pVHL acts as an Adapter to Promote the Inhibitory Phosphorylation of the NF-κB Agonist Card9 by CK2 (original) (raw)
. Author manuscript; available in PMC: 2008 Oct 12.
Summary
The VHL tumor suppressor protein (pVHL) is part of an E3 ubiquitin ligase that targets HIF for destruction. pVHL-defective renal carcinoma cells exhibit increased NF-κB activity but the mechanism is unclear. NF-κB affects tumorigenesis and therapeutic resistance in some settings. We found that pVHL associates with the NF-κB agonist Card9 but does not target Card9 for destruction. Instead, pVHL serves as an adaptor that promotes the phosphorylation of the Card9 C-terminus by CK2. Elimination of these sites markedly enhanced Card9's ability to activate NF-κB in VHL+/+ cells and Card9 siRNA normalized NF-κB activity in _VHL_−/− cells and restored their sensitivity to cytokine-induced apoptosis. Furthermore, downregulation of Card9 in _VHL_−/− cancer cells reduced their tumorigenic potential. Therefore pVHL can serve as an adaptor for both an ubiquitin conjugating enzyme and for a kinase. The latter activity, which promotes Card9 phosphorylation, links pVHL to control of NF-κB activity and tumorigenesis.
Inactivation of the VHL tumor suppressor gene plays a role in the pathogenesis of clear cell renal carcinomas, hemangioblastomas, and pheochromocytomas (Kim and Kaelin, 2004). The best understood function of the VHL tumor suppressor protein (pVHL) relates to its role as the substrate recognition unit of an E3 ubiquitin ligase complex that targets the alpha subunits of the heterodimeric transcription factor HIF (hypoxia-inducible factor) for destruction (Kaelin, 2007; Schofield and Ratcliffe, 2004). The interaction between pVHL and HIFα requires that HIFα be hydroxylated on either of two prolyl residues by members of the EglN family (also called PHDs or HPHs), which are oxygen-dependent enzymes that have evolved to respond to changes in oxygen over a physiologically relevant range in an intracellular milieu (Kaelin, 2007; Schofield and Ratcliffe, 2004). Increased HIF activity as a result of VHL inactivation contributes to renal carcinoma growth and probably plays a role in the development of hemangioblastomas (Kondo et al., 2003; Kondo et al., 2002; Maranchie et al., 2002; Raval et al., 2005; Zimmer et al., 2004).
Several lines of evidence, however, suggest that pVHL has functions in addition to regulating HIF. First, there are genotype-phenotype correlations with respect to site-specific tumor risk among individuals who carry a mutant VHL allele in their germline (VHL disease) (Kaelin, 2002). For example, some VHL alleles cause familial pheochromocytoma without an increased risk of hemangioblastoma and renal cell carcinoma. The products of these alleles appear to be normal with respect to HIF regulation but instead are compromised with respect to another pVHL target, aPKC (Clifford et al., 2001; Hoffman et al., 2001; Lee et al., 2005). Increased aPKC activity protects pheochromocytoma cells from apoptosis after growth factor withdrawal (Lee et al., 2005). It is probable that the other genotype-phenotype correlations in VHL disease are likewise manifestations of the degree to which different pVHL functions are quantitatively or qualitatively altered.
Second, chronic HIF activation does not appear to be sufficient for tumor development. Individuals who are chronically hypoxemic, such as occurs with life at high altitude or with various medical conditions, develop secondary polycythemia because of HIF accumulation and increased production of HIF-responsive gene products such as Erythropoietin (Golde and Hocking, 1981). However, the risk of developing the tumors seen in VHL disease is not conspicuously increased in these individuals. Similarly, individuals who are homozygous for a VHL allele that is hypomorphic with respect to HIF, or who carry a defective EglN1 allele, develop polycythemia but are not remarkably tumor prone (Gordeuk et al., 2004; Percy et al., 2006). Tumors are also not, so far, a feature of mice engineered to produce stabilized versions of HIFα (Kim et al., 2006)(WYK and WGK-unpublished data). These mice instead develop increased angiogenesis and, in some cases, polycythemia.
Finally, pVHL has been reported to bind to a dozen different proteins, has been found in multiple cellular compartments, and has been implicated in diverse cellular processes including cell division, apoptosis, differentiation, and control of extracellular matrix formation (Czyzyk-Krzeska and Meller, 2004; Kaelin, 2007). While some of these biological functions might relate to HIF, others, based on genetic experiments in human cells and model organisms, involve HIF-independent pVHL functions (Bishop et al., 2004; Calzada et al., 2006).
Clear cell renal carcinoma is the most common form of kidney cancer. Most of these tumors harbor biallelic VHL mutations or fail to produce VHL mRNA due to VHL promoter hypermethylation (Kim and Kaelin, 2004). Kidney cancers are difficult to treat medically and frequently exhibit increased NF-κB activity, which can promote resistance to chemotherapy or cytokines. Several studies have reported increased NF-κB activity in pVHL-defective renal carcinoma cells but how pVHL loss promotes NF-κB activity is unclear (An et al., 2005; An and Rettig, 2005; Oya et al., 2001; Oya et al., 2003; Qi and Ohh, 2003). Here we report that pVHL, bound to Casein Kinase 2, promotes the inhibitory phosphorylation of the NF-κB agonist Card9. Failure to phosphorylate Card9, which is known to interact with Bcl10 upstream of NF-κB (Bertin et al., 2000; Gross et al., 2006), leads to increased NF-κB activity and decreased apoptosis. Elimination of Card9 in pVHL-defective cells normalized NF-κB activity and restored sensitivity to a proapoptotic cytokine. Downregulation of Card9 in _VHL_−/− cancer cells retarded tumor growth. Therefore Card9 links pVHL to NF-κB biology and control of tumor growth.
Results
pVHL associates with Card9
To search for additional pVHL substrates we purified pVHL-associated proteins from PC12 pheochromocytoma cells that were engineered to produce HA-pVHL and treated with the proteasomal inhibitor MG132 (Figure 1A). Cell extracts were first fractionated by FPLC using a DEAE column. Fractions that were enriched for pVHL and the known pVHL substrate HIF1α, as determined by pVHL far western blotting (Figure 1B), were pooled, concentrated by ammonium sulfate precipitation, and subjected to alternating positive and negative affinity purification steps using anti-HA, control, and anti-pVHL antibodies. After the last step bound proteins were resolved by SDS-PAGE and analyzed by mass-spectrometry. Among the peptides identified, eight peptides from the NF-κB agonist Card9 were recovered (Figure 1C). Since we isolated multiple Card9 peptides and _VHL_-deficient cells display increased NF-κB activity (An et al., 2005; An and Rettig, 2005; Qi and Ohh, 2003), we pursued the interaction of pVHL with Card9 further.
Figure 1. pVHL binds to Card9.

(A) Purification schema.
(B) Farwestern blot analysis of DEAE sepharose fractions using recombinant pVHL as probe, which was detected with anti-pVHL antibody.
