Up-regulation of BDNF in Astrocytes by TNF-α: A Case for the Neuroprotective Role of Cytokine (original) (raw)
. Author manuscript; available in PMC: 2007 Dec 11.
Published in final edited form as: J Neuroimmune Pharmacol. 2006 May 16;1(3):212–222. doi: 10.1007/s11481-006-9020-8
Abstract
Tumor necrosis factor-alpha (TNF-α) is widely known to be involved in physiological and pathophysiological processes of the brain where this proinflammatory cytokine is implicated with regulation of inflammatory and survival components. We report that TNF-α up-regulates exon-IV-bdnf mRNA and brain-derived neurotrophic factor (BDNF) protein in primary astrocytes. The BDNF protein was detectable both in cellular lysate and in the extracellular medium. Activation of NF-κB by TNF-α and inhibition of TNF-α-induced BDNF expression by Δp65 (a dominant-negative mutant) and NEMO-binding domain peptide (an inhibitor of NF-κB) suggests that TNF-α induces BDNF expression through the activation of NF-κB. Similarly, TNF-α induced the activation of C/EBPβ and the expression of BDNF was sensitive to overexpression of ΔC/EBPβ (a dominant-negative mutant) and ETO (an inhibitor of C/EBPβ). Among three MAP kinases, TNF-α-induced BDNF up-regulation was sensitive only to inhibitors of ERK MAP kinase. However, the ERK MAP kinase pathway was coupled to activation of C/EBPβ but not NF-κB. Taken together, this study identifies a novel property of TNF-α in inducing the expression of BDNF via NF-κB and C/EBPβ in astrocytes that may be responsible for neurotrophic activity of the cytokine.
Keywords: astrocytes, BDNF, TNF-α, NF-κB, C/EBPβ, MAP kinase
Introduction
The brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family and is the most widely distributed trophic factor in the brain (McAllister et al. 1999). It participates in neuronal growth, maintenance, and in different aspects of activity-dependent synaptic physiology by acting across different spatial and temporal domains (Tyler et al. 2002). In the adult central nervous system (CNS), BDNF is expressed by multiple cell types including neurons and glia (Riley et al. 2004). Previous research has established that neuronal activity regulates bdnf transcription, where BDNF mRNA and protein are subsequently transported into neuronal processes followed by activity-dependent secretion of BDNF. However, much of this work has been performed in neurons whereas regulation of bdnf in glia has been studied with lesser enthusiasm. This lacuna in knowledge of glial bdnf regulation drew our attention to this project.
Interestingly, the bdnf gene has four differentially regulated promoter regions, each giving rise to an mRNA containing any one of the four noncoding 5′ exon (I–IV) and a common 3′ exon (V) that codes for mature BDNF protein. It has been suggested that various physiological stimuli may stimulate different 5′ promoter, thus ensuring bdnf regulation to a wide range of stimuli. In neurons, mRNA species containing any one of four 5′ exons have been recorded in response to various stimuli (Murer et al. 2001). In astrocytes, it has been shown that endothelins up-regulate exon-III- and exon-IV-containing transcripts of BDNF mRNA (Koyama et al. 2005). In addition, astroglial BDNF is up-regulated in response to several neuroprotective agents such as SR57746A (Labie et al. 1999), Riluzole (Mizuta et al. 2001), _R_-(−)-1-(benzo-furan-2-yl)-2-propylaminopentane (Ohta et al. 2002), and Levetiracetam (Cardile et al. 2003). Up-regulation of BDNF by these neurotrophic entities suggests a role of astroglial BDNF in undertaking neuroprotective measures during physiological and pathological conditions of the brain.
Contextually, it may be noted that during neurodegenerative diseases, the quantitative presence of BDNF and other neurotrophins decreases with a parallel increase in proinflammatory products (Nagatsu and Sawada 2005). Generally, these proinflammatory elements are considered to aggravate the degeneration process. Among these proinflammatory moieties, tumor necrosis factor-alpha (TNF-α) behaves alternately and is now widely recognized as a double-edged blade possessing both neuroprotective and neurodegenerative properties (Perry et al. 2002; Saha and Pahan 2003). Despite its duality, it is widely recognized as an excellent agent of neuronal preconditioning, where it up-regulates several survival-associated factors in them, thereby promoting their survival (Cheng et al. 1994; Tamatani et al. 1999). However, its effect on glial cells, the major volumetric component of brain, is not well elucidated in this regard. In the current study, we evaluated BDNF expression in rat primary astrocytes in response to TNF-α, where TNF-α signaling leads to up-regulation of exon IV containing bdnf transcript. Transcription factors NF-κB and C/EBPβ play important role in transducing this signal. Furthermore, we demonstrate that ERK MAP kinase pathway regulates TNF-α-induced expression of BDNF via C/EBPβ, but not NF-κB. To our knowledge, this is the first study to demonstrate positive regulation of bdnf by TNF-α in astroglia.
Material and methods
Reagents
Recombinant rat TNF-α and other cytokines were obtained from R&D Systems. Antibodies against BDNF and actin were obtained from SantaCruz Biotechnology. NEMO-binding domain (NBD) NEMO-binding domain peptides were procured from Biomol.
Cell culture
Astrocytes were prepared from rat cerebral tissue by a modified process described previously (McCarthy and de Vellis 1980). Cells were maintained in DMEM/F-12 medium (CellGro) containing 10% fetal bovine serum (Atlas). After 9 days of culture astrocytes were separated from microglia and oligodendrocytes by shaking for 4 h in an orbital shaker at 240 rpm. After 2 days, the shaking was repeated for 24 h before subculturing to ensure the complete removal of all nonastroglial cells, which were subsequently trypsinized, subcultured, and stimulated with TNF-α.
Plasmids and transfection
Reporter plasmids, pBIIX-Luc and C/EBPβ-Luc, have been described previously (Zhong et al. 1997; Jana et al. 2001). Expression vectors for ETO and its mutated form (ETO-AA) are a kind gift from Dr. Stephen O’Rahilly of Cambridge University, UK. These constructs have been described previously (Rochford et al. 2004). Primary astrocytes were transfected with Lipofectamine PLUS®(Invitrogen) as per manufacturer’s protocol. Briefly, each well of 12-well plate was transfected with 0.25 μg DNA complexed with Lipofectamine PLUS in serum and antibiotic-free DMEM/F-12 media. Serum and antibiotics was replaced after 5 h and cells were allowed to grow for 24 h before further experimentation.
