Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease (original) (raw)
Abstract
TGF-β1 functions as a negative regulator of T cell immune responses, signaling to target cells using the Smad family of proteins. We show here that Smad7, an inhibitor of TGF-β1 signaling, is overexpressed in inflammatory bowel disease (IBD) mucosa and purified mucosal T cells. Both whole tissue and isolated cells exhibit defective signaling through this pathway, as measured by phospho-Smad3 immunoreactivity. Specific antisense oligonucleotides for Smad7 reduce Smad7 protein expression in cells isolated from patients with IBD, permitting the cells to respond to exogenous TGF-β1. TGF-β1 cannot inhibit proinflammatory cytokine production in isolated lamina propria mononuclear cells from patients with Crohn disease (CD), but inhibition of Smad7 restores TGF-β1 signaling and enables TGF-β1 to inhibit cytokine production. In inflamed mucosal tissue explants from patients with CD, inhibition of Smad7 also restores p-Smad3 and decreases proinflammatory cytokine production, an effect that is partially blocked by anti–TGF-β1. These results show that Smad7 blockade of TGF-β1 signaling helps maintain the chronic production of proinflammatory cytokines that drives the inflammatory process in IBD and that inhibition of Smad7 enables endogenous TGF-β to downregulate this response.
Introduction
The etiology of Crohn disease (CD) and ulcerative colitis (UC), the two major forms of chronic inflammatory bowel disease (IBD), is still unknown. However, there is evidence that chronic intestinal inflammation is caused by an excessive immune response to mucosal antigens, inappropriately controlled by the normal counterregulatory mechanisms (1).
TGF-β1 is a multifunctional cytokine capable of regulating the growth, differentiation, and function of immune and nonimmune cells. Both in vitro and in vivo studies have demonstrated that TGF-β1 acts as a potent negative regulator of mucosal inflammation (2, 3). TGF-β1 initiates signaling through the ligand-dependent activation of a complex of heterodimeric transmembrane serine/threonine kinases, consisting of type I (TGF-β1 RI) and type II (TGF-β1 RII) receptors. Upon TGF-β1 binding, the receptors rotate relatively within the complex, resulting in phosphorylation and activation of TGF-β1 RI by the constitutively active and autophosphorylated TGF-β1 RII (4). TGF-β1 signals from the receptor to the nucleus using a set of proteins, termed Smads, based on their high homology to the Drosophila mad and the Caenorhabditis elegans Sma proteins. To date, nine different Smad genes that fall into three distinct functional sets have been identified: signal-transducing receptor-activated Smads, which include Smad 1, 2, 3, 5, 8, and 9; a single common mediator, Smad4; and inhibitory Smad 6 and 7 (5, 6). Activated TGF-β1 RI directly phosphorylates Smad2 and Smad3 at serine residues in the carboxy-terminal SSXS sequence (6, 7). Once activated, Smad2 and Smad3 associate with Smad4 and translocate to the nucleus, where Smad protein complexes participate in transcriptional control of target genes (6–9). Importantly, targeted disruption of the Smad3 gene is associated with diminished cell responsiveness to TGF-β1 (10). Mutant mice exhibit a massive infiltration of T cells and pyogenic abscess formation in the stomach and intestine, supporting the view that Smad3 is an essential mediator of the TGF-β1–induced anti-inflammatory and suppressive activities (10).
The inhibitory Smad7 acts by occupying ligand-activated TGF-β1 RI and interfering with the phosphorylation of Smad2/Smad3. Upregulation of Smad7 has been associated with an inhibition of TGF-β1–induced signaling (11, 12). Although the mechanisms that regulate Smad7 production are not fully known, recent studies have shown that activators of either NF-κB (e.g., TNF-α and IL-1β) or STAT-1 (e.g., IFN-γ) pathway can enhance Smad7 expression (13, 14). It is well known that the local production of TNF-α, IL-1β, and IFN-γ is increased in IBD tissue (1, 15–17), and studies in experimental models of IBD indicate that intestinal mucosal inflammation depends at least in part on a balance between proinflammatory cytokines and TGF-β1 (18–22). Paradoxically, however, TGF-β1 and other immunosuppressive cytokines (e.g., IL-10 or IL-1 receptor antagonist) are increased in IBD tissue (1, 23), and yet mucosal inflammation proceeds unchecked. The purpose of this study, therefore, was to examine TGF-β1 signaling in IBD mucosa.
Methods
Patients and samples.
Mucosal samples were taken from intestinal resection specimens of ten patients (median age, 31 years; range, 15–56) with moderate-to-severe CD. In six patients, the primary site of involvement of the disease was the terminal ileum and colon; in two patients, the colon was involved; and the remaining two had ileal disease. The disease duration in this group was 5 ± 3 years. All patients were receiving corticosteroids, although the indication for surgery was a chronic active course poorly responsive to medical treatment. Additional mucosal samples were taken during endoscopy from 11 patients with moderate-to-severe CD (median age, 18 years; range, 12–25). In all of these patients, the primary site of involvement of the disease was the terminal ileum and colon. The disease duration in this group was 7 ± 4 years. One patient was receiving mesalazine; another, mesalazine plus azathioprine; three were receiving corticosteroids; and the remaining six were untreated.
