-proximal CD81 labeling. Although the LSCM images indicated that most CD63 is located within cytoplasmic vesicles, Fig. 1B demonstrates that a proportion of the cellular pool of CD63 is also present at the cell surface. CD9 staining was more heterogeneous and shows a mixed population that contains mostly cells with weak surface CD9 expression but also a small population with higher surface expression. Double labeling of permeabilized cells confirmed the virtual absence of colocalization of CD63 with CD81 and CD9 (Fig. 1C).
FIG. 1.

Distribution of tetraspanins in uninfected Jurkat T cells. (A) Uninfected Jurkat T cells were adhered to poly-l-lysine-coated coverslips for 1 h at 37°C, fixed with paraformaldehyde, permeabilized, and stained with MAbs against CD63, CD81, or CD9 (red). Images shown are single confocal sections through the middle of a cell. (B) Cell surface expression of CD63, CD81, and CD9 was measured on uninfected Jurkat T cells by flow cytometry. The mean fluorescence intensity of CD63, CD81, and CD9 staining (solid line) is shown compared to a control MAb (broken line), and the mean fluorescence values (insert) are shown for each plot. Levels of surface expression of CD63 (white bars), CD81 (gray bars), and CD9 (black bars) are compared between HIV-1 infected Jurkat cells and uninfected Jurkat cells (bottom panel). Data are the means of three independent experiments, and error bars represent the standard error of the mean. (C) CD63 does not colocalize with CD81 or CD9 in Jurkat T cells. Jurkat cells were adhered to coverslips, fixed, permeabilized, and stained for CD63 (green) and either CD81 (left panel, red) or CD9 (right panel, red). Primary MAbs were detected with anti-IgG2b (CD63) and anti-IgG1 (CD81 and CD9) isotype-specific conjugated secondary antibodies. The three-dimensional reconstructed z series (top) and a single optical section through the middle of the cell (center) are shown. The bottom panels each show a magnified image of a region from the single optical section selected for strong staining (boxed).
HIV-1 Env and Gag colocalize with CD63 and CD81 in JurkatLAI cells.
Jurkat and primary CD4+ T cells infected with the X4 isolate HIV-1LAI (JurkatLAI and primaryLAI, respectively) were stained for CD63, CD81, and CD9 and either the Env gp120 subunit or the core proteins Gag p24 and p17. LSCM revealed readily detectable HIV-1 antigens in most cells, and viral proteins were frequently patched or capped towards one end of the cell, as reported previously (9, 26, 42). Surface costaining of live JurkatLAI (Fig. 2A) and primaryLAI (Fig. 2B) cells for HIV-1 Env and tetraspanins prior to fixation revealed Env localization in tetraspanin-enriched domains at the plasma membrane. This was particularly obvious for CD63 that was redistributed during HIV-1 infection from a principally intracellular location (Fig. 1A and C) into a tight cap (Fig. 2A) or polarized clusters (Fig. 2B) with Env at the plasma membrane. CD81-Env colocalization was also evident, whereas although regions of colocalization were observed between CD9 and Env on primaryLAI cells, no obvious colocalization was seen using JurkatLAI cells. However, the low levels of CD9 on Jurkat cells prevented an unequivocal interpretation of these data. Infection of Jurkat cells (Fig. 1B) or primary CD4+ T cells (not shown) did not significantly alter surface expression levels of CD63, CD81 or CD9, as measured by flow cytometry (P ≥ 0.05 by unpaired, two-tailed Student's t test). This suggests that any changes to the cellular localization of tetraspanins after HIV-1 infection are due to a redistribution of tetraspanin-containing compartments within the cytoplasm and/or coalescence of preexisting TEMs at the plasma membrane. Fixation and permeabilization of JurkatLAI (Fig. 2C) and primaryLAI (Fig. 2D) cells followed by labeling of Gag revealed similar labeling patterns to those observed for Env: strong colocalization of capped Gag was observed with CD63 and CD81. Similar to that seen with surface labeling, some colocalization was apparent between CD9 and Gag in primaryLAI cells and little or no colocalization of Gag and CD9 was observed in JurkatLAI cells, but the weak CD9 expression precluded a definitive conclusion. Very little specific colocalization was observed between Gag and the LE/lysosome marker Lamp2, indicating that the CD63/CD81/Gag-positive compartment was unlikely to be lysosomal in nature (Fig. 2E). The percentage of JurkatLAI cells showing colocalization of Env and Gag with CD63 and CD81 was quantified. We routinely detect cocapping of Env and Gag in about 50% of JurkatLAI cells examined by LSCM (26; data not shown), and of these capped cells, colocalization between HIV-1 proteins and CD63 or CD81 was as follows: CD63 or CD81 and Env, 67% ± 11% and 75% ± 9%, respectively; CD63 or CD81 and Gag, 73% ± 11% and 71% ± 9%, respectively. There was no statistically significant difference between colocalization of Env or Gag with CD63 or CD81 (P > 0.05 by one-way ANOVA). JurkatLAI cells without cocapping of tetraspanins with Env or Gag showed the same CD81 and CD63 staining pattern as uninfected cells: uniform surface staining of CD81 without any obvious polarization and a vesicular cytoplasmic distribution of CD63+ with weak surface expression. Why a percentage of cells do not show cocapping is unclear, but it may be influenced by what stage the infection is at and whether these cells were more recently infected than others in the culture.