RNA isolation and reverse transcriptase-polymerase chain reaction
Total RNA was isolated from astrocytes by using RNA-Easy Qiagen kit following the manufacturer’s protocol. To remove any contaminating genomic DNA, total RNA was digested with DNase. Semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) was carried out as previously described (Dasgupta et al. 2004), by using oligo (dT)12–18 as primer and MMLV reverse transcriptase (Clontech) in a 20-μL reaction mixture. The resulting cDNA was appropriately diluted and then amplified with Titanium Taq polymerase and following exon-specific primers (Zuccato et al. 2001):
| Rat-BDNF-I sense | 5′-GGT GGA TGA GAG TTG AAG CTT GCG A-3′ |
|---|---|
| Rat-BDNF-II sense | 5′-GGA GCG GAG CGT TTG GAG AGC CA-3′ |
| Rat-BDNF-III sense | 5′-CAG GAG TAC ATA TCG GCC ACC A-3′ |
| Rat-BDNF-IV sense | 5′-GGC TTT GAT GAG ACC GGG TTC CCT-3′ |
| Rat-BDNF-V antisense | 5′-GTA GGC CAA GTT GCC TTG TCC GT-3′ |
PCR products were separated on 1.2% agarose horizontal gel and viewed with Fluor Chem 8800 imaging system.
Immunoblotting
Cells were lysed in RIPA buffer [1× phosphate-buffered saline (PBS), 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS with freshly added 0.5% PIC]. The lysate was separated on Tris glycine gels by electrophoresis and were blotted to nitrocellulose membrane via standard Western blotting protocol. Immunoblots were probed with anti-BDNF and signal was detected by chemiluminescence (Perkin-Elmer). The same membrane was stripped and probed for with antiactin antibody in a similar manner.
Assay for BDNF synthesis
Concentration of BDNF was measured in culture supernatants by a high-sensitivity enzyme-linked immunosorbent assay (ELISA; Promega) according to the manufacturer’s instruction.
Immunocytochemistry
Transfected cells in 24-well plates were fixed with methanol at −20°C for 5 min and washed twice with PBS. Cells were then blocked with 3% bovine serum albumin (BSA)–PBS for 1 h at room temperature followed by incubation with anti-BDNF primary antibody in 1% BSA–PBS for 2 h at 37°C. Subsequently, samples were washed thrice with PBS–Tween solution, incubated with Cy5 tagged secondary antibody (Jackson ImmunoResearch) and observed under a BioRad MRC1024ES confocal laser scanning microscope. Negative controls were obtained by treating a set of samples similarly without incubating them with primary antibodies.
Electrophoretic mobility shift assay
Nuclear extract preparation and electrophoretic mobility shift assay (EMSA) were performed as previously described (Jana et al. 2005) with some modifications. Briefly, oligonucleotide containing the consensus binding sequence for NF-κB (Promega) was radiolabeled with [γ-32P]ATP by using polynucleotide T4 kinase. Labeled probe was purified with chroma spin column (BD Biosciences, San Jose, CA, USA). Nuclear extract (6 μg) was incubated with binding buffer and nonspecific oligonucleotides for 15 min in ice before incubation with labeled probe for another 15 min. For supershift assays, 2 μg antibody was added along with the binding buffer. Subsequently, samples were separated on a 6% polyacrylamide gel in 0.25× Tris–borate–EDTA buffer, which were then dried and exposed to generate autoradiograms.
Luciferase reporter assays
To assay the transcriptional activities of NF-κB and C/EBPβ, cells at 50–60% confluence were transfected with either pBIIX-Luc, an NF-κB-dependent reporter construct, or pC/EBPβ-Luc. All transfections included 50 ng/μg total DNA of pRL-TK (a plasmid encoding Renilla luciferase, used as transfection efficiency control; Promega). After 24 h of transfection, cells were treated with TNF-α for 6 h. Firefly and Renilla luciferase activities were analyzed with Dual-Glo™ Luciferase Assay System (Promega) as per the manufacturer’s protocol.
Results
TNF-α stimulates BDNF expression in primary astrocytes
TNF-α is neurotoxic as well as neuroprotective. Although TNF-α-induced neurotoxic signaling pathways are becoming clear, the molecular basis for its neuroprotective activity is poorly understood. We investigated if TNF-α is capable of inducing the expression of BDNF in astrocytes. Rat primary astrocytes were treated with various doses of TNF-α for 24 h. As shown in Fig. 1A, a TNF-α dose of 10 ng/mL was the most efficient in releasing immunoreactive BDNF in culture media. BDNF release with a higher or lower dose of TNF-α (50 or 1 ng/mL) was comparatively smaller. In addition to extracellular BDNF, we also found a similar pattern in intracellular BDNF level. As revealed via Western blot of whole-cell lysate obtained from astrocytes treated with TNF-α for 24 h, maximum immunoreactivity for BDNF was detected in cells treated with 10 ng/mL TNF-α (Fig. 1B). Thus, this dose was subsequently utilized in most experiments. Similarly, increase in cellular BDNF level was also observed via immunocytochemistry, where anti-BDNF primary antibody was detected with Cy5-conjugated secondary antibody (Fig. 1C). Next, we analyzed the mRNA level of BDNF in TNF-α-treated astrocytes. Because the transcription of BDNF gene may utilize one or more of the four 5′ promoters, we investigated the effect of TNF-α on the expression of BDNF mRNA by RT-PCR by using various exon-specific primers (Zuccato et al. 2001). As evident from Fig. 1D, various doses of TNF-α induced only exon-IV-specific bdnf amplicon of about 270 bp in size. No transcript was recorded with primers specific for exons I, II, or III (Fig. 1D). Furthermore, a time-course of TNF-α treatment was performed, followed by RT-PCR with exon-specific primers to detect the presence of any other exon at any time point. As shown in Fig. 1E, a single species of exon-IV-containing bdnf transcript was detected throughout (negative data for exons I, II, and III not shown). Interestingly, the transcript was detected as early as 3 h posttreatment, and its level was sustained throughout up to 18 h.
Fig. 1.
