Adiponectin protects against the development of systolic dysfunction following myocardial infarction (original) (raw)
. Author manuscript; available in PMC: 2008 Jun 1.
Published in final edited form as: J Mol Cell Cardiol. 2007 Mar 20;42(6):1065–1074. doi: 10.1016/j.yjmcc.2007.03.808
Abstract
Background
There is an association between obesity and heart failure associated with LV dysfunction. Adiponectin is an adipocyte-derived hormone that is downregulated in obesity. Here, we examined the role of adiponectin in cardiac remodeling after myocardial infarction with loss- and gain-of-function genetic manipulations in an experimental model.
Methods
Myocardial infarction was created in adiponectin-deficient (APN-KO) and wild-type (WT) mice by the permanent ligation of the left anterior descending (LAD) artery. For some experiments, adenoviral vectors expressing adiponectin or β-galactosidase were delivered systemically. Cardiac structure and function were assessed by echocardiographic and Millar catheter measurements. Myocardial capillary density was assessed by staining with anti-CD31 antibody. Myocyte apoptotic activity was determined by TUNEL-staining. Myocardial interstitial fibrosis was evaluated by Masson’s trichrome staining.
Results
APN-KO mice showed exacerbated left ventricular (LV) dilation, myocyte hypertrophy and contractile dysfunction compared with WT mice at 4 weeks after LAD ligation. Impaired LV function in APN-KO mice was coupled to myocyte hypertrophy, increased apoptotic activity and interstitial fibrosis in the remote zone, and reduced capillary density in the infarct border zone. No difference in infarct size was observed between WT and APN-KO mice. Administration of adenovirus-mediated adiponectin in WT mice resulted in decreased LV dilatation and improved LV function that was associated with increased capillary density in the infarct border zone and decreased myocyte hypertrophy, diminished myocardial apoptosis and decreased interstitial fibrosis in the remote zone.
Conclusions
These data suggest that adiponectin protects against the development of systolic dysfunction after myocardial infarction through its abilities to suppress cardiac hypertrophy and interstitial fibrosis, and protect against myocyte and capillary loss.
Keywords: heart failure, ischemia, myocytes, ventricular function
Introduction
Obesity is frequently associated with diabetes mellitus, dyslipidemia, hypertension, and vascular disease. These obesity-related disorders are implicated in the development of ischemic heart disease and cardiac hypertrophy [1, 2]. Obesity is also associated with increased left ventricular (LV) dilatation [3] and an increase in the incidence of heart failure [4].
Adiponectin is an adipocyte-derived secreted protein that appears to function as an endogenous modulator of obesity-linked complications [5]. Circulating adiponectin levels are decreased in obese patients [6] and are inversely correlated with cardiovascular risk factors [7]. Hypoadiponectinemia is an independent risk factor for the development of type 2 diabetes [8], coronary artery disease [9] and hypertension [10]. Experimental studies show that adiponectin inhibits the development of insulin resistance [11], atherosclerosis [12], hypertension [13] and chronic vascular insufficiency [14]. Recently, we have reported that adiponectin exerts beneficial actions on the heart after pressure overload [15] and ischemia-reperfusion injury [16] through its ability to activate protective signaling pathways within myocardial cells. It has also been reported that adiponectin suppresses the progress of viral myocarditis in diabetic obese mice [17].
In contrast to adiponectin’s recognized cardiovascular-protective actions, recent clinical studies have shown that high adiponectin levels are a predictor of mortality in patients with advanced heart failure [18, 19], while another study found that plasma adiponectin levels are not predictive of heart failure in men who were asymptomatic [20]. These epidemiological studies are difficult to interpret because systemic wasting, which is associated with high adiponectin levels, is a positive predictor of death in patients with heart failure [21]. Therefore, it is not clear whether adiponectin is beneficial or detrimental in the context of heart failure, and detailed studies in experimental models are required to better understand the mechanistic role of adiponectin in the development and progression of systolic dysfunction. Here, we examined the effect of adiponectin on the development of LV dysfunction in a mouse model of myocardial infarction (MI) with loss- and gain-of-function genetic manipulations. Our observations show that adiponectin has anti-apoptotic, anti-hypertrophic, anti-fibrotic and pro-angiogenic activities in this model, and that it protects the heart from chronic pathological remodeling.
Materials and Methods
Materials
The adenoviral vectors expressing β-galactosidase (Ad-βgal) or murine adiponectin (Ad-APN) from the cytomegalovirus promoter/enhancer have been described previously [11].
Mouse model of myocardial infarction
Male wild type (WT) and adiponectin deficient (APN-KO) mice in a C57/BL6 background were used for this study [22]. Study protocols were approved by the Institutional Animal Care and Use Committee at Boston University. Mice at the ages of 10 to 12 weeks were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally). The trachea was cannulated with a polyethylene tube connected to a respirator with a tidal volume of 0.6 mL (110/min). A left thoracotomy was performed between the fourth and fifth ribs. The pericardial tissue was removed, and the left anterior descending (LAD) artery was visualized under a microscope and permanently ligated with 8-0 silk suture [23]. Sham-treated mice underwent surgery but not LAD ligation. In some experiments, 2 × 108 plaque-forming units (pfu) of Ad-APN or Ad-β-gal were injected into the jugular vein of WT mice 3 days prior to the surgery. Survival of mice after MI was evaluated using the Kaplan-Meier method. Serum adiponectin levels were determined using an ELISA kit (Otsuka Pharmaceutical Co. Ltd) [15]. Serum creatinine levels were measured using a creatinine assay kit (Cayman chemical). Hemodynamic measurements were performed 4 weeks after surgery using a 1.4F catheter tip micromanometer (ARIA, Millar Instruments) as described previously [16]. Echocardiography was performed with an Acuson Sequoia C-256 machine using a 15-MHz probe as described previously [16].
Histology
Mice were sacrificed and left ventricular (LV) tissue was obtained at 4 weeks after MI. Tissue samples were embedded in OCT compound (Sakura Finetech USA Inc) and snap-frozen in liquid nitrogen. To determine the infarct size, sections were stained with Masson’s trichrome (MT). Total LV circumference was calculated as the sum of endocardial and epicardial segment lengths from all sections. Infarct size was calculated as total infarct circumference divided by total LV circumference. Myocyte cross-sectional area was determined in the remote zone as previously described [24]. Capillary density was assessed by CD31 staining of tissue sections in peri-infarct zone as described previously [25]. Myocardial apoptosis was qualitatively analyzed by TUNEL staining in the remote zone as previously described [26]. Myocyte identity was indicated by staining with anti-α-sarcomeric actin antibody (Sigma) [27]. Myocardial interstitial fibrosis was assessed by MT staining in the remote zone as previously described [28]. The extent of fibrosis was quantified by NIH Image software.