FIG. 2.

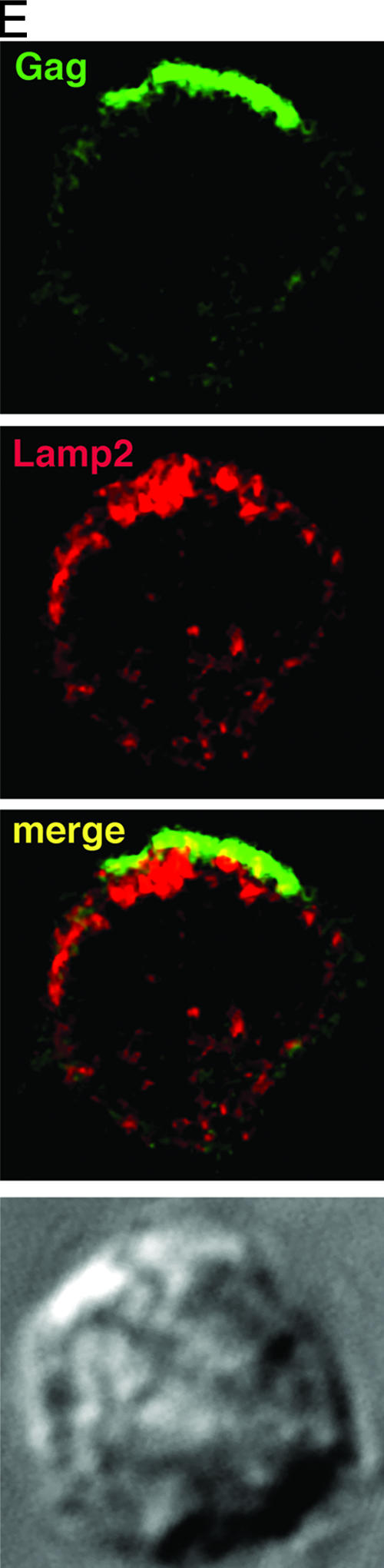
HIV-1 Env and Gag colocalize with tetraspanins in virus-infected T cells. HIV-1-infected JurkatLAI T cells (A) or primaryLAI cells (B) were surface stained for Env and tetraspanins. Cells were adhered to coverslips for 1 h at 37°C in the presence of the human Env-specific antibody 2G12 and then incubated at 4°C for 30 min with MAbs against CD63 (left panel), CD81 (center panel), or CD9 (right panel). Cells were fixed, and surface staining of Env (green) and tetraspanins (red) was analyzed by LSCM. Images are single sections through the middle of a cell, and areas of colocalization are yellow. JurkatLAI T cells (C) or primaryLAI cells (D) were permeabilized and stained for Gag and tetraspanins. Cells were adhered to coverslips, fixed and permeabilized, and incubated with rabbit antisera against Gag p17 and p24 and MAbs against CD63 (left panel), CD81 (center panel), or CD9 (right panel). Intracellular staining of Gag (green) and tetraspanins (red) was analyzed by LSCM. Images are single sections through the middle of the cell, and areas of colocalization are yellow. (E) Jurkat LAI cells were adhered to coverslips, fixed, permeabilized, and stained for intracellular Gag and Lamp2. Images are single sections through the middle of the cell, and areas of colocalization are yellow.
To confirm colocalization of CD63 and CD81 with HIV-1 proteins and to address the concept that the virus is targeting TEMs for viral assembly, we triple labeled the surface of live JurkatLAI and primaryLAI, cells and the corresponding uninfected T cells for CD63, CD81, and Env. Figure 3A and B show live cell surface staining of uninfected Jurkat cells and primary CD4+ T cells, respectively, of TEMs containing both CD63 and CD81. Regions of CD63 and CD81 overlap are limited and are less frequently observed on primary than Jurkat T cells. Consistent with the data shown in Fig. 2, these TEM domains also partially costain for HIV-1 Env (particularly evident on primary CD4+ T cells), indicating that TEMs may be sites of Env expression and/or virus assembly. The presence of double CD63/CD81-staining TEMs at the plasma membrane of live uninfected cells contrasts with their absence of colocalization in uninfected permeabilized cells (Fig. 1), indicating that although CD63 and CD81 are closely associated at the cell surface, they do not appear to reside in the same cytoplasmic compartment.
FIG. 3.

HIV-1 Env accumulates in CD63+ CD81+ TEMs at the plasma membrane of T cells. (A) Jurkat T cells and (B) primary CD4+ T cells were used uninfected (left panel) or infected with HIV-1 (right panel). Cells were adhered to coverslips for 1 h at 37°C in the presence of the human Env-specific antibody 2G12 and then incubated at 4°C for 30 min with MAbs against CD63 and CD81. Cells were fixed, and the surface staining of Env (blue), CD63 (green), and CD81 (red) was analyzed by LSCM. The top panels show a single section through the middle of the cell, and the bottom panels are magnified images of a region selected for strong staining from the single optical section (boxed). Regions of CD63 and CD81 colocalization are yellow, and regions of Env, CD63, and CD81 triple colocalization appear white.