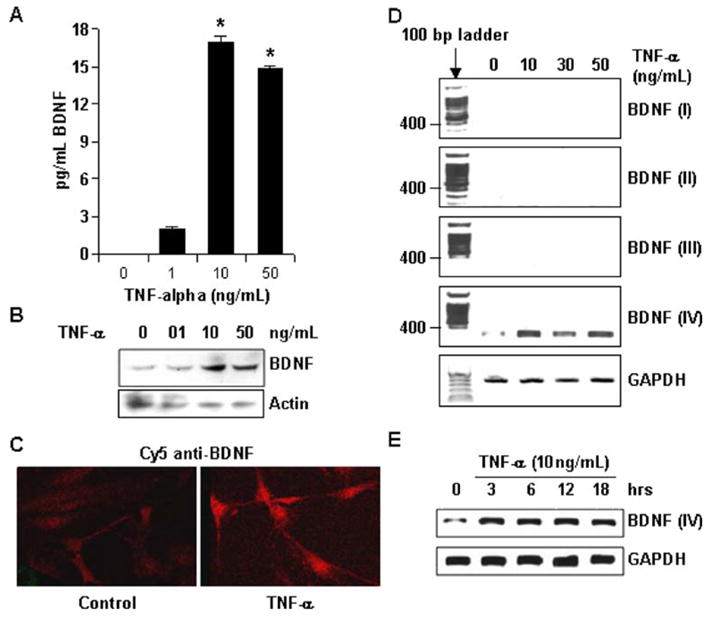
TNF-α induces BDNF in rat primary astrocytes. (A) Primary astrocytes were treated with indicated dose of TNF-α for 36 h and a fraction of culture medium was used to perform ELISA. Data analyzed with Student’s _t_-test and is expressed as mean ± SD of three separate experiments. *p < 0.001 (B) Cells from similar treatment as in (A), but only for 24 h, were used to obtain whole-cell lysate, which were then subjected to electrophoresis in degenerating condition. Subsequently, Western blot was performed and membrane was probed first with anti-BDNF antibody and then antiactin antibody. (C) Cells were also immunostained with anti-BDNF antibody. (D) Total RNA was extracted from primary astrocytes treated with indicated dose of TNF-α for 12 h. Subsequently, RT-PCR was performed with exon-specific primers of BDNF and primers for GAPDH. (E) Total RNA was extracted from primary astrocytes treated with 10 ng/mL TNF-α for the indicated number of hours. Subsequently, RT-PCR was performed with exon-specific primers of BDNF and primers for GAPDH. Only positive data with exon IV is shown.
IL-1β partially shares the BDNF-inducing role of TNF-α among several proinflammatory molecules
Similar to TNF-α, many other proinflammatory molecules associated to various pathological conditions are capable of stimulating astrocytes. We investigated if other proinflammatory molecules are also capable of inducing the expression of BDNF in astrocytes. Therefore, in addition to TNF-α, astrocytes were also treated with IL-1β, IFN-γ, LPS, and polyinosinic–polycytidylic acid (poly-IC, a synthetic dsRNA) in doses at which these compounds have been previously recorded to initiate a proinflammatory response in astrocytes (Auch et al. 2004; Jana et al. 2005). As shown in Fig. 2A, only TNF-α (but not IL-1β, IFN-γ, LPS, and poly-IC) was capable of inducing the expression of BDNF at mentioned doses. Similarly, only TNF-α treatment resulted in a rise in BDNF protein level, as revealed by ELISA assay in Fig. 2B. It may be noted here that LPS treatment by itself failed to up-regulate BDNF, thus negating any probability that bacterial contaminants in the recombinant TNF-α used in our assays may be responsible for the demonstrated effect.
Fig. 2.
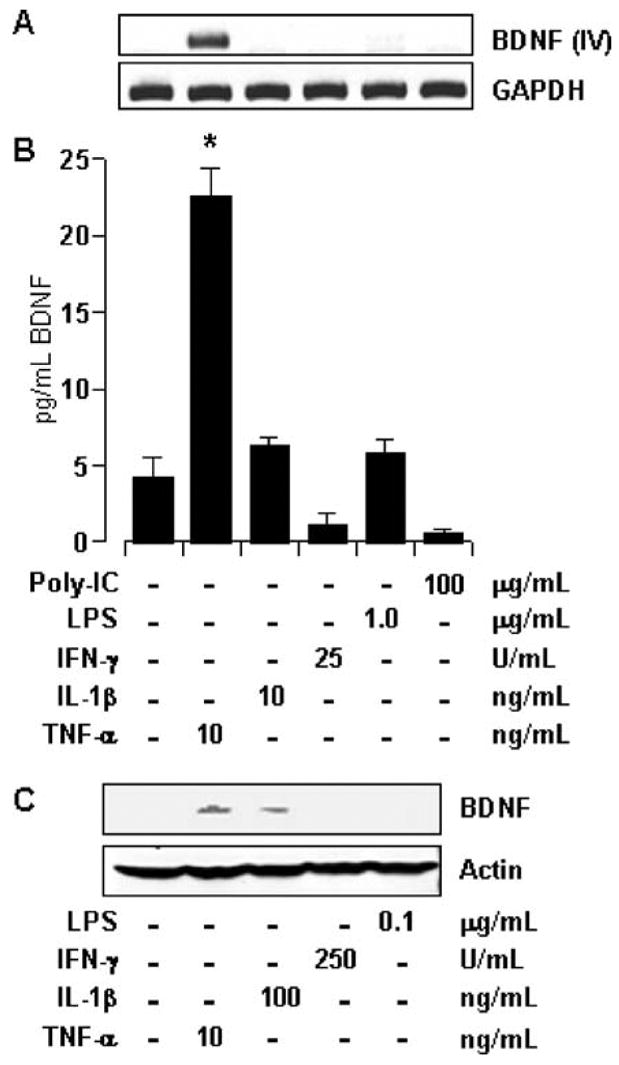
Effect of different proinflammatory molecules on the expression of BDNF. (A) Total RNA was extracted from primary astrocytes treated with indicated dose of different proinflammatory inducers for 12 h. Subsequently, RT-PCR was performed with exon-IV-specific primers of BDNF. (B) Samples of culture media obtained from cell treated as in (A) for 36 h were used to perform ELISA. Results were statistically analyzed with Student’s _t_-test and is expressed as mean ± SD of three separate experiments. *p < 0.001. (C) Whole-cell lysates obtained from cells treated with indicated components for 24 h, were subjected to SDS-PAGE and analyzed by Western blot using antibodies against BDNF and actin.
Because the ED50 of TNF-α is lower than that of IL-1β, we tested if IL-1β could induce BDNF at a higher dose. As shown in Fig. 2C, a 10× higher dose of IL-1β induced a detectable BDNF band in the Western blot of whole-cell lysate. However, IL-1β was less potent than TNF-α in inducing the expression of BDNF (Fig. 2C). In contrast, other proinflammatory molecules (like IFN-γ, LPS) failed to induce any such effect at higher or lower doses (Fig. 2C).
TNF-α stimulates BDNF through the classical NF-κB pathway
NF-κB refers to the dimeric transcription factor where the dimer is formed in various combinations from a pool of five subunits (i.e., p50, p52, p65, Rel-B, and cRel) (Li and Verma 2002). TNF-α is the prototypical inducer of NF-κB in several cell types, including astrocytes. To confirm the effectiveness of the system in our hands, we performed EMSA with the nuclear extract of astrocytes treated with TNF-α. Figure 3A shows a notable DNA–protein band in cytokine-treated extract (second lane), which was completely eliminated by preincubation of the binding mixture with anti-p50 and p65 antibodies (third and fourth lanes, respectively). This suggests that astrocytes respond to TNF-α by translocating a homogenous population of classical p65:p50 NF-κB dimer into the nucleus. Subsequently, the involvement of NF-κB in BDNF regulation was tested by pretreating cells with NBD peptide, a specific inhibitor of signal-stimulated NF-κB activation. NBD peptide blocks the association of IκB kinase (IKK) subunits, thereby preventing its activation (May et al. 2000). As indicated in Fig. 3B, both doses of NBD peptide effectively blocked bdnf-IV transcript formation, thereby reflecting the importance of IKK-NF-κB pathway in the process. Further confirmation was obtained by transfecting cells with Δp65 (dominant-negative mutant of p65). Cells transfected with Δp65 displayed significantly smaller amount of bdnf-IV transcript and BDNF protein as revealed by RT-PCR (Fig. 3C) and ELISA (Fig. 3D), respectively. Together, these observations reveal that NF-κB plays a pivotal role in BDNF regulation by TNF-α.
