E-cadherin regulates cell growth by modulating proliferation-dependent β-catenin transcriptional activity (original) (raw)
Abstract
β-Catenin is essential for E-cadherin–mediated cell adhesion in epithelial cells, but it also forms nuclear complexes with high mobility group transcription factors. Using a mouse mammary epithelial cell system, we have shown previously that conversion of epithelial cells to a fibroblastoid phenotype (epithelial-mesenchymal transition) involves downregulation of E-cadherin and upregulation of β-catenin transcriptional activity. Here, we demonstrate that transient expression of exogenous E-cadherin in both epithelial and fibroblastoid cells arrested cell growth or caused apoptosis, depending on the cellular E-cadherin levels. By expressing E-cadherin subdomains, we show that the growth-suppressive effect of E-cadherin required the presence of its cytoplasmic β-catenin interaction domain and/or correlated strictly with the ability to negatively interfere with β-catenin transcriptional activity. Furthermore, coexpression of β-catenin or lymphoid enhancer binding factor-1 or T cell factor 3 with E-cadherin rescued β-catenin transcriptional activity and counteracted E-cadherin–mediated cell cycle arrest. Stable expression of E-cadherin in fibroblastoid cells decreased β-catenin activity and reduced cell growth. Since proliferating cells had a higher β-catenin activity than G1 phase–arrested or contact-inhibited cells, we conclude that β-catenin transcriptional activity is essential for cell proliferation and can be controlled by E-cadherin in a cell adhesion-independent manner.
Keywords: carcinogenesis; catenins; cell proliferation; E-cadherin; LEF-1
Introduction
Cadherins represent a distinct family of single transmembrane domain glycoproteins that mediate calcium-dependent cell–cell adhesion via homophilic interactions of their NH2-terminal ectodomains (Shapiro et al., 1995; Gumbiner, 2000). The intracellular domain of E-cadherin associates with a protein family collectively termed catenins (Ozawa et al., 1989; Nathke et al., 1994). β-catenin and γ-catenin (plakoglobin) interact directly with E-cadherin's COOH-terminal domain in a mutually exclusive way, and both proteins associate with α-catenin, which links the cadherin complexes to the actin cytoskeleton and mediates stable cell adhesion. p120ctn is another catenin family member (Anastasiadis and Reynolds, 2000), which binds to the cytoplasmic juxtamembrane portion of E-cadherin and influences E-cadherin clustering and adhesive strength (Ozawa and Kemler, 1998; Yap et al., 1998; Aono et al., 1999; Ohkubo and Ozawa, 1999; Thoreson et al., 2000).
In addition to its adhesive functions, β-catenin has also been found to serve as a key component in signaling processes during embryonic development and adult tissue homeostasis. A series of genetic and biochemical studies have identified β-catenin as a central player of the wnt/wingless signal transduction cascade, which is important for pattern formation and axis determination during early development of invertebrate and vertebrate organisms (Kelly et al., 1995; Cox et al., 1996; Willert and Nusse, 1998; Huelsken et al., 2000). In the absence of wnt ligand, cytoplasmic β-catenin is part of a supramolecular complex containing axin/conductin and the adenomatous polyposis coli tumor suppressor protein and is NH2-terminally phosphorylated by glycogen synthase kinase 3β (Rubinfeld et al., 1996; Yost et al., 1996; Behrens et al., 1998; Ikeda et al., 1998), which targets the protein for proteosomal degradation (Aberle et al., 1997; Orford et al., 1997). The binding of wnt to its cognate receptor frizzled triggers a cascade of signaling events that led to the inhibition of glycogen synthase kinase 3β–dependent phosphorylation of β-catenin. Stabilized hypophosphorylated β-catenin accumulates in the cytoplasm and the nucleus and interacts with high mobility group box containing transcription factors of the lymphoid enhancer binding factor (LEF)*-1/T cell factor (TCF) family (Behrens et al., 1996; Huber et al., 1996; Molenaar et al., 1996). The nuclear β-catenin–LEF/TCF complex can transactivate transcription by binding to coactivators, such as histone acetylase p300/CBP (Hecht et al., 2000; Sun et al., 2000; Takemaru and Moon, 2000) and the TATA-binding protein TBP (Hecht et al., 1999), or can repress transcription by interaction with repressors such as groucho-related gene 5 (Roose et al., 1998). Both transactivation and repressive functions of the β-catenin–LEF/TCF complex are highly regulated and control the expression of a diverse set of target genes during development and tissue formation in invertebrates and vertebrates (for reviews see Roose and Clevers, 1999; Hecht and Kemler, 2000; Zhurinsky et al., 2000b).
