Distinct Roles for the p110α and hVPS34 Phosphatidylinositol 3′-Kinases in Vesicular Trafficking, Regulation of the Actin Cytoskeleton, and Mitogenesis (original) (raw)
Abstract
We have examined the roles of the p85/ p110α and hVPS34 phosphatidylinositol (PI) 3′-kinases in cellular signaling using inhibitory isoform-specific antibodies. We raised anti-hVPS34 and anti-p110α antibodies that specifically inhibit recombinant hVPS34 and p110α, respectively, in vitro. We used the antibodies to study cellular processes that are sensitive to low-dose wortmannin. The antibodies had distinct effects on the actin cytoskeleton; microinjection of anti-p110α antibodies blocked insulin-stimulated ruffling, whereas anti-hVPS34 antibodies had no effect. The antibodies also had different effects on vesicular trafficking. Microinjection of inhibitory anti-hVPS34 antibodies, but not anti-p110α antibodies, blocked the transit of internalized PDGF receptors to a perinuclear compartment, and disrupted the localization of the early endosomal protein EEA1. Microinjection of anti-p110α antibodies, and to a lesser extent anti-hVPS34 antibodies, reduced the rate of transferrin recycling in CHO cells. Surprisingly, both antibodies inhibited insulin-stimulated DNA synthesis by 80%. Injection of cells with antisense oligonucleotides derived from the hVPS34 sequence also blocked insulin-stimulated DNA synthesis, whereas scrambled oligonucleotides had no effect. Interestingly, the requirement for p110α and hVPS34 occurred at different times during the G1–S transition. Our data suggest that different PI 3′-kinases play distinct regulatory roles in the cell, and document an unexpected role for hVPS34 during insulin-stimulated mitogenesis.
Keywords: phosphatidylinositol 3′-kinase, signal transduction, endocytosis, vesicular trafficking, phosphoinositides
Phosphatidylinositol (PI)1 3′-kinases are lipid kinases implicated in a wide range of cellular phenomena (Fruman et al., 1998). PI 3′-kinases have been grouped into three classes based on their use of different phosphoinositide substrates (Vanhaesebroeck et al., 1997). Class I enzymes use PI, PI[4]P, and PI[4,5]P2 as substrates, producing PI[3]P, PI[3,4]P2, and PI[3,4,5]P3. They include the well-studied p85/p110 PI 3′-kinases, which are activated by receptor tyrosine kinases, and the PI3K-γ, which is activated by βγ-subunits from trimeric G proteins (Kapeller and Cantley, 1994; Stoyanov et al., 1995; Stephens et al., 1997). Class II enzymes are larger in size (170–210 kD) and contain a COOH-terminal C2 domain (MacDougall et al., 1995; Molz et al., 1996; Virbasius et al., 1996; Domin et al., 1997). These enzymes use both PI and PI[4]P as substrates, but will not produce PIP3 from PI[4,5]P2. Class III enzymes include the Saccharomyces cerevisiae enzyme VPS34 and its homologues in Schizosaccharomyces pombe, Dictyostelium discoideum, Drosophila, and humans (Herman and Emr, 1990; MacDougall et al., 1995; Volinia et al., 1995; Takegawa et al., 1995; Zhou et al., 1995; Linassier et al., 1997). VPS34 and its homologues are limited to production of PI[3]P from PI. In yeast, VPS34 associates in vivo with a myristylated serine kinase, VPS15 (Stack et al., 1993), and this association is critical for VPS34 function (Stack et al., 1993). The human homologue of VPS15, p150, is also a myristylated protein kinase that binds to hVPS34 in vivo and increases its lipid kinase activity twofold in vitro (Panaretou et al., 1997).
The lipid products of the PI 3′-kinases are not substrates for known phospholipases and appear to function as second messengers (Serunian et al., 1989). PI[3,4,5]P3 may directly activate its effector enzymes, such as calcium-independent protein kinase C isoforms (Toker et al., 1994, 1995). Alternatively, production of 3-phosphoinositides in cellular membranes may recruit signaling molecules that contain specialized lipid-binding domains (Lemmon et al., 1996). Examples include the serine kinase Akt/PKB and its upstream activator the 3-phosphoinositide–dependent kinase-1, which contain Pleckstrin homology domains that bind PI[3,4]P2 and PI[3,4,5]P3 (for review see Toker and Cantley, 1997), and the endosomal protein EEA1, which contains a zinc finger domain that binds to PI[3]P (Stenmark et al., 1996; Patki et al., 1997).
The functions of the different PI 3′-kinases in mammalian cells have been studied using tools that vary significantly in their specificity. Mutant p85 molecules or SH2 domains from p85 produce dominant-negative phenotypes in microinjected or transfected cells (Jhun et al., 1994; Hara et al., 1995), and mutagenesis of p85 binding sites in receptor tyrosine kinases blocks p85/p110 activation (for review see Cantley et al., 1991). However, these approaches do not distinguish between different p110 isoforms which should all bind p85. Less-specific approaches include overexpression of the p110α PI 3′-kinase, which produces PI[3]P, PI[3,4]P2, and PI[3,4,5]P3 and can feed into pathways normally regulated by class I, II, or III enzymes. Similarly, treatment of cells with wortmannin inhibits both the mammalian class I and class III enzymes at low nanomolar doses (Vanhaesebroeck et al., 1997). (Unlike the mammalian enzyme, the yeast VPS34 is relatively insensitive to wortmannin [Schu et al., 1993].) The use of wortmannin is further complicated by the fact that inhibition of mammalian class II PI 3′-kinases requires high nanomolar to micromolar concentrations (Domin et al., 1997). These doses also inhibit PI 4-kinases, and could affect cellular levels of PI[4,5]P2 (Meyers and Cantley, 1997).
The p85/p110 PI 3′-kinases have been implicated in mitogenic signaling, regulation of the actin cytoskeleton, resistance to apoptosis, and trafficking of the Glut 4 glucose transporter (Fruman et al., 1998). In contrast, little is known about the function of class III PI 3′-kinases in mammalian cells. In S. cerevisiae and S. pombe, VPS34-null strains show disruption of vacuolar sorting at permissive and nonpermissive temperatures, and reduced growth at elevated temperatures (Herman and Emr, 1990; Takegawa et al., 1995). The authors suggest that the combined stress of high temperature plus abnormal vacuolar function may inhibit growth at high temperature. Alternatively, they suggest that some vacuolar function might be required for high temperature growth (Takegawa et al., 1995). In D. discoideum, reduced expression of the DdPIK5 homologue leads to reduced growth on bacterial lawns but not in suspension culture (Zhou et al., 1995), suggesting a lysosomal defect that causes a reduced ability to utilize bacteria as food. On the other hand, a complete gene knockout of the D. discoideum VPS34 homologue is lethal, implying that VPS34 may be essential for growth.
Our approach has been to study the intracellular function of distinct PI 3′-kinase by developing isoform-specific inhibitory anti–PI 3′-kinase antibodies. We have focused on the role of p110α and hVPS34, which are wortmannin-sensitive class I and III enzymes, in a number of wortmannin-sensitive responses. We find that insulin-stimulated reorganization of filamentous actin is inhibited by microinjection of antibodies to p110α but not hVPS34. In contrast, antibodies to both enzymes inhibit vesicular trafficking: anti-hVPS34 antibodies interfere with the sorting of endocytosed PDGF receptors, disrupt the localization of the early endosomal protein EEA1, and modestly inhibit transferrin recycling, whereas anti-p110α antibodies strongly inhibit transferrin recycling. Surprisingly, antibodies to hVPS34 as well as p110α inhibit insulin-stimulated DNA synthesis. However, the requirement for p110α and hVPS34 occur at different times during the G1–S transition. These studies represent a first step in the assignment of distinct PI 3′-kinases to the regulation of distinct cellular events.
Materials and Methods
Cells
Growth of GRC-LR+73 cells, an insulin-responsive derivative of CHO cells, has been previously described (Pollard and Stanners, 1979; McIlroy et al., 1997). Hep G2 cells expressing the wild-type PDGF receptor (Valius and Kazlauskas, 1993) were generously provided by A. Kazlauskas (Harvard University, Cambridge, MA) and were grown in DME containing 10% fetal bovine serum. Trvb-1 cells, a CHO cell line expressing the human transferrin receptor (McGraw et al., 1987), were generously provided by T. McGraw (Cornell University School of Medicine, New York, NY) and were grown in α-MEM containing 10% fetal bovine serum.