Mucosal samples were also taken from nine patients with active UC (median age, 16 years; range, 9–21). Two patients were receiving mesalazine; one was being treated with corticosteroids; another, with mesalazine plus azathioprine; and the remaining five were untreated. Mucosal samples were also available from eight UC patients undergoing colectomy (median age, 38 years; range, 17–48). These patients were receiving mesalazine plus CS but had ongoing inflammation. Additional mucosal samples were available from two patients with indeterminate colitis and two patients with chronic pouchitis, none of whom were on medical therapy. Age-matched controls included normal colonic mucosal biopsies from eight subjects with irritable bowel syndrome, and colectomy specimens from six patients with colon cancer. In addition, specimens were taken from three patients with diverticular disease. These samples were taken at sites distant from the diverticuli and were normal, as verified by routine histology. Ethical approval was obtained from the City and East London Health Authority.
Purification of lamina propria mononuclear cells and mucosal tissue organ culture.
Lamina propria mononuclear cells (LPMCs) were prepared as described previously (15–17). In some experiments, cells were immediately resuspended in RPMI 1640 (Sigma Chemical, Aldrich Ltd., Dorset, United Kingdom) supplemented with a serum replacement reagent HL-1 (BioWhittaker, Wokingham, United Kingdom), and cultured in the presence or absence of TGF-β1 (1 ng/ml, Sigma Chemical, Aldrich Ltd.) for 1 hour. In other experiments, LPMCs were resuspended in RPMI 1640 supplemented with 10% FCS and cultured in the presence or absence of a Smad7 antisense or control sense oligonucleotide (both used at a final concentration of 4 μg/ml) for 24 hours. Details of the Smad7 antisense and sense oligonucleotides have previously been reported (14). Briefly, phosphorothioate single-stranded oligonucleotide matching the region 107–128 (5′-GCTGCGGGGAGAAGGGGCGAC-3′) of the human Smad7 complementary DNA sequence was synthesized in the sense and antisense orientation. LPMCs were then harvested and extensively washed, resuspended in RPMI 1640 plus HL-1, and cultured in the presence or absence of TGF-β1 (1 ng/ml) for 1 hour. Finally, in other experiments, LPMCs pretreated with the Smad7 antisense or sense oligonucleotides were resuspended in RPMI 1640 plus HL-1, and stimulated with SEB (1 μg/ml, Sigma Chemical, Aldrich Ltd.) in the presence or absence of graded doses of TGF-β1 (final concentration, 0.1–10 ng/ml) for 12 hours. LPMCs were then collected and RNA extracted as described previously (24). IFN-γ and TNF-α transcripts were determined by a competitive quantitative RT-PCR according to the method of Jung et al. (24). Competitive RT-PCR was performed after the standard RNA and test RNA were co–reverse transcribed (24). PCR amplification was carried out using 2 μl of cDNA per sample in 50 μl reaction volumes (10 mM Tris [pH 9.0], 50 mM KCl, 1.5 mM MgCl2, 200 μM dNTPs, 25 pmol 5′ and 3′ gene specific primers, and 1 U Taq polymerase (Pharmacia Biotech, Herts, United Kingdom). The thermal cycle was programmed with a hot start at 94°C for 5 minutes, followed by 33 cycles at 94°C for 1 minute, annealing at 58°C for IFN-γ, and 64°C for TNF-α for 2 minutes, followed by extension at 72°C for 2.5 minutes. PCR primers (Genosys, Cambridge, United Kingdom) were as follows: IFN-γ, FWD, 5′: ATGAAATATACAAGTTATATCTTGGCTTT-3′; REV, 5′: GATGCTCTTC-GACCTCGAAACAGCAT-3′; TNF-α, FWD, 5′: CGGGA-CGTGGAGCTGGCCGAGGAG-3′; REV, 5’: CACCAGCTGGTTATCTCTCAGCTC-3′. RT-PCR products were electrophoresed in 1% agarose gel containing 0.3 μg/ml ethidium bromide. Bands were visualized, and their intensities were quantified by densitometry. Numbers of cytokine transcripts were quantified as described elsewhere (24).
CD3+ lamina propria lymphocytes (LPLs) were purified by incubating LPMCs with immunomagnetic beads coated with monoclonal anti-CD3 antibody according the instructions of the manufacturer (Dynal A.S., Oslo, Norway). Purified LPLs were greater than 95% CD3+ as determined by FACS analysis (Becton Dickinson and Co., Franklin Lakes, New Jersey, USA).
Intestinal mucosa was dissected from the inflamed regions of the colon of seven patients undergoing resection for active CD. These were cut in small pieces (2–3 mm) and placed on steel grids in an organ culture chamber at 37°C in a 5% CO2/95% O2 atmosphere in serum-free media (25). Smad7 antisense or sense oligonucleotides (10 μg/ml) were added and, after 40 hours, proteins were extracted from the tissue. Organ culture supernatants were also collected and used to measure IFN-γ by ELISA (R&D Systems Ltd., Abingdon, United Kingdom). In three of seven patients with CD, the explants were also cultured with Smad7 antisense or sense with or without a neutralizing TGF-β antibody (15 μg/ml; R&D Systems Ltd.). After 40 hours, proteins were extracted from the tissue and analyzed for cytokine expression by Western blotting.
Determination of Smad3 and Smad3-bound Smad4.