Statistical Analysis
Data are presented as mean ± SD. The mean value was compared between 2 groups using an 2-tailed Student’s t test. The comparison among more than 3 groups was performed by analysis of variance (ANOVA) with Scheffe’s F test. A value of P < 0.05 was accepted as statistically significant.
Results
APN-KO mice exhibit enhanced LV dilatation, myocyte hypertrophy and contractile dysfunction after myocardial infarction
WT and APN-KO mice were subjected to MI by permanent LAD ligation (Figure 1A). No significant difference occurred in the survival frequencies after MI between WT and APN-KO mice (Figure 1A). Body weight (BW) did not differ between WT and APN-KO mice at 4 weeks after MI or sham treatment (Table 1). Serum creatine levels were not significantly different between any experimental groups at 4 weeks post-MI or -sham treatment (Table 2). No significant difference in heart rate was observed in the different experimental groups (Figure 1B). Systolic blood pressure was significantly decreased in both WT and APN-KO mice 4 weeks after MI compared with sham-treated mice, but there was no statistical difference between the WT and APN-KO of mice (Figure 1B).
Figure 1. Increased myocyte hypertrophy in APN-KO mice subjected to permanent LAD ligation.
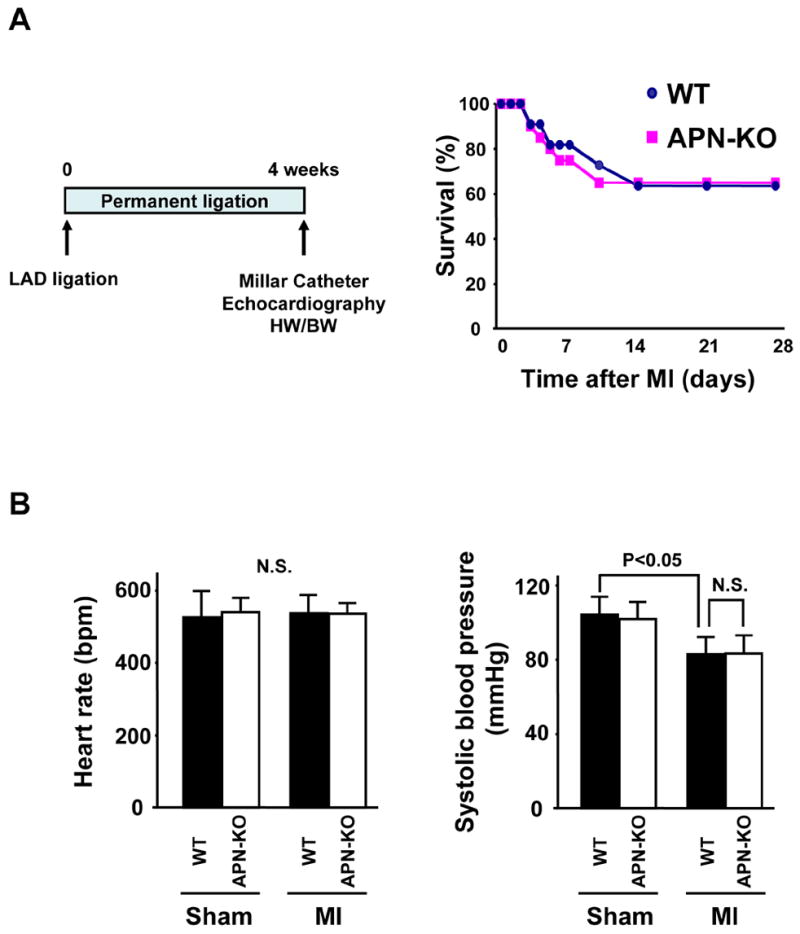
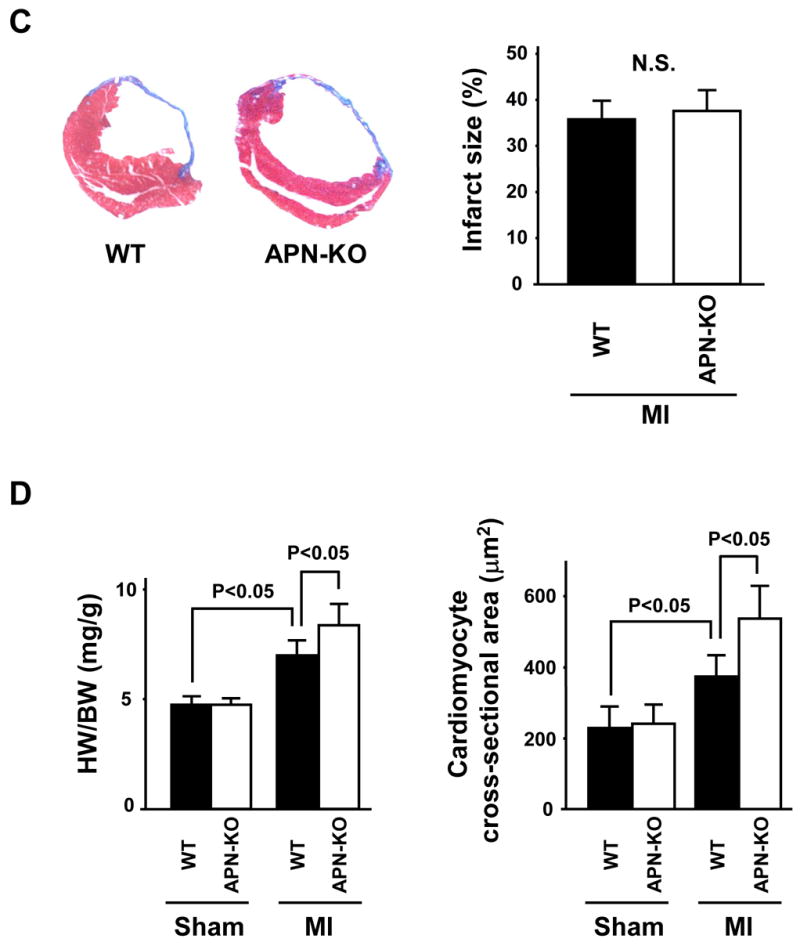
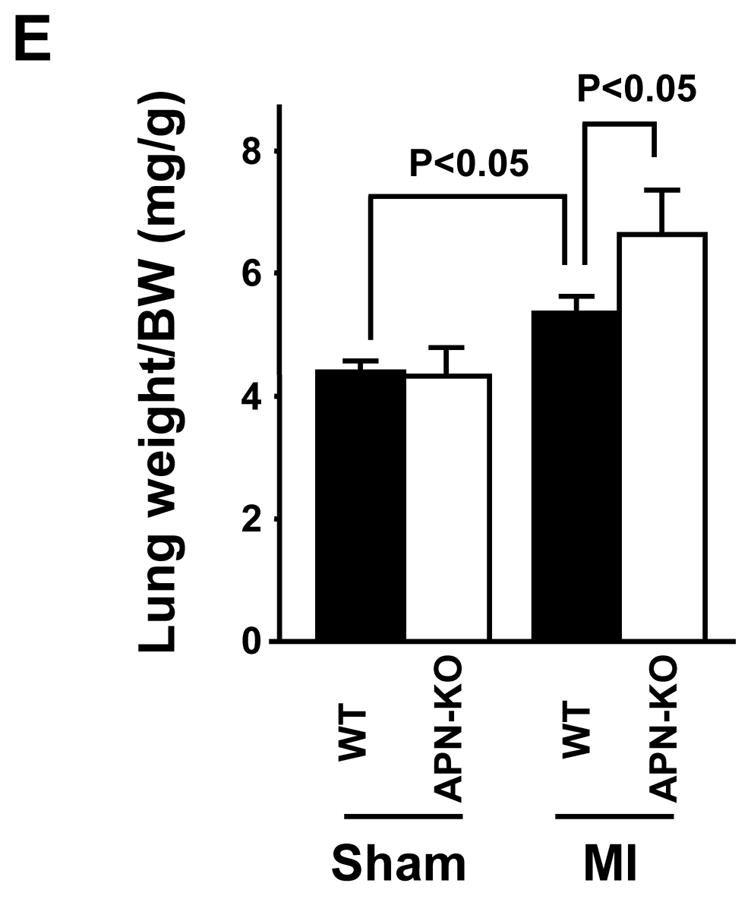
(A) Schematic illustration of experimental protocol, and survival curves of WT and APN-KO mice after MI. WT: n = 20, KO: n = 14. (B) Left: Heart rate, right: systolic blood pressure in WT and APN-KO mice 4 weeks after sham-operation or MI. (C) Left: Representative Masson’s trichrome-stained cross-sections of the heart from WT (left) and APN-KO (right) mice 4 weeks after MI. Right: Quantification of relative infarct size in WT and APN-KO mice. (D) Left: Heart weight/body weight (HW/BW) ratio in WT and APN-KO mice 4 weeks after sham-operation or MI. Right: Quantitative analysis of cardiomyocyte cross-sectional area in WT and APN-KO mice 4 weeks after sham-operation or MI. (E) Lung wet weight/BW ratio in WT and APN-KO mice 4 weeks after sham-operation or MI. Results are presented as mean ± s.d. (n = 7 mice per experimental group).
Table 1.
Body weight in WT and APN-KO mice at 4 weeks after sham-operation or MI.
| Sham | MI | |||
|---|---|---|---|---|
| WT | APN-KO | WT | APN-KO | |
| Body weight (g) | 27 ± 1.8 | 26 ± 1.8 | 26 ± 1.8 | 26 ± 1.6 |
Table 2.