Antibodies
Anti-hVPS34 antibodies were raised in New Zealand white rabbits against a peptide corresponding to residues 871–887 of the human VPS34 sequence, AVVEQIHKRAQYWRK (Volinia et al., 1995). Antibodies were purified using an affinity column made from the same peptide coupled to CNBr Sepharose (Pharmacia Biotech, Piscataway, NJ). Antibodies for micronjection were dialyzed into phosphate-buffered saline and concentrated to 3 mg/ml. Antibodies to p110α have been previously described (McIlroy et al., 1997). Anti-EEA1 antibodies were purchased from Transduction Laboratories (Lexington, KY).
Antisense Oligonucleotides
Phosphorothioate oligonucleotides were synthesized (Genelink, Thornwood, NY) so as to be anticomplimentary to sequences from hVPS34. AS1: TCCCCCCATCGCACCGTCTGC (based on nucleotides 36–56 in the hVPS34 GenBank/EMBL/DDBJ sequence Z46973). AS2: AAACTTCTCTGCTTCCCCCAT (based on nucleotides 48–68); AS3: TCTGATCCATCTGC-TTCTACA (based on nucleotides 491–511).
Production of Recombinant p110α and hVPS34
Sf-9 cells were infected with recombinant baculovirus for bovine p110α (cDNA provided by M. Waterfield, Ludwig Institute for Cancer Research, London, UK), hVPS34 (virus provided M. Waterfield) or p110β (virus provided by A. Morris, State University of New York at Stony Brook, Stony Brook, NY). After 2 d in culture, the cells were washed in ice-cold PBS and lysed by freeze-thawing in 10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 100 μg/ml aprotinin, 1 μg/ml leupeptin, and 350 μg/ ml PMSF. After removal of particulate material by centrifugation at 12,000 g, the lysates were assayed directly for PI 3′-kinase activity as described below.
In Vitro PI 3′-Kinase Assays
Sf-9 lysates containing recombinant p110α, p110β, or hVPS34 were incubated in the absence or presence of antibodies as described in the text. PI 3′-kinase activity was then assayed using sonicated bovine liver phosphatidylinositol (200 μg/ml), ATP (45 μM), and 10 mM MgCl2 or MnCl2 as previously described (Rordorf-Nikolic et al., 1995). Where indicated, p110α or hVPS34 was first immunoprecipitated from Sf-9 or CHO detergent lysates with anti-p110α or anti-hVPS34 antibodies. The protein A pellets were washed and assayed as described by Ruderman et al. (1990). In some cases, the enzymes were first eluted from the antibody by incubation for 30 min at 30°C in the presence of 100 μM hVPS34-derived peptide (Field et al., 1988).
[35S]Methionine
Cells were incubated for 5 h in cysteine/methionine-free medium containing 1 mCi/ml [35S]cysteine/methionine (Easy-tag, New England Nuclear, Boston, MA). The cells were then washed with phosphate-buffered saline, lysed in under non-denaturing (Yu et al., 1998) or denaturing (Martys et al., 1996) conditions, and then proteins were immunoprecipitated with anti-hVPS34 or anti-p110α antibodies and absorbed to protein A–Sepharose beads. The beads were washed five times in RIPA buffer, followed by an additional four washes in RIPA containing 1 M NaCl. After a final wash in PBS, the proteins were eluted, separated by SDS-PAGE, and then visualized by autoradiography.
Microinjection
Cells were grown on polylysine-coated glass coverslips. GRC-LR+73 cells were transferred to medium containing 1% fetal bovine serum for 48 h before injection. HepG2/PDGF-R cells were incubated in serum-free medium overnight before injection. Microinjections were conducted using an Eppendorf semiautomated microinjection system and needles pulled on a Sutter p-87 micropipette puller. Antibodies (2–4 mg/ml) or oligonucleotides (10 μM) were mixed with nonspecific rabbit IgG in PBS, pH 7.4, to a final antibody concentration of 3 mg/ml. Cells were allowed to recover for 2 h before further manipulation.
Actin Reorganization
GRC-LR+73 cells were injected with control or anti–PI 3′-kinase antibodies as indicated. After a 2-h recovery period, the cells were stimulated with insulin for 7 min, fixed with 3.7% formaldehyde, and stained with FITC anti-rabbit antibodies (to detect injected cells) or rhodamine-phalloidin (Molecular Probes, Eugene, OR), to visualize the actin cytoskeleton (Segall et al., 1996).
EEA1 Staining
Cells were injected with control IgG or anti–PI 3′-kinase antibodies, allowed to recover for 2 h, and then fixed with 3.7% formaldehyde for 20 min on ice. The cells were permeabilized with methanol on dry ice, blocked with 10% goat serum, and stained with FITC anti-rabbit IgG (to detect injected cells) or anti-EEA1 antibodies (Transduction Laboratories). EEA1 staining was visualized using the Renaissance signal amplification kit (New England Nuclear Life Science Products, Boston, MA), according to the manufacturer's instructions.
Transferrin Recycling
Trvb-1 cells were injected with control or anti–PI 3′-kinase antibodies as indicated, and allowed to recover for 2 h. The cells were loaded with Cy3-transferrin for 2 h, washed, and then fixed immediately or after an additional hour in transferrin-free medium as previously described (Martys et al., 1996). The cells were permeabilized with saponin as described (McGraw et al., 1987) and stained with FITC anti-rabbit antibodies to visualize injected cells.
PDGF Receptor Trafficking
PDGF receptor internalization was measured as described by Joly et al. (1994). HepG2/PDGF-R cells were incubated in serum-free medium overnight. The cells were injected with control or anti–PI 3′-kinase antibodies and allowed to recover for 2 h. The cells were then incubated for 70 min on ice with monoclonal anti-PDGF receptor antibody (20 μg/ml final; Calbiochem-Novabiochem, La Jolla, CA) and recombinant human PDGF-BB (20 ng/ml; Calbiochem-Novabiochem). The cells were either fixed immediately or rapidly warmed by immersion in medium at 37°C for 10 min before fixation. The cells were stained with FITC anti–rabbit antibodies to visualize injected cells, or Cy3–anti-mouse antibodies to visualize PDGF receptors.
BrdU Incorporation
After injection, cells were kept in medium containing 1% FBS or stimulated with 100 nM insulin as indicated. When indicated, the cells were injected at various times after insulin stimulation. 16 h after the onset of insulin stimulation, the cells were incubated with 100 μM BrdU for 2 h and fixed in 3.7% formaldehyde. Nuclear DNA was denatured by treating the cells with 4 N HCl for 3 min, and the cells were permeabilized in methanol at −20°C and stained with rhodamine-conjugated anti-BrdU antibody to measure DNA synthesis, and FITC-conjugated anti-rabbit IgG to determine microinjected cells. The percentage of microinjected cells that was positive for BrdU staining was determined. Data from each experiment reflects the counting of ∼100 injected cells per condition. The mean and SEM values were generated by pooling percentages from different experiments, where n = the number of separate experiments.
Results
Characterization of Anti-p110α and Anti-hVPS34 Antibodies
We have previously described antibodies to residues 1054– 1068 of p110α, which inhibit p110α in vitro and in microinjected cells (McIlroy et al., 1997). We also raised antibodies against residues 871–887 at the COOH terminus of the human VPS34 primary sequence. To test the specificity of the antibodies, we labeled three different cell lines for 5 h with a mixture of [35S]methionine and [35S]cysteine. The lines were: a CHO-derived line selected for tight quiescence during serum withdrawal (Pollard and Stanners, 1979) (GRC-LR+73), a CHO line expressing the human transferrin receptor (McGraw et al., 1987) (Trvb-1), and a HepG2 line expressing the human PDGF receptor (Valius and Kazlauskas, 1993). We then lysed the cells under nondenaturing and denaturing conditions, and performed immunoprecipitations with control IgG or the anti-PI kinase antibodies. The anti-p110α antibodies were ineffective under denaturing conditions, but under nondenaturing conditions precipitated a single 110-kD band from all three cell lines (Fig. 1, left). The p85 regulatory subunit of p85/ p110 PI 3′-kinase was not observed in these experiments, presumably because p110α has significantly more cysteine and methionine residues (70, versus 16 for p85) and turns over more rapidly than p85 (Yu et al., 1998). The anti-hVPS34 antibodies was effective under denaturing conditions and precipitated a major specific 100-kD band from all three cell lines (Fig. 1, right). Additional bands minor were observed but were also present after immunoprecipitation with control IgG, and therefore reflect nonspecific interactions.