(C) Primary sequence of Card9. Peptides recovered by mass spectrometry are underlined. Dots indicate potential CK2 sites.
(D) Autoradiogram of 35S-Card9 bound to GST-pVHL in the presence of hydroxylated (OH) or non-hydroxylated (WT) HIF1α peptide. Input = 10% of Card9 used for binding reaction.
(E) Immunoblot (IB) analysis of HeLa cells transfected to produce HA-pVHL and Myc-Card9 and immunoprecipitated (IP) with anti-HA antibody.
(F) Immunoblot analysis as in (E) of HeLa cells transfected to produce Myc-Card9 and indicated HA-pVHL mutants. FL= full-length.
(G) Autoradiogram of immunoprecipitates derived from metabolically labeled U937 cells. After initial immunoprecipitation (‘First IP’) under native conditions bound proteins were released under denaturing conditions and reimmunoprecipitated with the indicated antibodies. The identity of the intense 50 kD band in the sequential anti-pVHL, anti-pVHL lane is not known.
In vitro translated Myc-Card9 bound to bacterially produced GST-pVHL (complexed with elongin B and elongin C) in vitro (Figure 1D). This interaction was specifically blocked by a hydroxylated HIF1α peptide that docks with the pVHL β-domain (Figure 1D) (Hon et al., 2002; Min et al., 2002), suggesting that the pVHL β-domain is also important for binding to Card9. HA-pVHL and Myc-Card9 also bound to one another in cotransfection assays with HeLa cells (Figure 1E). Interestingly, Myc-Card9 migrated as a doublet in these assays and the lower (or ‘bottom’) band consistently displayed a higher affinity for HA-pVHL than the upper (or ‘top’) band. Consistent with the peptide competition assays, mutational analysis revealed that pVHL beta domain residues 92−121, which are critical for recognition of hydroxylated HIF1α, contribute to Card9 binding (Figure 1F). To ask if pVHL can bind to Card9 under endogenous conditions, U937 VHL+/+ leukemia cells were metabolically labeled with 35S and immunoprecipitated with control, anti-pVHL, or anti-Card9 antibodies. After extensive washing the bound proteins were eluted by boiling in SDS and, after dilution of the SDS, reimmunoprecipitated with the same or a different antibody. The initial anti-pVHL immunoprecipitate contained a protein doublet of the expected molecular weight for Card9 that was recognized by an anti-Card9 antibody, but not a control antibody, in the second immunoprecipitation. Moreover, this doublet comigrated with the doublet captured when proteins released from the initial anti-Card9 immunoprecipitation reaction were reimmunoprecipitated with the same anti-Card9 antibody under denaturing conditions (Figure 1G and data not shown). This suggests that pVHL can interact with Card9 under physiological conditions. Note that Fig 1G cannot be directly compared to Fig 1E because different electrophoretic conditions were used (15% vs. 10% polyacrylamide, respectively) and because Fig 1G, in contrast to Fig 1E, depicts only newly synthesized Card9.
pVHL promotes the phosphorylation of the Card9 C-terminus by CK2
In pilot experiments we determined that Card9 abundance was not altered in VHL+/+ cells treated with proteasomal inhibitors or hydroxylase inhibitors, in contrast to the canonical pVHL substrate HIFα (data not shown). Instead, we noted that the Card9 top band was virtually absent in VHL−/− pRC3 cells (Figure 2A). Similar results were observed in isogenic A498 renal carcinoma cells that do or do not contain wild-type pVHL (Figure S1). WT8 cells and pRC3 cells are 786-O _VHL_−/− renal carcinoma cell subclones that were stably transfected with a plasmid encoding wild-type pVHL or with the backbone vector, respectively (Iliopoulos et al., 1995). The top band and bottom band isolated from cells that contain wild-type pVHL (Hep3B and WT8) collapsed into a faster migrating form when phosphatase treated, suggesting that pVHL promotes the (hyper)phosphorylation of Card9 (Figure 2B).
Figure 2. pVHL promotes Card9 hyperphosphorylation.

(A) Anti-Card9 immunoblot analysis of whole cell extracts (left) or anti-Card9 immunoprecipitates (right) prepared from 786-O _VHL_−/− subclones stably producing wild-type pVHL (WT8) or transfected with empty vector (pRC3). (*)= background band.
(B) Anti-Card9 immunoblot analysis of anti-Card9 immunoprecipitates derived from the indicated cells. Immunoprecipitates were treated with alkaline or lambda phosphatase in vitro where indicated.
(C) Anti-Myc immunoblot analysis of the indicated cell lines transfected to produce Myc-Card9. Transfection mix contained VHL or control (SCR) siRNA where indicated.
(D) Anti-Myc immunoblot analysis of anti-Myc immunoprecipitates prepared from HeLa cells producing Myc-Card9. Immunoprecipitates were dephosphorylated in vitro and then incubated with WT8 or pRC3 cytosolic extract in the absence (−) or presence (+) of an energy regenerating system (ERS). Immunoprecipitates were retreated with lambda phosphatase where indicated prior to SDS-PAGE.
To pursue this idea further, we transiently transfected cells that do or do not contain wild-type pVHL with a plasmid encoding Myc-Card9. Under these experimental conditions only the top band was detected in cells containing wild-type pVHL (WT8 and HeLa cells) while both the top and bottom bands were detected in the _VHL_−/− pRC3 cells (Figure 2C). The appearance of an exogenous top band of Card9 in _VHL_−/− cells, in contrast to the behavior of endogenous Card9, might be due to the higher concentrations of Card9 achieved after transient transfection. For example, these conditions might promote the interaction of Card9 with the relevant kinase(s) and hence partially obviate the need for an adaptor (see below). Nonetheless, the Card9 bottom band reappeared in HeLa cells producing Myc-tagged Card9 and treated with VHL siRNA (Figure 2C and Figure S2). Conversely, incubation of dephosphorylated Myc-Card9 with a pVHL-containing cell extract led to the appearance of the top band in vitro, which disappeared upon lambda phosphatase treatment (Figure 2D).
These results indicated that pVHL promotes Card9 phosphorylation. In parallel we discovered that pVHL associates with a kinase that can phosphorylate Card9 in vitro (Figure 3A). pVHL was recently reported to be a CK2 substrate (Lolkema et al., 2005). We independently discovered that pVHL is a CK2 substrate (data not shown), which led us to ask if CK2 binds to pVHL. We found that CK2 and pVHL can be coimmunoprecipitated (Figure 3B), raising the possibility that pVHL acts as an adaptor to promote the phosphorylation of Card9 by CK2. In support of this, pVHL enhanced the binding of Card9 and CK2 in vivo (Figure 3C).
Figure 3. pVHL promotes the phosphorylation of the Card9 C-terminus by CK2.

