Death-effector Filaments: Novel Cytoplasmic Structures that Recruit Caspases and Trigger Apoptosis (original) (raw)
Abstract
The death-effector domain (DED) is a critical protein interaction domain that recruits caspases into complexes with members of the TNF-receptor superfamily. Apoptosis can also be induced by expressing certain DED-containing proteins without surface receptor cross-linking. Using Green Fluorescent Protein to examine DED-containing proteins in living cells, we show that these proteins cause apoptosis by forming novel cytoplasmic filaments that recruit and activate pro-caspase zymogens. Formation of these filaments, which we term death-effector filaments, was blocked by coexpression of viral antiapoptotic DED-containing proteins, but not by bcl-2 family proteins. Thus, formation of death-effector filaments allows a regulated intracellular assembly of apoptosis-signaling complexes that can initiate or amplify apoptotic stimuli independently of receptors at the plasma membrane.
Activation of caspases, a family of cysteine proteases, is an essential step in many forms of apoptosis (Nicholson and Thornberry, 1997). Caspases have been found in many multicellular organisms, and their role in programmed cell death is highly conserved. Among cysteine proteases, caspases are unique in requiring aspartate at the cleavage site in their substrates. Caspase activation occurs through the limited proteolysis of a proenzyme form. Cleavage of the proenzyme occurs at aspartates, suggesting that autoprocessing or processing by other caspases plays a prominent role in generating the active enzyme. Each precursor contains a prodomain, a large subunit, and a small subunit. Evidence suggests that the fully active enzyme is a tetramer containing two large and two small subunits, and lacking the prodomain. The substrates of the active enzyme are molecules whose cleavage is believed to be necessary for orderly disintegration of the cell (Rosen and Casciola-Rosen, 1997). In addition, certain caspases, such as caspase 1, play an important role in cytokine processing (Fantuzzi et al., 1997; Ghayur et al., 1997).
A principal means of caspase activation is through death domain–containing transmembrane receptors such as CD95/FAS/APO-1 or other members of the tumor necrosis factor receptor (TNFR) superfamily. The death domain is a 60–amino acid portion of the cytoplasmic tail necessary for apoptosis signaling. Although widely expressed, these receptors have been best characterized in the immune system, where they play a critical role in lymphocyte homeostasis, tolerance, and immune protection (Chinnaiyan and Dixit, 1997; Lenardo et al., 1995; Nagata and Golstein, 1995). Both B and T lymphocytes are regulated by Fas-induced apoptosis. Mice or humans with genetic defects in the Fas death receptor can develop lymphoproliferation and severe humoral autoimmunity (Fisher et al., 1996; Rieux-Laucat et al., 1995; Singer et al., 1994). During immune responses, cytotoxic T cells can induce target cell lysis through Fas/Fas ligand interactions, and can also directly activate caspases in target cells by releasing perforin and granzyme B (Chinnaiyan et al., 1996; Kagi et al., 1994; Schroter et al., 1995). Moreover, there are other forms of nonreceptor apoptosis involving caspase action, implying that additional as yet undiscovered cytoplasmic mechanisms of caspase activation must exist.
Apoptosis induction by the TNFR family receptors is due to formation of a protein-signaling complex that involves the physical association of caspases followed by their activation (Kischkel et al., 1995; Medema et al., 1997). Since no other posttranslational modification has been shown to be required for apoptotic signaling, the oligomerization state of the receptor and associated proteins appears to be the critical factor in activating the signaling cascade. In the case of Fas, the interaction and activation of caspase-8 (FLICE/MACHα1/MCH5) is thought to be the first step in a cascade of caspase activation. (Boldin et al., 1995; Medema et al., 1997). The caspase-8 proenzyme contains a long amino-terminal prodomain of 209 amino acids, followed by the coding sequences for the p18 and p11 subunits of the active enzyme. Activation of this pivotal caspase is a consequence of oligomerization and autoprocessing after recruitment to Fas. The apoptosis-signaling complex assembles very rapidly after receptor trimerization following Fas ligand engagement. FADD (MORT1), an adapter molecule that contains a death domain in its COOH terminus, homotypically associates with the Fas death domain (Kischkel et al., 1995; Medema et al., 1997). The crucial interaction for caspase recruitment, however, is a second homotypic interaction between the NH2 terminus of FADD and the prodomain of caspase 8 through a conserved 80–amino acid domain termed the death effector domain (DED).1
The DED was originally defined by the minimal portion of the FADD molecule capable of inducing apoptosis in a transient transfection assay (Chinnaiyan et al., 1995). The long prodomain of caspase-8 harbors two highly homologous DED domains, termed DED-A and DED-B (Boldin et al., 1995; Muzio et al., 1996). Yeast two-hybrid experiments have shown that the DED domains are essential for binding the caspase-8 prodomain to FADD. When expressed in certain cell types, the caspase 8 prodomain can potently induce apoptosis without any requirement for FAS cross-linking (Boldin et al., 1995; Muzio et al., 1996). How the DED initiates this nonreceptor form of death is unknown.
Paradoxically, DEDs have also been identified recently in several viral proteins, such as the molluscum contagiosum virus MC159 protein or the equine herpes virus type 2 E8 protein, that block apoptosis induced by Fas cross-linking. Overexpression of these proteins, termed FLIPs (FLICE inhibitory proteins), does not produce apoptosis, but instead interferes with Fas signaling by inhibiting the DED-mediated interaction of FADD and Caspase-8 (Bertin et al., 1997; Hu et al., 1997; Thome et al., 1997). In addition, a cellular homologue of Caspase-8 harboring tandem DEDs but a nonfunctional caspase domain has been identified. Overexpression of this molecule, termed c-FLIP (or variously: Casper, MRIT, FLAME, Usurpin, or I-FLICE), or its DED-containing prodomain, can induce apoptosis when highly overexpressed, or can block Fas-induced apoptosis at lower expression levels (Han et al., 1997; Hu et al., 1997; Irmler et al., 1997; Rasper et al., 1998; Shu et al., 1997; Srinivasula et al., 1997). Thus, proteins containing the same critical protein interaction domain can have pro- and antiapoptotic effects.
What could account for the divergent behavior of proteins bearing the DED domain? Since subcellular localization is often important for regulating signaling molecules (Mochly-Rosen, 1995), we examined the localization of various DED proteins using Green Fluorescent Protein and other epitope tags. We find that apoptosis triggered by the DED protein motif is in all cases associated with the formation of intracellular filaments, which we term death-effector filaments (DEF). Procaspases are efficiently recruited to these structures. Viral DED-containing proteins that block Fas-induced apoptosis lack the ability to form these filaments, and can antagonize apoptosis by inhibiting formation of the DEF. These data provide a mechanism for the initiation of apoptosis by DED-containing proteins such as FADD, and show that assembly of apoptosis-signaling complexes can occur intracellularly as well as at the plasma membrane.