Serum creatine in WT and APN-KO mice at 4 weeks after sham-operation or MI
| Sham | MI | |||
|---|---|---|---|---|
| WT | APN-KO | WT | APN-KO | |
| Creatine (mg/dL) | 0.79 ± 0.20 | 0.88 ± 0.18 | 0.72 ± 0.11 | 0.97 ± 0.18 |
Myocardial tissue sections were stained with Masson’s trichrome to determine infarct size in WT and APN-KO mice at 4 weeks post-MI (Figure 1C). Exaggerated LV dilatation was observed in APN-KO mice as compared to WT mice. However, the ratio of total infarct length to total LV circumference was the same in APN-KO and WT mice. As shown in Figure 1D, the increase in heart weight/BW (HW/BW) ratio at 4 weeks after MI was significantly greater in APN-KO mice than that in WT mice. No significant difference in HW/BW ratio was observed in sham-operated WT and APN-KO mice. Analysis of cardiomyocyte cross-sectional area in histological sections of areas remote to the infarct revealed that cardiomyocyte hypertrophy in response to MI was greater in APN-KO mice than in WT mice (Figure 1D). Lung wet weight/BW ratio at 4 weeks after MI was significantly increased in APN-KO mice compared with that in WT mice (Figure 1E), indicating an increase in pulmonary congestion.
Echocardiographic measurements revealed that LV diastolic dimension (LVDd) was increased in both APN-KO and WT mice 4 weeks after MI, but the APN-KO mice showed more deterioration in this parameter (Figure 2A). Hemodynamic measurements revealed that ventricular end-diastolic pressure (LVEDP) was markedly elevated in APN-KO mice compared with WT mice 4 weeks after MI, and decreased d_P_/d_t_max and increased d_P_/d_t_min was observed in APN-KO mice compared with WT mice after MI (Figure 2B), indicating greater LV dysfunction in APN-KO mice. No differences in LVDd, dP/d_t_max and min or LVEDP could be detected between sham-operated WT and APN-KO mice.
Figure 2. Increased LV dilatation and LV dysfunction in APN-KO mice 4 weeks after MI.
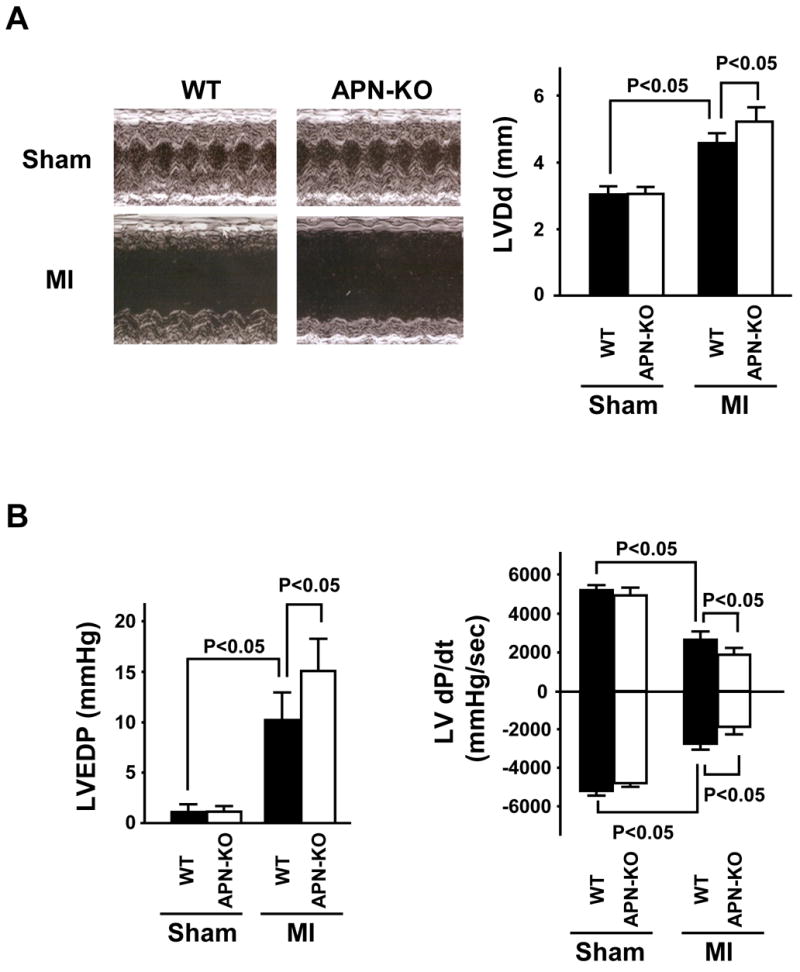
(A) Left: Representative M-mode echocardiogram for WT and APN-KO mice 4 weeks after sham-operation or MI. Right: LV diastolic dimension (LVDd) in WT and APN-KO mice 4 weeks after sham-operation or MI (n = 7 mice per experimental group). (B) Left: LV end-diastolic pressure (LVEDP) in WT and APN-KO mice 4 weeks after sham-operation or MI. Right: LV d_P_/d_t_ in WT and APN-KO mice 4 weeks after sham-operation or MI (n = 5 mice per experimental group). Results are presented as mean ± s.d.