Figure 1.

Immunoprecipitation of p110α and hVPS34 from metabolically labeled cells. Hep G2 cells expressing the human PDGF receptor, Trvb-1, and GRC-LR+73 cells were labeled for 5 h in medium containing 0.2 mCi/ml [35S]methionine/[35S]cysteine. The cells were lysed as described in the text, and immunoprecipitates were prepared using control IgG, anti-p110α (left) or anti-hVPS34 antibody (right). The samples were washed and then proteins were eluted and separated by SDS-PAGE and visualized by autoradiography. The data is representative of three experiments.
We further characterized the new anti-hVPS34 antibodies. They could immunoprecipitate recombinant human hVPS34 from Sf-9 lysates (Fig. 2 A). The antibody also precipitate a PI 3′-kinase activity from CHO lysates; this activity was manganese dependent, consistent with the ion specificity of hVPS34 (Fig. 2 B) (Volinia et al., 1995). Although we could measure PI kinase activity in the anti-hVPS34 immunoprecipitates, the antibody was in fact inhibitory toward hVPS34. Fig. 2 C shows that the activity of immunoprecipitated recombinant hVPS34 was increased when eluted from antibody–protein A beads by incubation with the antigen peptide. Inhibition of hVPS34 could be directly measured in a soluble in vitro assay with recombinant hVPS34. hVPS34 activity was inhibited 75% by anti-hVPS34 antibodies, but was unaffected by a previously described antibody that binds and inhibits the p110α PI 3′-kinase (Fig. 2 D, left). Importantly, the anti–hVPS34-1 antibody had no effect on the activity of recombinant p110α (Fig. 2 D, right). In contrast, anti-p110α antibody inhibited p110α by 80%. Neither antibody inhibited the activity of recombinant p110β (data not shown). Inhibition of hVPS34 by the anti-hVPS34 antibody was dose dependent (Fig. 2 E).
Figure 2.

Characterization of anti-hVPS34 antibodies. (A) Lysates from Sf-9 cells expressing hVPS34 were immunoprecipitated with anti-hVPS34 antibody or control IgG. Immune complexes were immobilized on protein A beads, washed, and then assayed for PI 3′-kinase activity. (B) CHO cells were lysed and proteins were immunoprecipitated with anti-hVPS34 antibodies. The immune complexes were immobilized on protein A–Sepharose, washed, and then assayed for PI 3′-kinase activity in the presence of 10 mM MgCl2 or 10 mM MnCl2. (C) Recombinant hVPS34 was immunoprecipitated with anti-hVPS34 antibody and immune complexes were absorbed to protein A–Sepharose. The washed beads were incubated in the absence or presence of hVPS34 peptide at 30°C for 30 min and then assayed for PI 3′-kinase activity. (D) Recombinant hVPS34 (left) or p110α (right) was incubated with rabbit IgG, anti-hVPS34 antibody, or anti-p110α antibody (20 μg/ml final) at 4°C for 2 h, and then assayed for PI 3′-kinase activity. (E) Recombinant hVPS34 was incubated with various concentrations of anti-hVPS34 antibody for 2 h at 4°C and then assayed for PI 3′-kinase activity.
Role of p110α and hVPS34 in Insulin-stimulated Membrane Ruffling
We used the isoform-specific antibodies to examine the roles of p110α and hVPS34 in the regulation of the actin cytoskeleton. GRC-LR+73 cells were microinjected with control IgG or anti-PI kinase antibody, incubated in the absence or presence of 100 nM insulin for 7 min, and then fixed. Cells were stained with FITC-labeled anti-rabbit antibodies to identify microinjected cells (Fig. 3, left), or rhodamine-phalloidin, to visualize filamentous actin (Fig. 3, right; asterisks, injected cells). In quiescent cells injected with control IgG, rhodamine-phalloidin–stained cells appeared flat with prominent stress fibers (Fig. 3, A and B). Quiescent cells injected with anti-hVPS34 antibodies (Fig. 3, E and F) or anti-p110α antibodies (Fig. 3, I and J) were similar to the IgG-injected cells in appearance. When IgG-injected cells were stimulated with insulin for 7 min, prominent actin-rich projections appeared at the periphery of the cells (Fig. 3, C and D). These projections appear somewhat blurry, as they extend out of the plane of focus used to visualize the stress fibers. In cells injected with anti-hVPS34 antibodies, insulin-stimulation of actin-rich projections was also apparent (Fig. 3, G and H). However, injection of cells with antibodies against p110α completely blocked the insulin-stimulated appearance of actin-rich projections (Fig. 3, K and L). These data implicate p110α, rather than hVPS34, as critical for insulin-stimulated rearrangement of the actin cytoskeleton.
Figure 3.
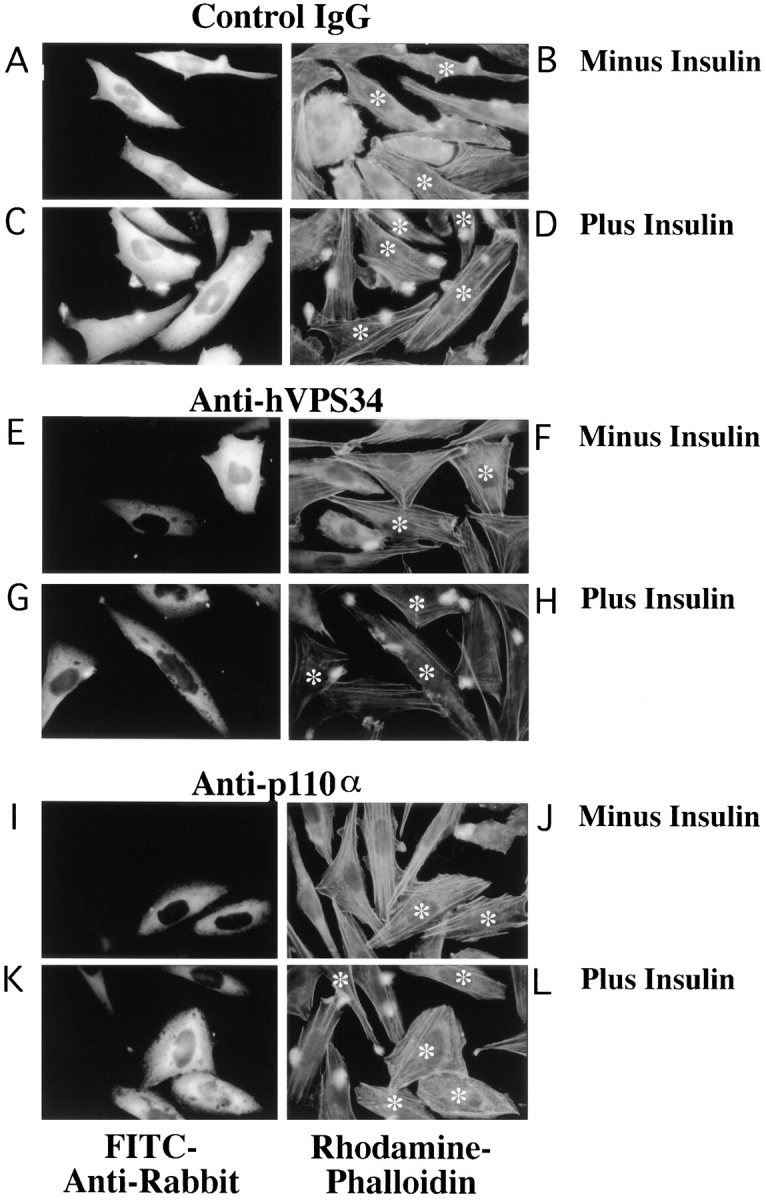
Insulin regulation of the actin cytoskeleton requires p110α but not hVPS34. Quiescent GRC-LR+73 cells were injected with 3 mg/ml rabbit IgG (A–D), anti-hVPS34 (E–H) or anti-p110α (I–L). The cells were fixed in the absence (A, B, E, F, I, and J) or presence (C, D, G, H, K, and L) of 100 nM insulin for 7 min. The cells were fixed and stained with FITC anti-rabbit IgG (left) or rhodamine-phalloidin (right). Asterisk, injected cells on the right. Images were collected using a Nikon Diaphot inverted microscope with Nikon 100× NA 1.25 Achromat optics. The data is representative of two experiments.