Fig. 3.
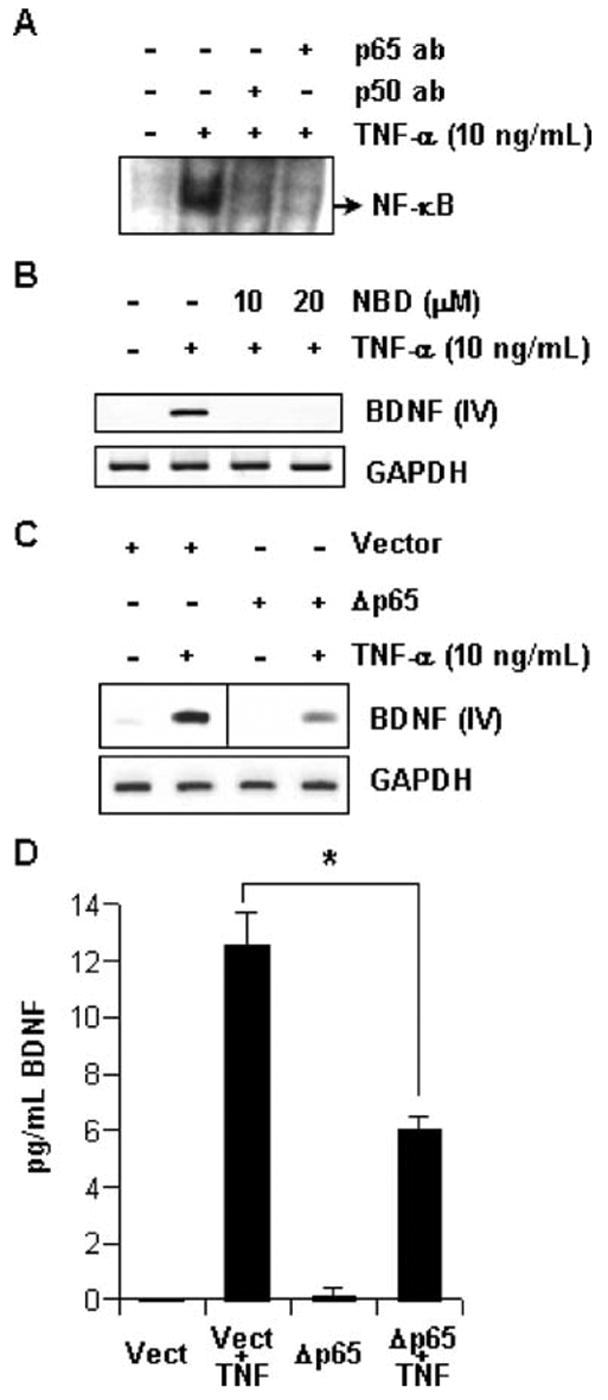
TNF-α induces BDNF via NF-κB. (A) Nuclear extracts were prepared from primary astrocytes treated with 10 ng/mL TNF-α for 60 min. EMSA and supershift assay was performed with labeled NF-κB consensus probe. (B) Primary astrocytes pretreated with indicated dose of NBD peptides for 3 h before treatment with 10 ng/mL TNF-α for 12 h. Subsequently, total RNA was extracted from these cells and RT-PCR was performed with exon-IV-specific primers of BDNF and primers for GAPDH. (C) After 24 h of transfecting primary astrocytes with empty vector or Δp65, these cells were treated with 10 ng/mL TNF-α for 12 h. Subsequently, total RNA was extracted from these cells and RT-PCR was performed with exon-IV-specific primers of BDNF and primers for GAPDH. (D) Samples of culture media were obtained from cells treated as in (C) for 36 h were used to perform ELISA. Data was analyzed by ANOVA statistics and is expressed as mean ± SD of three separate experiments. *p < 0.001.
BDNF up-regulation by TNF-α is contingent on C/EBPβ
Transcription factor C/EBPβ has been previously shown to regulate bdnf-IV promoter in neuronal NG108-15 cells (Takeuchi et al. 2002). We therefore hypothesized that the same transcription factor could be involved in our case. To test this hypothesis, we performed EMSA with C/EBP consensus oligonucleotides. As shown in Fig. 4A, TNF-α treatment induced a significant DNA–protein complex, which was completely eliminated by C/EBPβ antibody. This suggests the involvement of C/EBPβ dimers in the process. To test whether C/EBPβ is involved in BDNF up-regulation, we transfected astrocytes with ΔC/EBPβ, the dominant-negative mutant of C/EBPβ. It is evident from Fig. 4B that ΔC/EBPβ, but not the empty vector, suppressed TNF-α-induced expression of bdnf-IV transcript. To reconfirm the involvement of C/EBPβ in the process, we utilized ETO (MTG8), an endogenous transcriptional corepressor of C/EBPβ (Rochford et al. 2004). Dimeric wild-type ETO is ordinarily localized to the nucleus where it selectively inhibits the activity of C/EBPβ. However, the mutation of K238 and R239 to alanine (ETO-AA) in the nuclear localization sequence of ETO restricts it to cytosol, where its C/EBPβ inhibitory properties are compromised (Rochford et al. 2004). We overexpressed wild-type GFP-ETO and mutated GFP-ETO-AA in astrocytes, and observed the nuclear and cytosolic localization of the respective proteins [Fig. 4C (e,f)]. Subsequently, these cells were subjected to TNF-α treatment. Interestingly, as represented in Fig. 4C(c,g), cells expressing GFP-ETO (arrow), exhibited weaker anti-BDNF signal than adjacent untransfected cells lacking GFP-ETO transfection. However, this restriction in BDNF expression is negated in cells transfected with GFP-ETO-AA construct [smaller arrows in Fig. 4C(d,h)]. Considering the inhibitory effect of ETO on C/EBPβ, these observations strongly indicate the involvement of this transcription factor in regulation of BDNF by TNF-α.
Fig. 4.