Materials and Methods
Vectors, Plasmid Construction, and Reagents
Caspase-8-GFP fusion proteins were constructed using Pwo polymerase to amplify fragments of a pCMV-Caspase-8 plasmid whose sequence had been confirmed to be identical to the published MACHα1/FLICE sequences. Primers were designed to contain unique sites that were ligated into pEGFPN1 (CLONTECH Laboratories, Inc., Palo Alto, CA) cut with compatible restriction enzymes. pEGFPN contains the enhanced GFP sequence with multiple point mutations as well as codon optimization for mammalian expression. Caspase-8 103-209 (DED-B) and 210-479 (CD) were generated using additional 5′ primers containing unique BamHI sites. FADD 1-79 (DED) and 80-220 (DD) were cloned using an endogenous SalI site at the AA 79 codon and unique 5′Hind III and 3′ BamHI sites in pcDNA FADD (a gift from Dr. Vishva Dixit, Genentech Inc., South San Francisco, CA). FADD 1-79 was cloned into pEGFP N3, and FADD 80-220 was cloned into the COOH-terminal multiple cloning site of pEGFP C2. The C360S mutation was introduced using the QuickChange™ kit (Stratagene, La Jolla, CA) with appropriate mutagenesis primers, and candidate clones were screened by automated fluorescent sequencing. HA-tagged MC159 and E8 fusion protein constructs were made by amplifying the coding sequences of these proteins with a 5′ oligonucleotide containing an EcoRI site and the HA tag sequence (YPYDVPDYA) in frame with the first methionine codon, and a 3′ oligonucleotide containing the COOH terminal sequences followed by an XbaI site. Digested PCR products were cloned into EcoRI/XbaI-digested pCI vector (Promega Corp., Madison, WI). Pbcl-2CMV and pBcl-x CMV were gifts from Dr. Charles Zacharchuk. Plasmids were prepared for transfection with the Monster™ prep system (Bio 101, La Jolla, CA). zVAD FMK was obtained from Enzyme System Products (Livermore, CA) or Kamiya Biochemical Co. (Seattle, WA). DEVD-Rhodamine (PhiPhiLux) was obtained from Oncoimmunin (College Park, MD). HeLa and 293T cell lines were obtained from American Type Culture Collection (Rockville, MD). The Jurkat Tag cell line expressing the SV40 large T antigen was a gift from Dr. Gerry Crabtree (Stanford University, Stanford, CA).
Transient Transfection and Apoptosis Assays
1–2 × 107 Jurkat Tag cells were transfected with 20 μg of each DNA construct using a BTX EC600 electroporator (260V, 1040 μF, 720 Ohm). Jurkat Tag cells stably express the SV40 large T antigen, which allows the transfected plasmids containing the SV40 ori to replicate, increasing protein expression by ∼50%. Apoptosis induced by caspases or Fas cross-linking was not affected by SV40 T antigen expression. In control experiments, 50 μM zVAD fmk was added directly after transfection. 16 h later, cells were harvested and analyzed in parallel for apoptosis by two separate methods using flow cytometry. Phosphaditylserine exposure on the outer leaflet of the plasma membrane was quantitated by incubating 2.5–5 × 105 cells with 1:50 diluted Annexin-V Cy3 (CLONTECH Laboratories) according to the manufacturer's instructions. DEVD cleavage was assayed by incubating 1 × 106 transfected cells with 50 μl of PhiPhiLux DEVD-rhodamine substrate (Oncoimmunin) and 5 μl FCS for 1 h at 37°C. This substrate is relatively nonfluorescent due to quenching interactions between the two rhodamine molecules on either side of the peptide until cleavage by caspases occurs. Generation of fluorescent cleavage products has been shown to be specific for caspases (Pierre Henkart, personal communication). Cells were washed 1× with FACS buffer and analyzed on a FACSscan™ cytometer (Becton Dickinson & Co., Franklin Lakes, NJ). To exclude necrotic cells that may be nonspecifically positive for Annexin or DEVD cleavage, only cells that fell into a viable FSC/SSC gate were quantitated. In parallel experiments, these cells were found to exclude propidium iodide uniformly. Thus, the percentages obtained, although specific for apoptosis, are indicative of early apoptotic cells only, and are probably an underestimate of the total percentage of cells induced to undergo apoptosis in the culture. HeLa cell apoptosis was quantitated by visual inspection of Green Fluorescent Protein (GFP)-expressing cells with an inverted fluorescent microscope. Cells with a shrunken or blebbed appearance were scored as apoptotic. Counts were done in duplicate, and at least 100 cells per data point were counted.
Immunofluorescence
293T or HeLa cells were grown on glass coverslips (pretreated with 1% poylysine K for 293T cells), and expression vectors were transfected using the Superfect™ reagent (Qiagen Inc., Chatsworth, CA) according to the manufacturer's instructions. 16–24 h later, cells were labeled with 1 μg/ml Hoechst 33342 for 30 min at 37°C, and were fixed with 100% methanol at −20°C for 15 min. Coverslips were then washed with PBS and blocked in IFA buffer (PBS with 0.1% BSA and 0.01% Tween-20). Primary monoclonal antibodies were added at 1 μg/ml in IFA buffer for 1 h, and secondary antibodies were added for 30 min with 2 washes between each step. Sources of antibodies were antitubulin and antivimentin (Amersham Corp., Arlington Heights, IL), anti Bcl-x (Trevigen, Gaithersburg, MD), anti-FADD (Signal Transduction Labs, Lexington, KY), and anti-AU1 and HA.11 antihemagglutinin (Berkeley Antibody Co., Inc., Richmond, CA). After rinsing briefly in H2O, coverslips were mounted with Fluoromount G (Electron Microscopy Sciences). For mitochondrial staining, 25 nM Mitotracker Red (OR R7512; Molecular Probes, Inc., Eugene, OR) was added to cultures for 15 min at 37°C before fixation. Cells were examined with an Axiophot microscope (63× or 100× objectives; Carl Zeiss Inc., Thornwood, NY), and images were acquired with a CCD camera (Princeton Digital Instruments, Princeton, NJ) and appropriate filters and then processed with Adobe Photoshop software.