Role of p110α and hVPS34 in Early Endosomal Events
Recent data from Corvera, Stenmark, and their colleagues have shown that the early endosomal protein EEA1 is displaced from endosomes by wortmannin and binds specifically to vesicles containing PI[3]P (Patki et al., 1997, 1998; Gaullier et al., 1998). Although the production of PI(3)P in the endosomal membrane could be the result of any of the known PI 3′-kinases, the exclusive production of PI(3)P by hVPS34 would make it a likely candidate. We therefore tested the effects of the inhibitory antibodies on the localization of EEA1. In control Trvb-1 cells or cells injected with rabbit IgG (Fig. 4 A), EEA1 is located in large vesicular structures scattered throughout the cytoplasm. Injection of Trvb-1 cells with anti-p110α antibodies (Fig. 4 B) had little effect on EEA1. In contrast, in cells injected with anti-hVPS34 antibodies, EEA1 was no longer associated with small vesicles but concentrated in large perinuclear structures (Fig. 4 C). This change in the subcellular localization of EEA1 was similar to that seen in Trvb-1 cells treated with 100 nM wortmannin (Fig. 4, D and E). These data suggest that hVPS34 is required for the localization of EEA1, presumably by producing PI(3)P in the early endosomal membrane.
Figure 4.
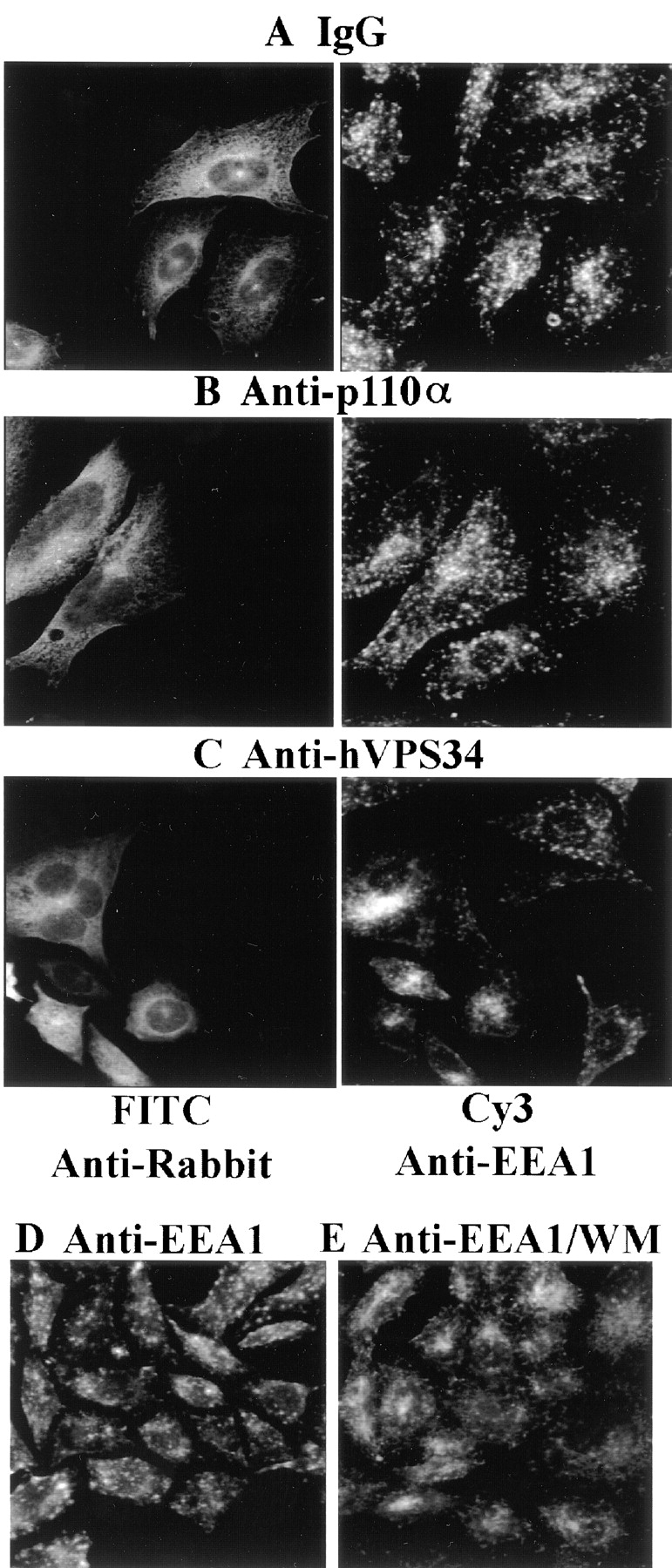
Disruption of EEA1 localization by anti-hVPS34 antibodies. Cells were injected with control IgG (A), anti-p110α (B), or anti-hVPS34 (C) antibodies. After 2 h the cells were fixed and stained with FITC anti-rabbit antibodies to identify injected cells (left) or anti-EEA1 antibodies (right). The Renaissance signal amplification kit (New England Nuclear) was used to enhance detection of EEA1. Images were collected using a Bio-Rad MRC laser scanning confocal microscope (Hercules, CA) with Nikon 60× NA 1.4 Planapo optics (Tokyo, Japan). The data are representative of three experiments. Alternatively, cells were incubated in the absence (D) or presence (E) of 100 nM wortmannin for 30 min, fixed, and then stained for EEA1 as above.
We also examined the postendocytic sorting of internalized PDGF receptors to a perinuclear compartment, which is blocked by wortmannin (Joly et al., 1995). HepG2 cells expressing the wild-type PDGF receptor were incubated at 4°C with mouse anti-PDGF receptor antibodies and 20 ng/ml PDGF, then rapidly warmed to 37°C for 10 min to initiate endocytosis. After fixation, injected cells were visualized with FITC-labeled anti-rabbit antibodies (Fig. 5, left) and PDGF receptors were visualized with Cy3-labeled anti-mouse antibodies (Fig. 5, right; asterisks, injected cells). In cells that were fixed before the 10 min incubation 37°C, anti-PDGF receptor staining was entirely in the plasma membrane and was unaffected by injection with control or anti–PI 3′-kinase antibodies (data not shown). In cells that were warmed to 37°C for 10 min, internalized receptors accumulated in small peripheral vesicles as well as a prominent ring of larger perinuclear vesicles (Fig. 5 B). This perinuclear accumulation of PDGF receptors was not affected by injection with control IgG (Fig. 5, A and B) or anti-p110α antibodies (Fig. 5, C and D). In both cases, the distribution of PDGF receptors in injected cells is the same as that seen in noninjected cells in the same field. In contrast, injection of cells with inhibitory anti-hVPS34 antibodies almost completely blocked the perinuclear accumulation of internalized PDGF receptors (Fig. 5, E and F). The staining in these cells was similar to that seen in cells treated with 100 nM wortmannin (data not shown).
Figure 5.

PDGF receptor trafficking requires hVPS34, but not p110α. HepG2 cells expressing the human PDGF-β receptor were injected with 3 mg/ml rabbit IgG (A and B), anti-p110α (C and D) or anti-hVPS34 (E and F). The cells incubated at 4°C with anti-PDGF receptor antibodies and 20 ng/ml PDGF, warmed rapidly to 37°C for 10 min, and then fixed. The cells were stained with FITC anti-rabbit IgG to labeled injected cells (left) or Cy3–anti-mouse IgG to label PDGF receptors (right). Asterisk, injected cells on the right. The data is representative of six experiments. Images were collected using a Nikon Diaphot inverted microscope with Nikon 100× NA 1.25 Achromat optics.
The effects of inhibitory anti-hVPS34 antibodies on the sorting of PDGF receptors was more clearly seen in cells fixed after 5 min at 37°C (Fig. 6). In control cells or cells injected with rabbit IgG (Fig. 6 A), PDGF receptors could be seen in small vesicles scattered throughout the cytoplasm, as well as large vesicles near the nucleus. Injection of anti-hVPS34 antibodies (Fig. 6 B) does not block internalization of PDGF receptors, but the internalized receptors are located in smaller vesicles that are primarily located at the cell periphery. These data would be consistent with a role for hVPS34 in the fusion and/or maturation of early endosomal vesicles.
Figure 6.