APN-KO mice displayed reduced myocardial capillary density and increased apoptosis after infarction
Capillary status was evaluated by histological analysis of CD31-positive cells in the peri-infarct areas (Figure 3A). Capillary density was significantly decreased in APN-KO mice compared with WT mice at 4 weeks after MI, but not in sham-operated mice. To investigate the extent of apoptosis, TUNEL staining was performed on histological sections from areas remote to the infarct in the different experimental groups (Figure 3B). Quantitative analysis showed a significantly higher proportion of TUNEL-positive cardiac myocytes in APN-KO mice compared with WT mice, whereas little or no TUNEL-positive cells could be detected in the hearts of WT or APN-KO mice after sham-operation. Myocardial interstitial fibrosis in the remote zone was assessed in MT-stained tissue sections (Figure 3C). Interstitial fibrosis was significantly increased in APN-KO mice compared with WT mice at 4 weeks after MI, whereas very little fibrosis lesion could be detected in either APN-KO or WT mice that underwent sham surgeries.
Figure 3. Reduced capillary density and increased myocardial apoptosis in APN-KO mice after MI.
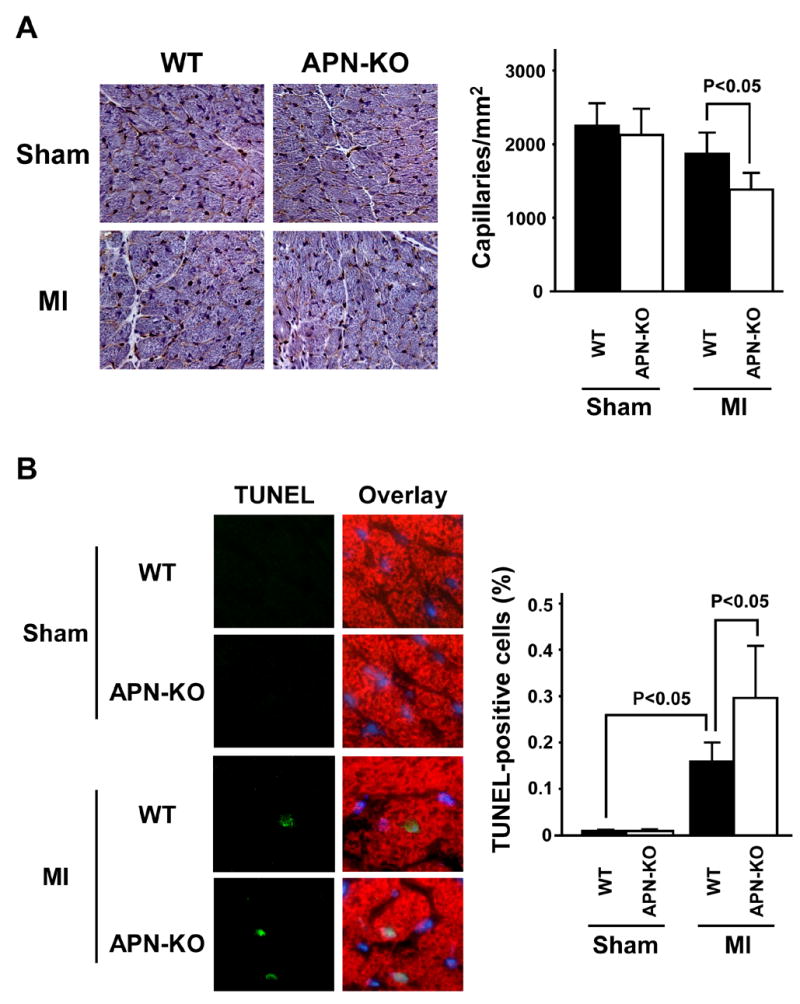
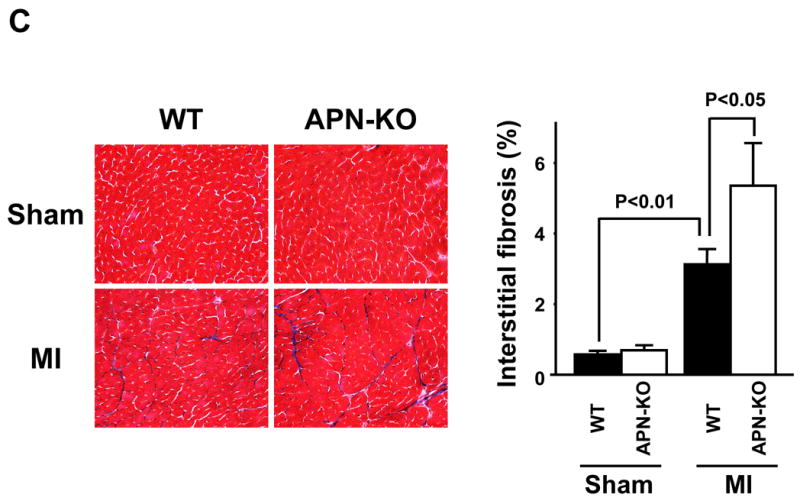
(A) Left: Representative images of anti-CD31-stained heart sections. Right: Quantitative analysis of CD31-positive capillary density in WT and APN-KO mice 4 weeks after sham-operation or MI. (B) Left: Representative images of TUNEL-stained heart sections from WT and APN-KO mice 4 weeks after sham-operation or MI. Apoptotic nuclei were identified by TUNEL staining (green), cardiac myocytes by staining with anti-α-sarcomeric actin antibody (red), and total nuclei by DAPI counterstaining (blue). Right: Quantitative analysis of apoptotic nuclei in cardiac myocytes from WT and APN-KO mice 4 weeks after sham-operation or MI. TUNEL-positive nuclei are expressed as a percentage of the total number of DAPI-positive nuclei. (C) Left: Representative images of Masson’s trichrome (MT)-stainined heart sections. Right: Quantitative analysis of myocardial interstitial fibrosis in WT and APN-KO mice 4 weeks after sham-operation or MI. Results are presented as mean ± s.d. (n = 5 mice per experimental group).