(A) Autoradiogram of proteins phosphorylated in vitro by anti-HA immunoprecipitates derived from HeLa cells transfected with the indicated plasmids. In vitro kinase assays were performed in the presence of 32P-γ-ATP using anti-HA immunoprecipitates that were immobilized on Protein A sepharose. Bound proteins were released by boiling in SDS-containing buffer and, after SDS dilution, recaptured with anti-HA or anti-Myc antibody.
(B and C) Immunoblot analysis with indicated antibodies of control or CK2α immunoprecipitates derived from HeLa cells that were (+) or were not (−) transfected to produce HA-pVHL. In (C) cells were also transfected to produce Myc-Card9.
(D) Coomassie stained gel of anti-Myc immunoprecipitates from HeLa cells transiently transfected to produce Myc-Card9. Immunoprecipitated Myc-Card9 was dephosphorylated in vitro with lambda phosphatase and then incubated with recombinant CK2, where indicated, prior to SDS-PAGE. Also shown are Card9 residues phosphorylated by CK2 under these conditions as determined by mass spectrometry.
(E) Immunoblot analysis with the indicated antibodies of HeLa cells producing Myc-Card9 and treated with siRNA.
(F) Immunoblot analysis of endogenous Card9 immunoprecipitated from Hep3B cells treated with DMSO, CK2 inhibitor TBB (4, 20, 100 μM), or Cdk inhibitor 3-Amino-1H-pyrazolo[3.4-b]quinoxaline (100 μM).
(G) Immunoblot analysis with the indicated antibodies of anti-Myc immunoprecipitates derived from HeLa cells transiently transfected to produce Myc-Card9 and treated with lambda phosphatase where indicated.
(H) Immunoblot analysis with the indicated antibodies of anti-HA immunoprecipitates prepared from WT8 or pRC3 cells and incubated with Myc-Card9 in vitro (see methods).
(I) Immunoblot analysis with the indicated antibodies of anti-Card9 immunoprecipitates of mouse liver extracts prepared from VHL flox/flox mice or VHL flox/flox; Cre-ER mice. 2-week old mice were treated with tamoxifen on day 1 and 3 and livers were harvested 1 week later.
The Card9 C-terminus contains multiple serine and threonine residues that resemble CK2 phosphorylation sites. Mass spectrometry analysis of Myc-Card9 recovered from HeLa cells revealed that these sites, including T531 and T533, were phosphorylated in vivo (Figure S3). These sites were also phosphorylated by CK2 in vitro, as determined by mass spectrometry, which coincided with the appearance of the Card9 top band (Figure 3D). Conversely, the top band/bottom band ratio was markedly decreased in HeLa cells transfected to produce epitope-tagged Card9 and treated with siRNA directed against the CK2 catalytic (CK2α) or regulatory (CK2β) subunits (Figure 3E). In addition, the top band of endogenous Card9 in Hep3B cells was eliminated with the CK2 inhibitor TBB but not a Cdk inhibitor (Figure 3F).
To study this further, we generated a polyclonal antibody that recognizes a Card9 peptide phosphorylated on T531 and T533 (Figure S4A). This antibody preferentially recognized the Card9 top band and did not react with exogenous or endogenous Card9 following phosphatase treatment (Figure 3G and Figure S4B). Using this reagent, we confirmed that anti-pVHL immunoprecipitates direct the phosphorylation of these two residues in vitro (Figure 3H) and that this reaction is specifically inhibited by TBB (Figure S4C). Moreover, Card9 phosphorylation was diminished following somatic VHL inactivation in vivo (Figure 3I).
Phosphorylation of the Card9 C-terminus Inhibits Its Ability to Activate NF-κB
To decipher the functional relevance of Card9 phosphorylation by CK2 we created C-terminal Card9 truncation and missense mutants. In pilot experiments, we confirmed that Card9 is an NF-κB agonist (Figure 4A). Deletion of the Card9 C-terminus, and hence all of the Card9 C-terminal CK2 sites, eliminated the top band, as expected (Figure 4B). Moreover, deletion of the Card9 C-terminus markedly increased its ability to enhance NF-κB activity (Figure 4B) (note different y-axes in Figure 4A and Figure 4B). Elimination of 4 C-terminal CK2 sites within Card9, alone or in combination, by alanine substitutions also increased reporter activity and replacement of all 4 sites with alanines markedly decreased top band formation (Figure 4C). Likewise, the Card9 phosphosite mutants were more potent than wild-type Card9 with respect to the induction of the NF-κB target gene TNFα (Figure 4D) and NF-κB DNA-binding activity (Figure S5A). Consistent with this latter observation, NF-κB DNA-binding activity was also increased in cells treated with a CK2 inhibitor (Figure S5B). These results indicate that phosphorylation of the Card9 C-terminus impairs its ability to activate NF-κB.
Figure 4. Phosphorylation of the Card9 C-terminus by CK2 inhibits NF-κB activity.

(A) Normalized luciferase values of 293T cells transfected with an NF-κB-responsive luciferase reporter plasmid and plasmids encoding GFP, Card9, or an NF-κB antagonist (DN-IκBα). Error bars = 1 std. dev.
(B and C) Normalized luciferase values and immunoblot of 293T cells transfected to produce the indicated Card9 mutants along with an NF-κB responsive luciferase reporter. On a lighter exposure the Card9 (1−476) top band/bottom band ratio resembled the other truncation mutants (data not shown).
(D) TNFα mRNA levels, normalized to Actin mRNA, of 293T cells transfected to produce the indicated Card9 mutants (measured by RT-PCR).
Increased NF-κB activity in _VHL_−/− cells requires Card9
In keeping with earlier studies (An et al., 2005; An and Rettig, 2005; Qi and Ohh, 2003), we observed that the activity of artificial and naturally occurring NF-κB responsive promoters was increased in _VHL_−/− renal carcinoma cells relative to their wild-type pVHL counterparts (Figure 5A and B). In keeping with this, the induction of the endogenous NF-κB targets TNFα and IκBα by the NF-κB agonist TNFα was much more pronounced in _VHL_−/− cells compared to their wild-type counterparts (Figure 5C and D). The changes in IκBα mRNA were mirrored by changes in the accumulation of IκBα protein (Figure S6), which again supports the earlier conclusion of others that endogenous NF-κB activity is enhanced in _VHL_−/− cells. Likewise, a number of NF-κB target genes were transcriptionally activated after conditional inactivation of VHL in VHL flox/flox mice (Haase et al., 2001) that ubiquitously express a Cre-ER fusion protein (Vooijs et al., 2001) (Table 1). These effects were seen in liver (Table 1) but not spleen (data not shown), suggesting that regulation of Card9 and NF-κB by pVHL is tissue-specific. Similarly, endogenous NF-kB targets were activated in livers of VHL flox/flox mice expressing Cre recombinase under the control of the Albumin promoter (Haase et al., 2001; Kim et al., 2006) (Figure S7).
Figure 5. Increased NF-κB activity in _VHL_−/− Renal Carcinoma Cells Requires Card9.