Subcellular Fractionation and Western Blotting
Subconfluent six-well dishes of 293T cells were transfected with 4 μg of the indicated constructs using Superfect™ (Stratagene) according to the manufacturer's instructions. 50 μM zVAD FMK was added when active caspase-containing constructs were used. 24 h after transfection, 293T cells were lysed for 30 min on ice in buffer containing 140 mM NaCl, 10 mM Tris (pH 7.2), 2 mM EDTA, 1% NP-40, complete protease inhibitor mix (Boerhinger Mannheim Corp., Indianapolis, IN), and 10 mM iodoacetamide. The detergent-insoluble fraction was pelleted by centrifugation at 14,000 rpm in an Eppendorf centrifuge for 10 min. Pellets were washed three times with lysis buffer before boiling in SDS sample buffer. Samples containing lysates and pellets from equal cell numbers were electrophoresed on 4–20% Tris/glycine/SDS gels and blotted onto nitrocellulose using a semidry transfer apparatus. Blots were blocked with 5% nonfat dry milk for 30 min, and were probed with 1:1,000 dilution of a mixture of anti-GFP mAb (Boerhinger Mannheim Corp.) or 1:1,000 diluted anti-HA mAb (Berkeley Antibody Co., Inc.) followed by 1:10,000 dilution of donkey anti–mouse HRP (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), with three washes after each incubation. Incubations and washes were performed with 0.05% PBS TritonX-100. Bands were imaged with SuperSignal™ HRP substrate (Pierce Chemical Co., Rockford, IL).
Results
The DED Domains From FADD and Caspase-8 Can Induce Apoptosis Independently of Fas Signaling
To study the intracellular location of DED-containing proteins, full-length and truncated versions of FADD and Caspase-8 were fused with GFP (Fig. 1). GFP allowed us to examine the subcellular localization of these proteins in living cells under various conditions. Transient transfection of the GFP-tagged version of full-length caspase-8 efficiently induced morphological changes, consistent with apoptosis in HeLa and Jurkat cell lines (Figs. 1 and 2, wild-type). However, fusions of GFP with the caspase-8 prodomain, containing the two tandem DEDs and FADD containing a single DED, also induced cell death (Fig. 1). Because FADD and the caspase-8 prodomain contain no caspase moiety, we wanted to determine if they triggered caspase activation and apoptosis. We confirmed that DED-transfected cells were dying via apoptosis by the morphological appearance of Hoechst-stained nuclei (data not shown), and by exposure of phosphaditylserine (PS) as detected by Annexin-V surface staining (Fig. 2, A and B). To demonstrate caspase activation directly in GFP-expressing cells, we used a new cell-permeable DEVD–rhodamine caspase substrate that exhibits red fluorescence only after cleavage (Packard et al., 1996). Flow cytometry after exposure to this substrate revealed that cells expressing the GFP-DED proteins fluoresced red, indicating proteolysis by activated caspases. Flow cytometry of the intact cells revealed that caspase activation is tightly regulated by the intracellular concentration of DED proteins. Above a threshold value of fluorescence, which is proportional to the amount of protein expressed, both PS exposure and DEVD-rhodamine fluorescence were observed, indicating that apoptosis commences in most cells (Fig. 2 A, bottom). Control experiments confirmed that endogenous caspase activation was involved, since death was prevented by the inhibitor zVAD-fmk (Fig. 2 B).
Figure 1.

Structure and function of fusion proteins used in this study. The hatched bars denote DED, and the checkered bars denote the death domain (DD) in FADD homologous to that in death domain–containing receptors. Processing sites at aspartate residues (D) and the active-site cysteine (C360*) are shown. HA denotes the influenza hemagglutinin epitope tag. p18 and p11 are the large and small subunits of the mature processed caspase-8 enzyme. The numbers indicate the amino acids at the junction of the GFP fusion proteins, numbered according to the human MACH α1 and FADD sequences available on Genbank. The + and − symbols indicate whether transfection of the molecule in HeLa or Jurkat cells induced apoptosis (>20%) or filament formation (>50%). CD, caspase domain.
Figure 2.

Apoptosis induction by DED and caspase proteins. (A) FACS profiles demonstrating apoptosis induced by transient transfection of the caspase-8 prodomain. Jurkat Tag cells (expressing the SV40 Tag for higher expression of replicating plasmids) were transiently transfected with the indicated GFP fusion protein constructs. 16 h after transfection, cells were analyzed by flow cytometry for Annexin-V or DEVD-rhodamine staining (y-axis) and GFP fluorescence (x-axis) as described in the experimental procedures. Percentages in each quadrant are shown by the numbers in the diagram at the top right corner of each plot. The apoptotic cells that are GFP-negative but Annexin-V–positive are nonspecifically induced by electroporation. (B) Summary of Annexin-V binding and DEVD rhodamine cleavage induced by the different fusion proteins and inhibition by zVAD FMK. Jurkat cells were transfected with 20 μg of the indicated constructs and analyzed by flow cytometry 16 hours after transfection. Percentages are the percentage of GFPhi cells gated as in A, positive for DEVD-rhodamine or Annexin-V binding. Data are representative of at least three independent transfection experiments for each data point. Constructs that produced <20% apoptotic GFP-positive cells were considered to be negative for apoptosis.
DED Proteins Form Cytoplasmic Filaments Associated with Apoptosis
When we examined cells expressing GFP fusion proteins with caspase-8, FADD, and the various truncation mutants under the microscope, we found two dramatically different patterns of fluorescence distribution (Fig. 3). The first pattern, seen with the full-length caspase-8 protein, was a diffuse distribution throughout the cytoplasm (C360S; Fig. 3 A, top left and data not shown). A similar diffuse pattern was observed when the caspase domain alone was expressed (CD; Fig. 3 A, middle left). In these analyses, apoptosis was inhibited with zVAD-fmk or by using the C360S active site mutant. Without blocking apoptosis, we observed packaging of the caspase-GFP proteins into apoptotic blebs in dying cells (data not shown).
Figure 3.

Localization and solubility of DED-containing proteins. (A and C) HeLa cells were transiently transfected with 0.5–1 μg of the indicated portions of Caspase-8 (A) and FADD (C) fused to GFP in the presence of 50 μM zVAD-FMK to inhibit apoptosis. Cells were selected for photography 16–24 h after transfection. The blue counterstain indicates nuclei. B shows the predominantly apoptotic morphology of cells transfected with caspase-8 DED-AB (left), which can be inhibited by adding of 50 μM zVAD-FMK at the time of transfection (right). The middle right photograph in A and bottom left in C are conventional immunofluorescence pictures of representative cells transfected with AU-1-epitope–tagged Caspase-8 prodomain stained with anti-AU1 and untagged FADD full-length protein stained with anti-FADD mAb, followed by Texas Red–conjugated anti-mouse IgG. (D and E) Solubility of DED proteins in detergent correlates with their morphological appearance. 1% NP40 lysate supernatants (S) and pellets (P) from equal numbers of transfected cells were separated on 4–20% gradient polyacrylamide gels and blotted with anti-GFP mAbs. In each case, the top band corresponds to the predicted size of the fusion protein, and lower bands represent degradation products that were variably seen in different experiments. Similar results were obtained with 1% Triton-X100 detergent lysates. Fusion proteins of the correct predicted sizes were found for the other Caspase-8 fusion proteins (not shown).