Effects of inhibitory anti-hVPS34 antibodies on PDGF receptor trafficking. HepG2 cells expressing the human PDGF-β receptor were incubated with anti-receptor antibodies and PDGF at 4°C as above, then warmed to 37°C for 5 min. The cells were fixed and stained as in Fig. 5. The data is representative of three separate experiments. Images were collected using a Nikon Eclipse E-400 microscope with Nikon 60× NA 1.4 Planapo optics.
Role of p110α and hVPS34 in Transferrin Recycling
We next examined the recycling of endocytosed transferrin, which is significantly slowed in cells treated with 100 nM wortmannin (Martys et al., 1996; Spiro et al., 1996). Trvb-1 cells were injected with control IgG or anti-PI kinase antibodies, labeled for 2 h with Cy3-transferrin, and washed into transferrin-free medium. We visualized the injected cells with FITC-labeled anti-rabbit IgG (Fig. 7, left) and compared their content of Cy3 transferrin (Fig. 7, right). None of the antibodies affected the steady state labeling of CHO cells, observed after the 2-h incubation with Cy3-transferrin (Fig. 7, A, C, and E); the internalized transferrin accumulated in a diffuse perinuclear compartment as previously described (McGraw et al., 1987; Martys et al., 1996). After 60 min in transferrin-free medium, most of the internalized Cy3-transferrin had effluxed from cells injected with control IgG (Fig. 7 B). Cells injected with anti-hVPS34 antibodies showed a slightly higher level of residual Cy3-transferrin after 60 min (Fig. 7 D). In contrast, cells injected with anti-p110α antibodies showed a persistent labeling with Cy3 after the 60-min incubation in transferrin-free medium (Fig. 7 F), reflecting a delay in the efflux of internalized Cy3-transferrin. These data are similar to those obtained from cells treated with 100 nM wortmannin (Martys et al., 1996), and suggest that p110α is the primary wortmannin-sensitive PI 3′-kinase involved in regulation of transferrin recycling.
Figure 7.

Role of PI 3′-kinases in transferrin receptor recycling. CHO cells expressing the human transferrin receptor (Trvb-1) were injected with 3 mg/ml rabbit IgG (A and B), anti-hVPS34 (C, D, and G) or anti-p110α (E and F). The cells were labeled with Cy3-transferrin for 2 h, and then fixed immediately (A, C, and E) or fixed after a 30- (G) or 60-min (B, D, and F) incubation in transferrin-free medium. The cells were stained with FITC-labeled anti-rabbit IgG to label injected cells (left). Right, Cy3-transferrin. The data is representative of three experiments. Images were collected using a Nikon Eclipse E-400 microscope with Nikon 60× NA 1.4 Planapo optics.
A 60-min chase in transferrin-free medium was chosen to maximize the differences between cells injected with anti–PI 3′-kinase antibodies versus control IgG. Under these conditions, hVPS34 antibodies had only a slight effect. However, when we examined the cells after a 30-min chase in transferrin-free medium, it was clear that hVPS34 antibodies did reduce the rate of transferrin recycling (Fig. 7 G). To summarize, these data show that both anti-p110α and anti-hVPS34 antibodies inhibit recycling. Inhibition of p110α had a greater effect on recycling than inhibition of hVPS34.
Role of p110α and hVPS34 in Insulin-stimulated Mitogenic Signaling
Roche et al. (1994) and ourselves have previously shown that inhibitory antibodies to p110α block mitogen-stimulated DNA synthesis. To compare the role of p110α and hVPS34 in mitogenic signaling, GRC-LR+73 cells were injected with control IgG or anti–PI 3′-kinase antibodies, stimulated with insulin for 16 h and labeled with BrdU. Surprisingly, injection of anti-hVPS34 as well as anti-p110α antibodies inhibited insulin-stimulated DNA synthesis by 70% (Fig. 8).
Figure 8.

Inhibition of insulin-stimulated DNA synthesis by anti-PI kinase antibodies. GRC-LR+73 cells were quiesced in serum containing 1% FBS for 48 h, then injected with 3 mg/ml rabbit IgG, anti-p110α, or anti-hVPS34. The cells were then incubated in the absence or presence of insulin for 16 h plus an additional 2 h in 100 μM BrdU, fixed, and then stained with FITC anti-rabbit IgG to labeled injected cells, or mouse anti-BrdU antibodies and Cy3–anti-mouse antibodies to label cells in S phase. The percentage of injected cells that were labeled with BrdU was calculated; greater than 100 cells were counted per condition. The data are the mean ± SEM from five experiments.
To confirm the unexpected requirement for hVPS34 in mitogenic signaling, we designed three antisense phosphorothioate oligonucleotides derived from NH2-terminal or internal nucleotide sequences from hVPS34 (Materials and Methods). We initially tested these oligonucleotides in HeLa cells, as they were derived from a human sequence and were more likely to work in a human line. HeLa cells were difficult to render quiescent; after 2 d without serum the cells were still 28% BrdU positive, and insulin stimulation caused only a twofold increase in BrdU labeling to 58%. Nonetheless, this increase was completely blocked by injection of anti-hVPS34 antibody, and was reduced by 60–100% by injection of antisense oligonucleotides (data not shown).
We repeated the experiments in GRC-LR+73 cells, which are more easily acquiesced (Pollard and Stanners, 1979). Insulin stimulated DNA synthesis by more than 10-fold. Injection of AS1 and AS2 inhibited this insulin-stimulated DNA synthesis by 80 and 90%, respectively, as compared with a 70% inhibition achieved with anti-hVPS34 antibodies (Fig. 9 A). AS3 was less effective, inhibiting DNA synthesis by ∼50% (Fig. 9 A). To test the specificity of these effects, we synthesized scrambled versions of the more effective AS1 and AS2 oligonucleotides. These were only slightly inhibitory in comparison to the effects seen with the unscrambled oligonucleotides; AS1 and AS2 inhibited DNA synthesis by 87 and 91%, as opposed to 35 and 13% inhibition for their scrambled counterparts (Fig. 9 B). These antisense experiments provide a confirmation of the antibody injection data, using entirely distinct reagents.
Figure 9.

Inhibition of insulin-stimulated DNA synthesis by antisense oligonucleotides to hVPS34. (A) GRC-LR+73 cells were incubated in medium containing 1% FBS for 36 h, injected with 3 mg/ml rabbit IgG, anti-hVPS34 antibody, or rabbit IgG plus 10 μM antisense oligonucleotide (AS-1, AS-2, or AS-3). 8 h later, the cells were stimulated without or with 100 nM insulin for 16 h followed by 2 h in 100 μM BrdU. The cells were fixed and BrdU incorporation in injected cells was analyzed as in Fig. 8. The data are the mean ± SEM from four experiments. (B) Cells were treated as above, but were injected with antisense oligonucleotides (AS-1 and AS-2) or their scrambled counterparts (SCR-1 and SCR-2). Insulin-stimulated BrdU incorporation was measured as above. The data are the mean ± SEM from two (AS-2 and SRC-2) or four (AS-1 and SCR-1) experiments.
To better define the requirement for hVPS34 in the insulin-stimulated G1–S transition, we injected cells with anti-p110α or anti-hVPS34 antibodies before stimulating the cells or at various times after insulin stimulation. Consistent with Roche et al. (1994), we find that p110α is required throughout the first 6 h of insulin stimulation (Fig. 10). By 9 h of insulin stimulation, the cells become largely independent of p110α. Interestingly, the temporal requirement for hVPS34 was different than that for p110α. Although injection of anti-hVPS34 antibodies into cells after 3 h of insulin stimulation inhibited DNA synthesis by over 85%, injection at 6 h inhibited by only 50%, and injection at 9 h inhibited by only 20%. These data suggest that both hVPS34 and p110α are required for the insulin-stimulated G1–S transition. However, the requirement for hVPS34 is limited to an earlier period within G1 than the requirement for p110α.
Figure 10.

Time-dependent inhibition of insulin-stimulated DNA synthesis by anti–PI 3′-kinase antibodies. Quiescent GRC-LR+73 cells were injected with 3 mg/ml rabbit IgG, anti-p110α, or anti-hVPS34 antibodies, and then stimulated with insulin. Alternatively, the cells were injected at various times after insulin stimulation. After 16 h of insulin stimulation and an additional 2-h incubation in 100 μM BrdU, the cells were fixed. BrdU incorporation was calculated as in Fig. 8. The data are the mean ± SEM from three experiments.