Although the LSCM colocalization studies imply spatial overlap between HIV-1 antigens and CD63 and CD81, the resolution of this technique is insufficient to unequivocally determine whether they are in the same membrane or in a proximal but distinct cellular compartment. If they are indeed within the same membrane, it would be expected that tetraspanins would be incorporated into plasma membrane budding virions, as has been shown in other cell types (5, 51, 54). CEM of immunostained ultrathin sections revealed 100-nm virion-like structures budding from the plasma membrane of JurkatLAI cells that colabeled for HIV-1 Gag and CD63 or CD81 (Fig. 4A and B and Table 1). Consistent with the LCSM data showing minimal Gag-Lamp2 colocalization (Fig. 2E), we did not observe incorporation of the lysosomal marker Lamp2 into a budding virion (Fig. 4C and Table 1). To confirm these results, we immunoprecipitated from virus-containing supernatants using a mixture of HIV-1 Env-specific MAbs 2G12 and IgG1b12, pooled human serum from HIV-1-infected individuals (HIVIg), or MAbs to CD63, CD81, or CD9, according to established methods (43, 51), and quantified the precipitated material for Gag p24 by ELISA. Figure 4D shows that antibodies specific for HIV-1 proteins and CD63 or CD81 precipitated 5- to 16-fold more p24 than an irrelevant control antibody. CD63 and CD81 precipitated approximately two- and threefold more virus than Env-specific MAbs, suggesting that these cellular proteins may be more abundant on the virion envelope than Env, although we cannot exclude effects of antibody avidity. Precipitation with CD9 gave more variable results that probably reflect the low level and heterogeneous expression of CD9 expression on CD4+ T cells, although an average fourfold increase above the control was observed.
FIG. 4.

CD63 and CD81 are incorporated into budding virions. Ultrathin cryosections of HIV-1-infected JurkatLAI cells were labeled with rabbit antiserum against HIV Gag p17 and a mouse MAb specific for either CD63 (A) or CD81 (B). Primary antibodies were labeled with mouse-specific 5-nm and rabbit-specific 10-nm gold colloids. CD63 and CD81 labeling (5-nm gold colloid) is highlighted with arrowheads. (C) Cryosections were labeled with a mouse MAb against Gag and rabbit antiserum specific for Lamp2. Here Gag was visualized with mouse-specific 5-nm gold colloids and Lamp2 with rabbit-specific 10-nm colloids. Lamp2 labeling is highlighted with arrowheads, and a budding virion (Lamp− Gag+) is highlighted with an asterisk. Bar, 100 nm. (D) Virus precipitation with antibodies against HIV-1 proteins or tetraspanins. Cell-free viral supernatants were precipitated with Env-specific antibodies (MAbs 2G12 and IgG1b12), tetraspanin-specific MAbs, or a control antibody, and the Gag p24 content of the precipitated material was measured by ELISA. The nonspecific background signal was determined by incubating viral supernatants with preblocked Pansorbin cells without antibody, and these values were subtracted to calculate the concentration of p24 in ng/ml. Bars represent means of two independent experiments, and variability is represented by the standard error of the mean.
TABLE 1.
Quantification of tetraspanin incorporation into HIV-1 virions
| Antigen | % of positive virionsa | Avg no. of colloids/virionb | No. of budding virionsc |
|---|---|---|---|
| Gag | 100 | 5.6 | 63 |
| CD63 | 52 | 0.65 | 23 |
| CD81 | 82 | 1.4 | 28 |
| Lamp2 | 0 | 0 | 12 |
Actin depolymerization disrupts polarized TEM-HIV-1 assembly platforms.
Since tetraspanins are regulators of actin organization (21), we wished to investigate the existence of physical and/or functional associations between the T-cell actin cytoskeleton and HIV-1 assembly and budding platforms at the plasma membrane. To do this, JurkatLAI cells untreated or treated with latrunculin A for 1 h were fixed, labeled, and imaged by LCSM (Fig. 5A and B). In untreated cells, the polar cap of Gag, Env, and tetraspanins CD63 and CD81 is evident, with strong overlap between the antigens. Latrunculin A destroyed HIV-1 antigen polarization: Gag was dispersed and Env staining was eliminated, probably by dilution within the plane of the membrane, as we have observed previously (27). CD63 labeling was confined to intracellular clusters with no obvious staining at the plasma membrane, whereas CD81 staining was dispersed within or proximal to the plasma membrane in small patches reminiscent of uninfected Jurkat cells. Similar results were obtained using primaryLAI cells (data not shown). Quantification of the colocalization before and after latrunculin A treatment revealed an 85% decrease in the number of cells with cocapping of HIV-1 antigens and CD63 (P = 0.001) and a 69% decrease with CD81 (P = 0.01). These data are consistent with a role for actin association with and stabilization of tetraspanin-HIV-1 protein-enriched plasma membrane domains. To determine if actin depolymerization disrupts the tetraspanin-HIV-1 association in budding virions, we treated JurkatLAI cells with latrunculin A for 3 h at 37°C, washed them, collected the supernatants over 3 h, and performed an immunoprecipitation assay to measure the effect of latrunculin A on p24 capture by anti-HIV-1 and antitetraspanin antibodies. We did not detect any significant difference between the untreated and latrunculin A-treated cells in the amount of p24 captured and precipitated (data not shown), indicating that although depolymerization of the actin cytoskeleton disperses HIV-1 and tetraspanin staining and colocalization, tetraspanins still associate with budding virions. The most likely explanation for this is that actin depolymerization reverses macroscopic viral antigen clustering with tetraspanins but does not eliminate microscopic clustering sufficient for viral budding, allowing tetraspanin-HIV-1 association. This is in agreement with other data showing that latrunculin A treatment reduces, but does not prevent, HIV-1 budding (29).