Adiponectin supplementation improves cardiac remodeling in WT mice
To test whether increased expression of adiponectin could minimize cardiac remodeling, WT mice were treated with adenoviral vectors expressing either adiponectin (Ad-APN) or β-galactosidase (Ad-βgal) as a control at 3 days prior to MI (Figure 4A). Plasma APN levels were significantly increased in Ad-APN-treated mice 7 and 14 days after surgery, but returned to control levels by day 28 (Table 3). Infarct size did not differ between Ad-βgal- and Ad-APN-treated mice at 28 days after MI (Figure 4B). However, there was a statistically significant decrease in HW/BW ratio in Ad-APN-treated mice compared to Ad-βgal-treated animals at 4 weeks after MI (Figure 4C). No significant difference in HW/BW was observed in sham-operated Ad-βgal- and Ad-APN-treated mice. Furthermore, there were no significant differences in overall BW in each experimental group at 4 weeks after MI or sham treatment (Table 4). Myocyte cross-sectional area in remote areas of post-MI hearts was reduced in Ad-APN-treated compared to Ad-βgal-treated mice, whereas Ad-APN had no effect on myocyte size in sham-treated mice (Figure 4C). Finally, lung wet weight/BW ratio in Ad-APN-treated mice at 4 weeks after MI was smaller that that in Ad-βgal-treated mice (Figure 4D), suggesting that pulmonary congestion was attenuated by adiponectin overexpression.
Figure 4. Adenovirus-mediated expression of adiponectin inhibits hypertrophy and cardiac remodeling in WT mice at 4 weeks after MI.
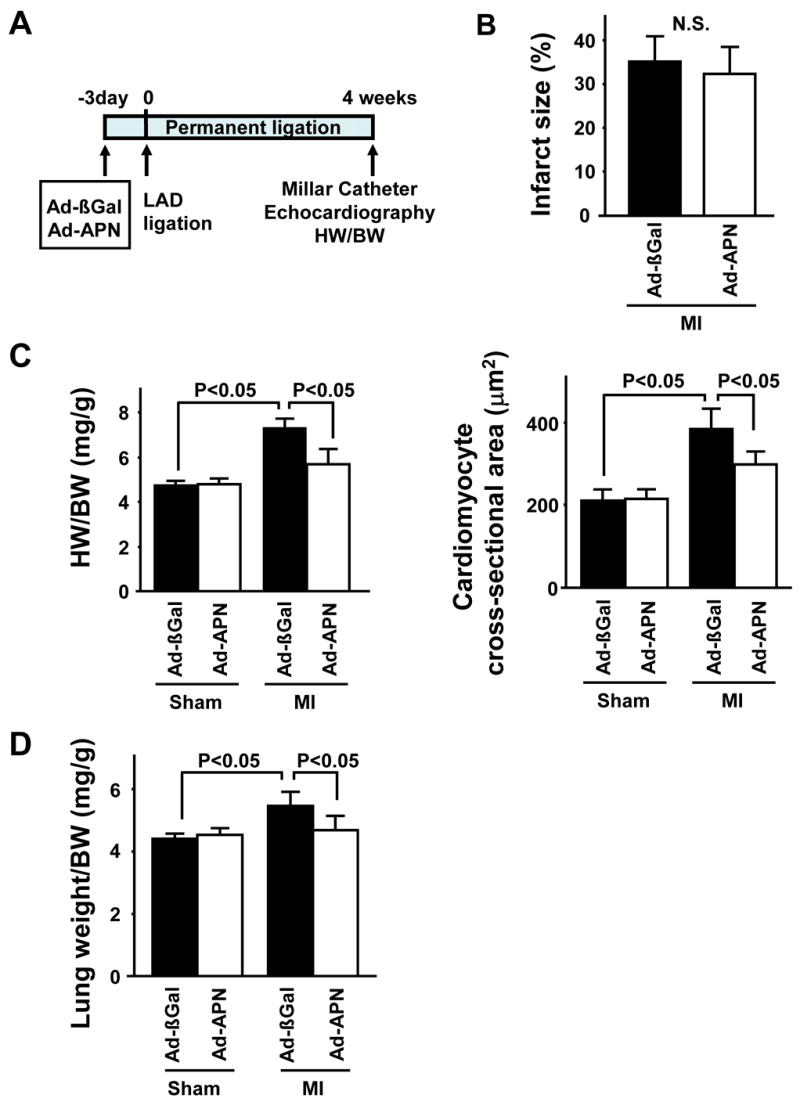
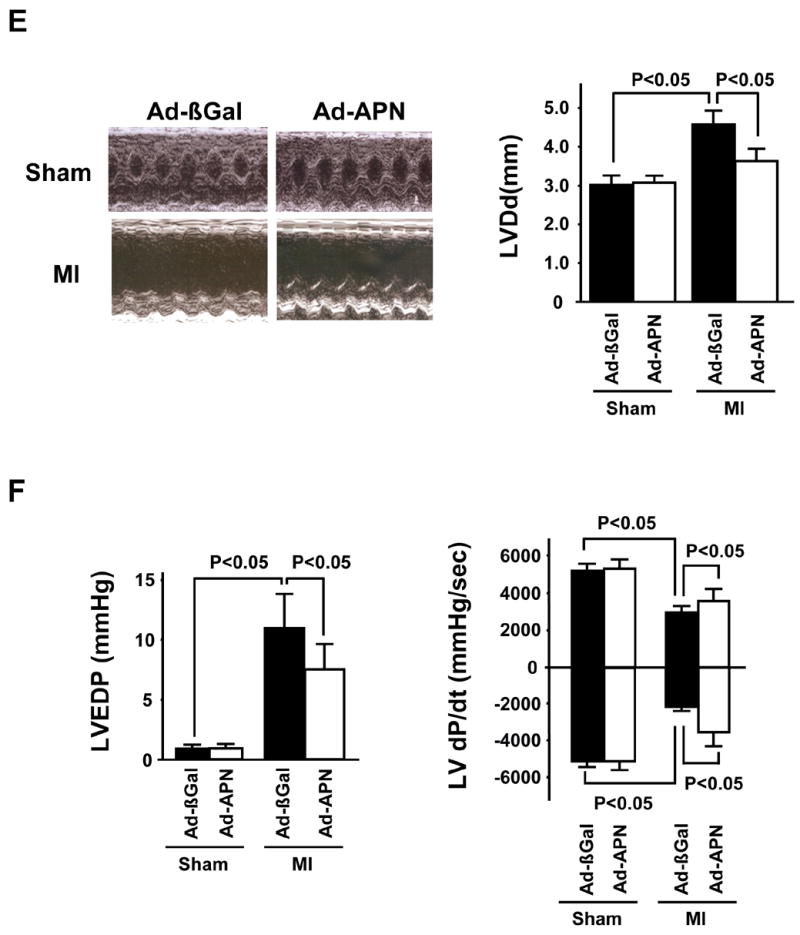
(A) Schematic illustration of experimental protocol. (B) Quantification of relative infarct size in Ad-βgal- or Ad-APN-treated mice 4 weeks after MI. (C) Left: HW/BW ratio in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. Right: Quantitative analysis of cardiomyocyte cross-sectional area in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. (D) Lung wet weight/BW ratio in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. (E) Left: Representative M-mode echocardiogram for Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. Right: LVDd in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. (F) Left: LVEDP in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. Right: Left ventricular d_P_/d_t_ in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. Results are presented as mean ± s.d. (n = 6 mice per experimental group).