Total proteins were immunoprecipitated with an anti-Smad3 antibody (2 μg/ml; Santa Cruz Biotechnology Inc., Santa Cruz, California, USA) and analyzed by Western blotting. An mAb against phosphoserine (1:50 final dilution; Biomeda Corp., Foster City, California, USA), followed by a horseradish peroxidase–conjugated goat anti-mouse IgG mAb (1:2,000 dilution; DAKO Ltd., High Wycombe, United Kingdom), was used. Immunoreactivity was visualized using an ECL kit (Amersham International, Amersham, United Kingdom). After the analysis of phosphorylated Smad3, blots were stripped and then incubated with a goat anti-human Smad3 antibody (1:300 dilution; Santa Cruz Biotechnology Inc.), followed by a rabbit anti-goat antibody conjugated to horseradish peroxidase (1:2,500 dilution) and detection by ECL.
To show that the phosphoserine antibody specifically recognizes phosphorylated residues, total proteins immunoprecipitated with Smad3 antibody were incubated in the presence or absence of 1U of protein phosphatase-2A catalytic subunit (PP-2A) (Promega Corp., Madison, Wisconsin, USA). Western blotting for p-Smad3 and total Smad3 was performed as just described.
To examine the interaction between Smad3 and Smad4, proteins immunoprecipitated with Smad3 antibody were analyzed by Western blotting using a rabbit anti-human antibody against Smad4 (1:300 dilution; Santa Cruz Biotechnology Inc.). Goat anti-rabbit antibody conjugated to horseradish peroxidase (1:2,500 dilution) was used as secondary antibody and detected by ECL. Computer-assisted scanning densitometry (Seescan, Cambridge, United Kingdom) was used to analyze the intensity of the immunoreactive bands.
Determination of Smad7, IFN-γ, and TNF-α protein by Western blotting.
Smad7, IFN-γ, and TNF-α were analyzed using specific goat anti-human Smad7, IFN-γ, or anti-TNF-α antibodies (1:300 final dilution; Santa Cruz Biotechnology Inc.). Rabbit anti-goat antibody conjugated to horseradish peroxidase (1:2,500 dilution) was used to detect primary antibody binding and immunoreactivity visualized by ECL. Ponceau S staining (Sigma Chemical, Aldrich Ltd.) was performed to confirm the equal loading and transfer of proteins. In addition, each blot was stripped and analyzed for β-actin, as internal loading control, using a specific rabbit anti-human β-actin antibody (1:500 final dilution; Santa Cruz Biotechnology Inc.), followed by a goat anti-rabbit antibody conjugated to horseradish peroxidase (1:2,500 dilution).
Statistical analysis.
Differences between groups were compared using either the Mann-Whitney U test, if the data were not normally distributed, or the Student’s t test, if the observations were consistent with a sample from a normally distributed population. The correlation between Smad7 and active/inactive Smad3 was determined by regression analysis.
Results
Smad7 in active lesions of patients with IBD.
The TGF-β1/Smad signaling system is notable for an autoinhibitory loop that involves Smad7 (11, 12). Total proteins extracted from intestinal mucosal samples were first analyzed by Western blotting for Smad7. Smad7 protein expression was increased in all patients with active CD and most UC patients compared with normal controls (Figure 1a). Elevated levels of Smad7 were seen in IBD regardless of whether proteins were extracted from surgically derived mucosal tissues or endoscopic biopsies with the same degree of inflammation. Patients with active CD had a median of 7.17 densitometry arbitrary units (range, 3.9–16.08) and patients with UC a median of 4.985 (range, 1.9–8.22). Both CD and UC patients exhibited levels of Smad7 significantly higher than those found in normal controls (median, 1.06; range, 0.5–2.5) (P < 0.0003 and 0.0006, respectively) (Figure 1a, middle panel). Increased expression of Smad7 was also seen in the mucosal samples of patients with indeterminate colitis and pouchitis (data not shown).
Figure 1.

Smad 7 in IBD. (a) Representative expression of Smad7 (upper blot) and β-actin (lower blot) protein in colonic mucosal samples taken from three normal controls, three patients with active CD, and three with UC. The example is representative of five separate experiments analyzing in total mucosal samples from eight patients with CD, eight with UC, and eight normal controls. Quantitative data are shown in the middle panel as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.). Each point represents the value (a.u.) of Smad7 in mucosal samples taken from a single subject. Horizontal bars indicate the median value. Shown in the bottom panel is expression of Smad7 protein in both CD3-depleted LPMCs and positively selected CD3+ T lymphocytes (T-LPL) from intestinal mucosal samples taken from three patients with CD, three patients with UC, and three normal controls. The example is representative of three separate experiments analyzing Smad7 in both CD3-depleted LPMCs and CD3+ T-LPLs from five patients with CD, four with UC, and six normal controls. (b) Expression of Smad7 (upper blot) and β-actin (lower blot) protein in CD: effect of disease activity. Colonic mucosal biopsy specimens were taken from two normal controls and from inflamed (I) and uninflamed (U) areas of three patients with CD. Arrows indicate Smad7 and β-actin detected by specific polyclonal antibodies. The example is representative of two separate experiments using in total samples from four controls and five CD patients.
Five patients with CD and three with UC had paired biopsy specimens taken from endoscopically abnormal (gross lesions) and endoscopically normal areas. Smad7 was expressed at high levels in all areas of endoscopically abnormal mucosa compared with endoscopically normal mucosa (Figure 1b). The expression of Smad7 in normal mucosal areas of patients with IBD was not different from that seen in normal controls.