(A and B) Normalized luciferase values of WT8 and pRC3 cells transfected with artificial (A) or naturally-occurring (B) NF-κB -responsive reporter. IL-6 = Interleukin 6 promoter. Mut-Luc = same reporter with NF-κB site mutated. In (A) cells were treated with the NF-κB agonists TPA or transfected to produce the NF-κB agonist LMP1, where indicated. Error bars = 1 std. dev.
(C and D) TNFα and IκBα mRNA levels, normalized to Actin mRNA, in indicated cell lines stimulated with 10ng/ml TNFα for indicated time (measured by RT-PCR). EV = empty vector. SCR = scrambled shRNA.
(E and F) Normalized luciferase values of WT8 and pRC3 cells (E) and Hep3B (F) cells transfected with NF-κB-responsive reporter in the presence of the indicated siRNAs. SCR = scrambled siRNA.
Table 1.
Induction of NF-κB targets in liver after VHL inactivation.
The increased NF-κB activity observed in _VHL_−/− renal carcinoma cells is, at least partly, due to Card9 because it was effectively neutralized with several independent Card9 siRNAs/shRNA (Figure 5C-E and Figures S8 and S9). The behavior of the Card9 shRNA in these assays was similar to that of an siRNA directed against Bcl10, which together with its partner Card9, stimulates NF-κB activity (Figure S10). Similarly, VHL siRNA increased NF-κB activity in VHL+/+ Hep3B cells, which was partially blocked by coadministration of Card9 siRNA (Figure 5F). This suggests that Card9 acts downstream or parallel of pVHL and supports the idea that increased NF-κB activity in _VHL_−/− renal carcinoma cells reflects a failure to properly phosphorylate Card9.
Increased NF-κB activity in _VHL_−/− renal carcinoma cells is not due to HIF dysregulation
We confirmed the recent finding that HIF2α, at least when artificially overproduced in some cell types, can also activate NF-κB (data not shown) (An and Rettig, 2005). This led us to ask whether NF-κB activation after pVHL loss was primarily due to Card9, HIFα, or both. 786-O cells produce HIF2α but not HIF1α (Maxwell et al., 1999). We identified 3 different HIF2α siRNAs that reduced HIF transcriptional activity in _VHL_−/− pRC3 cells to the levels seen in pVHL-proficient WT8 cells (Figure 6A and data not shown). These HIF2α siRNAs did not, however, diminish NF-κB activity (Figure 6B). ARNT1 is the major DNA-binding partner for HIFα. As expected, HIF transcriptional activity was markedly increased in ARNT1+/+ mouse hepatoma cells (Hoffman et al., 1991), but not in their _ARNT1_−/− counterparts, by VHL siRNA (Figure 6C). In stark contrast, however, NF-κB activation by VHL siRNA was _ARNT1_-independent (Figure 6D). Overproduction of a stabilized version of HIF2α (Kondo et al., 2003) in mouse hepatoma cells activated a HIF-responsive promoter (Figure 6E) but did not activate NF-κB, regardless of ARNT1 status (Figure 6F), which argues against the possibility that NF-κB activation by VHL siRNA in these cells reflects an _ARNT1_-independent function of HIF2α. Finally, we have not observed NF-κB activation in cells in which HIF is activated by hypoxia or hypoxia mimetics (data not shown). Collectively, these results suggest that pVHL inhibits NF-κB activity primarily by promoting the inhibitory phosphorylation of Card9 by CK2.
Figure 6. Increased NF-κB activity in _VHL_−/− Renal Carcinoma Cells is Not Due to HIF Dysregulation.

(A and B). Normalized Luciferase values of VHL −/− pRC3 renal carcinoma cells transfected with a HIF responsive (A) or NF-κB-responsive (B) reporter in the presence of the indicated siRNAs. Error bars = 1 std. dev.
(C-F). Normalized Luciferase values of mouse hepatoma cells (ARNT1+/+ or −/−) transfected with HIF responsive (C and E) or NF-κB-responsive (D and F) reporter in the absence or presence of a plasmid encoding stabilized HIF2α (P405A; P531A) (E and F) or the indicated siRNAs (C and D).
Deregulation of Card9 in _VHL_−/− Tumor Cells Leads to Cytokine Resistance and Enhanced Tumorigenesis
Enhanced NF-κB activity protects against TNFα–induced cell death (Beg and Baltimore, 1996; Van Antwerp et al., 1996; Wang et al., 1996) and _VHL_−/− renal carcinoma cells are relatively resistant to this cytokine (Caldwell et al., 2002). Stable downregulation of Card9 in _VHL_−/− renal carcinoma cells with a Card9 shRNA restored their sensitivity to the proapoptotic cytokine TNFα to levels seen in cells producing wild-type pVHL (Figure 7A). TNFα can, itself, induce NF-κB. In order to investigate basal levels of NF-κB, we blocked the induction with cycloheximide, as done by others before (Brummelkamp et al., 2003) (Wang et al., 1996). The effect of the Card9 shRNA was specifically due to Card9 downregulation because it was largely reversed by coexpression of a Card9 cDNA bearing silent mutations that destroy the shRNA recognition sequence without altering the Card9 protein sequence (Figure S11).
Figure 7. Deregulation of Card9 in _VHL_−/− Tumor Cells Leads to Cytokine Resistance and Enhanced Tumorigenesis.