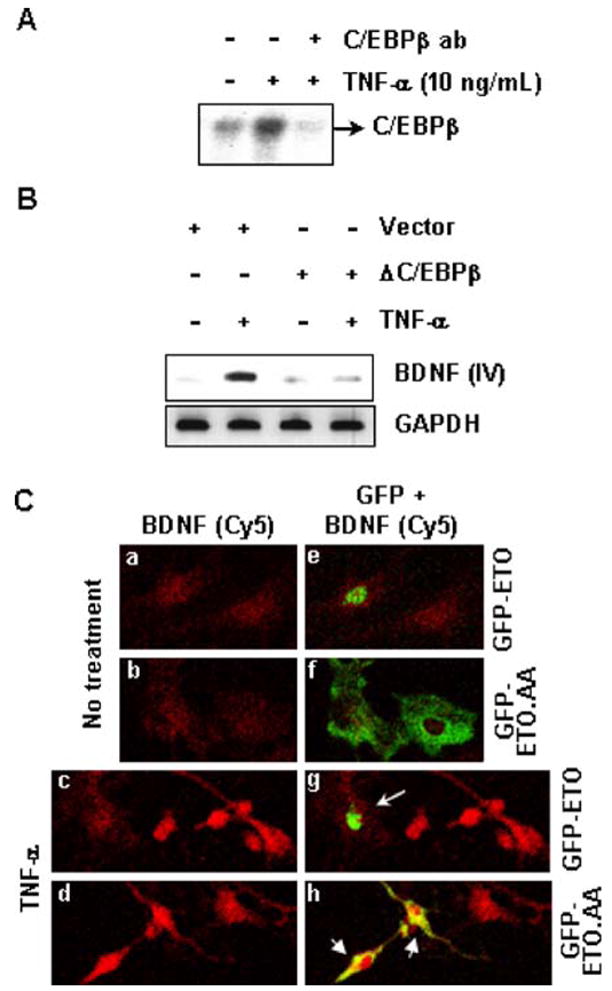
TNF-α induces BDNF via C/EBPβ. (A) Nuclear extracts were prepared from primary astrocytes treated with 10 ng/mL TNF-α for 60 min. This was used to perform EMSA and supershift assay for C/EBPβ. (B) After 24 h of transfecting primary astrocytes with empty vector or ΔC/EBPβ, these cells were treated with 10 ng/mL TNF-α for 12 h. Subsequently, total RNA was extracted from these cells and RT-PCR was performed with exon-IV-specific primers of BDNF and primers for GAPDH. (C) After 24 h of transfecting primary astrocytes with GFP-ETO or GFP-ETO-AA, these cells were treated with 10 ng/mL TNF-α for 24 h. Subsequently, cells were fixed and observed under microscope. Frames on the left panel represent Cy5 channel (red) signal of anti-BDNF immunoreactivity and are overlaid on the right panel with Cy2 channel (green) autosignal of GFP-ETO or GFP-ETO-AA. Note the nuclear or nonnuclear localization of ETO and ETO-AA, respectively (green in “g” and yellow in “h” due to overlay with Cy5 red signal). Frames represent sections of microscopic fields observed at 40× in 1:1 ratio.
BDNF regulation by TNF-α is sensitive to inhibitors of MEK-ERK pathway
Signals from TNF-α receptors converge to induce two lines of signal transduction, the NF-κB pathway and mitogen-activated protein (MAP) kinase pathways (Gaur and Aggarwal 2003). Several studies have now identified the involvement of all three MAP kinase (i.e., JNK, ERK, and p38) pathways in astrocyte signal transduction. To delineate their involvement in bdnf regulation by TNF-α, we pretreated astrocytes with Jun kinase inhibitor (JNKi), PD98059 (a specific pharmacological inhibitor of ERK), and SB203580 (a specific pharmacological inhibitor of p38) prior to cytokine treatment. Subsequently, RT-PCR was performed with RNA obtained from these cells. As presented in Fig. 5A, bdnf-IV expression remained largely unaffected by inhibitors of JNK and p38, but was sensitive to PD98059, suggesting the involvement of ERK MAP kinase in the process. This injunction was further confirmed by utilizing U0126, another specific pharmacological inhibitor of MEK, which is the upstream kinase of ERK. As shown in Fig. 5B, U0126 also inhibits the expression of bdnf-IV in a dose-dependent manner. Furthermore, ELISA was performed to assess any quantitative alteration in secreted BDNF level. Figure 5C demonstrates that TNF-α-induced BDNF secretion was significantly down-regulated by both U0126 and PD98059 in a dose-dependent manner. Taken together, among three MAP kinase pathways, these observations suggest the exclusive commitment of MEK-ERK pathway in BDNF regulation by TNF-α.
Fig. 5.
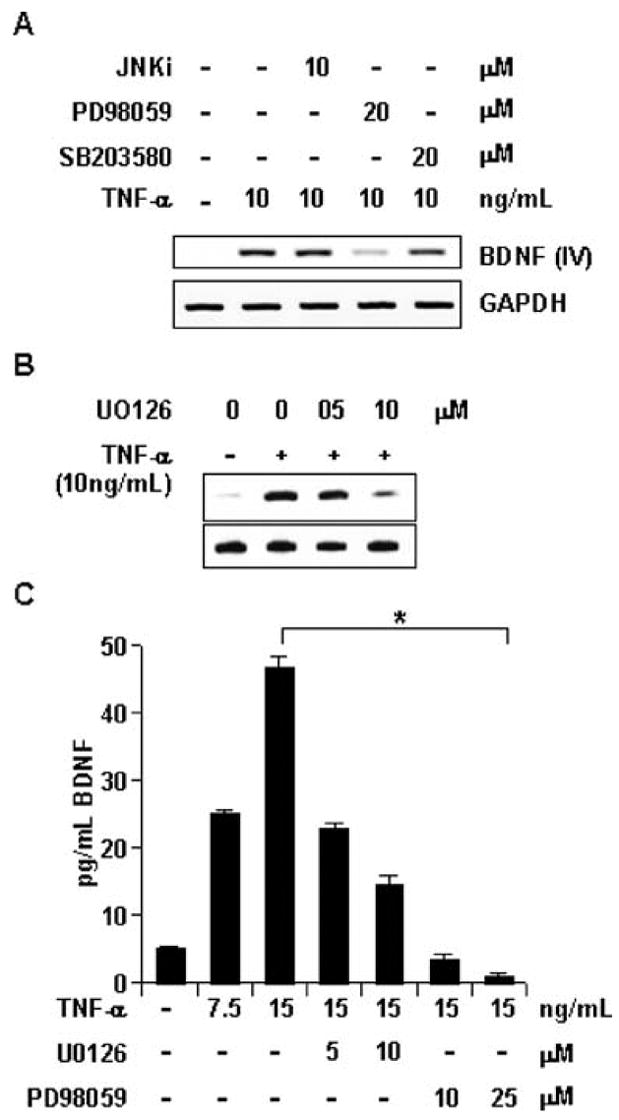
TNF-α induces BDNF via MEK-ERK pathway. (A) Primary astrocytes pretreated with indicated doses of JNKi peptide, PD98059, and SB203580 for 1 h were treated with 10 ng/mL TNF-α for 12 h. Then total RNA was extracted from these cells and RT-PCR was performed with exon-IV-specific primers of BDNF and primers for GAPDH. (B) Similar study as in (A), except that astrocytes were pretreated for 1 h with indicated doses of U0126. (C) Primary astrocytes pretreated with indicated dose of U0126 and PD98059 for 1 h were treated with 10 ng/mL TNF-α for 36 h. Subsequently, samples of culture media were obtained from these cells and used to perform ELISA. Data was analyzed by Student’s _t_-test and is expressed as mean ± SD of three separate experiments. *p < 0.001.