FIG. 5.

Actin depolymerization disrupts TEM-HIV-1 polarized assembly platforms. JurkatLAI cells (5 × 105) were washed, resuspended in RPMI 1640-1% FCS, and adhered to poly-l-lysine-coated coverslips at 37°C. The cells were either untreated (left panel) or treated with latrunculin A (right panels) and stained for HIV-1 Env with the MAb 50-69 (blue). Cells were fixed, permeabilized, and stained for HIV-1 Gag (green) and either CD63 (A [red]) or CD81 (B [red]). Images are single sections through the middle of the cell with the corresponding Nomarski image: areas of red/green colocalization appear yellow, and areas of red/green/blue colocalization appear white.
Tetraspanins are enriched at the VS.
HIV-1 can spread directly between T cells via formation of a VS (26, 28). The HIV-1 CD4+ T-cell VS is characterized by actin-dependent recruitment of adhesion molecules and viral receptors on the uninfected target cell and HIV-1 Env and Gag in the infected cells to the intercellular junction. Polarized budding of virions occurs across the synaptic cleft, and because directed budding of virus at the HIV-1 VS is reminiscent of regulated exocytosis at the cytotoxic T-cell-target interface and because HIV-1 proteins colocalize with tetraspanins at or close to the plasma membrane, we hypothesized that CD63 and CD81 might also be recruited to the VS. As shown in Fig. 6, this is indeed the case. Both CD63 and CD81 colocalized strongly with HIV-1 Env and Gag in JurkatLAI cells and were highly enriched in the effector cell at the cell-cell interface (Fig. 6). Clustering of CD63 and CD81 at the VS occurred at approximately the same frequency as cocapping of tetraspanins with HIV-1 proteins in unconjugated JurkatLAI cells (67 to 75% and 75 to 80%, respectively), and no significant increase in colocalization of HIV-1 with CD63 or CD81 was observed in response to VS formation. CD9 also colocalized at the VS in 50% of synapses examined, but the intensity of staining was appreciably less than those of CD63 and CD81, reflecting the weak expression of CD9 on Jurkat cells. As expected, given the undetectable association of Lamp2 with assembling HIV-1 proteins, there is little colocalization of Lamp2 with HIV-1 Env and Gag in infected cells and no obvious enrichment at the VS. When taken together, our LSCM, CEM, and immunoprecipitation data demonstrate the presence of tetraspanins at the HIV-1 exit site from infected cells and within virions. However, we cannot rule out a contribution of tetraspanin labeling in the VS from the target cell in effector-target cell conjugates. With relevance to this possibility, it has recently been reported that CD81 and CD9 on target cells can play a role in HIV-1-induced fusion (17). To investigate whether tetraspanin enrichment at the intercellular contact surface has a functional effect on VS formation, we introduced antibodies against tetraspanins during conjugate formation between HIV-1-infected T cells and uninfected T cells (Table 2). Our data show that MAbs to CD63, CD81, and CD9 all independently reduced VS formation by approximately 50%, and the CD81-specific MAb had the additional property of reducing conjugate formation by approximately 50%. These data did not reach statistical significance (P > 0.05 measured by one-way ANOVA) but showed a clear trend.
FIG. 6.

Tetraspanins are components of the T-cell VS. HIV-1-infected JurkatLAI cells (effectors) were mixed with an equal number of freshly-isolated primary CD4+ T cells (targets) and incubated on poly-l-lysine-treated coverslips in the presence of the Env-specific MAb 50-69 (blue) for 1 h at 37°C. Conjugate evolution was arrested by fixing, and cells were permeabilized and stained using rabbit antisera directed against Gag p17 and p24 (green) and mouse MAbs specific for either CD63 (red, left panel), CD81 (red, second panel from left), or CD9 (red, second panel from right). Alternatively, cells were stained with a mouse MAb against Gag (green) and rabbit antisera against Lamp2 (red, right panel). CD81 and CD63, and to a lesser extent CD9, but not Lamp2, are enriched in the effector cell at the conjugate interface and colocalize with HIV-1 Env and Gag. Images are single sections through the middle of a conjugate, and the target cell is labeled with an asterisk. Areas of colocalization appear white, and the corresponding Nomarski image is shown.
TABLE 2.