The second pattern, illustrated using the DED-containing prodomain of caspase-8 fused to GFP, was a distinctive cytoplasmic filament network (DED-AB; Fig. 3 A, top right). Greater than 90% of the fluorescence was concentrated into what appeared to be a small number of interconnecting filaments with a perinuclear localization, sometimes appearing as a cage or lariat structure around the nucleus (see Fig. 7). The filaments were variable in thickness, and could be clearly identified as round when seen on end with confocal microscopy (data not shown). The filaments were observed in unfixed cells in culture as well as after methanol or formaldehyde fixation, and did not break down or reorganize in mitotic cells (data not shown). When observed over time, the filament assemblies first formed in healthy-appearing cells, and did not depend on caspase activity since zVAD-fmk treatment did not prevent their formation. In fact, zVAD-fmk promoted formation of a more complex filamentous network (Fig. 3 B, right). To show that GFP was not required for the formation of these structures, the filaments were also visualized with conventional immunostaining using an AU-1 epitope tag (Fig. 3 A, middle right). Unlike many other proteins that are typically packaged into apoptotic blebs, the filaments remained intact in a collapsed configuration in the perinuclear cytoplasm of apoptotic cells (Fig. 3 B, left).
Figure 7.
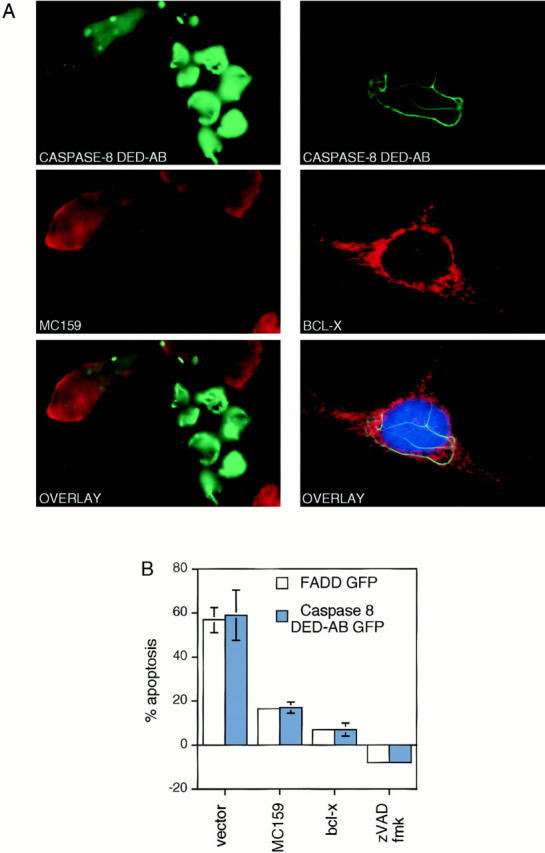
Protection from apoptosis by MC159, but not Bcl-x, correlates with inhibition of DEF formation. (A) In the left panels, Hela cells were cotransfected with Caspase-8–DED-AB–GFP and HA-MC159, and were analyzed by anti-HA immunofluorescence. A representative field is shown with apoptotic cells with DEFs transfected with Caspase-8 DED-AB alone (green) and two viable cells in the upper left that are cotransfected with MC159 (red). Note the shortening or disappearance of the DEFs in the cotransfected cells. Similar results were obtained with FADD WT and FADD DED cotransfections. The right panels show a representative cell cotransfected with Bcl-x (stained with anti-Bclx; red) and Caspase-8 DED-AB GFP. Note the preservation of the filamentous pattern with the DED-GFP. Similar results were obtained with FADD-GFP as well as Bcl-2. (B) Quantitation of the protective effect of MC159 and Bcl-x on DED-induced apoptosis. HeLa cells were cotransfected with 0.5 μg of the Caspase-8 DED GFP construct, and 1.5 μg of MC159 HA or P CMV Bcl-x. 16 h later, cells were harvested and fixed with formaldehyde, and apoptotic cells were scored with an inverted fluorescence microscope. At least 100 cells/sample were counted for each data point. Percentages are the average of two independently transfected wells. Similar results were obtained in two other experiments.
To determine if filament formation is a general property of apoptosis-inducing DED-domain proteins, we performed similar experiments with FADD. A GFP fusion protein with the death domain–containing COOH terminal of FADD, which does not induce apoptosis, was dispersed throughout the cytoplasm (Fig. 3 C, top left). However, the full-length GFP-FADD fusion protein that contains a single DED and induces apoptosis, formed filaments similar to the caspase-8 prodomain (Fig. 3 C, bottom right). The minimal 79–amino acid DED domain of FADD that also induces apoptosis, produced shorter, more numerous filaments with some diffuse staining, especially in the more viable cells (Fig. 3 C, top right). The FADD filaments could also be detected by immunostaining in the absence of GFP or any epitope tag revealing that these structures form as an intrinsic property of the proteins (Fig. 3 C, bottom left). These data suggest a strong correlation between filament formation and apoptosis induction.
When we tested the A and B DEDs of caspase-8 individually, we found additional evidence associating apoptosis with filament formation. The DED-B fusion with GFP formed fine filaments with a beads-on-a-string morphology (Fig. 3 A, bottom right) and induced apoptosis (Fig. 2 B). By contrast, the DED-A-GFP protein neither formed filaments nor induced apoptosis (Fig. 3 A, bottom left). It is notable that although full-length caspase-8 induces apoptosis and contains the filament-forming DED-B, it does not assemble into filaments (Fig. 3 A, top left). Overexpression of the full-length or the caspase domain of caspases initiates apoptosis by direct autoactivation of its enzymatic function, and thus may bypass the requirement for filament formation (Nicholson and Thornberry, 1997). In the full-length caspase-8 protein, the domain that initiates filament formation may be shielded by the caspase domain. Thus, the DED-A and DED-B motifs in the prodomain of caspase-8 are functionally distinct, and support the conclusion that filament assembly is required for the lethal effect of DEDs. We therefore propose the name death-effector filament (DEF) for these apoptosis-associated cytoplasmic structures.