Discussion
These data provide new insights into the role of different classes of PI 3′-kinase in distinct cellular events. We have examined a number of cellular responses that have been previously shown to be inhibited by low doses of the PI 3′-kinase inhibitor wortmannin. Our studies have focused on the wortmannin-sensitive class I (p110α) and class III (hVPS34) PI 3′-kinases. The class II PI 3′-kinase are significantly less sensitive to wortmannin and are unlikely to be involved in responses that are inhibited by 50–100 nM wortmannin (Vanhaesebroeck et al., 1997).
p110α can catalyze the production of PI[3]P, PI[3,4]P2, and PI[3,4,5]P3, whereas hVPS34 is limited to the production of PI[3]P (Vanhaesebroeck et al., 1997). Previous studies have shown that cellular levels of PI[3,4]P2 and PI[3,4,5]P3 increase in mitogen-stimulated cells, whereas levels of PI[3]P remain unchanged (Auger et al., 1989; Kapeller et al., 1991). Furthermore, the yeast VPS34 plays a role in the regulation of biosynthetic vacuolar sorting (Schu et al., 1993). Thus, our expectations before beginning these studies was that p110α would be involved in mitogen-stimulated responses such as rearrangement of the cytoskeleton and DNA synthesis, whereas hVPS34 would be involved in activities such as endocytic sorting. In fact, the situation is more complex. Both hVPS34 and p110α are involved in vesicular trafficking and mitogenic signaling, albeit at different steps. In contrast, insulin-stimulated actin rearrangement requires p110α, but does not require hVPS34.
PI 3′-Kinases and the Organization of the Actin Cytoskeleton
Previous studies have shown that microinjection of Δp85 (a mutant p85 that lacks p110 binding sites) inhibits insulin-stimulated membrane ruffling in KB cells (Kotani et al., 1994). Furthermore, Martin et al. (1996_b_) have shown that microinjection of an activated p110α construct is sufficient to induce ruffling and stress fiber breakdown in 3T3-L1 adipocytes or rat-1 fibroblasts, whereas SH2 domains from p85 were inhibitory. Our finding that p110α is required for insulin-stimulated actin rearrangement is not surprising, but serves as a useful control for the specificity of the antibodies with regard to inhibition of p110α- and hVPS34-dependent responses in intact cells.
PI 3′-Kinases in Early Endosomal Function
Inhibition of hVPS34, but not p110α, has pronounced effects on two early endosomal events: the targeting of EEA1 to the early endosome, and the postendocytic sorting of the PDGF receptor. These data suggest that the early endosome is a major site of hVPS34 action. Our finding that hVPS34 is responsible for the targeting of EEA1 is consistent with several recent studies showing that the FYVE finger of EEA1 binds specifically to the product of hVPS34, PI(3)P (Gaullier et al., 1998; Patki et al., 1998). Moreover, expression of EEA1 in S. cerevisiae leads to a VPS34-dependent accumulation in intracellular membranes (Burd and Emr, 1998). EEA1 in turn is required for homotypic fusion of early endosomes, and may function by recruitment of rab5 to the endosomal membrane (Mills et al., 1998; Simonsen et al., 1998). Thus, the hVPS34-dependent recruitment of EEA1 plays a critical role in early endosomal function. It remains to be seen how hVPS34 itself is targeted to the endosomal membrane, perhaps via binding to the myristylated p150 (Panaretou et al., 1997). We also cannot yet explain the perinuclear localization of EEA1 in cells injected with anti-hVPS34 antibodies or treated with wortmannin. It is possible that this reflects the continued activity of a class II PI 3′-kinase, which would not be inhibited by either our antibodies or 100 nM wortmannin.
Inhibitory antibodies to hVPS34 also disrupt the postendocytic trafficking of the PDGF receptor, consistent with the ability of low-dose wortmannin to block PDGF receptor targeting and degradation (Joly et al., 1995). Although this could be due to an early endosomal defect, we cannot rule out additional sites of action at the late endosome or lysosome. The the involvement of hVPS34 in PDGF receptor trafficking may be analogous to the role of yeast VPS34 in vacuolar targeting (Herman et al., 1992), and is consistent with data from numerous laboratories showing that wortmannin inhibits both early and late endosomal fusion events and lysosomal delivery (for review see De Camilli et al., 1996).
Although our data suggest that p110α is not involved in early endosomal function, it should be noted that CHO cells contain both p110α and p110β. p110β also couples to p85 and should be similarly regulated by insulin or PDGF (Hu et al., 1993). Thus, it is possible that p85/p110β complexes play a role in the regulation of PDGF receptor sorting or EEA1 localization that augments the role of hVPS34. Such a requirement would be consistent with the finding that mutation of the p85 binding sites in the PDGF receptor mimic the effects of wortmannin on receptor sorting (Joly et al., 1994). The testing of anti-p110β antibodies is ongoing in the laboratory and will help to resolve this question.
PI 3′-Kinases in Transferrin Recycling
The recycling of transferrin receptors in Trvb-1 cells was significantly slowed by treatment of cells with wortmannin (Martys et al., 1996). The effects of this drug on the distribution of transferrin receptors was half-maximal at 30 nM wortmannin, consistent with the involvement of a mammalian class I or III PI 3′-kinase (Vanhaesebroeck et al., 1997). We now find that inhibition of both p110α and to a lesser extent hVPS34 replicates the wortmannin phenotype with regard to transferrin recycling.
The role of the p85/p110α PI 3′-kinase as a positive regulatory of receptor recycling is consistent the fact that insulin, which activates p85/p110 (Ruderman et al., 1990), enhances the recycling of a number of constitutively recycling receptors (Wardzala et al., 1984; Davis et al., 1986). Moreover, the p85/p110 PI 3′-kinase is required for the insulin stimulation of Glut-4 recycling to the plasma membrane (Okada et al., 1994; Cheatham et al., 1994) and overexpression of p110α increases Glut 4 recycling in several systems (Martin et al., 1996_a_ ; Frevert and Kahn, 1997). Inhibition of hVPS34 also delays transferrin recycling, but to a much smaller extent. The different degrees of inhibition observed with anti-hVPS34 versus anti p110α antibodies could reflect differential recruitment of a regulatory protein that binds to PI[3,4,5]P3 with higher affinity than to PI[3]P. Alternatively, hVPS34 and p110α could act on distinct proteins within the recycling compartment.
PI 3′-Kinases in Mitogenic Signaling
The most striking result in this study is the requirement for the hVPS34 in insulin-stimulated DNA synthesis. This result has been verified using two entirely different approaches to reduce hVPS34 activity and/or expression. Our data suggest that hVPS34 is specifically required for entry of insulin-stimulated cells into S phase.
Previous studies have shown that the levels of PI[3,4]P2 and PI[3,4,5]P3 increase acutely in response to mitogens such as insulin or PDGF, whereas the levels of PI[3]P does not change (Auger et al., 1989; Kapeller et al., 1991). hVPS34 is restricted to the production of PI[3]P, and it is therefore surprising that its activity is required for mitogenic signaling. It may be that constitutive levels of PI[3]P are required for cellular systems that are needed during the transition to S phase. Alternatively, PI[3]P levels may be acutely regulated in specific intracellular locations that are not detectable when whole cell lipid production is measured. Although we do not yet know the function of PI[3]P in mitogenic signaling, our data suggests that the products of hVPS34 are required only during the first 6 h of insulin stimulation. In contrast, the products of p110α are still critical at 6–9 h of insulin stimulation.
One can propose three general mechanisms by which hVPS34 could act during mitogenic signaling. First, as suggested above, the constitutive production of PI[3]P could be required for a cellular process that is needed during early G1. For example, it is possible that normal trafficking of tyrosine kinase receptors is required for efficient signal transduction. However, insulin receptor signaling is relatively normal in cells expressing a dominant-negative mutant of dynamin, which blocks coated pit-mediated endocytosis (Ceresa et al., 1998). Second, insulin could regulate the activity and/or subcellular distribution of hVPS34, leading to the production of PI[3]P at specific intracellular locales. This PI[3]P could in turn recruit specific PI[3]P-binding proteins involved in mitogenic signaling. Finally, a third hypothesis takes note of the potential conversion of PI[3]P to higher-order polyphosphoinositides. A recent finding by Anderson and colleagues (Zhang et al., 1997) shows that PI[4]-5 kinases can convert PI[3]P to PI[3,4,5]P3. If this pathway is a significant source of PI[3,4]P2 and PI[3,4,5]P3 in mitogen-stimulated cells, then reductions in the PI[3]P pool could affect intracellular levels of PI[3,4]P2 and PI[3,4,5]P3. In this way, changes in PI[3]P levels could influence the production of the 3-phosphoinositides that have known signaling functions (Toker and Cantley, 1997). Alternatively, two groups have identified a novel lipid, PI[3,5]P2, in yeast and mammalian cells (Dove et al., 1997; Tolias et al., 1998). In S. cerevisiae, PI[3,5]P2 is produced in response to osmotic stress by a pathway that requires VPS34 but is independent of the Hog1 mitogen-activated protein kinases (Dove et al., 1997). These data suggest a role for VPS34 in signal transduction, which would be consistent with our findings regarding hVPS34 and mitogenesis.