Effect of tetraspanin antibodies on VS formation
| Treatment | % Conjugateda | % VSb | No. of Env+ Gag+ effector cellsc | P valued |
|---|---|---|---|---|
| Untreated | 21 | 32 | 191 | |
| Anti-CD63 | 16 | 14 | 183 | >0.05 |
| Anti-CD81 | 11 | 15 | 183 | >0.05 |
| Anti-CD9 | 15 | 17 | 216 | >0.05 |
DISCUSSION
Since CD4+ T cells are the major targets for HIV-1 infection in vivo, understanding the mechanisms that govern the trafficking of viral proteins and the production of infectious virions in these cells is paramount. Increasing evidence indicates that the temporal and spatial control of HIV-1 assembly is complex, involving many components of the cellular transport machinery. By identifying the molecules involved in the trafficking of HIV-1 proteins, it is anticipated that novel targets may be identified for potential therapeutic intervention. Here we concentrated on the association of HIV-1 proteins with tetraspanins, markers commonly used to identify compartments within the secretory complex. We show that in uninfected T cells most CD63 occupies an intracellular compartment distinct from CD81 and CD9, a large proportion of CD81 expression is at the cell surface, and none of these tetraspanins colocalizes with the LE marker Lamp2. Upon infection with HIV-1, most CD63 staining relocalizes from a dispersed vesicular cytoplasmic distribution towards a more condensed arrangement proximal to and at the plasma membrane. This results in CD63 being brought into close proximity with CD81, where these markers colocalize with patches or caps of Env and Gag. These data imply that HIV-1 infection is altering the relative distribution of tetraspanins within the cell, altering their trafficking spatially or kinetically, or both.
In macrophages, HIV-1 accumulates in structures resembling MVBs but recently reported to be continuous with the plasma membrane (63). These compartments are positive for CD63, CD81, and CD82, and the tetraspanins are incorporated into virions (51, 54). This differs from HIV-1-pulsed but uninfected dendritic cells, in which surface-bound HIV-1 particles are internalized into a CD81+ CD82+ CD9+ but CD63− endocytic compartment (14) that is then trafficked to the cell surface and presented to T cells at the dendritic cell-T-cell VS (37). There are conflicting reports about whether Gag colocalizes with CD63 in cells that are not of the myeloid lineage. This uncertainty reflects differences in the cell types used but may also be a consequence of expressing HIV-1 Gag in the absence of other viral proteins such as Env, Nef, and Vpu (5, 9, 20, 33, 34, 59, 62). Our findings in Jurkat T cells agree with those of another recent study in which viral antigen-associated TEMs were observed in Jurkat cells and HIV-1 Env colocalized with CD63 and CD9 at the plasma membrane (45). In addition, Booth et al. (5) have recently reported colocalization of Gag with CD63 and CD81 in Jurkat cells expressing recombinant Gag protein. Our observations that CD63, CD81, and, to a lesser extent, CD9 all colocalize with both Env and Gag in primary CD4+ and Jurkat T cells infected with replication-competent virus support and extend these previous studies and provides strong evidence for a universal HIV-1 exit strategy via T-cell tetraspanin-enriched membranes. In addition, we have shown for the first time that the HIV-1 assembly process intersects the tetraspanin sorting pathway in primary T cells, that CD81 and CD63 are incorporated into virions that bud from CD4+ T cells, that a functional actin cytoskeleton is required to maintain the integrity of HIV-1 antigen-containing TEMs, and that TEMs are recruited to the VS. Thus, a consensus is emerging that HIV-1 intersects elements of the secretory pathway to coordinate viral egress in cells that are the natural targets for HIV-1 in vivo.
We did not observe any colocalization of Lamp2 with Env or Gag in virus-infected T cells, nor was Lamp2 incorporated into budding virions, similar to results reported for macrophages (51). Thus, it appears that the compartments through which HIV-1 proteins traffic and the membranes from which virions bud contain similar tetraspan markers in both T cells and macrophages but are distinct from the conventional CD63+ Lamp+ vesicle characteristic of LEs. The association of HIV-1 proteins with tetraspanins such as CD63 has been taken as evidence that HIV-1 budding is targeted to endosomal membranes, be they intracellular vesicles or plasma membrane regions with inserted endosomal membrane patches (5, 44, 51) and that endosomal membranes are the final target in all cell types of the HIV-1 transport pathway. It has been proposed that targeting endosomal membranes would allow HIV-1 to access the ESCRT machinery that is necessary for membrane vesiculation and viral budding (5, 41). An alternate hypothesis is that newly synthesized HIV-1 Gag is targeted directly to the plasma membrane in all cell types, including macrophages (30, 45, 57, 63). The detection of ESCRT proteins at the plasma membrane (63) adds credibility to this proposal. Subsequent plasma membrane endocytosis may then transport viral proteins to LE in some cell types, where they could accumulate and their detection therein would give the impression that HIV-1 proteins are trafficked directly to these organelles (30). In support of a “single-membrane model” of HIV-1 budding it has been reported that a significant fraction of the cellular CD63 (and MHC class II) traffics via the plasma membrane en route to lysosomes (25). The colocalization of Env and Gag with CD63 at the plasma membrane may therefore simply reflect the utilization by these proteins of similar transport pathways, with plasma membrane as the predominant site of HIV-1 assembly in all cell types.