DEFs Have Solubility Properties of the Cytoskeleton, but Do Not Colocalize With Other Known Cytoskeletal Elements or Mitochondria
Because DEFs display a morphology similar to that of cytoskeletal filaments, we examined the solubility properties of the GFP fusion proteins by extracting transfected cells with various detergents. Polymerized cytoskeletal proteins are uniformly insoluble in nonionic detergents (Aamodt and Williams, 1986; Steinert et al., 1982). Filament formation by the DED proteins led to a similar insolubility in 1% NP-40 or Triton X-100 (Fig. 3, D and E). The DEF formed by the caspase 8 DED-AB was almost exclusively detergent-insoluble (Fig. 3 D, lanes 1 and 2). Conversely, the WT caspase-8 protein was predominantly in the soluble fraction, corresponding to its diffuse intracellular distribution (Fig. 3 D, lanes 3 and 4). FADD and its derivatives behaved similarly. The death domain of FADD, which does not induce apoptosis and is diffuse throughout the cytoplasm, was fully detergent-soluble (Fig. 3 E, lanes 1 and 2). The FADD DED domain, which forms shorter filaments with some diffuse distribution, was partially soluble (Fig. 3 E, lanes 3 and 4). The full-length FADD protein, which exclusively localized to long filaments, was almost fully insoluble in NP-40 (Fig. 3 E, lanes 5 and 6). The fact that the full-length FADD protein formed more extensive filaments and was more insoluble than the DED alone suggests that additional domains in the FADD protein may facilitate its aggregation into DEFs.
We also examined the relationship of death-effector filaments to known cytoskeletal structures by double-labeling experiments. We found that the DEF formed by the caspase-8 prodomain was independent of either microtubules or intermediate filaments (Fig. 4). There was some resemblance to the vimentin network, including identical perinuclear localization and hyperaggregation with colchicine (data not shown), but little direct overlap was seen. Staining with a monoclonal antibody that recognizes all types of intermediate filaments (Pruss et al., 1981) or rhodamine-phalloidin for polymerized actin also showed no colocalization with DEFs (data not shown). Since caspases have been reported to coprecipitate with mitochondrially located bcl-2 family proteins (Chinnaiyan et al., 1997; Han et al., 1997), and mitochondria can have filament-like patterns in some cells, we also examined DED-transfected cells with mitochondrial dyes. No overlap was seen between the pattern of mitochondrial staining and the DEF. (Fig. 4, right). Thus, the DEF is a novel cytoskeleton-like structure that could result either from self- assembly of DEDs or their binding to an as-yet unidentified cytoskeletal element.
Figure 4.

Comparison of DEFs with other cellular structures and costaining with anti-Caspase-8 antisera. HeLa cells (β Tubulin) or 293T cells (Vimentin and Mitotracker Red) were transfected with 1 μg of the Caspase-8 DED-AB GFP construct, and then fixed and prepared for immunofluorescence analysis with antitubulin or antivimentin antibodies followed by Texas-Red–conjugated secondary antibodies as described in experimental procedures. For mitochondrial staining, coverslip cultures were preincubated with 25 nM of Mitotracker Red 7512 before fixation. The top row shows the pattern of staining with these reagents in the red channel; the middle row show the pattern of DED-AB GFP expression, and the lower row shows the overlay of the two channels, with blue indicating the Hoechst-stained nuclei. Overlapping fluorescence would be yellow.
Pro-caspase-8 is Efficiently Recruited to the DEF
DED-containing proteins can aggregate and activate caspase-8 at the plasma membrane during Fas-stimulated apoptosis. We therefore tested the hypothesis that the DEF may function similarly in the cytoplasm, and may cause apoptosis by the recruitment and activation of endogenous pro-caspases. We cotransfected the GFP-tagged full-length caspase-8 molecule that is normally diffuse in the cell with the filament-forming FADD or the caspase-8 prodomains. Because apoptosis distorts the DEF, we used the C360S inactive version of caspase 8 and added the caspase inhibitor zVAD-fmk. We found that expression of the caspase 8 prodomain (visualized by an AU-1 tag; Fig. 5 C) caused the normally diffuse expression pattern of the caspase 8-C360S-GFP molecule to relocalize to the DEF (Fig. 5, A, C, and E). The relocalization was remarkably efficient. The overlay picture (Fig. 5 E) reveals little residual green fluorescence, indicating that essentially all of the caspase-8 has translocated to the DEF. Recruitment of caspase-8 required the DEDs, since the caspase domain only, without the prodomain (caspase 8 CD), did not relocalize to the DEF (Fig. 5, B, D, and F). FADD also efficiently recruited caspase-8 into DEFs (data not shown). These data are consistent with yeast two-hybrid experiments in which the caspase-8 prodomain and FADD were found to interact with each other and with full-length caspase-8 (Boldin et al., 1995; L. Zheng and M.J. Lenardo, unpublished data). Taken together, these findings suggest that the cytoplasmic mechanism for nonreceptor apoptosis induced by DED proteins involves recruitment of procaspases into the DEF. To verify this hypothesis, it was important to inhibit DEF formation to determine whether this could prevent apoptosis.
Figure 5.
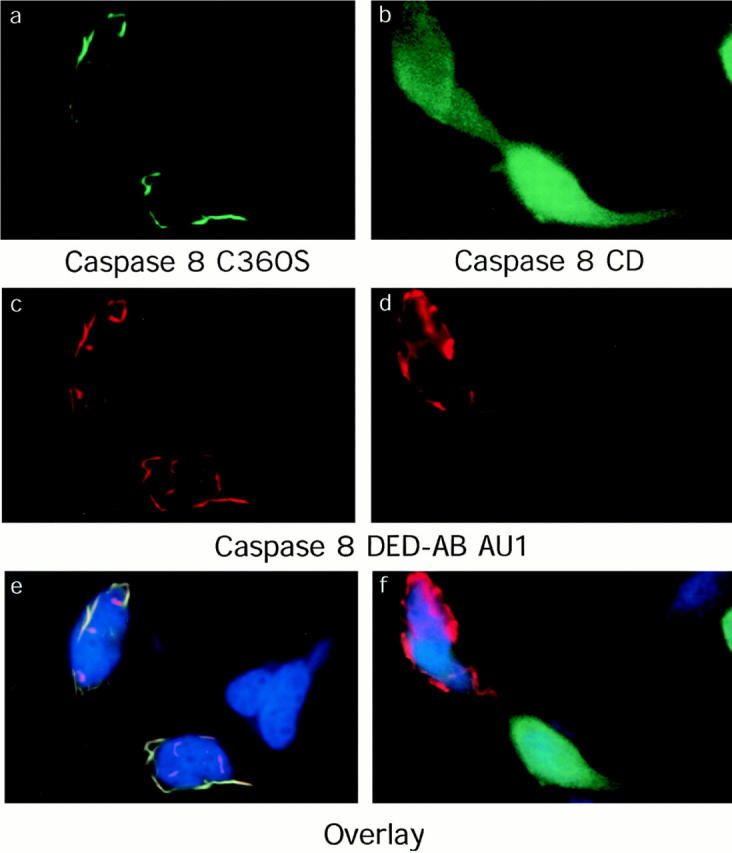
Recruitment of procaspase-8 into DEF. HeLa cells were cotransfected with 1.5 μg of AU1-tagged caspase-8 prodomain molecule and 0.5 μg of GFP-tagged full-length caspase-8 C360S (a, c, and e) or the caspase domain only (amino acids 210–479) of caspase-8 (b, d, and f). 50 μM zVADfmk was added to inhibit apoptosis. Coverslips were incubated with anti-AU1, followed by Texas-Red antimouse IgG. Green fluorescence is shown in a and b, red is shown in c and d, and the overlay of the two panels is shown in f and g. The overlap of AU1 and GFP is shown in yellow. In the left panels, clear overlap of the caspase-8 and DED signals are seen, showing recruitment of Caspase-8 to the DEF. In the right panels, the cell at the top right expresses high levels of the DED in red, but a diffuse pattern staining is still seen with Caspase-8 caspase domain (CD), indicating that recruitment to the DED occurs through DED–DED interactions.