In summary, we have examined the roles of type I and type III PI 3′-kinases in mammalian cells. p110α and hVPS34 PI 3′-kinases both effect the endoctyic system: hVPS34 regulates events in the early endosome and to a lesser extent the recycling compartment, whereas p110α primarily regulates the recycling pathway. Furthermore, both p110α and hVPS34 are necessary for insulin-stimulated DNA synthesis. However, the requirement for hVPS34 occurs during a narrower time window in G1 than the requirement for p110α, suggesting that these enzymes perform different functions in mitogenic signaling. The determination of the distinct roles played by different PI 3′-kinase isoforms adds a new layer of complexity to the functions of these lipid kinases in cellular signaling.
Acknowledgments
We thank T. McGraw (Cornell University School of Medicine), G. Orr, and T. Meier (both from Albert Einstein College of Medicine [AECOM]), and B. Vanhaesebroeck (Ludwig Institute for Cancer Research) for helpful discussions. We thank M. Waterfield for the hVPS34 baculovirus and p110α cDNA, and A. Morris for the p110β baculovirus. We thank M. Cammer (Analytical Imaging Facility, AECOM) for his help with the confocal microscopy.
This work was supported by grants to J.M. Backer from the American Diabetes Association and National Institutes of Health (GM-55692). J.M. Backer is an Established Scientist of the American Heart Association, New York Affiliate, and is a recipient of a Scholar Awards from the Irma T. Hirschl Trust. J. McIlroy was supported by a fellowship from the Juvenile Diabetes Foundation.
Footnotes
1. Abbreviation used in this paper: PI, phosphatidylinositol.
References
- Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R, Soltoff S. Oncogenes and signal transduction. Cell. 1991;64:231–302. doi: 10.1016/0092-8674(91)90639-g. [DOI] [PubMed] [Google Scholar]
- Ceresa BP, Kao AW, Santeler SR, Pessin JE. Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol. 1998;18:3862–3870. doi: 10.1128/mcb.18.7.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol. 1994;14:4902–4911. doi: 10.1128/mcb.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ, Corvera S, Czech MP. Insulin stimulates cellular iron uptake and causes the redistribution of intracellular transferrin receptors to the plasma membrane. J Biol Chem. 1986;261:8708–8711. [PubMed] [Google Scholar]
- De Camilli P, Emr SD, McPherson PS, Novick P. Phosphoinositides as regulators in membrane traffic. Science. 1996;271:1533–1539. doi: 10.1126/science.271.5255.1533. [DOI] [PubMed] [Google Scholar]
- Domin J, Pages F, Volinia S, Rittenhouse SE, Zvelebil MJ, Stein RC, Waterfield MD. Cloning of a human phosphoinositide 3-kinase with a C2 domain that displays reduced sensitivity to the inhibitor wortmannin. Biochem J. 1997;326:139–147. doi: 10.1042/bj3260139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997;390:187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- Field J, Nikawa J-I, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purifcation of a RAS-responsive adenylyl cyclase complex from Saccharomyces cervisiaeby use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frevert EU, Kahn BB. Differential effects of constitutively active phosphatidylinositol 3-kinase on glucose transport, glycogen synthase activity, and DNA synthesis in 3T3-L1 adipocytes. Mol Cell Biol. 1997;17:190–198. doi: 10.1128/mcb.17.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Gaullier J-M, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Sakue H, Kotani K, Kojima A, Waterfield MD, Kasuga M. Normal activation of p70 S6 kinase by insulin in cells overexpressing dominant negative 85kD subunit of phosphoinositide 3-kinase. Biochem Biophys Res Commun. 1995;208:735–741. doi: 10.1006/bbrc.1995.1399. [DOI] [PubMed] [Google Scholar]
- Herman PK, Emr SD. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Sarccharomyces cerevisiae. . Mol Cell Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman PK, Stack JH, Emr SD. An essential role for a protein and lipid kinase complex in secretory protein sorting. Trends Cell Biol. 1992;2:363–368. doi: 10.1016/0962-8924(92)90048-r. [DOI] [PubMed] [Google Scholar]
- Hu P, Mondino A, Skolnik EY, Schlessinger J. Cloning of a novel, ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol Cell Biol. 1993;13:7677–7688. doi: 10.1128/mcb.13.12.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun BH, Rose DW, Seely BL, Rameh L, Cantley L, Saltiel AR, Olefsky JM. Microinjection of the SH2 domain of the 85-kilodalton subunit of phosphatidylinositol 3-kinase inhibits insulin-induced DNA synthesis and c-fos expression. Mol Cell Biol. 1994;14:7466–7475. doi: 10.1128/mcb.14.11.7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly M, Kazlauskas A, Fay FS, Corvera S. Disruption of PDGF receptor trafficking by mutation of its PI-3 kinase binding sites. Science. 1994;263:684–687. doi: 10.1126/science.8303278. [DOI] [PubMed] [Google Scholar]
- Joly M, Kazlauskas A, Corvera S. Phosphatidylinositol 3-kinase activity is required at a postendocytic step in platelet-derived growth factor receptor trafficking. J Biol Chem. 1995;270:13225–13230. doi: 10.1074/jbc.270.22.13225. [DOI] [PubMed] [Google Scholar]
- Kapeller R, Cantley LC. Phosphatidylinositol 3-kinase. Bioessays. 1994;16:565–576. doi: 10.1002/bies.950160810. [DOI] [PubMed] [Google Scholar]
- Kapeller R, Chem KS, Yoakim M, Schaffhausen BS, Backer JM, White MF, Cantley LC, Ruderman NB. Mutations in the juxtamembrane region of the insulin receptor impair activation of phosphatidylinositol 3-kinase by insulin. Mol Endocrinol. 1991;5:769–777. doi: 10.1210/mend-5-6-769. [DOI] [PubMed] [Google Scholar]
- Kotani K, Yonezawa K, Hara K, Ueda H, Kitamura Y, Sakaue H, Ando A, Chavanieu A, Calas B, Grigorescu F, et al. Involvement of phosphoinositide 3-kinase in insulin- or IGF-1-induced membrane ruffling. EMBO (Eur Mol Biol Organ) J. 1994;13:2313–2321. doi: 10.1002/j.1460-2075.1994.tb06515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, Schlessinger J. PH domains: diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 1996;85:621–624. doi: 10.1016/s0092-8674(00)81022-3. [DOI] [PubMed] [Google Scholar]
- Linassier C, MacDougall LK, Domin J, Waterfield MD. Molecular cloning and biochemical characterization of a Drosophilaphosphatidylinositol-specific phosphoinositide 3-kinase. Biochem J. 1997;321:849–856. doi: 10.1042/bj3210849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall LK, Domin J, Waterfield MD. A family of phosphoinositide 3-kinases in Drosophilaidentifies a new mediator of signal transduction. Curr Biol. 1995;5995:1404–1414. doi: 10.1016/s0960-9822(95)00278-8. [DOI] [PubMed] [Google Scholar]
- Martin SS, Haruta T, Morris AJ, Klippel A, Williams LT, Olefsky JM. Activated phosphatidylinositol 3-kinase is sufficient to mediate actin rearrangement and GLUT4 translocation in 3T3- L1 adipocytes. J Biol Chem. 1996a;271:17605–17608. doi: 10.1074/jbc.271.30.17605. [DOI] [PubMed] [Google Scholar]
- Martin SS, Rose DW, Saltiel AR, Klippel A, Williams LT, Olefsky JM. Phosphatidylinositol 3-kinase is necessary and sufficient for insulin-stimulated stress fiber breakdown. Endocrinology. 1996b;137:5045–5054. doi: 10.1210/endo.137.11.8895379. [DOI] [PubMed] [Google Scholar]
- Martys JL, Wjasow C, Gangi DM, Kielian MC, McGraw TE, Backer JM. Wortmannin sensitive trafficking pathways in CHO cells: differential effects on endocytosis and lysosomal sorting. J Biol Chem. 1996;271:10953–10962. doi: 10.1074/jbc.271.18.10953. [DOI] [PubMed] [Google Scholar]