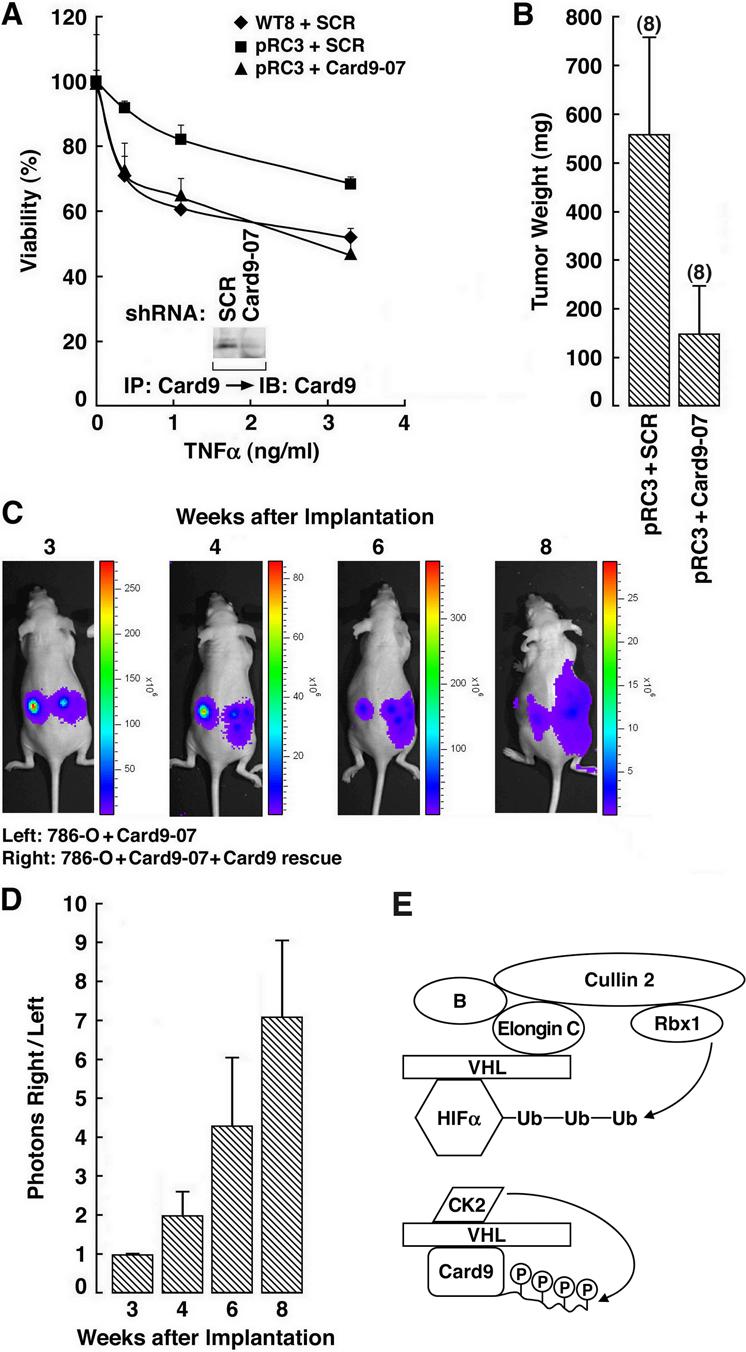
(A) Viability of the indicated cell lines after exposure to TNFα. Error bars = 1 std. dev, (B) Tumor weights ∼15 weeks after orthotopic implantation of pRC3 VHL renal carcinoma cells infected with retroviruses expressing the indicated shRNA in nude mice. Number of tumors analyzed is shown in parentheses. Error bars = one standard error. Error bars = 1 std. dev.
(C) Representative bioluminescent images of nude mice after orthotopic implantation of luciferase-positive 786-O cells expressing a Card9 shRNA in the absence (left) or presence (right) of a rescue retrovirus encoding an exogenous Card9 mRNA that escapes recognition by the shRNA.
(D) Quantification of data in (C) (right/left) from five mice. Error bars = one standard error.
(E) Model for Regulation of HIF and Card9 by pVHL. For simplicity pVHL is shown binding directly to Card9. Whether pVHL binds directly to Card9, or requires an accessory protein, is not known.
Next we performed orthotopic xenograft assays with _VHL_−/− kidney cancer cells expressing either a scrambled shRNA or Card9 shRNA. Downregulation of Card9 substantially decreased tumor growth (Figure 7B). The Card9 rescue cDNA described above nullified the effect of the Card9 shRNA, indicating that the observed suppression of tumor formation was due specifically to downregulation of Card9 and not an off-target effect (Figure 7C and D).
Discussion
We found that pVHL associates with the NF-κB agonist Card9, which is reported to act in concert with Bcl10 upstream of NF-κB (Bertin et al., 2000; Gross et al., 2006). pVHL does not direct the destruction of Card9. Instead pVHL facilitates the phosphorylation of Card9 by CK2 on C-terminal residues that regulate Card9's ability to activate NF-κB. Card9 mutants lacking these phosphorylation sites are NF-κB superagonists while elimination of Card9 in _VHL_−/− renal carcinoma cells normalizes NF-κB activity and promotes apoptosis and suppresses tumorigenesis. In contrast, deregulation of HIF does not, based on our genetic experiments, appear to play a prominent role in promoting NF-κB activity when pVHL function is compromised.
Our biochemical studies suggest that pVHL, bound to CK2, brings CK2 into proximity with Card9 and thereby increases the local concentration of the substrate. Therefore pVHL can serve as an adaptor for a kinase, CK2, as well as for the ubiquitin conjugating machinery that directs the polyubiquitination of HIFα subunits (Figure 7E). Phosphorylation of the Card9 C-terminus by CK2 might promote an inhibitory intra or intermolecular interaction or prevent the binding of Card9 to an accessory protein required for its activity. In this regard, we have obtained preliminary data that Card9 can interact with NEMO (also called IKKγ)(data not shown), which might conceivably play a role here.
We observed decreased Card9 phosphorylation, and increased NF-κB activity, in mouse livers after VHL inactivation, indicating that regulation of Card9 and NF-κB by pVHL occurs under physiological conditions. To date the role of Card9 in NF-κB activation has been most clearly documented during the myeloid immune response to particular infections (Colonna, 2007). Clearly it will be important to determine the physiological and pathological signals that govern the interaction of pVHL and Card9.
VHL mutations play a causal role in renal carcinomas, which are notoriously resistant to chemotherapy, radiotherapy, and proapoptotic cytokines. Increased NF-κB activity, which has been documented in renal carcinomas before (Oya et al., 2001; Oya et al., 2003), is likely to play a role here (Karin et al., 2002; Nakanishi and Toi, 2005). Indeed, there is already preclinical evidence that suppression of NF-κB in renal carcinomas with interferon alpha, which is widely used in the treatment of this disease, can increase their sensitivity to chemotherapeutic agents or other cytokines (Bukowski, 2000; Steiner et al., 2001). Further elucidation of Card9 regulation and function might reveal additional opportunities to manipulate NF-κB activity in tumor cells.
All renal carcinoma-associated missense VHL mutants are defective for the regulation of HIF but some preserve the ability to regulate Card9 (data not shown). This is consistent with HIF playing a dominant role in _VHL_−/− renal tumors. Nonetheless, many of the VHL mutations found in renal carcinomas are deletion, truncation, or nonsense mutations that would predictably eliminate pVHL function (Gnarra et al., 1996). Our data suggests that, in this setting, deregulation of Card9 would confer a more aggressive phenotype and resistance to therapy.
A number of HIF-responsive genes implicated in renal carcinogenesis, including VEGF, cyclin D1, and MMP2/9, are also NF-κB targets, suggesting that HIF and NF-κB might act cooperatively (Karin et al., 2002; Nakanishi and Toi, 2005). Agents that inhibit HIF-responsive growth factors such as VEGF have demonstrated activity in kidney cancer clinical trials (Kim and Kaelin, 2004) and might now be rationally combined with agents that modulate NF-κB function.