Inappropriate activation of the wnt pathway was shown to drive cell proliferation and tumor formation (Korinek et al., 1997; Morin et al., 1997; Rubinfeld et al., 1997; for reviews see Barker and Clevers, 2000; Bienz and Clevers, 2000; Polakis, 2000). Increased β-catenin transcriptional activity upon overexpression of stable activated β-catenin in the intestine (Harada et al., 1999) or of truncated β-catenin in the skin (Gat et al., 1998) of transgenic mice led to the development of intestinal polyps and hair tumors, respectively.
In addition, the development of invasive tumors from epithelial tissues frequently involves the conversion of epithelial cells to a mesenchymal fibroblastoid phenotype (epithelial-mesenchymal transition [EMT]), which is characterized by downregulation of E-cadherin (Birchmeier et al., 1996; Christofori and Semb, 1999). In line with this hypothesis, ectopic expression of E-cadherin in a transgenic mouse model of pancreatic β-cell carcinogenesis arrested tumor development (Perl et al., 1998), and E-cadherin expression in mammary carcinoma cells was found to inhibit cell growth (St. Croix et al., 1998). We have found recently that EMT was also accompanied by an upregulation of β-catenin/LEF-1 activity (Eger et al., 2000). Since E-cadherin is known to negatively regulate β-catenin transcriptional activity by recruiting β-catenin from transcriptional complexes (Heasman et al., 1994; Fagotto et al., 1996; Sadot et al., 1998; Simcha et al., 1998; Orsulic et al., 1999; Eger et al., 2000), cellular E-cadherin levels might regulate LEF-1/TCF activity and thus be crucial for regulating cell proliferation and neoplastic transformation. However, a direct relationship between E-cadherin expression and β-catenin signaling in the control of cell proliferation has not been demonstrated yet.
Mouse mammary epithelial cells expressing an estrogen-inducible cFos estrogen receptor fusion protein (FosER) are epithelial and express E-cadherin but upon estrogen treatment convert to fibroblastoid cells that are devoid of E-cadherin (Reichmann et al., 1992). Using FosER cells before and after estradiol-induced EMT (Eger et al., 2000), we show here that ectopic expression of E-cadherin in both epithelial and fibroblastoid cells resulted in retardation of cell growth and G1 phase arrest, depending on the expression level of exogenous E-cadherin. Expression of E-cadherin subdomains revealed that the growth suppressive effect is cell adhesion-independent and strictly correlated with the downregulation of β-catenin/LEF activity.
Results
Ectopic expression of E-cadherin blocks cell cycle progression in epithelial and fibroblastoid cells
To test the relationship between E-cadherin expression and β-catenin/LEF signaling during cell cycle progression, we used the FosER cell system (Reichmann et al., 1992) for two reasons. First, the cellular phenotype is controlled by estradiol-dependent activity of a constitutively expressed cFos–estrogen receptor chimeric protooncogene. Whereas cells form highly polarized epithelial cell sheets in the absence of estradiol, estradiol-induced activation of FosER induces EMT, leading to fibroblastoid cells that lack E-cadherin and exhibit increased β-catenin/LEF-1 transcriptional activity (Eger et al., 2000). This allowed us to analyze the effects of ectopic E-cadherin expression on cell proliferation in two different cellular phenotypes of a single cell clone, expressing or lacking endogenous E-cadherin. Second, we have shown previously that transient expression of E-cadherin in fibroblastoid and epithelial FosER cells significantly reduced β-catenin/LEF transcriptional activity (Eger et al., 2000).
Upon transient expression of myc- or GFP-tagged full-length E-cadherin in epithelial FosER cells, we observed two major phenotypes. Cells expressing exogenous E-cadherin at a very low level revealed a nearly exclusive localization of the tagged protein at the lateral plasma membrane (Fig. 1A), similar to the localization of the endogenous E-cadherin. Higher expression levels led to an additional cytoplasmic localization of the ectopic E-cadherin, presumably representing E-cadherin localized in the ER, the Golgi, or in vesicles on their way to the plasma membrane (Fig. 1 B; Adams et al., 1998). Ectopically expressed E-cadherin colocalized with endogenous β-catenin at the plasma membrane, suggesting that it is incorporated into functional cell–cell adhesion complexes (Fig. 1 A′), but high levels of ectopic E-cadherin led to cytoplasmic accumulation of endogenous β-catenin most likely due to the formation of cytoplasmic E-cadherin–β-catenin complexes (Fig. 1 B′). In fibroblastoid FosER cells, expression of exogenous E-cadherin at moderate levels resulted in its predominantly peripheral localization at the plasma membrane and a partial recruitment of β-catenin to the cellular periphery (Fig. 1 C). Higher expression levels resulted in increased cytoplasmic E-cadherin and a strong upregulation of endogenous β-catenin levels in the cytoplasm (Fig. 1 D). In agreement with previous studies (Gottardi et al., 2001) showing that E-cadherin expression in colon cancer cells did not change β-catenin's nuclear localization, although it interfered with β-catenin transcriptional activity, we did not observe a clear depletion of β-catenin from the nucleus in E-cadherin–expressing cells.