Viral-FLIP Proteins Block DEF Formation and Cell Death Induced by Proapoptotic DED Proteins
We and others have shown that viral DED-containing proteins (v-FLIPs) can block apoptosis induced by multiple death receptors, rather than stimulating apoptosis themselves (Bertin et al., 1997; Hu et al., 1997; Thome et al., 1997). We therefore tested the ability of these viral proteins to form death-effector filaments. The patterns of the v-FLIP proteins MC159 and E8 were examined in HeLa cells using anti-HA antibodies directed against amino terminal epitope tags. By immunofluorescence, both proteins were diffusely distributed throughout the cytoplasm, with a small amount of nuclear staining (Fig. 6 A). Thus, neither protein formed DEFs, and as we and others have previously reported, neither protein induced apoptosis. Western blot analysis showed that HA-tagged MC159 and E8 were completely soluble in nonionic detergents (Fig. 6 B). These data suggest that v-FLIPs do not cause apoptosis because they cannot form the DEF, and therefore cannot recruit and activate endogenous caspases in the cytoplasm.
Figure 6.
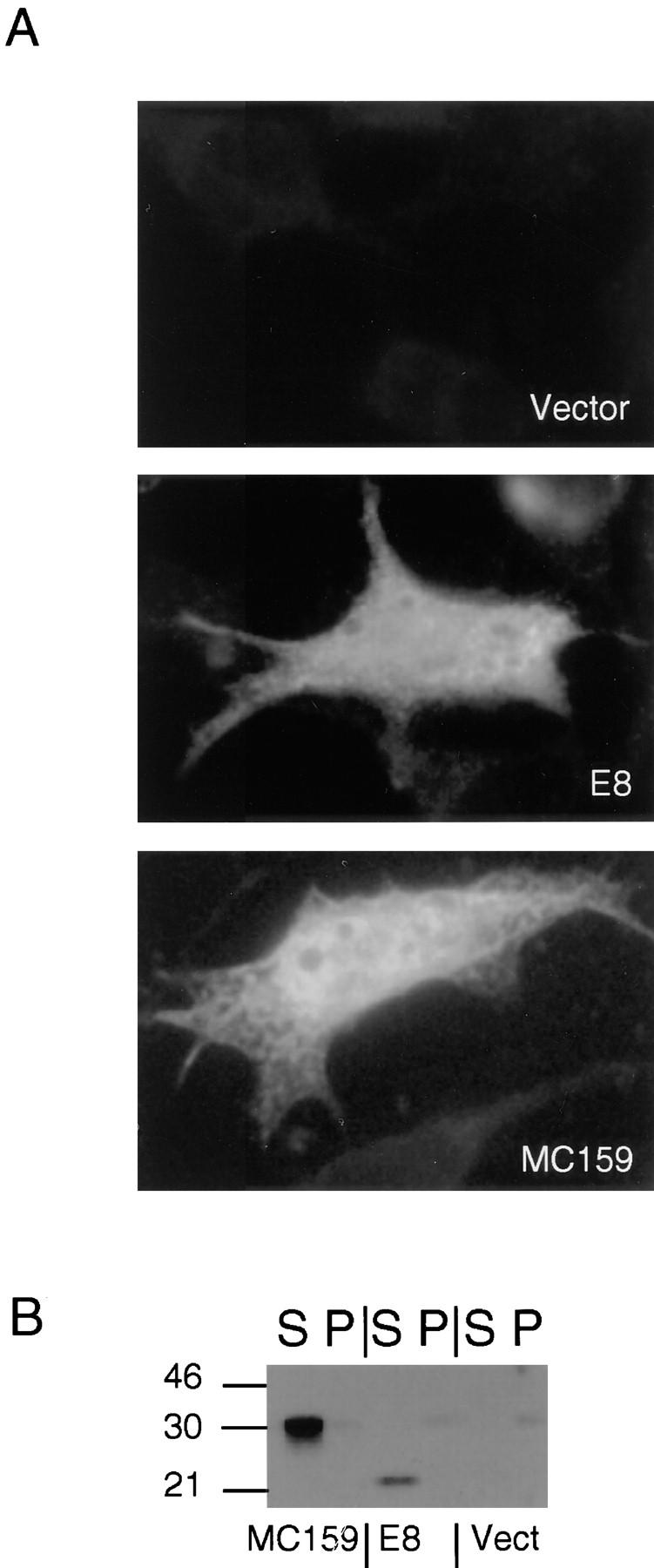
Viral DED-containing proteins are soluble, and do not form filaments. (A) 0.5 μg of HA-tagged MC159 and E8 fusion protein constructs were transfected into HeLa cells, and were then analyzed by immunofluorescence 24 h later with anti-HA mAb, followed by antimouse IgG Texas Red. Diffuse staining above background can be seen with both MC159 and E8. (B) Western blots: HA-tagged MC159 and E8 were transfected into 293T cells, and equivalent cell numbers from supernatants and pellets of NP-40 lysates were analyzed by Western blotting with anti-HA mAb.