In IBD tissue, both positively selected CD3+ T lymphocytes and CD3-depleted LPMCs contained high levels of Smad7 compared with those of controls (Figure 1a, bottom panel).
Downregulation of phosphorylated Smad3 in active lesions of patients with IBD.
Phosphorylation of Smad3 by the activated TGF-β1 RI is an important step in the initiation of TGF-β1 signal transduction (5, 6, 10). We therefore analyzed Smad3 phosphorylation in the intestinal mucosa of patients with IBD. Total proteins extracted from intestinal mucosal samples were immunoprecipitated with anti-Smad3 and subsequently incubated with a phosphoserine antibody. In mucosal samples from normal controls, abundant phosphorylated Smad3 was seen. In contrast, Smad3 phosphorylation was markedly reduced in mucosal samples from CD and, to a lesser degree, UC patients (Figure 2a). p-Smad3 was also detected in mucosal samples from patients with diverticular disease (data not shown).
Figure 2.

(a) Reduced phosphorylation of Smad3 in IBD tissue. Active (phosphorylated, p-Smad3) and inactive Smad3 in mucosal samples of normal controls and patients with CD or UC. Total proteins were immunoprecipitated with a specific Smad3 antibody and subsequently incubated with a phosphoserine antibody (upper blot). After detection of p-Smad3, the membrane was stripped and incubated with a second Smad3 antibody (lower blot) to ascertain equivalent loading of the lanes. To investigate interactions between Smad3 and Smad4, proteins were immunoprecipitated with an anti-Smad3 antibody and subsequently incubated with a Smad4 antibody (middle blot). The example is representative of three separate experiments analyzing in total mucosal samples from eight patients with CD, eight with UC, and eight normal controls. The bottom right panel of a shows quantitative analysis of active/inactive Smad3 ratio in colonic mucosal samples from eight controls, eight patients with CD, and 8 with UC, as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.). Each point represents the value (a.u.) of active/inactive Smad3 ratio in mucosal samples taken from a single subject. Horizontal bars indicate the mean. The bottom left panel of a shows a lack of phosphorylated-Smad3 (p-Smad3) signal after treatment with protein phosphatase-2A (PP-2A). Total protein was extracted from three normal controls immunoprecipitated with a specific Smad3 antibody and incubated in the absence or presence of PP-2A. Proteins were then incubated with a phosphoserine antibody. After detection of p-Smad3, the membrane was stripped and incubated with a second Smad3 antibody to show that PP-2A does not affect the content of total Smad3 proteins. The example is representative of two separate experiments in which mucosal samples from six normal controls were used. (b) Relation between active/inactive Smad3 ratio and Smad7 in mucosal samples taken from patients with CD (left) and UC (right). Both Smad3 and Smad7 levels were measured by densitometry scanning of Western blots and expressed in arbitrary units (a.u.). The expression of Smad7 is inversely related to the active/inactive Smad3 ratio in both CD (r = 0.818; P < 0.003) and UC (r = 0.616; P < 0.03).
To demonstrate that the phosphoserine antibody specifically recognizes phosphorylated residues, total proteins extracted from normal controls and immunoprecipitated with Smad3 antibody were incubated with or without PP-2A. Treatment with PP-2A abolished the p-Smad3 signal with no change in the total Smad3 protein content (Figure 2a, bottom left panel).
Analysis of active (phosphorylated)/inactive Smad3 ratios showed a significantly decrease in patients with active CD and UC compared with controls (P < 0.0001 and P < 0.0032, respectively) (Figure 2a, bottom right panel). Once activated, Smad3 associates with Smad4 and translocate to the nucleus (5, 6). To investigate whether downregulation of active Smad3 was associated with a reduced expression of Smad3-bound Smad4, proteins were immunoprecipitated with anti-Smad3 and then probed with a Smad4 antibody. Mucosal samples taken from active IBD exhibited a reduced expression of Smad3-bound Smad4 in comparison to that observed in normal controls (Figure 2a). There was a clear inverse relationship in individual samples between the expression of Smad7 and the amount of p-SMAD3 (Figure 2b).
To further analyze TGF-β1/Smad signaling in IBD tissue, LPMCs extracted from normal or IBD patients were stimulated with TGF-β1, and both total and phosphorylated Smad3 analyzed by Western blotting. p-Smad3 signal was detected in unstimulated normal LPMCs and enhanced by TGF-β1 stimulation (Figure 3). In contrast, p-Smad3 immunoreactivity was barely detectable in unstimulated LPMCs from both CD and UC and not increased by TGF-β1 stimulation (Figure 3).
Figure 3.

Stimulation of CD and UC LPMCs with TGF-β1 does not result in enhanced p-Smad3. LPMCs isolated from the colon of one normal control, one CD patient, or one UC patient were cultured in the presence or absence of TGF-β1 (1 ng/ml) for 1 hour. Phosphorylated (top blot, p-Smad3) and total (bottom blot) Smad3 were analyzed in proteins extracted from LPMCs as indicated in Figure 2. The example is representative of four separate experiments, in each of which, LPMCs from one normal control, one patient with CD, and one patient with UC were analyzed. In all cases, LPMCs from normal patients showed endogenous p-Smad3 that was enhanced by TGF-β1, whereas in all IBD patients, endogenous p-Smad3 was barely detectable after TGF-β1 stimulation. The bottom panel shows quantitative analysis of active/inactive Smad3 ratio in TGF-β1–stimulated LPMCs from four controls, four patients with CD, and four with UC, as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.). Each point represents the value (a.u.) of active/inactive Smad3 ratio in LPMC samples taken from a single subject. Horizontal bars indicate the mean.