MEK-ERK pathway regulates transcriptional activity of C/EBPβ, but not NF-κB
To correlate the activation of transcription factors (NF-κB and C/EBPβ) with the MEK-ERK pathway, we next scrutinized the effect of PD98059 and U0126 on NF-κB and C/EBPβ activation by using NF-κB- and C/EBPβ-driven luciferase reporter constructs. Interestingly, neither PD98059 nor U0126 had any effect on NF-κB transcriptional activity (Fig. 6A). In contrast, cells pretreated with these inhibitors demonstrated a significant reduction in transcriptional activity of C/EBPβ (Fig. 6B). These observations suggest that TNF-α-induced astroglial activation of C/EBPβ, but not NF-κB, is dependent on the MEK-ERK cascade.
Fig. 6.
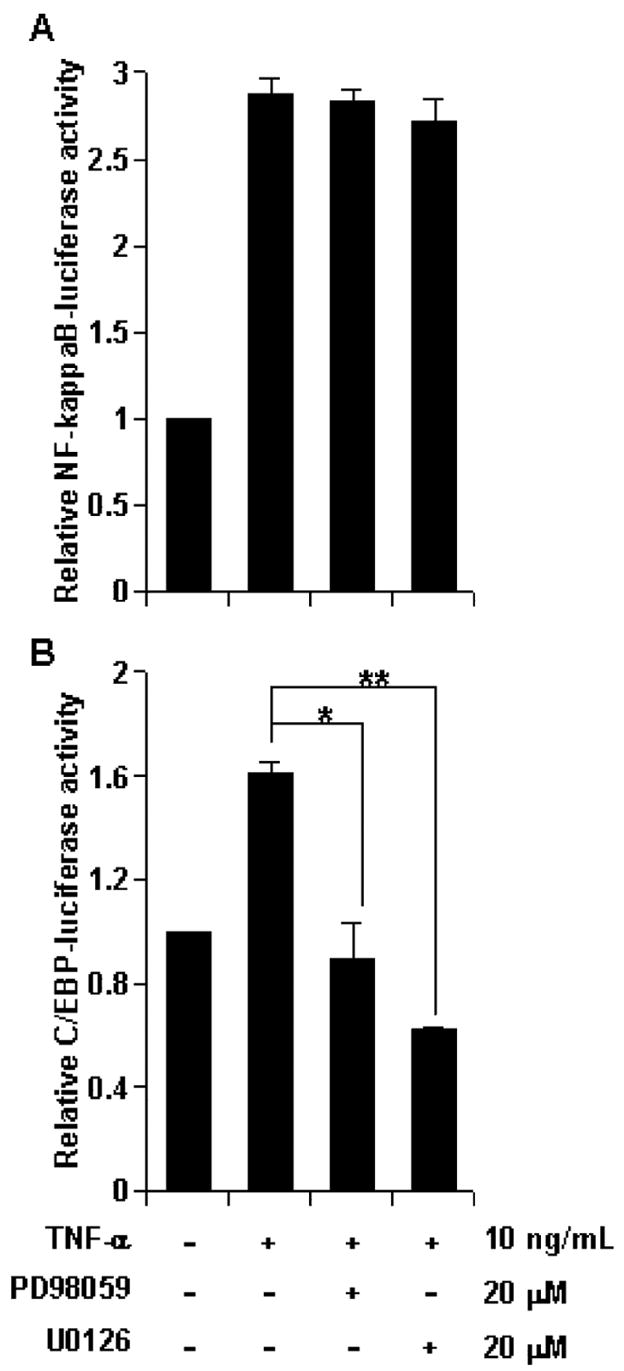
MEK-ERK pathway regulates the activation of C/EBPβ but not NF-κB. Primary astrocytes were cotransfected with Renilla reporter plasmid and either (A) NF-κB-Luc or (B) C/EBP-Luc reporter. After 24 h, cells treated with 10 ng/mL TNF-α for 6 h and dual Renilla/Luciferase assay was performed. Data obtained as ratio of Luciferase/Renilla activity is represented in a relative scale. Data were analyzed by Student’s _t_-test and is expressed as mean ± SD of three separate experiments. *p < 0.01, **p < 0.001.
Discussion
Proinflammatory cytokines are considered neuroinflammatory and neurotoxic agents. Despite being a proinflammatory cytokine, TNF-α is regarded as a neurotoxic as well as a neuroprotective agent (Perry et al. 2002; Saha and Pahan 2003). It has been proposed that this cytokine acts as a neurodegenerative agent by silencing survival signals in neurons (Venters et al. 2000). However, contrasting observations in vitro and in vivo are present in the literature that suggest a neuroprotective role for this cytokine, where it has been shown to precondition neurons to resist neurotoxins or injury (Cheng et al. 1994; Barger et al. 1995; Bruce et al. 1996; Bruce-Keller et al. 1999; Heldmann et al. 2005), and assist in their repair or recovery processes after injury (Schwartz et al. 1991; Arnett et al. 2001). Such a pleotrophic and diametric role of TNF-α may depend on, among other things, the involved cell type (Saha and Pahan 2003). In lieu of such a consideration, the effect of TNF-α on astrocyte is considered primarily proinflammatory in nature (resulting in astrogliosis). However, as TNF-α is neuroprotective in vivo, deleterious proinflammatory products should not be the exclusive consequence of astroglial activation by this cytokine. Logically, therefore, expression of proinflammatory products must be balanced with expression of neurotrophic elements in order for TNF-α to be able to protect from neuronal injury and death in vivo. However, such mechanisms are still not well understood. In this work, we report that TNF-α is capable of inducing the expression of neurotrophin BDNF from astrocytes.
There are several reports in the literature suggesting that astrocytes are capable of BDNF production in response to various stimuli (Miklic et al. 2004; Sasaki et al. 2004; Toyomoto et al. 2004; Wu et al. 2004; Aharoni et al. 2005). One of these studies (Miklic et al. 2004) reported the neutral effect of TNF-α on astroglial BDNF regulation. Contrarily, the present study provides strong evidence to prove that BDNF is up-regulated by TNF-α in astrocytes. The difference in reports between these two studies may be attributed to experimental limitations. Although the current study investigates the event with various experimental techniques (RT-PCR, ELISA, Western blot, and immunocytochemical detection), the former study relied on solitary enzyme immunoassays for their inference.