Experimental Procedures
Partial Purification of pVHL-associated Proteins
PC12 cells stably transfected to produce human HA-VHL (generated by Michael Hoffman) were incubated overnight with 10μM MG132 and lysed in EBC buffer (50mM Tris·HCl, [pH8.0], 120mM NaCl, 0.5% Nonidet [NP]-40). Approximately 1 gm of clarified cell extract was resolved by FPLC using a DEAE sepharose column (Amersham-Pharmacia). Aliquots of column fractions were farwestern blotted with recombinant pVHL, elongin B, elongin C complexes as described before (Yang et al., 2004). Fractions enriched for pVHL and HIF1α were pooled, precipitated with 60% ammonium sulfate (final conc.), resolubilized in ∼ 1 ml of EBC, and dialyzed overnight against EBC to remove excess salt prior to immunoprecipitation with ∼50 μl of protein A sepharose crosslinked to 12CA5 (∼1 μg/μl) for ∼5 hours at 4 °C. After 3 washes with NETN (20mM Tris·HCl, [pH8.0], 100mM NaCl, 1mM EDTA, 0.5% NP-40) the bound proteins were eluted with 1 μg/μl HA peptide in PBS buffer. The eluate (total volume ∼ 600 μl) was then precleared by incubation with ∼ 30 μl sepharose cross-linked to control antibody for ∼5 hours at 4 °C. The supernatant was then immunoprecipitated with anti-VHL antibody (IG32) sepharose for ∼5 hours at 4 °C. Bound proteins were eluted by boiling in SDS, resolved by SDS-PAGE, and analyzed by Mass spectrometry at Taplin Mass Spectrometry Center at Harvard Medical School.
Antibodies
Monoclonal anti-HA antibodies (12CA5 and cross-linked 3F10) were from Roche Applied Science. Anti-Myc antibodies (9B11, 9E10) were from Cell Signaling Technology and Zymed respectively. Monoclonal anti-pVHL (IG32) was described before (Kibel et al., 1995). For preparative immunoprecipitation of HA-pVHL complexes 12CA5, 9E10, IG32 were crosslinked to Protein A sepharose (Amersham Pharmacia) using dimethyl pimelimidate (Sigma). Polyclonal Anti-Card9 antibody was from Prosci. Polyclonal anti-CK2α antibodies (BL753, BL754) were from Bethyl Laboratories. Monoclonal anti-CK2β(6D5) and polyclonal anti-HA (Y11) were from Santa Cruz Biotechnology. Monoclonal anti-Card9 (TJ24) was made by the DFCI antibody core facility using mice that were immunized with GST-Card9 (1−200). Polyclonal antibodies recognizing T531T533 Card9 phosphopeptide (524−536) was made by Bethyl Laboratories.
Plasmids
A full length Card9 cDNA clone (NM_052813) was purchased from Origene. The open reading frame was PCR amplified with primers that introduced a 5'BamHI site and 3'EcoRI site to facilitate subcloning into a pcDNA3 plasmid (Invitrogen) that introduced an N-terminal Myc-epitope tag. Truncation and point mutants were made similarly using PCR primers that introduced the desired mutations. All Card9 plasmids were confirmed by DNA sequencing. Plasmids encoding LMP1 and DN-IκBα were gifts from Eliott Kieff and Ballad Dean, respectively. pcDNA3-HIF2α P405A; P531A was described before (Kondo et al., 2003).
Phosphatase Assays
Card9 was immunoprecipitated from EBC cell extracts using polyclonal anti-Card9 (endogenous) or anti-Myc (exogenous) antibodies. Immunoprecipitates were washed 3 times with NETN and once with lambda phosphatase or alkaline phosphatase buffer (provided by the manufacturer). The residual wash buffer was aspirated and the sepharose was resuspended in 100 μl of lambda or alkaline phosphatase buffer containing 400 units or 40 units of the corresponding enzymes. After 1 hour incubation at 30 °C the sepharose was washed once with NETN and bound Card9 was eluted by boiling in SDS-containing sample buffer. Alternatively, the sepharose was washed once with CK2 kinase buffer (20mM Hepes·NaOH, [pH7.9], 20mM MgCl2, 150mM NaCl) and the bound Card9 used as a substrate for in vitro kinase assays (see below). To prepare soluble, dephosphorylated, Myc-Card9, the above procedure was performed using a Catch and Release kit (Upstate Biotechnology) to immunoprecipitate Myc-Card9 and lambda phosphatase. Dephosphorylated Myc-Card9 was eluted according to the manufacturer's instructions.
Chemicals and Recombinant Enzymes
TPA, recombinant CK2, CK2 inhibitor 4, 5, 6, 7-Tetrabromobenzotriazole (TBB), and 3-Amino-1H-pyrazolo[3.4-b]quinoxaline were from EMD-Calbiochem. Alkaline and Lambda phosphatase was from New England Biolabs. TNFα was from R&D Systems.
siRNA
Cells grown in six-well plates were transfected with 200 nM siRNA (except for CK2 siRNA used at 100 nM) using Lipofectamine2000 (for mixtures of plasmids and siRNA oligos) or Oligofectamine (for siRNA alone). SiRNAs were purchased from Dharmacon unless otherwise indicated. Sense strands were as follows: VHLC 5'GCCUGAGAAUUACAGGAGATT3', and VHLD 5'AGACCUGGAGCGGCUGACACTT3', Card9−03 5'CUACGUAAGGACUCCAAGATT3', Card9−06 5'GGAGAAGGAGAUGUUCGAGTT3' (Ambion), and Card9−07 5'GGACUCCAAGAUGUACAAGTT3' (Ambion). HIF2A01 5'GACAAGGUCUGCAAAGGGUTT3', HIF2A02 5'GGAGACGGAGGUGUUCUAUTT3', and HIF2A03 5'CAGCAUCUUUGAUAGCAGUTT3', Bcl10 5'GATGAAGTGCTGAAACTTAGA3'. SCR 5'GCGCGCUUUGUAGGAUUCGTT3' was used as control. siRNA oligos against CK2α (sc-29918) and CK2β (sc-29916) were purchased from Santa Cruz Biotechnology. Mouse VHL siRNA smart pool was purchased from Dharmacon (M-004916−00).
Metabolic Labeling
To detect endogenous complexes ∼20 ml of U937 leukemic suspension cells (∼2 × 107 cells) were starved in Cysteine and Methionine-free media for 1 hour and then labeled with 2mCi [35S] Methionine for four hours (0.2 mCi/ml). The cells were lysed in 3ml EBC buffer, split into 3 aliquots, and immunoprecipitated with control, pVHL or Card9 antibodies. The immunoprecipitates, bound to sepharose, were washed four times with NETN, eluted by boiling in 50 μl 2% SDS for 5 minutes, and transferred to a second tube to which was added 1 ml EBC buffer. Aliquots of the eluate were then reimmunoprecipitated with the indicated antibodies (Figure 1G).