- McGraw TE, Greenfield L, Maxfield FR. Functional expression of the human transferrin receptor cDNA in Chinese Hamster Ovary cells deficient in endogenous transferrin receptor. J Cell Biol. 1987;105:207–214. doi: 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy J, Chen DX, Wjasow C, Michaeli T, Backer JM. Specific activation of p85-p110 phosphatidylinositol 3′-kinase stimulates DNA synthesis by ras- and p70 S6 kinase-dependent pathways. Mol Cell Biol. 1997;17:248–255. doi: 10.1128/mcb.17.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers R, Cantley LC. Cloning and characterization of a wortmannin-sensitive human phosphatidylinositol 4-kinase. J Biol Chem. 1997;272:4384–4390. doi: 10.1074/jbc.272.7.4384. [DOI] [PubMed] [Google Scholar]
- Mills IG, Jones AT, Clague MJ. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr Biol. 1998;8:881–884. doi: 10.1016/s0960-9822(07)00351-x. [DOI] [PubMed] [Google Scholar]
- Molz L, Chen YW, Hirano M, Williams LT. Cpk is a novel class of DrosophilaPtdIns 3-kinase containing a C2 domain. J Biol Chem. 1996;271:13892–13899. doi: 10.1074/jbc.271.23.13892. [DOI] [PubMed] [Google Scholar]
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem. 1994;269:3568–3573. [PubMed] [Google Scholar]
- Panaretou C, Domin J, Cockcroft S, Waterfield MD. Characterization of p150, an adaptor protein for the human phosphatidylinositol (PtdIns) 5-kinase—substrate presentation by phosphatedylinositol transfer protein to the p150-PtdIns 3-kinase complex. J Biol Chem. 1997;272:2477–2485. doi: 10.1074/jbc.272.4.2477. [DOI] [PubMed] [Google Scholar]
- Patki V, Virbasius J, Lane WS, Toh BH, Shpetner HS, Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki V, Lawe DC, Corvera S, Virbasius JV, Chawla A. A functional PtdIns(3)P-binding motif. Nature. 1998;394:433–434. doi: 10.1038/28771. [DOI] [PubMed] [Google Scholar]
- Pollard JW, Stanners CP. Characterization of cell lines showing growth control isolated from both the wild-type and a leucyl-tRNA synthetase mutant of Chinese hamster ovary cells. J Cell Physiol. 1979;98:571–585. doi: 10.1002/jcp.1040980315. [DOI] [PubMed] [Google Scholar]
- Roche S, Koegl M, Courtneidge SA. The phosphatidylinositol 3-kinase a is required for DNA synthesis induced by some, but not all, growth factors. Proc Natl Acad Sci USA. 1994;91:9185–9189. doi: 10.1073/pnas.91.19.9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM. Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem. 1995;270:3662–3666. doi: 10.1074/jbc.270.8.3662. [DOI] [PubMed] [Google Scholar]
- Ruderman N, Kapeller R, White MF, Cantley LC. Activation of phosphatidylinositol-3-kinase by insulin. Proc Natl Acad Sci USA. 1990;87:1411–1415. doi: 10.1073/pnas.87.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Segall JE, Tyerch S, Boselli L, Masseling S, Helft J, Chan A, Jones J, Condeelis J. EGF stimulates lamellipod extension in metastatic mammary adenocarcenoma cells by an actin-dependent mechanism. Clin Exp Met. 1996;14:61–72. doi: 10.1007/BF00157687. [DOI] [PubMed] [Google Scholar]
- Serunian LA, Haber MT, Fukui T, Kim JW, Rhee SG, Lowenstein JM, Cantley LC. Polyphosphoinositides produced by phosphatidylinositol 3-kinase are poor substrates for phospholipase C from rat liver and bovine brain. J Biol Chem. 1989;264:17809–17815. [PubMed] [Google Scholar]
- Simonsen A, Lippé R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh B-H, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- Spiro DJ, Boll W, Kirchhausen T, Wessling-Resnick M. Wortmannin alters the transferrin receptor endocytic pathway in vivo and in vitro. Mol Biol Cell. 1996;7:355–367. doi: 10.1091/mbc.7.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Herman PK, Schu PV, Emr SD. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3′-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO (Eur Mol Biol Organ) J. 1993;12:2195–2204. doi: 10.1002/j.1460-2075.1993.tb05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Aasland R, Toh B-H, Arrigo A. Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The Gβgamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Stoyanov B, Volinia S, Hanck T, Rubio I, Moubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B, et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269:690–693. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- Takegawa K, DeWald DB, Emr SD. Schizosaccharomyces pombeVps34p, a phosphatidylinositol-specific PI 3-kinase essential for normal cell growth and vacuole morphology. J Cell Sci. 1995;108:3745–3756. doi: 10.1242/jcs.108.12.3745. [DOI] [PubMed] [Google Scholar]
- Toker A, Meyer M, Reddy KK, Falck JR, Aneja R, Aneja S, Parra A, Burns DJ, Ballas LM, Cantley LC. Activation of protein kinase C family members by the novel polyphosphoinositides PtdIns-3,4-P2 and PtdIns-3,4,5-P3. J Biol Chem. 1994;269:32358–32367. [PubMed] [Google Scholar]
- Toker A, Bachelot C, Chen CS, Falck JR, Hartwig H, Cantley LC, Kovacsovics TJ. Phosphorylation of the platelet p47 phosphoprotein is mediated by the lipid products of phosphoinositide 3-kinase. J Biol Chem. 1995;270:29525–29531. doi: 10.1074/jbc.270.49.29525. [DOI] [PubMed] [Google Scholar]
- Toker A, Cantley LC. Signaling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Rameh LE, Ishihara H, Shibisaki Y, Chen J, Prestwich GD, Cantley LC, Carpenter CL. Type I phosphatidylinositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol 3,5-bisphosphate and phosphatidylinositol 5-phosphate. J Biol Chem. 1998;273:18040–18046. doi: 10.1074/jbc.273.29.18040. [DOI] [PubMed] [Google Scholar]
- Valius M, Kazlauskas A. Phospholipase C-gamma and phosphatidylinositol 3-kinase are downstream mediators of the PDGF receptor's mitogenic signal. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- Virbasius JV, Guilherme A, Czech MP. Mouse p170 is a novel phosphatidylinositol 3-kinase containing a C2 domain. J Biol Chem. 1996;271:13304–13307. doi: 10.1074/jbc.271.23.13304. [DOI] [PubMed] [Google Scholar]
- Volinia S, Dhand R, Vanhaesebroeck B, MacDougall LK, Stein R, Zvelebil MJ, Domin J, Panaretou C, Waterfield MD. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO (Eur Mol Biol Organ) J. 1995;14:3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardzala LJ, Simpson IA, Rechler MM, Cushman SW. Potential mechanism of the stimulatory action of insulin on insulin-like growth factor II binding to the isolated rat adipose cell. Apparent redistribution of receptors cycling between a large intracellular pool and the plasma membrane. J Biol Chem. 1984;259:8378–8383. [PubMed] [Google Scholar]
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110-alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Loijens JC, Boronenkov IV, Parker GJ, Norris FA, Chen J, Thum O, Prestwich GD, Majerus PW, Anderson RA. Phosphatidylinositol-4-phosphate 5-kinase isozymes catalyze the synthesis of 3-phosphate-containing phosphatidylinositol signaling molecules. J Biol Chem. 1997;272:17756–17761. doi: 10.1074/jbc.272.28.17756. [DOI] [PubMed] [Google Scholar]
- Zhou KM, Takegawa K, Emr SD, Firtel RA. A phosphatidylinositol (PI) kinase gene family in Dictyostelium discoideum: Biological roles of putative mammalian p110 and yeast Vps34p PI 3′-kinase homologs during growth and development. Mol Cell Biol. 1995;15:5645–5656. doi: 10.1128/mcb.15.10.5645. [DOI] [PMC free article] [PubMed] [Google Scholar]