Table 3.
Plasma APN levels in Ad-βgal- or Ad-APN-treated WT mice after surgery.
| Ad-βgal | Ad-APN | |
|---|---|---|
| Pre | 12.8 ± 1.2 | 12.5 ± 0.6 |
| Day 7 | 13.4 ± 1.8 | 21.3 ± 2.5* |
| Day 14 | 13.9 ± 2.2 | 18.1 ± 2.3* |
| Day 28 | 12.5 ± 1.6 | 15.6 ± 1.4 |
Table 4.
Body weight in Ad-βgal- or Ad-APN-treated WT mice at 4 weeks after MI.
| Sham | MI | |||
|---|---|---|---|---|
| Ad-βgal | Ad-APN | Ad-βgal | Ad-APN | |
| Body weight (g) | 26 ± 1.6 | 26 ± 1.2 | 26 ± 1.7 | 26 ± 1.5 |
At 28 days after surgery, cardiac function was assessed by echocardiography and Millar catheter in the different experimental groups. Echocardiographic measurements revealed that Ad-APN-treated mice displayed decreased LV dilatation (Figure 4E). The increase in LVEDP following MI was minimized by Ad-APN treatment (Figure 4F). Ad-APN treatment also increased d_P_/d_t_max and decreased d_P_/d_t_min compared to Ad-βgal treatment in response to permanent coronary ischemia (Figure 4F). No differences in LVDd, LVEDP, dP/d_t_max and min could be detected between sham-operated Ad-βgal- and Ad-APN treated mice. Collectively, these data show thatadiponectin overexpression can minimize MI-induced LV dysfunction.
As revealed by histology, capillary density in the peri-infarct area was maintained in Ad-APN-treated mice at 4 weeks after MI compared to Ad-βgal-treated mice (Figure 5A). Myocyte apoptosis in areas remote to the infarct was reduced by overexpression of adiponectin, whereas little or no TUNEL positive cells could be detected in either Ad-βgal- or Ad-APN-treated mice that underwent sham surgeries (Figure 5B). Myocardial interstitial fibrosis in the remote zone was significantly reduced in Ad-APN-treated mice compared with Ad-βgal-treated mice at 4 weeks after MI, whereas minimal interstitial fibrosis was present in hearts after sham-operation (Figure 5C).
Figure 5. Adenovirus-mediated expression of adiponectin increases capillary density and diminishes apoptosis in WT mice 4 weeks after MI.
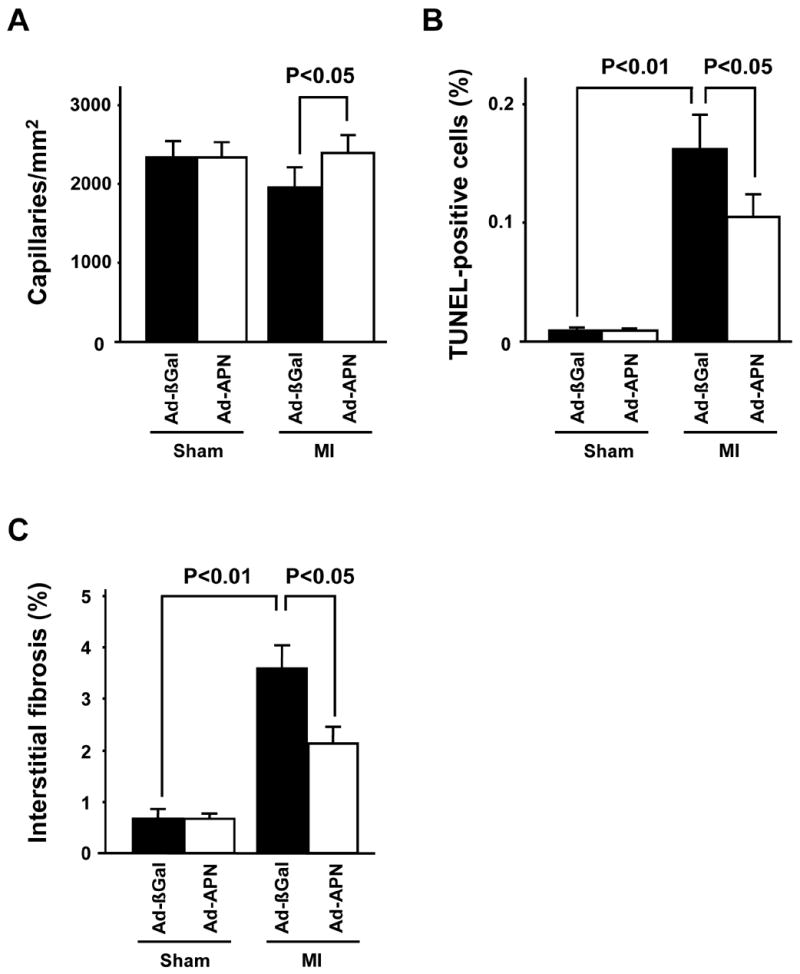
(A) Quantitative analysis of capillary density in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. (B) Quantitative analysis of apoptotic nuclei in cardiac myocytes from Ad-βgal- or Ad-APN-treated wild-type mice 4 weeks after sham-operation or MI. (C) Quantitative analysis of myocardial interstitial fibrosis in Ad-βgal- or Ad-APN-treated mice 4 weeks after sham-operation or MI. Results are presented as mean ± s.d. (n = 6 mice per experimental group).