In vitro kinase assays
Hypotonic cell extracts
∼ 20 15cm plates of nearly confluent cells were washed once with PBS and resuspended in 10 ml cold hypotonic lysis buffer (HEB) (20mM Tris·HCl, [pH7.5], 5mM KCl, 1.5 mM MgCl2, 1mM dithiothreitol) by mechanical scraping. The cells were disrupted using a dounce homogenizer with the ‘B’ pestle for 30 to 40 strokes on ice. Crude extract was centrifuged twice (once at 10,000 × g for 10 minutes and then at 10,000 × g for 30 minutes) to remove cell debris and nuclei. Aliquots were flash frozen and stored at −80°C before use (∼3−5 mg/ml as determined by Bradford Assay). 81μl of cell lysate and 9μl of 10X Energy Regenerating System (ERS, 20mM Tris·HCl, [pH7.5], 10mM ATP, 10mM magnesium acetate, 300mM creatine phosphate, 0.5mg/ml creating phosphokinase) was added to 20 μl of protein A sepharose preloaded with dephosphorylated Myc-Card9 (see above) and gently mixed on a platform shaker for two hours at 30°C (Figure 2D).
Recombinant CK2
500 units recombinant E. coli CK2 (α2β2) was added to the 20 μl of protein A sepharose preloaded with dephosphorylated Myc-Card9 (see above) in 100μl CK2 kinase buffer with 4μM ATP and gently mixed on a platform shaker for mixed for two hours at 30°C (Figure 3D).
Immunoprecipitates
WT8 and pRC3 cells (∼2 15cm plates each) were immunoprecipitated with 20μl anti-HA 3F10 beads. The sepharose was washed 3 times with NETN and once with CK2 buffer. The residual wash buffer was aspirated and the sepharose was resuspended in 74μl CK2 kinase buffer to which was added 4μM ATP and 4 μl of soluble, dephosphorylated, Myc-Card9 (see above). At various timepoints aliquots (sepharose and solution) were boiled and resolved by SDS-PAGE (Figure 3H).
Alternatively, HeLa cells were transfected with plasmids encoding HA-VHL and/or Myc-Card9, lysed in EBC buffer, and immunoprecipitated with 20μl anti-HA 3F10 beads. After 3 washes with NETN the residual wash buffer was aspirated and the sepharose was resuspended in 100μl CK2 kinase buffer containing 10μCi [32P] ATP. After 2 hour incubation at 30°C with gentle agitation bound proteins were eluted by incubation at 95°C in 1% SDS for 5 minutes. The eluates were collected, diluted 10-fold, split to two fresh tubes, and immunoprecipitated with anti-HA or anti-Myc. Phosphorylated proteins were resolved by SDS-PAGE and detected by autoradiography (Figure 3A).
Luciferase Assays
Cells grown in six-well plates were transfected with an NF-κB firefly luciferase reporter plasmid [3X-κB-Luc (a gift from Elliot Kieff) or IL-6 promoter (a gift of BCCM/LMBP (Plaisance et al., 1997)) and a renilla luciferase control plasmid (pRL-CMV; Promega) using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions. Twenty-four hours later firefly luciferase activity was measured using the Dual Luciferase Assay System and normalized to renilla luciferase activity. For experiments where siRNA was used the cells were split 1:2 24 hours after transfection and analyzed 24 hours later.
TNFα Killing Curves
10,000 cells/well were plated in 96 well plates. The next day the cells were treated with 9 μg/ml cycloheximide together with increasing amounts (0, 0.37, 1.1, 3.3 ng/ml) of TNFα. Twenty hours later, the media was replaced with 100 μl 0.05% crystal violet in 20% Methanol. After a 20 minute incubation the plates were gently rinsed with water and allowed to air dry. The remaining dye was dissolved with 100 μl acetic acid:methanol (1:3) and measured based on absorbance at 590 nm.
Real Time RT-PCR
Cell Lines
Total RNA was isolated using RNeasy mini kit with on-column DNase digestion (Qiagen). First-strand cDNA was generated using StrataScript First-Stand Synthesis System (Statagene). Real-time PCR was performed in triplicate using QuantiTect SYBR Green PCR master mix (Qiagen) and the Mx3000P QPCR system (Stratagene). Primers for TNFα and Iκbα were purchased from Qiagen (Cat No. QT01079561 and QT00014266, respectively). All values were normalized to the level of Actin mRNA abundance.
Tissues
Three week old mice of indicated genotypes were injected with 2 mg Tamoxifen intraperitoneally weekly for 3 weeks. RNA was extracted using Trizol and chloroform followed by RNeasy mini kit (Qiagen). 2 μg of RNA was used to generate the first strand cDNA using a SuperArray kit (SuperArray Bioscience), which was analyzed by real-time RT-PCR using the SuperArray's RT2 Profiler PCR Array Mouse NF-κB Signaling Pathway plates according to the manufacturer's instructions.
Orthotopic xenograft tumor growth assays and bioluminescence
Mice were anesthetized with ketamine hydrochloride and Xyla-Ject. A 2 cm incision was made along the middle of the mouse. After the kidney was exposed, 2×106 viable cells were injected near the lower pole into the renal parenchyma. After both kidneys were injected, wounds were closed with absorbable suture or autoclip (Becton Dickinson) and subsequent renal tumors were monitored by palpation. The mice were sacrificed ∼15 weeks after injection. Tumor mass was calculated by subtracting the mass of the normal kidneys from the mass of the injected kidneys. Results are presented as mean ± standard error of the mean (SEM).
For bioluminescent detection and quantification of cancer cells noninvasively, mice were given a single i.p. injection of a mixture of luciferin (50 mg/kg) ketamine (150 mg/kg) and xylazine (12 mg/kg) in sterile PBS buffer. Fifteen minutes later, mice were placed in a light-tight chamber equipped with a charge-coupled device IVIS imaging camera (Xenogen, Alameda, CA). Photons were collected for a period of 1–60 s, and images were obtained by using LIVING IMAGE 2.60.1 software (Xenogen) and quantified using IGOR Pro 4.09A image analysis software (WaveMatrics, Lake Oswego, OR). The total photons from the right were divided with the total photons from the left, and the number at week three was arbitrarily set as 1. The relative ratios of tumor mass at other time points were derived by dividing the calculated numbers with the above number at week three, and the results were presented as mean ± standard error of the mean (SEM).