In addition to highlighting the up-regulation of BDNF by TNF-α, the current study also delineates the signaling mechanism involved in the process. TNF-α treatment, presumably via receptor engagement, activates two nonoverlapping pathways that finally activate two transcription factors, NF-κB, and C/EBPβ (Fig. 7A). TNF-α-induced NF-κB is activated by the canonical pathway involving the IKK complex (Li and Verma 2002). It may be noted that the IKK complex is known to directly interact with TNF receptor1 via TRAF2 and get activated without the involvement of MAP kinase pathways (Saha and Pahan 2003). Therefore, reversal of BDNF up-regulation by NBD peptide (an inhibitor of IKK complex assembly) and over-expression of dominant-negative Δp65 suggest a transcriptional role for NF-κB in regulating the neurotrophin. Because only bdnf-IV was up-regulated by TNF-α among four probable noncoding bdnf exons, we performed a MatInspector® promoter search for probable NF-κB binding sites within the promoter region of rat bdnf-IV (Takeuchi et al. 2002). This search revealed two consensus NF-κB binding sites between −238 to −223 (#1) and −505 to −493 (#2) regions (Fig. 7B). Further investigation is required to reveal the importance of these two κB sites in regulating bdnf-IV.
Fig. 7.
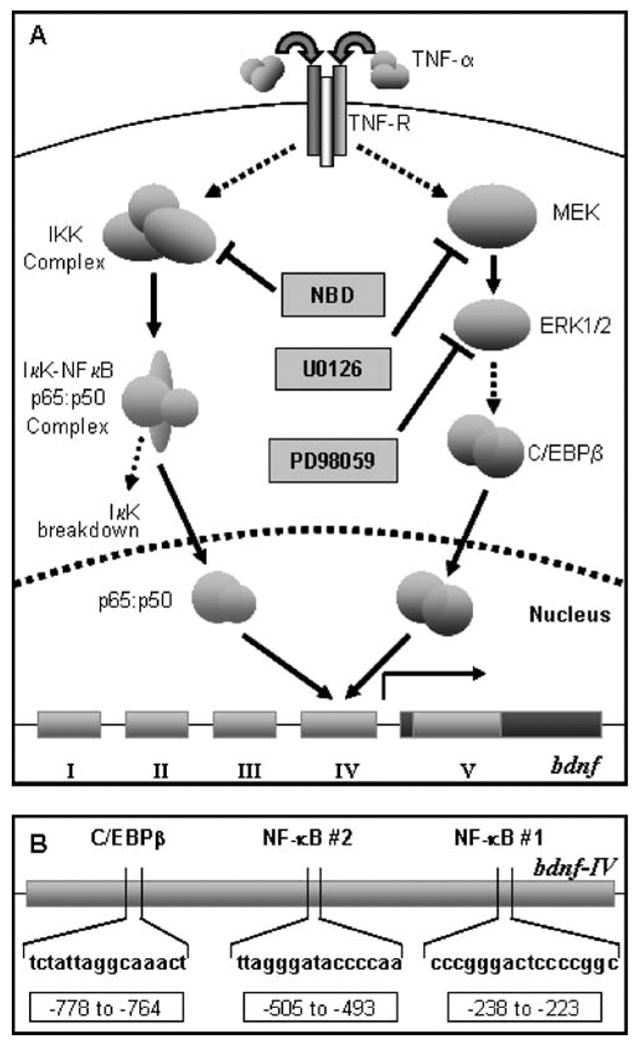
Overview. (A) Diagrammatic representation of signaling pathways elicited in the current article. TNF-α induces NF-κB p65:p50 via IKK and C/EBPβ via the MEK-ERK pathway. Once recruited, these two transcription factors then localize to the nucleus and mediate the activation of bdnf-IV. (B) Representation of potential C/EBPβ and NF-κB binding sites in the bdnf-IV promoter as revealed by MatInspector© analysis.
On the other hand, C/EBPβ is known to act downstream of the MEK-ERK1/2 pathway in several instances in astrocytes (Liu et al. 2002; Jana et al. 2003). Accordingly, restriction of C/EBPβ activity by inhibitors of MEK (by U0126) and ERK1/2 (by PD98059) suggests that TNF-α-induced C/EBPβ activation eventuates downstream of the MEK-ERK1/2 MAP kinase pathway. Furthermore, restricting C/EBPβ activity via overexpression of ΔC/EBP or ETO led to down-regulation of BDNF expression, thereby advocating a direct role for the transcription factor in regulating bdnf-IV. Accordingly, a MatInspector® search of bdnf-IV promoter revealed one probable C/EBP binding site in distal promoter region (−778 to −764) (Fig. 7B). Whether this site is responsible for regulation of TNF-α-induced bdnf-IV is subject to further verification.
In addition to TNF-α, IL-1β also induces both NF-κB and C/EBPβ in astrocytes (Jana et al. 2005). Thus, it is quite pragmatic to expect BDNF induction by IL-1β such as TNF-α. Although it fails to induce BDNF at a lower dose, it does so at a higher dose. Considering the high ED50 for IL-1β, it is perhaps natural for the higher dose to be effective.
Among several neurotrophins in the CNS, BDNF is an important modulator of neuronal function and survival. In addition to their normal physiological function, enhanced expression of BDNF in the CNS after various insults suggests a neuroprotective and neurorestorative role for this neurotrophin. Accordingly, BDNF can rescue degenerating neurons and promote axonal outgrowth, remyelination, and regeneration (Murer et al. 2001; Riley et al. 2004). One basic criterion to fulfill these functions is proposed to be the activity-dependent secretory nature of the neurotrophin, which aids its local diffusion in brain milieu (Thomas and Davies 2005). In our current study, we were able to detect cellular as well as secreted BDNF (by Western blot of whole-cell lysate and ELISA, respectively). Thus, a population TNF-α-induced astroglial BDNF is secreted while the rest is retained in the cell. It is quite imperative to draw a correlation of the situation with neuronal BDNF, where the latter is retained in neurons unless directed by activity for secretion. Potentially, therefore, the secreted population of astroglial BDNF can diffuse and act on neurons, thus aiding their survival, while the cellular BDNF remains in reserve to undertake similar neuroprotective assignments in the future.
In conclusion, our current findings conjure the neuroprotective nature of TNF-α in a new light. TNF-α acts on neurons to up-regulate several survival factors in them. But there is a certain amount of apprehension that this good work is nullified by the proinflammatory effect of TNF-α on glial cells, whereby the proinflammatory products override the survival posts of neurons, leading to their death. However, in this work we present evidence to illuminate a neuroprotective facet of astroglial interaction with TNF-α. Considering the secretory component of TNF-α-induced astroglial BDNF, it is expected that the neurotrophin will bestow neuroprotection as a consequence of TNF-α-mediated stimulation of astrocytes. Taken together, this study suggests a greater functional purview for TNF-α beyond regulation of proinflammatory products, whereby it may mediate neuroprotection indirectly by promoting astroglial BDNF expression in adverse inflamed situations.