Tetraspanin function is diverse and incompletely characterized due to a lack of defined ligand-receptor interactions and a striking functional redundancy between members (21, 22). Despite this, they are known to exert pleiotropic effects on cell migration, signaling, adhesion, fusion, and cytoskeletal reorganization and are thought to function as adaptor molecules to modulate the function of protein complexes, the so-called “tetraspan web” (reviewed in references 21 and 22). Although almost ubiquitously expressed (21, 22), CD81 and CD9 sorting is less well studied than that of CD63. CD81 is a component of the immunological synapse (40), a multimolecular complex that shares many features with the retrovirus-induced VS (28, 53). By analogy with the immunological synapse therefore, recruitment of CD81 to the VS may contribute to VS stability (61). Alternatively, given the ability of tetraspanins to regulate actin cytoskeleton organization and actin-associated lipid rafts (2, 8), they may be involved in recruitment and/or assembly of HIV-1 proteins. Our observation that actin depolymerization disrupts the polar cap of CD81 and CD63 associated with HIV-1 Gag and Env is consistent with a role for actin-TEM interactions. Likewise, the association of CD81 and CD9 with PIP2 (21), which has been demonstrated to play a role in HIV-1 Gag trafficking (46, 58, 60), may also help direct HIV-1 assembly to the plasma membrane. Once at the plasma membrane, tetraspanins may enhance cell-cell spread of HIV-1 across the VS. Further analysis will shed light on whether tetraspanins confer a significant functional advantage to the virus or whether they are simply passengers during HIV-1 trafficking, assembly, and spread.
Acknowledgments
We thank Mark Marsh and Gillian Griffiths for useful discussion and Mark Marsh, Gillian Griffiths, Fedor Berditchevski, and Joanna Miller for the gifts of antibodies. Mike Shaw provided assistance with cryoimmunoelectron microscopy.
This work was funded by grants G0400453 and G0100137 from the Medical Research Council, UK.
Footnotes
▿
Published ahead of print on 23 May 2007.
REFERENCES
- 1.Batonick, M., M. Favre, M. Boge, P. Spearman, S. Honing, and M. Thali. 2005. Interaction of HIV-1 Gag with the clathrin-associated adaptor AP-2. Virology 342**:**190-200. [DOI] [PubMed] [Google Scholar]
- 2.Berditchevski, F., and E. Odintsova. 1999. Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J. Cell Biol. 146**:**477-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharya, J., P. J. Peters, and P. R. Clapham. 2004. Human immunodeficiency virus type 1 envelope glycoproteins that lack cytoplasmic domain cysteines: impact on association with membrane lipid rafts and incorporation onto budding virus particles. J. Virol. 78**:**5500-5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boge, M., S. Wyss, J. S. Bonifacino, and M. Thali. 1998. A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J. Biol. Chem. 273**:**15773-15778. [DOI] [PubMed] [Google Scholar]
- 5.Booth, A. M., Y. Fang, J. K. Fallon, J. M. Yang, J. E. Hildreth, and S. J. Gould. 2006. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 172**:**923-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell, S. M., S. M. Crowe, and J. Mak. 2001. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J. Clin. Virol. 22**:**217-227. [DOI] [PubMed] [Google Scholar]
- 7.Chan, W.-E., H.-H. Lin, and S. S.-L. Chen. 2005. Wild-type-like viral replication potential of human immunodeficiency virus type 1 envelope mutants lacking palmitoylation signals. J. Virol. 79**:**8374-8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaguillaumie, A., J. Harriague, S. Kohanna, G. Bismuth, E. Rubinstein, M. Seigneuret, and H. Conjeaud. 2004. Tetraspanin CD82 controls the association of cholesterol-dependent microdomains with the actin cytoskeleton in T lymphocytes: relevance to co-stimulation. J. Cell Sci. 117**:**5269-5282. [DOI] [PubMed] [Google Scholar]
- 9.Deschambeault, J., J. P. Lalonde, G. Cervantes-Acosta, R. Lodge, E. A. Cohen, and G. Lemay. 1999. Polarized human immunodeficiency virus budding in lymphocytes involves a tyrosine-based signal and favors cell-to-cell viral transmission. J. Virol. 73**:**5010-5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding, L., A. Derdowski, J.-J. Wang, and P. Spearman. 2003. Independent segregation of human immunodeficiency virus type 1 Gag protein complexes and lipid rafts. J. Virol. 77**:**1916-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong, X., H. Li, A. Derdowski, L. Ding, A. Burnett, X. Chen, T. R. Peters, T. S. Dermody, E. Woodruff, J. J. Wang, and P. Spearman. 2005. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 120**:**663-674. [DOI] [PubMed] [Google Scholar]
- 12.Escola, J. M., M. J. Kleijmeer, W. Stoorvogel, J. M. Griffith, O. Yoshie, and H. J. Geuze. 1998. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 273**:**20121-20127. [DOI] [PubMed] [Google Scholar]
- 13.Frank, I., H. Stoiber, S. Godar, H. Stockinger, F. Steindl, H. W. Katinger, and M. P. Dierich. 1996. Acquisition of host cell-surface-derived molecules by HIV-1. AIDS 10**:**1611-1620. [DOI] [PubMed] [Google Scholar]