Acknowledgements
We thank Volker Haase for the VHL flox/flox mice and Eliot Kieff for NFkB reporter plasmids. Supported by grants from the NIH and the Murray Foundation. W.G.K. is an HHMI Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
01
References
- An J, Fisher M, Rettig MB. VHL expression in renal cell carcinoma sensitizes to bortezomib (PS-341) through an NF-kappaB-dependent mechanism. Oncogene. 2005;24:1563–1570. doi: 10.1038/sj.onc.1208348. [DOI] [PubMed] [Google Scholar]
- An J, Rettig MB. Mechanism of von Hippel-Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol Cell Biol. 2005;25:7546–7556. doi: 10.1128/MCB.25.17.7546-7556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Bertin J, Guo Y, Wang L, Srinivasula SM, Jacobson MD, Poyet JL, Merriam S, Du MQ, Dyer MJ, Robison KE, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J Biol Chem. 2000;275:41082–41086. doi: 10.1074/jbc.C000726200. [DOI] [PubMed] [Google Scholar]
- Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O'Rourke D, Pugh CW, Gleadle JM, Taylor MS, Hodgkin J, et al. Genetic analysis of pathways regulated by the von hippel-lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2:e289. doi: 10.1371/journal.pbio.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- Bukowski RM. Cytokine combinations: therapeutic use in patients with advanced renal cell carcinoma. Semin Oncol. 2000;27:204–212. [PubMed] [Google Scholar]
- Caldwell MC, Hough C, Furer S, Linehan WM, Morin PJ, Gorospe M. Serial analysis of gene expression in renal carcinoma cells reveals VHL-dependent sensitivity to TNFalpha cytotoxicity. Oncogene. 2002;21:929–936. doi: 10.1038/sj.onc.1205140. [DOI] [PubMed] [Google Scholar]
- Calzada MJ, Esteban MA, Feijoo-Cuaresma M, Castellanos MC, Naranjo-Suarez S, Temes E, Mendez F, Yanez-Mo M, Ohh M, Landazuri MO. von Hippel-Lindau tumor suppressor protein regulates the assembly of intercellular junctions in renal cancer cells through hypoxia-inducible factor-independent mechanisms. Cancer Res. 2006;66:1553–1560. doi: 10.1158/0008-5472.CAN-05-3236. [DOI] [PubMed] [Google Scholar]
- Clifford S, Cockman M, Smallwood A, Mole D, Woodward E, Maxwell P, Ratcliffe P, Maher E. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10:1029–1038. doi: 10.1093/hmg/10.10.1029. [DOI] [PubMed] [Google Scholar]
- Colonna M. All Roads Lead to Card9. Nat Immunol. 2007;8:554–555. doi: 10.1038/ni0607-554. [DOI] [PubMed] [Google Scholar]
- Czyzyk-Krzeska MF, Meller J. von Hippel-Lindau tumor suppressor: not only HIF's executioner. Trends Mol Med. 2004;10:146–149. doi: 10.1016/j.molmed.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Duan DR, Weng Y, Humphrey JS, Chen DYT, Lee S, Pause A, Dudley CF, Latif F, Kuzmin I, et al. Molecular cloning of the von Hippel-Lindau tumor suppressor gene and its role in renal cell carcinoma (Review). Biochimica et Biophysica Acta. 1996;1242:201–210. doi: 10.1016/0304-419x(95)00012-5. [DOI] [PubMed] [Google Scholar]
- Golde DW, Hocking WG. Polycythemia: mechanisms and management. Ann Intern Med. 1981;95:71–87. doi: 10.7326/0003-4819-95-1-71. [DOI] [PubMed] [Google Scholar]
- Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, Butman JA, Jedlickova K, Prchal JT, Polyakova LA. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924–3932. doi: 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006 doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- Haase V, Glickman J, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinata K, Gervin AM, Jennifer Zhang Y, Khavari PA. Divergent gene regulation and growth effects by NF-kappa B in epithelial and mesenchymal cells of human skin. Oncogene. 2003;22:1955–1964. doi: 10.1038/sj.onc.1206198. [DOI] [PubMed] [Google Scholar]
- Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- Hoffman M, Ohh M, Yang H, Klco J, Ivan M, Kaelin WJ. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–1027. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Structural basis for the recognition of hydroxyproline in HIF-1alpha by pVHL. Nature. 2002 doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, Gray S, Kaelin WG. Tumor Suppression by the Human von Hippel-Lindau Gene Product. Nature Medicine. 1995;1:822–826. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr von Hippel-Lindau Disease. Annual Review of Pathology: Mechanisms of Disease. 2007;2:145–173. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kibel A, Iliopoulos O, DeCaprio JD, Kaelin WG. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science. 1995;269:1444–1446. doi: 10.1126/science.7660130. [DOI] [PubMed] [Google Scholar]
- Kim WY, Kaelin WG. Role of VHL Gene Mutation in Human Cancer. J Clin Onc. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M, Regan M, Kaelin WG., Jr. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. Embo J. 2006;25:4650–4662. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr. Inhibition of HIF2alpha Is Sufficient to Suppress pVHL-Defective Tumor Growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG,, Jr., et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Lolkema MP, Gervais ML, Snijckers CM, Hill RP, Giles RH, Voest EE, Ohh M. Tumor suppression by the von Hippel-Lindau protein requires phosphorylation of the acidic domain. J Biol Chem. 2005;280:22205–22211. doi: 10.1074/jbc.M503220200. [DOI] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–255. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- Maxwell P, Weisner M, Chang G-W, Clifford S, Vaux E, Pugh C, Maher E, Ratcliffe P. The von Hippel-Lindau gene product is necessary for oxgyen-dependent proteolysis of hypoxia-inducible factor α subunits. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr., Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- Oya M, Ohtsubo M, Takayanagi A, Tachibana M, Shimizu N, Murai M. Constitutive activation of nuclear factor-kappaB prevents TRAIL-induced apoptosis in renal cancer cells. Oncogene. 2001;20:3888–3896. doi: 10.1038/sj.onc.1204525. [DOI] [PubMed] [Google Scholar]
- Oya M, Takayanagi A, Horiguchi A, Mizuno R, Ohtsubo M, Marumo K, Shimizu N, Murai M. Increased nuclear factor-kappa B activation is related to the tumor development of renal cell carcinoma. Carcinogenesis. 2003;24:377–384. doi: 10.1093/carcin/24.3.377. [DOI] [PubMed] [Google Scholar]
- Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103:654–659. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisance S, Vanden Berghe W, Boone E, Fiers W, Haegeman G. Recombination signal sequence binding protein Jkappa is constitutively bound to the NF-kappaB site of the interleukin-6 promoter and acts as a negative regulatory factor. Mol Cell Biol. 1997;17:3733–3743. doi: 10.1128/mcb.17.7.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Ohh M. The von Hippel-Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res. 2003;63:7076–7080. [PubMed] [Google Scholar]
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Steiner T, Junker U, Henzgen B, Nuske K, Durum SK, Schubert J. Interferon-alpha suppresses the antiapoptotic effect of NF-kB and sensitizes renal cell carcinoma cells in vitro to chemotherapeutic drugs. Eur Urol. 2001;39:478–483. doi: 10.1159/000052489. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Xie X, Wu S, Lam KM, Yan H. PromoterExplorer: an effective promoter identification method based on the AdaBoost algorithm. Bioinformatics. 2006;22:2722–2728. doi: 10.1093/bioinformatics/btl482. [DOI] [PubMed] [Google Scholar]
- Yang H, Ivan M, Min J, Kim W, Kaelin W. Analysis of von Hippel-Lindau hereditary cancer syndrome: implications of oxygen sensing. Methods Enzymol. 2004;381:320–335. doi: 10.1016/S0076-6879(04)81022-X. [DOI] [PubMed] [Google Scholar]
- Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01