Inhibition of Smad7 enables cells to respond to TGF-β1 and downregulates proinflammatory cytokines in IBD.
To examine whether Smad7 is directly responsible for inhibiting TGF-β1/Smad3 signaling in IBD, we analyzed the IBD LPMC response to TGF-β1 after treating the cells with specific Smad7 antisense or sense oligonucleotides. Treatment of both CD and UC LPMCs with the Smad7 antisense but not with the sense oligonucleotide inhibited Smad7 expression (Figure 4a), and this was associated with the restoration of Smad3 phosphorylation in response to TGF-β1 (Figure 4b). Densitometric analysis of the Western blots in five CD patients and five UC patients showed clearly that the antisense markedly reduced Smad7 protein and also markedly increased p-Smad3 in the same cells. Taken together, these observations indicate that in both CD and UC intestinal mucosa, overexpression of Smad7 is associated with defective TGF-β1 signaling.
Figure 4.

Antisense to Smad7 restores TGF-β1 signaling in both CD and UC LPMCs. (a) Treatment of CD and UC LPMCs with a specific Smad7 antisense but not a sense oligonucleotide inhibits Smad7 expression. CD and UC LPMCs were cultured in the absence (U, unstimulated) or presence of a specific Smad7 antisense (AS) or control sense (S) oligonucleotide for 24 hours. Arrows indicate Smad7 detected by a specific polyclonal antibody. The example is representative of three separate experiments analyzing in total LPMCs from five CD or five UC patients. Quantitative data are shown in the right panel as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.). Each point represents the value (a.u.) of Smad7 in LPMCs taken from a single subject. Horizontal bars indicate the median value. (b) Inhibition of Smad7 restores the TGF-β1–induced Smad3 phosphorylation in both CD and UC LPMCs. LPMCs were cultured in the absence (U, unstimulated) or presence of a specific Smad7 antisense (AS) or control sense (S) oligonucleotide, and then stimulated with 1 ng/ml TGF-β1 for 1 hour. The right panel shows quantitative analysis of active/inactive Smad3 ratio in LPMC from five patients with CD and five with UC, as measured by densitometry scanning of Western blots. Values are expressed in arbitrary units (a.u.). Each point represents the value (a.u.) of active/inactive Smad3 ratio in LPMCs taken from a single subject. Horizontal bars indicate the mean.
There is a large body of evidence that intestinal inflammation is in part dependent on the balance between inflammatory cytokines, especially IFN-γ, and TGF-β1 activity (18–22). Therefore, we examined whether restoring TGF-β1 signaling by Smad7 antisense resulted in an inhibition of inflammatory cytokines. Given that in preliminary experiments we determined that IFN-γ was produced at high levels in CD but not UC in comparison to controls, we focused on CD samples. CD LPMCs pretreated with medium alone or Smad7 antisense or sense oligonucleotides were stimulated with SEB in the presence or absence of graded doses of TGF-β1. LPMCs pretreated with or without oligonucleotides and then cultured in medium alone showed only low numbers of transcripts for both IFN-γ (4,293 ± 839/μg RNA in LPMCs pretreated with medium; 5,748 ± 958/μg RNA in LPMCs pretreated with sense; and 1,239 ± 543/μg RNA in LPMCs pretreated with antisense) and TNF-α (3,939 ± 1,245/μg RNA in LPMCs pretreated with medium; 4,983 ± 981/μg RNA in LPMCs pretreated with sense; and 2,019 ± 765/μg RNA in LPMCs pretreated with antisense). Stimulation of LPMCs with SEB resulted in a large increase in the number of transcripts for both IFN-γ and TNF-α (P < 0.0001), and there was no effect of the oligonucleotides on this response (Figure 5). The addition of graded doses of TGF-β1 to CD LPMCs precultured in medium alone or with sense oligonucleotides had only a marginal effect on TNF-α and IFN-γ transcripts, although there was modest inhibition at the highest dose used (Figure 5; P ≤ 0.05) (Table 1). In contrast, CD LPMCs pretreated with the Smad7 antisense oligonucleotide showed markedly reduced TNF-α and IFN-γ transcripts at all doses of TGF-β1 tested (Figure 5; P ≤ 0.002) (Table 1). The ability of TGF-β1 to inhibit cytokine production of LPMCs from three normal controls was also studied (Figure 5, insets). Unstimulated LPMCs cultured in medium alone or with the sense or antisense oligonucleotides showed less than 1,000 TNF-α and IFN-γ transcripts per microgram of RNA. After SEB stimulation in the presence of oligonucleotides, there was a 10- to 12-fold increase in the number of cytokine transcripts. This was inhibited in a dose-dependent fashion by TGF-β1, regardless of whether the cells had been pretreated with sense or antisense oligonucleotides.
Figure 5.