Discussion
The present study provides in vivo evidence that adiponectin plays a protective role in the development of systolic dysfunction following MI. APN-KO mice exhibit greater LV systolic dysfunction following permanent myocardial ischemia compared with WT mice. Conversely, adenovirus-mediated overexpression of adiponectin led to increase plasma APN levels at 7 and 14 days post-MI and attenuated detrimental LV dysfunction at 28 days in WT mice. Under the conditions of these assays, adiponectin gain- or loss-of-function had no impact on infarct size. Thus we conclude that adiponectin protects against the development of LV dysfunction, primarily by inhibiting the pathological remodeling process that occurs in the surviving myocardium.
In contrast with what might be expected based on these experimental findings, it has been difficult to document a correlation between adiponectin levels and better clinical outcomes in heart failure patients. Two studies have found that high adiponectin levels are associated with increased mortality in heart failure patients [18, 19], perhaps because high levels of adiponectin are indicative of systemic wasting in this population [6]. In this regard, low body mass index, which leads to elevated adiponectin levels, is associated with increased mortality following the onset of heart failure, whereas elevated body mass index, which leads to reduced adiponectin levels, increases the risk for developing heart failure [29, 30]. Similarly, hypercholesterolemia is predictive of improved survival in heart failure patients presumably because it is a surrogate marker of body mass [31]. Therefore, results of epidemiological studies can be counterintuitive due to the complex pathology associated with heart failure. It should also be noted that renal dysfunction caused by heart failure could also influence adiponectin levels because adiponectin is cleared by the kidney [32]. In contrast, controlled studies in genetically modified mice can rigorously assess the role of adiponectin in heart failure, and these models can be conducted in a manner whereby they are subject to fewer confounding variables. In the current study, we examined the consequences of adiponectin levels on the development of heart failure following permanent LAD ligation in an experimental model that is not associated with weight loss or renal dysfunction. Adiponectin was found to protect against the development of heart failure in this model, and this beneficial effect was associated with a reduction in cardiac hypertrophy and protection against capillary and myocyte loss. These conclusions are consistent with the observations of Liao et al. [33] who observed a greater degree of cardiac failure in APN-KO mice three weeks after transverse aortic constriction. However, because adiponectin deficiency leads to a greater degree of hypertrophy, it cannot be determined whether the exacerbated heart failure in this model is the indirect consequence of a greater degree pressure overload-induced hypertrophy, direct effects on the remodeling processes that contribute to LV dysfunction, or a combination of these effects.
The ratio of myocyte size to capillary density has been shown to be a critical determinant in the transition between compensatory hypertrophy and heart failure in experimental animals [34–36]. Furthermore, analyses of autopsy samples reveals that patients with end-stage heart failure, due to ischemic, inflammatory or idiopathic dilated cardiomyopathy, exhibit diminished densities and irregular patterns of myocardial capillaries [37] Indeed, agents that promote angiogenesis have been shown to be beneficial for cardiac remodeling after chronic myocardial ischemia [38–40]. In this study, the greater impaired LV function in adiponectin KO mice was accompanied by greater reductions in capillary density, whereas adenovirus-mediated overexpression of adiponectin maintained capillary density in the infarct border zone. In accordance with these observations, we have previously shown that adiponectin can function as a survival factor for endothelial cells [41] and that APN-KO mice exhibit impaired angiogenic repair in ischemic hindlimbs whereas adiponectin overexpression stimulates blood vessel growth [14, 42]. Thus, the ability of adiponectin to directly affect endothelial cell survival and stimulate vascularization may contribute to the maintenance of myocardial capillary density in response to chronic myocardial ischemia, resulting in the preservation of LV function.
Progressive cardiac hypertrophy that occurs in response to MI is known to increase risk of heart failure, although it is believed to be compensatory at the initial stages of remodeling [43]. In this regard, some experimental studies have shown that the development of heart failure can be inhibited by genetic alterations that inhibit cardiac hypertrophy [44]. In the present study, adiponectin KO mice displayed a small but statistically significant increase in HW/BW ratio and exaggerated hypertrophy of cardiac myocytes at 4 weeks after MI. Conversely, adiponectin overexpression inhibited cardiac hypertrophy in WT mice after MI. Adiponectin directly acts on cardiac cells to inhibit pathological growth and adiponectin overexpression attenuates hypertrophy induced by pressure overload or other hypertrophic stimuli [15, 33, 45]. Therefore, the beneficial actions of adiponectin on chronic heart failure could be due, at least in part, to its anti-hypertrophic effects.
Apoptosis in cardiomyocytes is thought to contribute to the progression of heart failure after MI [46], and chronic cardiac remodeling with chamber dilation and impaired systolic function is associated with increased myocyte apoptosis in the infarct border zone after MI [47]. In this study, the extent of apoptosis was significantly higher in the hearts of APN-KO mice compared to WT mice at 28 days post-MI. Conversely, transient overexpression of adiponectin diminished myocardial apoptosis in the hearts of WT mice. The adiponectin-dependent reduction in apoptotic cells may result from the direct pro-survival effects of adiponectin on myocytes. It has been shown that adiponectin suppresses apoptosis of cardiac myocytes and fibroblasts under conditions of hypoxia-reoxygenation [16].