Since v-FLIPs interact with DED-containing proteins (Bertin et al., 1997; Hu et al., 1997; Thome et al., 1997), we were able to ask whether this interaction could block DED-induced apoptosis by affecting the DEF. In our experimental system, the viral MC159 protein could suppress apoptosis induced by either FADD or the prodomain containing DEDs A and B from caspase 8 (Fig. 7 B). When cells coexpressing the v-FLIP and the GFP-tagged caspase-8 prodomains or FADD DED were examined microscopically, we observed shorter or absent filaments, and the cell morphology was not apoptotic (Fig. 7 A, left). The E8 v-FLIP had similar effects (data not shown). Our cell transfection assay also generated some cells that only take up and express one plasmid, thereby allowing us to examine cells in the same sample that expressed DED protein in the absence of the v-FLIP. These cells exhibited DEFs with a collapsed morphology, indicating that apoptosis was occurring (Fig. 7 A, left bottom, green filaments). From these data we conclude that the DEF is essential for the nonreceptor cytoplasmic mechanism of apoptosis caused by DEDs, and that v-FLIPs abrogate this mode of death by blocking DEF formation.
To gain additional insight into the regulation of apoptosis caused by the DEF, we asked whether blocking apoptosis with bcl-2 family proteins could disrupt the DEF. Bcl-x potently blocked apoptosis induced by transfection of FADD and the caspase-8 prodomain (Fig. 7 B). However, examination of cotransfected cells did not reveal any disruption of the death-effector filaments (Fig. 7 A, right). Taken together, these data demonstrate that the v-FLIPS and Bcl-x block apoptosis induced by DEFs, but exert their effects through distinct mechanisms.
Discussion
In these studies we have characterized a cytoplasmic nonreceptor mechanism of apoptosis that is induced by proteins containing certain forms of the death-effector domain, and involves the formation of a striking cytoskeletal-like protein assembly. Several observations allow us to suggest how this type of programmed cell death occurs. First, we find that DEDs from either FADD or caspase 8 can induce classic apoptosis that involves activation of caspases, and can be blocked by the peptide inhibitor zVAD-fmk for these proteases. Second, we find that apoptosis by these DED proteins is associated with the assembly of these proteins into cytoplasmic filamentous structures that we have called DEFs. Third, DEFs are detergent-insoluble, and do not correspond to other known cellular structures. Fourth, we have shown that soluble caspase-8 has the property of being recruited to the DEF via its DED domains. Fifth, viral DED proteins that prevent apoptosis do not form the DEF, and cause dissolution of the DEF formed by apoptosis-promoting DEDs. Finally, although Bcl-x prevents this form of death, it does not prevent DEF assembly, and therefore blocks a downstream or parallel step in the death pathway. Thus, we propose that DEF assembly leads to recruitment and activation of DED-containing caspases, and initiation of the molecular cascade of apoptosis. Recruitment to the DEF is highly efficient, and would cause a dramatic increase in local concentration of procaspases, which in other systems has resulted in autocatalytic processing and release of the active enzyme into the cytoplasm. We have shown that proximity-induced oligomerization of caspases is critical for their activation (Martin et al., 1998). The DEVD-rhodamine substrate used in these experiments primarily measures Caspase-3/CPP32 activity, so our conclusions about caspase-8 activation are based on indirect evidence at this time. Nevertheless, the DEF represents a novel cytoplasmic protein assembly that has an essential role in apoptosis caused by DEDs. Our findings suggest that assembly of higher order structures enhances the efficiency of caspase activation. This type of macromolecular assembly may also play a role in the Fas death-signaling complex at the membrane where the stoichiometry of the components of the complex is not presently known.
Why cellular and not viral DED proteins aggregate into apoptosis-inducing structures is not clear. Computer modeling predicts that the DED domain folds into an alpha-helix–rich structure, with some similarity to the Fas/CD95 death domain (Hofmann et al., 1997; Huang et al., 1996). The fact that the full-length pro-caspase-8 protein contains both DED domains but does not aggregate into filaments, suggests that there may be major conformational differences between the DED fragment and the whole protein. Posttranslational modification may also play a role in determining the subcellular localization of DED-containing proteins, as it does in other signaling systems. The spacer region between the DED and caspase domains of caspase-8 (amino acids 178–210) is not homologous with the viral FLIPs, and may also contain domains important in filament formation. Three-dimensional structures of DED domains or unprocessed pro-caspases may shed light on these differences in functional properties.
The DEF is one of several intracellular sites at which apoptosis signaling is now known to be regulated. The fact that Bcl-2 family proteins block DED-induced apoptosis without blocking filament formation suggests that their mechanism of action is downstream or in a parallel pathway. Since Bcl-x has been shown to bind pro-caspase-8, presumably through a ced-4–like adapter molecule (Chinnaiyan et al., 1997), one possibility for its mechanism of action may be the sequestration of procaspases to mitochondrial and internal cell membranes where Bcl-x is located. However, in cotransfection experiments we did not find significant colocalization of pro-caspase-8 with overexpressed Bcl-x or Bcl-2. Whether this result is due to insufficient quantities of adapter molecules, or to other requirements for such an interaction is not clear. Bcl-2 family proteins also have been shown to inhibit release of proapoptotic molecules such as cytochrome c from mitochondria into the cytoplasm (Kluck et al., 1997; Yang et al., 1997), and can also block apoptosis after release of cytochrome c into the cytoplasm (Li et al., 1997; Rosse et al., 1998). Through either of these pathways, Bcl-x could block the downstream activation of the effector caspases (which include caspase 3) without affecting Caspase-8 autoactivation or DEF formation.
Our data show that DEF formation is a regulated mechanism of apoptosis induction that might occur in various physiological situations. The filaments are not dependent on the transient transfection system, as 293T cells stably expressing the caspase-8 DED filaments have been grown in tissue culture for >3 mo (R.M. Siegel and M.J. Lenardo, unpublished data). 293T cells are resistant to the proapoptotic effects of the DEF, perhaps because of their expression of the adenovirus E1B19K protein. It is not clear at this time whether DEFs are formed after Fas cross-linking. It has been shown that caspase processing within the Fas signaling complex causes release of the caspase 8 DEDs into the cytoplasm (Medema et al., 1997). The released prodomains could form a DEF, and amplify the death signal in the cytoplasm. However, preliminary experiments with a caspase-8 construct tagged separately at the amino and carboxy termini have thus far failed to show a filamentous pattern of expression for the DED domain after Fas cross-linking or staurosporine treatment. The concentration of apoptosis inhibitory proteins such as c-FLIP may also regulate the threshold for filament formation by DED proteins. The ability of viral proteins that block cell death to disrupt the DEF suggests a role for these proteins in the outcome of virus infection. It is also possible that the DEF is the prototype for cytoplasmic assemblies that lead to caspase activation and apoptosis induction under pathological circumstances. Filament formation by normally soluble proteins has been proposed as an important mechanism in the pathogenesis of neurodegenerative diseases. The paired helical filaments in Alzheimer's disease and the cytoplasmic aggregates formed by pathogenic prion protein are composed of conformationally altered soluble proteins, and have been reported to be involved in causing apoptotic death of neurons (Forloni et al., 1993; LaFerla et al., 1995). In light of our data, it will be important to determine if formation of these filaments can also recruit or activate caspases in the manner we have observed.