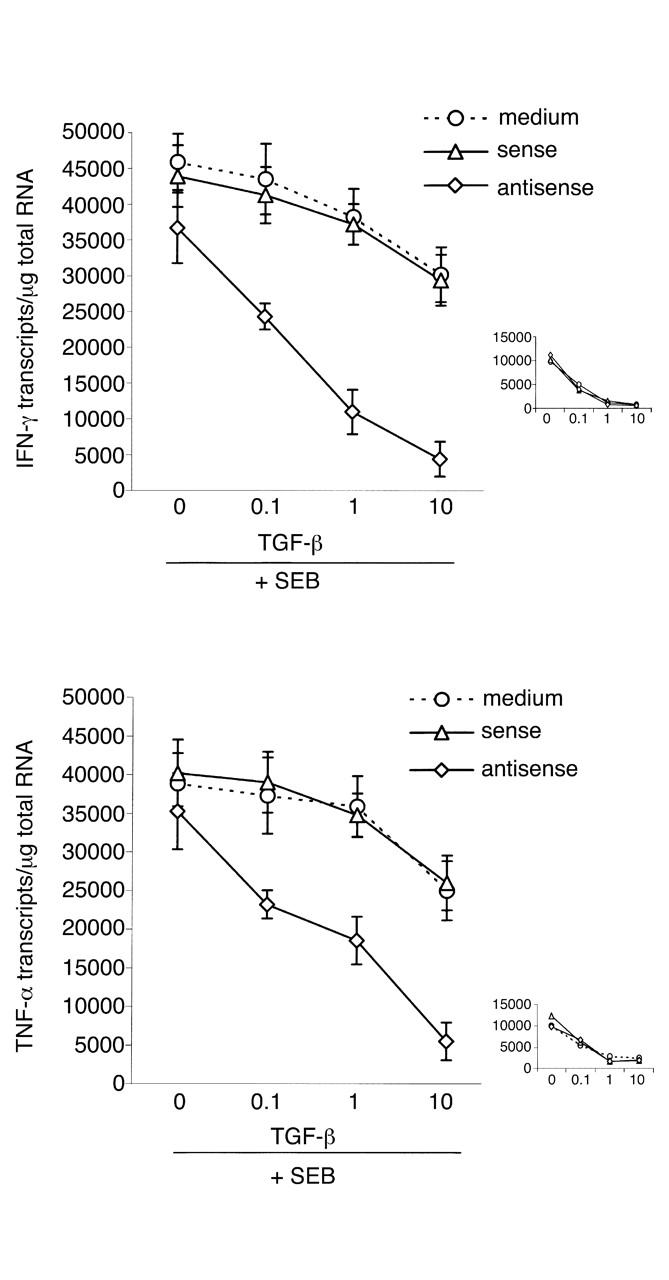
Antisense to Smad7 enables TGF-β1 to inhibit CD LPMC proinflammatory cytokine production. CD LPMCs were cultured in the absence or presence of a specific Smad7 antisense or sense oligonucleotide for 24 hours. The cells were washed with PBS, resuspended in RPMI 1640 supplemented with HL-1 and cultured with or without SEB in the presence or absence of graded doses of TGF-β1 for 12 hours. In CD LPMCs pretreated with the Smad7 antisense oligonucleotide, as little as 0.1 ng/ml of TGF-β1 significantly decreased transcripts for both IFN-γ and TNF-α (P < 0.002). In CD LPMCs precultured with medium or sense Smad7 oligonucleotide, only a modest decrease in IFN-γ and TNF-α transcripts was seen with 10 ng/ml TGF-β1 (P ≤ 0.05). Data are expressed as mean ± SEM of four separate experiments, in each of which, LPMCs from one CD patient were used. The inset shows that, as expected, in LPMCs from normal controls, TGF-β1 dose-dependently inhibited SEB-stimulated cytokine transcripts, regardless of whether LPMCs were pretreated or not with oligonucleotides. Data are expressed as mean ± SEM of three separate experiments, in each of which, LPMCs from one normal control were used.
Table 1.
Percentage of inhibition of SEB-stimulated IFN-γ and TNF-α RNA transcripts in CD LPMCs by TGF-β1A

We next used ex vivo organ culture of inflamed mucosal samples from CD patients to determine whether antagonizing Smad7 also reduced proinflammatory cytokines in tissue. After 40 hours of culture in medium or with the sense oligonucleotide, Smad7 was readily detectable; p-Smad3 was present but barely detectable; and TNF-α and IFN-γ proteins were clearly present in the tissue (Figure 6a). Smad7 antisense reduced Smad7 protein and also resulted in increased p-Smad3; importantly, it also reduced TNF-α and IFN-γ in the tissues (Figure 6a). To confirm these results, we analyzed IFN-γ protein in the organ culture supernatants. After 40 hours in medium alone, the amount of IFN-γ in the supernatants was 439 ± 221 pg/mg tissue. With the sense oligonucleotide, there was no decrease (421 ± 181.2 pg/mg tissue). However with antisense to Smad7, IFN-γ concentrations decreased 60% and 80% in all samples to a mean of 108 ± 38.7 pg/mg tissue (P < 0.02), also showing that the cytokine Western blotting did reflect cytokine concentrations in the supernatants. Finally, to examine whether the downregulation of cytokines in ex vivo organ culture of inflamed mucosal samples from CD treated with the Smad7 antisense was due to the endogenous TGF-β1, Smad7 antisense was added to the organ cultures with or without a neutralizing TGF-β1 antibody. Anti–TGF-β1 had no effect on the decreased expression of Smad 7 protein induced by the antisense oligonucleotide (Figure 6b). However in the presence of the antibody, there was clearly less endogenous p-Smad3 in the tissue, and cytokine expression remained elevated.
Figure 6.