Acknowledgments
This study was supported by grants from NIH (NS39940 and NS48923), National Multiple Sclerosis Society (RG3422A1/1), and Michael J. Fox Foundation for Parkinson’s research. The authors would like to acknowledge Marian Schmidt of UNMC for her excellent work in animal handling, Dr. Steve O’Rahilly and Dr. Justin Rochford of the University of Cambridge for ETO constructs, and Avik Roy of Pahan Laboratory for his help in the initial stages of this study.
References
- Aharoni R, Eilam R, Domev H, Labunskay G, Sela M, Arnon R. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci USA. 2005;102:19045–19050. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Auch CJ, Saha RN, Sheikh FG, Liu X, Jacobs BL, Pahan K. Role of protein kinase R in double-stranded RNA-induced expression of nitric oxide synthase in human astroglia. FEBS Lett. 2004;563:223–228. doi: 10.1016/S0014-5793(04)00302-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, Parthasarathy S, Steiner SM, Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol. 1999;93:53–71. doi: 10.1016/s0165-5728(98)00190-8. [DOI] [PubMed] [Google Scholar]
- Cardile V, Pavone A, Gulino R, Renis M, Scifo C, Perciavalle V. Expression of brain-derived neurotrophic factor (BDNF) and inducible nitric oxide synthase (iNOS) in rat astrocyte cultures treated with Levetiracetam. Brain Res. 2003;976:227–233. doi: 10.1016/s0006-8993(03)02720-3. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic–excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. Antineuroinflammatory effect of NF-kappaB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol. 2004;173:1344–1354. doi: 10.4049/jimmunol.173.2.1344. [DOI] [PubMed] [Google Scholar]
- Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66:1403–1408. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- Heldmann U, Thored P, Claasen JH, Arvidsson A, Kokaia Z, Lindvall O. TNF-alpha antibody infusion impairs survival of stroke-generated neuroblasts in adult rat brain. Exp Neurol. 2005;196:204–208. doi: 10.1016/j.expneurol.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Jana M, Liu X, Koka S, Ghosh S, Petro TM, Pahan K. Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem. 2001;276:44527–44533. doi: 10.1074/jbc.M106771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Dasgupta S, Saha RN, Liu X, Pahan K. Induction of tumor necrosis factor-alpha (TNF-alpha) by interleukin-12 p40 monomer and homodimer in microglia and macrophages. J Neurochem. 2003;86:519–528. doi: 10.1046/j.1471-4159.2003.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Tsujikawa K, Matsuda T, Baba A. Endothelin increases expression of exon III- and exon IV-containing brain-derived neurotrophic factor transcripts in cultured astrocytes and rat brain. J Neurosci Res. 2005;80:809–816. doi: 10.1002/jnr.20512. [DOI] [PubMed] [Google Scholar]
- Labie C, Lafon C, Marmouget C, Saubusse P, Fournier J, Keane PE, Le Fur G, Soubrie P. Effect of the neuroprotective compound SR57746A on nerve growth factor synthesis in cultured astrocytes from neonatal rat cortex. Br J Pharmacol. 1999;127:139–144. doi: 10.1038/sj.bjp.0702545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklic S, Juric DM, Carman-Krzan M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci. 2004;22:119–130. doi: 10.1016/j.ijdevneu.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Kuno S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neurosci Lett. 2001;310:117–120. doi: 10.1016/s0304-3940(01)02098-5. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- Ohta K, Ohta M, Mizuta I, Fujinami A, Shimazu S, Sato N, Yoneda F, Hayashi K, Kuno S. The novel catecholaminergic and serotoninergic activity enhancer R-(−)-1-(benzofuran-2-yl)-2-propylaminopentane up-regulates neurotrophic factor synthesis in mouse astrocytes. Neurosci Lett. 2002;328:205–208. doi: 10.1016/s0304-3940(02)00461-5. [DOI] [PubMed] [Google Scholar]
- Perry SW, Dewhurst S, Bellizzi MJ, Gelbard HA. Tumor necrosis factor-alpha in normal and diseased brain: Conflicting effects via intraneuronal receptor crosstalk? J Neurovirol. 2002;8:611–624. doi: 10.1080/13550280290101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley CP, Cope TC, Buck CR. CNS neurotrophins are biologically active and expressed by multiple cell types. J Mol Histol. 2004;35:771–783. doi: 10.1007/s10735-004-0778-9. [DOI] [PubMed] [Google Scholar]
- Rochford JJ, Semple RK, Laudes M, Boyle KB, Christodoulides C, Mulligan C, Lelliott CJ, Schinner S, Hadaschik D, Mahadevan M, Sethi JK, Vidal-Puig A, O’Rahilly S. ETO/MTG8 is an inhibitor of C/EBPbeta activity and a regulator of early adipogenesis. Mol Cell Biol. 2004;24:9863–9872. doi: 10.1128/MCB.24.22.9863-9872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Tumor necrosis factor-alpha at the crossroads of neuronal life and death during HIV-associated dementia. J Neurochem. 2003;86:1057–1071. doi: 10.1046/j.1471-4159.2003.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Hirabayashi J, Manya H, Kasai K, Endo T. Galectin-1 induces astrocyte differentiation, which leads to production of brain-derived neurotrophic factor. Glycobiology. 2004;14:357–363. doi: 10.1093/glycob/cwh043. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Solomon A, Lavie V, Ben-Bassat S, Belkin M, Cohen A. Tumor necrosis factor facilitates regeneration of injured central nervous system axons. Brain Res. 1991;545:334–338. doi: 10.1016/0006-8993(91)91309-o. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Miyamoto E, Fukunaga K. Analysis on the promoter region of exon IV brain-derived neurotrophic factor in NG108-15 cells. J Neurochem. 2002;83:67–79. doi: 10.1046/j.1471-4159.2002.01096.x. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Thomas K, Davies A. Neurotrophins: a ticket to ride for BDNF. Curr Biol. 2005;15:R262–R264. doi: 10.1016/j.cub.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Toyomoto M, Ohta M, Okumura K, Yano H, Matsumoto K, Inoue S, Hayashi K, Ikeda K. Prostaglandins are powerful inducers of NGF and BDNF production in mouse astrocyte cultures. FEBS Lett. 2004;562:211–215. doi: 10.1016/S0014-5793(04)00246-7. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002;9:224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venters HD, Dantzer R, Kelley KW. A new concept in neurodegeneration: TNFalpha is a silencer of survival signals. Trends Neurosci. 2000;23:175–180. doi: 10.1016/s0166-2236(99)01533-7. [DOI] [PubMed] [Google Scholar]
- Wu H, Friedman WJ, Dreyfus CF. Differential regulation of neurotrophin expression in basal forebrain astrocytes by neuronal signals. J Neurosci Res. 2004;76:76–85. doi: 10.1002/jnr.20060. [DOI] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]