- 14.Garcia, E., M. Pion, A. Pelchen-Matthews, L. Collinson, J. F. Arrighi, G. Blot, F. Leuba, J. M. Escola, N. Demaurex, M. Marsh, and V. Piguet. 2005. HIV-1 trafficking to the dendritic cell-T-cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic 6**:**488-501. [DOI] [PubMed] [Google Scholar]
- 15.Gelderblom, H. R., E. H. Hausmann, M. Ozel, G. Pauli, and M. A. Koch. 1987. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology 156**:**171-176. [DOI] [PubMed] [Google Scholar]
- 16.Gluschankof, P., I. Mondor, H. R. Gelderblom, and Q. J. Sattentau. 1997. Cell membrane vesicles are a major contaminant of gradient-enriched human immunodeficiency virus type-1 preparations. Virology 230**:**125-133. [DOI] [PubMed] [Google Scholar]
- 17.Gordon-Alonso, M., M. Yanez-Mo, O. Barreiro, S. Alvarez, M. A. Munoz-Fernandez, A. Valenzuela-Fernandez, and F. Sanchez-Madrid. 2006. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J. Immunol. 177**:**5129-5137. [DOI] [PubMed] [Google Scholar]
- 18.Graham, D. R. M., E. Chertova, J. M. Hilburn, L. O. Arthur, and J. E. K. Hildreth. 2003. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with β-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J. Virol. 77**:**8237-8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grigorov, B., F. Arcanger, P. Roingeard, J. L. Darlix, and D. Muriaux. 2006. Assembly of infectious HIV-1 in human epithelial and T-lymphoblastic cell lines. J. Mol. Biol. 359**:**848-862. [DOI] [PubMed] [Google Scholar]
- 20.Harila, K., I. Prior, M. Sjöberg, A. Salminen, J. Hinkula, and M. Suomalainen. 2006. Vpu and Tsg101 regulate intracellular targeting of the human immunodeficiency virus type 1 core protein precursor Pr55gag. J. Virol. 80**:**3765-3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemler, M. E. 2005. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6**:**801-811. [DOI] [PubMed] [Google Scholar]
- 22.Hemler, M. E. 2003. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 19**:**397-422. [DOI] [PubMed] [Google Scholar]
- 23.Hockley, D. J., R. D. Wood, J. P. Jacobs, and A. J. Garrett. 1988. Electron microscopy of human immunodeficiency virus. J. Gen. Virol. 69**:**2455-2469. [DOI] [PubMed] [Google Scholar]
- 24.Holm, K., K. Weclewicz, R. Hewson, and M. Suomalainen. 2003. Human immunodeficiency virus type 1 assembly and lipid rafts: Pr55gag associates with membrane domains that are largely resistant to Brij98 but sensitive to Triton X-100. J. Virol. 77**:**4805-4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janvier, K., and J. S. Bonifacino. 2005. Role of the endocytic machinery in the sorting of lysosome-associated membrane proteins. Mol. Biol. Cell 16**:**4231-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolly, C., K. Kashefi, M. Hollinshead, and Q. J. Sattentau. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199**:**283-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jolly, C., and Q. J. Sattentau. 2005. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 79**:**12088-12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolly, C., and Q. J. Sattentau. 2004. Retroviral spread by induction of virological synapses. Traffic 5**:**643-650. [DOI] [PubMed] [Google Scholar]
- 29.Jolly, C., I. Mitar, and Q. J. Sattentau. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J. Virol. 81**:**5547-5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jouvenet, N., S. J. Neil, C. Bess, M. C. Johnson, C. A. Virgen, S. M. Simon, and P. D. Bieniasz. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4**:**e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kramer, B., A. Pelchen-Matthews, M. Deneka, E. Garcia, V. Piguet, and M. Marsh. 2005. HIV interaction with endosomes in macrophages and dendritic cells. Blood Cells Mol. Dis. 35**:**136-142. [DOI] [PubMed] [Google Scholar]
- 32.Lindwasser, O. W., and M. D. Resh. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75**:**7913-7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lodge, R., H. Gottlinger, D. Gabuzda, E. A. Cohen, and G. Lemay. 1994. The intracytoplasmic domain of gp41 mediates polarized budding of human immunodeficiency virus type 1 in MDCK cells. J. Virol. 68**:**4857-4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lodge, R., J. P. Lalonde, G. Lemay, and E. A. Cohen. 1997. The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J. 16**:**695-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mantegazza, A. R., M. M. Barrio, S. Moutel, L. Bover, M. Weck, P. Brossart, J. L. Teillaud, and J. Mordoh. 2004. CD63 tetraspanin slows down cell migration and translocates to the endosomal-lysosomal-MIICs route after extracellular stimuli in human immature dendritic cells. Blood 104**:**1183-1190. [DOI] [PubMed] [Google Scholar]
- 36.Marsh, M., and M. Thali. 2003. HIV's great escape. Nat. Med. 9**:**1262-1263. [DOI] [PubMed] [Google Scholar]
- 37.McDonald, D., L. Wu, S. M. Bohks, V. N. KewalRamani, D. Unutmaz, and T. J. Hope. 2003. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300**:**1295-1297. [DOI] [PubMed] [Google Scholar]
- 38.Meerloo, T., H. K. Parmentier, A. D. Osterhaus, J. Goudsmit, and H. J. Schuurman. 1992. Modulation of cell surface molecules during HIV-1 infection of H9 cells. An immunoelectron microscopic study. AIDS 6**:**1105-1116. [DOI] [PubMed] [Google Scholar]