(a) Inhibition of Smad7 protein by a specific Smad7 antisense oligonucleotide in CD mucosal tissue results in increased p-Smad3 and decreased tissue TNF-α and IFN-γ. The example is representative of four separate experiments, in each of which inflamed mucosal tissue from one CD patient was used. Identical results were seen in each patient. (b) Addition of a neutralizing TGF-β1 antibody decreases but does not abrogate the Smad7 antisense effect on both p-Smad3 and cytokine expression. The example is representative of three separate experiments, in each of which, inflamed mucosal tissue from one CD patient was used. Identical results were seen in each patient.
Discussion
The results presented in this study demonstrate that in the inflamed intestine of patients with IBD, there is marked overexpression of Smad7. This is associated with a reduction in Smad3 phosphorylation, a crucial step in the TGF-β1–mediated signal transduction. In contrast, abundant phosphorylated Smad3 and Smad3/Smad4 complexes are seen in the normal intestinal mucosa, consistent with constitutively active TGF-β1 signaling. We also demonstrate that blocking Smad7 with a specific antisense oligonucleotide restores TGF-β1 signaling and importantly allows TGF-β1 to inhibit proinflammatory cytokine production by isolated mucosal lamina propria mononuclear cells. Finally, in CD tissue, Smad7 antisense restored p-Smad3 and importantly resulted in decreased cytokine production. We believe that this is due to the activity of endogenous TGF-β, given that addition of a neutralizing TGF-β1 to the organ cultures decreased the Smad7 antisense effect on cytokine expression. Together, these results clearly demonstrate that modulating Smad7 expression has profound effects on proinflammatory cytokine production in IBD. Although we only carried out functional studies with CD samples, we believe that the overexpression of Smad7 in UC will also compromise the ability of TGF-β1 to downregulate immune responses in this condition.
TGF-β1 is one of the most widely distributed cytokines that acts on virtually all cell types and mediates highly pleiotropic functions. In particular, TGF-β1 plays an important role in the control of immune homeostasis and prevention of mucosal inflammation (2, 3). TGF-β1–knockout mice develop a severe multiple organ inflammatory disease, in which the lymphocytic infiltration of the affected organs is associated with increased production of TNF-α and IFN-γ (26, 27). Abrogation of TGF-β1 signaling in T cells alone is sufficient to break T and B cell homeostasis and induce T cell–mediated inflammatory lesions in various organs, including the intestine (28). Finally, it has been widely demonstrated that neutralization of TGF-β1 results in the induction and/or amplification of pathogenic responses responsible for the development of experimental colitis resembling either CD or UC (19–22). A diminished ability to mount an efficient counterregulatory TGF-β1 response to inflammatory stimuli is therefore believed to be relevant in the pathogenesis of IBD (18). The diminished Smad3 phosphorylation documented in CD and UC further supports this notion, given that Smad3 activation is essential in the TGF-β1–mediated control of mucosal immunity (10).
Smad3 activation can be inhibited through various mechanisms (6). In this study, however, we focused on the expression of the inhibitory Smad7. In contrast to the TGF-β1R–associated Smads, Smad7 has no carboxy-terminal SSXS sequence and thus does not require any phosphorylation to be active. Smad7 forms a stable complex with activated TGF-β1 RI, thereby blocking the association and phosphorylation of Smad2 and Smad3 (11, 12). We show that all CD and most UC patients exhibit high levels of Smad7 compared with normal controls. Proteins extracted from both positively selected CD3+ T lymphocytes and non–CD3+ LPMCs from IBD patients showed enhanced Smad7 immunoreactivity on Western blotting in comparison to controls. To the best of our knowledge, this is the first study showing an abnormal expression of Smad7 in a human inflammatory disease. We were also able to show that inhibition of Smad7 protein with Smad7 antisense restored TGF-β1 signaling in LPMCs and enabled exogenous TGF-β1 to inhibit proinflammatory cytokine production; even in inflamed CD tissues, antisense to Smad7 restored TGF-β1 signaling and decreased proinflammatory cytokine production. Although we do not know the mechanisms operating in the tissue, this result nonetheless clearly indicates that Smad7 inhibition may be a novel therapeutic approach in IBD, enabling endogenous TGF-β to dampen local inflammation.
Consistent with our human data, it was recently reported that Smad7 transgenic mice exhibit defective T cell responses to TGF-β1, show markedly greater cytokine production in vitro, and show enhanced antigen-induced airway inflammation (29). Together, these observations suggest that, despite enhanced TGF-β1 production, defective TGF-β1/Smad signaling contributes to maintaining the chronic inflammatory state in IBD.