In summary, we show that adiponectin protects against adverse cardiac remodeling following MI. The favorable effects of adiponectin are associated with maintained capillary density, increased myocyte survival and attenuated myocardial hypertrophy and fibrosis. Therefore, therapeutic approaches aimed at increasing adiponectin production or promoting the sensitivity of the heart to adiponectin could be useful for treating LV dysfunction following MI.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL86785, AG15052, HL77774, and HL81587) to K.W. R.S. was supported by grants from the American Heart Association Postdoctoral Fellowship Award, Northeast Affiliate. Y.I. was supported by grants from the American Heart Association Postdoctoral Fellowship Award, Northeast Affiliate. N.O. was supported by a Department of Medicine Pilot Project Grant from Boston University and American Heart Association Scientist Development Grant, Northeast Affiliate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function: sex-related differences in the Framingham Heart Study. Circulation. 2003;107:448–54. doi: 10.1161/01.cir.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- 2.Wolk R, Berger P, Lennon RJ, Brilakis ES, Somers VK. Body mass index: a risk factor for unstable angina and myocardial infarction in patients with angiographically confirmed coronary artery disease. Circulation. 2003;108:2206–11. doi: 10.1161/01.CIR.0000095270.85646.E8. [DOI] [PubMed] [Google Scholar]
- 3.Lauer MS, Anderson KM, Kannel WB, Levy D. The impact of obesity on left ventricular mass and geometry. The Framingham Heart Study Jama. 1991;266:231–6. [PubMed] [Google Scholar]
- 4.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–13. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 5.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–9. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 6.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 7.Ouchi N, Kihara S, Funahashi T, Nakamura T, Nishida M, Kumada M, et al. Reciprocal association of C-reactive protein with adiponectin in blood stream and adipose tissue. Circulation. 2003;107:671–4. doi: 10.1161/01.cir.0000055188.83694.b3. [DOI] [PubMed] [Google Scholar]
- 8.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–9. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 9.Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N, et al. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol. 2003;23:85–9. doi: 10.1161/01.atv.0000048856.22331.50. [DOI] [PubMed] [Google Scholar]
- 10.Iwashima Y, Katsuya T, Ishikawa K, Ouchi N, Ohishi M, Sugimoto K, et al. Hypoadiponectinemia is an independent risk factor for hypertension. Hypertension. 2004;43:1318–23. doi: 10.1161/01.HYP.0000129281.03801.4b. [DOI] [PubMed] [Google Scholar]
- 11.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–7. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 12.Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–70. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- 13.Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension. 2006;47:1108–16. doi: 10.1161/01.HYP.0000222368.43759.a1. [DOI] [PubMed] [Google Scholar]
- 14.Shibata R, Ouchi N, Kihara S, Sato K, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis in response to tissue ischemia through stimulation of amp-activated protein kinase signaling. J Biol Chem. 2004;279:28670–4. doi: 10.1074/jbc.M402558200. [DOI] [PubMed] [Google Scholar]
- 15.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–9. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, et al. Adiponectin replacement therapy attenuates myocardial damage in leptin-deficient mice with viral myocarditis. J Int Med Res. 2005;33:207–14. doi: 10.1177/147323000503300208. [DOI] [PubMed] [Google Scholar]
- 18.George J, Patal S, Wexler D, Sharabi Y, Peleg E, Kamari Y, et al. Circulating adiponectin concentrations in patients with congestive heart failure. Heart. 2006;92:1420–4. doi: 10.1136/hrt.2005.083345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, et al. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation. 2005;112:1756–62. doi: 10.1161/CIRCULATIONAHA.104.530972. [DOI] [PubMed] [Google Scholar]
- 20.Ingelsson E, Riserus U, Berne C, Frystyk J, Flyvbjerg A, Axelsson T, et al. Adiponectin and risk of congestive heart failure. Jama. 2006;295:1772–4. doi: 10.1001/jama.295.15.1772-c. [DOI] [PubMed] [Google Scholar]
- 21.Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–3. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 22.Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation. 1999;100:2473–6. doi: 10.1161/01.cir.100.25.2473. [DOI] [PubMed] [Google Scholar]
- 23.Pfeffer JM, Pfeffer MA, Fletcher PJ, Braunwald E. Progressive ventricular remodeling in rat with myocardial infarction. Am J Physiol. 1991;260:H1406–H14. doi: 10.1152/ajpheart.1991.260.5.H1406. [DOI] [PubMed] [Google Scholar]
- 24.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–9. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, et al. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–91. doi: 10.1161/hc3601.094298. [DOI] [PubMed] [Google Scholar]
- 26.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–93. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anker SD, Rauchhaus M. Insights into the pathogenesis of chronic heart failure: immune activation and cachexia. Curr Opin Cardiol. 1999;14:211–6. doi: 10.1097/00001573-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Mosterd A, Cost B, Hoes AW, de Bruijne MC, Deckers JW, Hofman A, et al. The prognosis of heart failure in the general population: The Rotterdam Study. Eur Heart J. 2001;22:1318–27. doi: 10.1053/euhj.2000.2533. [DOI] [PubMed] [Google Scholar]
- 31.Rauchhaus M, Koloczek V, Volk H, Kemp M, Niebauer J, Francis DP, et al. Inflammatory cytokines and the possible immunological role for lipoproteins in chronic heart failure. Int J Cardiol. 2000;76:125–33. doi: 10.1016/s0167-5273(00)00224-2. [DOI] [PubMed] [Google Scholar]
- 32.Zoccali C, Mallamaci F, Tripepi G, Benedetto FA, Cutrupi S, Parlongo S, et al. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J Am Soc Nephrol. 2002;13:134–41. doi: 10.1681/ASN.V131134. [DOI] [PubMed] [Google Scholar]
- 33.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, et al. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res. 2005;67:705–13. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–18. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–93. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–65. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 37.Karch R, Neumann F, Ullrich R, Neumuller J, Podesser BK, Neumann M, et al. The spatial pattern of coronary capillaries in patients with dilated, ischemic, or inflammatory cardiomyopathy. Cardiovasc Pathol. 2005;14:135–44. doi: 10.1016/j.carpath.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation. 2001;104:982–5. doi: 10.1161/hc3401.095946. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Takemura G, Kosai K, Yuge K, Nagano S, Esaki M, et al. Postinfarction treatment with an adenoviral vector expressing hepatocyte growth factor relieves chronic left ventricular remodeling and dysfunction in mice. Circulation. 2003;107:2499–506. doi: 10.1161/01.CIR.0000065579.19126.B8. [DOI] [PubMed] [Google Scholar]
- 40.Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–4. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi H, Ouchi N, Kihara S, Walsh K, Kumada M, Abe Y, et al. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ Res. 2004;94:e27–e31. doi: 10.1161/01.RES.0000119921.86460.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, et al. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–9. doi: 10.1074/jbc.M310389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 44.Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 45.Fujioka D, Kawabata K, Saito Y, Kobayashi T, Nakamura T, Kodama Y, et al. Role of adiponectin receptors in endothelin-induced cellular hypertrophy in cultured cardiomyocytes and their expression in infarcted heart. Am J Physiol Heart Circ Physiol. 2006;290:H2409–16. doi: 10.1152/ajpheart.00987.2005. [DOI] [PubMed] [Google Scholar]
- 46.MacLellan WR, Schneider MD. Death by design. Programmed cell death in cardiovascular biology and disease. Circ Res. 1997;81:137–44. doi: 10.1161/01.res.81.2.137. [DOI] [PubMed] [Google Scholar]
- 47.Sam F, Sawyer DB, Chang DL, Eberli FR, Ngoy S, Jain M, et al. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol. 2000;279:H422–8. doi: 10.1152/ajpheart.2000.279.1.H422. [DOI] [PubMed] [Google Scholar]