Acknowledgments
We wish to thank Drs. Lou Staudt and S. Venkatesan for use of microscopes, Drs. Charles Zacharchuk and Vishva Dixit for plasmids, and Dr. Emad Alnemri for anti-MCH-5 antisera. We would also like to thank John Yewdell, Pierre Henkart, Luciano D'Adamio, and Julie Donaldson for advice and critical reading of the manuscript.
Abbreviations used in this paper
DED
death-effector domain
DEF
death-effector filaments
FLIP
FLICE (caspase-8) inhibitory proteins
inhibitory proteins
GFP, green fluorescent protein
v-FLIP
viral DED-containing proteins
Footnotes
D.A. Martin was a research scholar with the Howard Hughes Medical Institute.
The present address of John Bertin is Millennium Pharmaceuticals, 640 Memorial Drive, Cambridge, MA 02139.
Address all correspondence to Michael J. Lenardo, Laboratory of Immunology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10, Room 11N311, Bethesda, MD 20892. E-mail: mlenardo@atlas.niaid.nih.gov
References
- Aamodtd EJ, Williams RC., Jr Methods to study the microtuble-eurofilament nework in vitro. Methods Enzymol. 1986;134:544–555. doi: 10.1016/0076-6879(86)34119-3. [DOI] [PubMed] [Google Scholar]
- Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1995;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Dixit VM. Portrait of an executioner: the molecular mechanism of FAS/APO-1-induced apoptosis. Semin Immunol. 1997;9:69–76. doi: 10.1006/smim.1996.0055. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Hanna WL, Orth K, Duan H, Poirier GG, Froelich CJ, Dixit VM. Cytotoxic T-cell–derived granzyme B activates the apoptotic protease ICE-LAP3. Curr Biol. 1996;6:897–899. doi: 10.1016/s0960-9822(02)00614-0. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G, Ku G, Harding MW, Livingston DJ, Sipe JD, Kuida K, Flavell RA, Dinarello CA. Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. J Immunol. 1997;158:1818–1824. [PubMed] [Google Scholar]
- Fisher GH, Lenardo MJ, Zuniga-Pflucker JC. Synergy between T cell Receptor and Fas (CD95/APO-1) signaling in mouse thymocyte death. Cell Immunol. 1996;169:99–106. doi: 10.1006/cimm.1996.0096. [DOI] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L, Talanian R, Paskind M, et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- Han D, Chaudhary PM, Wright ME, Friedman C, Trask BJ, Riedel R, Baskin D, Schwartz SM, Hood L. MRIT, a novel death-effector domain-containing protain, interacts with caspases abd BclXl and initiates cell death. Proc Natl Acad Sci USA. 1997;94:11333–11338. doi: 10.1073/pnas.94.21.11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signaling motif. Trends Biochem Sci. 1997;22:155–156. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- Hu S, Vincenz C, Buller M, Dixit VM. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J Biol Chem. 1997;272:9621–9624. doi: 10.1074/jbc.272.15.9621. [DOI] [PubMed] [Google Scholar]
- Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 1996;384:638–641. doi: 10.1038/384638a0. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)- associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur Mol Biol Organ) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lenardo MJ, Boehme S, Chen L, Combadiere B, Fisher G, Freedman M, McFarland H, Pelfrey C, Zheng L. Autocrine feedback death and the regulation of mature T lymphocyte antigen responses. Int Rev Immunol. 1995;13:115–134. doi: 10.3109/08830189509061742. [DOI] [PubMed] [Google Scholar]
- Li F, Srinivasan A, Wang Y, Armstrong RC, Tomaselli KJ, Fritz LC. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release. J Biol Chem. 1997;272:30299–30305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- Martin DA, Siegel RM, Zheng L, Lenardo MJ. Membrane oligomerization and release activates the caspase 8 (FLICE/MACHα1) death signal. J Biol Chem. 1998;273:4345–4349. doi: 10.1074/jbc.273.8.4345. [DOI] [PubMed] [Google Scholar]
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO (Eur Mol Biol Organ) J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Nicholson D, Thornberry N. Caspases: killer proteases. TIBS (Trends Biochem Sci) 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Packard BZ, Toptygin DD, Komoriya A, Brand L. Profluorescent protease substrates: intramolecular dimers described by the exciton model. Proc Natl Acad Sci USA. 1996;93:11640–11645. doi: 10.1073/pnas.93.21.11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruss RM, Mirsky R, Raff MC, Thorpe R, Dowding AJ, Anderton BH. All classes of intermediate filaments share a common antigenic determinant defined by a monoclonal antibody. Cell. 1981;27:419–428. doi: 10.1016/0092-8674(81)90383-4. [DOI] [PubMed] [Google Scholar]
- Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SLC, Tawa P, Xanthoudakis S, Nasir J, Martindale D, et al. Cell death attenuation by Usurpin, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death and Differentiation. 1998;5:271–288. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F, Diest FL, Roberts IA, Debatin KM, Fisher A, Villartay JP. Mutations in Fas-associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1351. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- Rosen A, Casciola-Rosen L. Macromolecular substrates for the ICE-like proteases during apoptosis. J Cell Biochem. 1997;64:50–54. doi: 10.1002/(sici)1097-4644(199701)64:1<50::aid-jcb8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Schroter M, Lowin B, Borner C, Tschopp J. Regulation of Fas(Apo-1/CD95)- and perforin-mediated lytic pathways of primary cytotoxic T lymphocytes by the protooncogene bcl-2. Eur J Immunol. 1995;25:3509–3513. doi: 10.1002/eji.1830251245. [DOI] [PubMed] [Google Scholar]
- Shu HB, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- Singer GG, Carrera AC, Marshak-Rothstein A, Martinez C, Abbas AK. Apoptosis, Fas and systemic autoimmunity: the MRL-lpr/lpr model. Curr Opin Immunol. 1994;6:913–920. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Ottilie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, et al. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/ TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- Steinert P, Zackroff R, Aynardi-Whitman M, Goldman RD. Isolation and characterization of intermediate filaments. Methods Cell Biol. 1982;24:399–419. doi: 10.1016/s0091-679x(08)60667-6. [DOI] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]