The mechanisms/factors that regulate the expression of Smad7 are not fully known. Smad7 was first identified as a gene induced by shear stress in vascular endothelial cells (30). Neither exposure to TNF-α nor to TGF-β1 altered Smad7 RNA expression in these cells. More recently, Bitzer et al. have shown that activation of NF-κB/RelA by TNF-α, IL-1β, and LPS, but not IFN-γ, increases transcription of the Smad7 gene and elevates intracellular levels of Smad7 protein in fibroblast cell lines (13). In contrast, Ulloa et al. have demonstrated that activation of the transcription factor STAT1 by IFN-γ enhances Smad7 in human monocytic leukemia U937 cells (14). It is noticeable that Smad7 is detectable in normal intestine; it will be of interest to determine which cells express this molecule and whether they are unresponsive to TGF-β1. In IBD mucosa, IFN-γ and proinflammatory cytokines are upregulated (15–17), so it is likely that these molecules contribute to enhance Smad7 expression in the intestine of patients with IBD and provide a basis for the known antagonism between TGF-β1 and IFN-γ/TNF-α in the regulation of immune cell functions in the gut (18). In preliminary experiments, however, we have not been able to reduce Smad 7 expression in IBD LPMCs by the addition to the cultures of IFNR-IgG and TNFR-IgG, although negative cytokine neutralization experiments should always be treated with caution. Nor do we believe that these preliminary results exclude a role for both IFN-γ and TNF-α in the regulation of Smad7 in IBD. Given that Smad7 expression is largely dependent on activation of STAT1 and NF-κβ and, in IBD, STAT1 and NF-κβ may be induced by several locally produced molecules, it is possible that blocking IFN-γ or TNF-α is not sufficient to completely inhibit STAT1 and NF-κβ activation and downregulate Smad7. Studies are now in progress to address this issue. Moreover, it is important to point out that regulation of Smad7 induction is a complex phenomenon dependent on the cell type. Therefore we are now carrying on experiments to examine the effect of various cytokines, including IFN-γ and TNF-α, on Smad7 expression in single cell populations isolated from the human intestine.
Acknowledgments
This work was supported by the British Biotechnology, Science and Research Council and the European Union (grant ERBFMRXCT9).
Footnotes
See the related Commentary beginning on page 523.
References
- 1.Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 1998;115:182–205. doi: 10.1016/s0016-5085(98)70381-6. [DOI] [PubMed] [Google Scholar]
- 2.Wahl SM. Transforming growth factor β: the good, the bad, and the ugly. J Exp Med. 1994;180:1587–1590. doi: 10.1084/jem.180.5.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 4.Piek E, Heldin C-H, ten Dijke P. Specificity, diversity, and regulation in TGF-β superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- 5.Heldin C-H, Kohei M, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Zhang Y, Feng X-H. Smads: transcriptional activators of TGF-β responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- 7.Abdollah S, et al. TGFβRI phosphorylation of Smad2 on Ser465 and 467 is required for Smad2/Smad4 complex formation and signalling. J Biol Chem. 1997;272:27678–27685. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- 8.Baker JC, Harland RM. A novel mesoderm inducer, Madr2, functions in the activin signal transduction pathway. Genes Dev. 1996;10:1880–1889. doi: 10.1101/gad.10.15.1880. [DOI] [PubMed] [Google Scholar]
- 9.Dennler S, et al. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi H, et al. The Mad-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 12.Nakao A, et al. Identification of Smad7, a TGFβ-inducible antagonist of TGFβ signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 13.Bitzer M, et al. A mechanism of suppression of TGF-β/SMAD signaling by NF-kB/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- 14.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 15.Breese EJ, et al. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–1466. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 16.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-γ secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131. [PMC free article] [PubMed] [Google Scholar]
- 17.Fais S, et al. Spontaneous release of interferon gamma by intestinal lamina propria lymphocytes in Crohn’s disease. Kinetics of in vitro response to interferon gamma inducers. Gut. 1991;32:403–407. doi: 10.1136/gut.32.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strober W, et al. Reciprocal IFN-γ and TGF-β responses regulate the occurrence of mucosal inflammation. Immunol Today. 1997;18:61–64. doi: 10.1016/s0167-5699(97)01000-1. [DOI] [PubMed] [Google Scholar]
- 19.Neurath MF, et al. Experimental granulomatous colitis in mice is abrogated by induction of TGF-—mediated oral tolerance. J Exp Med. 1996;183:2605–2616. doi: 10.1084/jem.183.6.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ludviksson BR, Ehrhardt RO, Strober W. TGF-β production regulates the development of the 2,4,6-trinitrophenol-conjugated keyhole lipet hemocyanin-induced colonic inflammation in IL-2-deficient mice. J Immunol. 1997;159:3622–3628. [PubMed] [Google Scholar]
- 21.Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med. 1998;188:1929–1939. doi: 10.1084/jem.188.10.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL. A critical role for transforming growth factor-β but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RBlow CD4+ T cells. J Exp Med. 1996;183:2669–2674. doi: 10.1084/jem.183.6.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors α and β in colonic mucosa in inflammatory bowel disease. Gastroenterology. 1996;110:975–984. doi: 10.1053/gast.1996.v110.pm8613031. [DOI] [PubMed] [Google Scholar]
- 24.Jung HC, et al. A distinct array of proinflammatory cytokines is expressed in human colonic epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browning TH, Trier JS. Organ culture of mucosal biopsies of human small intestine. J Clin Invest. 1969;48:1423–1432. doi: 10.1172/JCI106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christ M, et al. Immune dysregulation in TGF-β1-deficient mice. J Immunol. 1994;153:1936–1946. [PubMed] [Google Scholar]
- 27.Shull MM, et al. Targeted disruption of the mouse TGF-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorelik L, Flavell RA. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 29.Nakao A, et al. Blockade of transforming growth factor β/Smad signaling in T cells by overexpression of Smad7 enhances antigen-induced inflammation and airway reactivity. J Exp Med. 2000;192:151–158. doi: 10.1084/jem.192.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topper JN, et al. Vascular MADs: two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc Natl Acad Sci. 1997;94:9314–9319. doi: 10.1073/pnas.94.17.9314. [DOI] [PMC free article] [PubMed] [Google Scholar]