- 39.Meerloo, T., M. A. Sheikh, A. C. Bloem, A. de Ronde, M. Schutten, C. A. van Els, P. J. Roholl, P. Joling, J. Goudsmit, and H. J. Schuurman. 1993. Host cell membrane proteins on human immunodeficiency virus type 1 after in vitro infection of H9 cells and blood mononuclear cells. An immuno-electron microscopic study. J. Gen. Virol. 74**:**129-135. [DOI] [PubMed] [Google Scholar]
- 40.Mittelbrunn, M., M. Yanez-Mo, D. Sancho, A. Ursa, and F. Sanchez- Madrid. 2002. Cutting edge: dynamic redistribution of tetraspanin CD81 at the central zone of the immune synapse in both T lymphocytes and APC. J. Immunol. 169**:**6691-6695. [DOI] [PubMed] [Google Scholar]
- 41.Morita, E., and W. I. Sundquist. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20**:**395-425. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen, D. G., A. Booth, S. J. Gould, and J. E. Hildreth. 2003. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 278**:**52347-52354. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen, D. H., and J. E. Hildreth. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74**:**3264-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nydegger, S., M. Foti, A. Derdowski, P. Spearman, and M. Thali. 2003. HIV-1 egress is gated through late endosomal membranes. Traffic 4**:**902-910. [DOI] [PubMed] [Google Scholar]
- 45.Nydegger, S., S. Khurana, D. N. Krementsov, M. Foti, and M. Thali. 2006. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 173**:**795-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono, A., S. D. Ablan, S. J. Lockett, K. Nagashima, and E. O. Freed. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 101**:**14889-14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ono, A., and E. O. Freed. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 78**:**1552-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 98**:**13925-13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Orentas, R. J., and J. E. Hildreth. 1993. Association of host cell surface adhesion receptors and other membrane proteins with HIV and SIV. AIDS Res. Hum. Retrovir. 9**:**1157-1165. [DOI] [PubMed] [Google Scholar]
- 50.Pearce-Pratt, R., D. Malamud, and D. M. Phillips. 1994. Role of the cytoskeleton in cell-to-cell transmission of human immunodeficiency virus. J. Virol. 68**:**2898-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelchen-Matthews, A., B. Kramer, and M. Marsh. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162**:**443-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perlman, M., and M. D. Resh. 2006. Identification of an intracellular trafficking and assembly pathway for HIV-1 Gag. Traffic 7**:**731-745. [DOI] [PubMed] [Google Scholar]
- 53.Piguet, V., and Q. Sattentau. 2004. Dangerous liaisons at the virological synapse. J. Clin. Investig. 114**:**605-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raposo, G., M. Moore, D. Innes, R. Leijendekker, A. Leigh-Brown, P. Benaroch, and H. Geuze. 2002. Human macrophages accumulate HIV-1 particles in MHC II compartments. Traffic 3**:**718-729. [DOI] [PubMed] [Google Scholar]
- 55.Resh, M. D. 2005. Intracellular trafficking of HIV-1 Gag: how Gag interacts with cell membranes and makes viral particles. AIDS Rev. 7**:**84-91. [PubMed] [Google Scholar]
- 56.Rousso, I., M. B. Mixon, B. K. Chen, and P. S. Kim. 2000. Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc. Natl. Acad. Sci. USA 97**:**13523-13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rudner, L., S. Nydegger, L. V. Coren, K. Nagashima, M. Thali, and D. E. Ott. 2005. Dynamic fluorescent imaging of human immunodeficiency virus type 1 Gag in live cells by biarsenical labeling. J. Virol. 79**:**4055-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saad, J. S., J. Miller, J. Tai, A. Kim, R. H. Ghanam, and M. F. Summers. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 103**:**11364-11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanfridson, A., S. Hester, and C. Doyle. 1997. Nef proteins encoded by human and simian immunodeficiency viruses induce the accumulation of endosomes and lysosomes in human T cells. Proc. Natl. Acad. Sci. USA 94**:**873-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shkriabai, N., S. A. Datta, Z. Zhao, S. Hess, A. Rein, and M. Kvaratskhelia. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45**:**4077-4083. [DOI] [PubMed] [Google Scholar]
- 61.VanCompernolle, S. E., S. Levy, and S. C. Todd. 2001. Anti-CD81 activates LFA-1 on T cells and promotes T cell-B cell collaboration. Eur. J. Immunol. 31**:**823-831. [DOI] [PubMed] [Google Scholar]
- 62.Varthakavi, V., R. M. Smith, K. L. Martin, A. Derdowski, L. A. Lapierre, J. R. Goldenring, and P. Spearman. 2006. The pericentriolar recycling endosome plays a key role in Vpu-mediated enhancement of HIV-1 particle release. Traffic 7**:**298-307. [DOI] [PubMed] [Google Scholar]
- 63.Welsch, S., O. T. Keppler, A. Habermann, I. Allespach, J. Krijnse-Locker, and H. G. Krausslich. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3**:**e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wyss, S., C. Berlioz-Torrent, M. Boge, G. Blot, S. Höning, R. Benarous, and M. Thali. 2001. The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 envelope glycoprotein is critical for its association with the AP-1 clathrin adapter. J. Virol. 75**:**2982-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]