Figure 1.

Transient expression of ectopic E-cadherin. Epithelial and fibroblastoid FosER cells were transfected with an expression plasmid encoding full-length myc-tagged E-cadherin and processed for double immunofluorescence microscopy 24 h after transfection using antibodies to the myc tag (A–D) and β-catenin (A′–D′). Confocal images are shown. Bar, 10 μm.
To analyze the effect of ectopic E-cadherin expression on cell cycle progression, we determined the percentage of transfected cells in S phase by testing for the incorporation of BrdU into cellular DNA. 48 h after transfection of cells with E-cadherin constructs or with GFP vectors alone, nuclei of most cells expressing high levels of E-cadherin did not stain for BrdU, whereas a large number of nuclei in nontransfected cells in the same preparation (Fig. 2A, top) or in GFP-expressing control cells (unpublished data) were stained brightly with the BrdU antibody. Statistical analysis of these data revealed that ∼70% of epithelial and fibroblastoid cells transfected with the GFP construct alone incorporated BrdU independent of GFP expression levels (Fig. 2 B). In contrast, upon moderate expression of E-cadherin in epithelial cells (defined by mostly peripheral localization of E-cadherin; Fig. 1 A) only 20% of cells exhibited BrdU-specific staining (Fig. 2 B, Epithelial FosER, white bar), and almost no BrdU incorporation could be detected in epithelial cells expressing high levels of the protein in the cytoplasm (Fig. 2 B, Epithelial FosER, black bar). In fibroblastoid cells, the decrease in BrdU incorporation was less efficient than in epithelial cells, yielding a reduction of BrdU-positive cells to 40% upon low level expression of E-cadherin and to ∼5% in highly expressing cells (Fig. 2 B, Fibroblastoid FosER, white and black bars). Thus, E-cadherin expression led to a significant reduction in DNA synthesis, suggesting that increased cellular E-cadherin levels might cause a growth arrest in the G1 phase of the cell cycle. To directly prove G1 phase arrest, we stained transfected cells with an antibody to the cyclin-dependent kinase inhibitor p27KIP1, which has been found to be upregulated upon E-cadherin–induced growth suppression (St. Croix et al., 1998). As shown in Fig. 2 A (bottom), p27KIP1 was significantly upregulated exclusively in nuclei of cells expressing high levels of E-cadherin.
Figure 2.
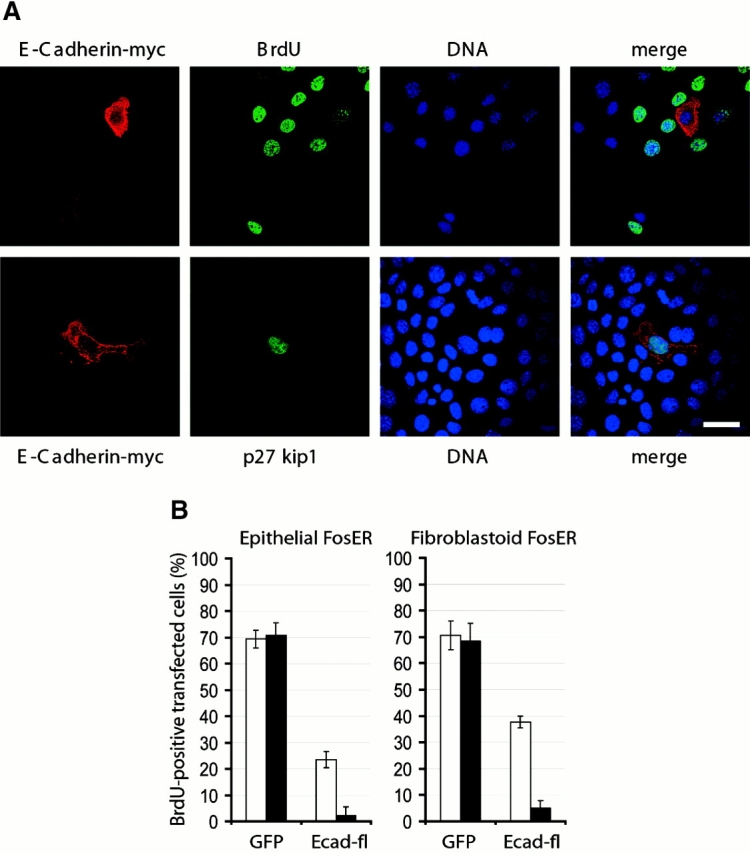
Expression of E-cadherin leads to G1-S phase arrest. Epithelial and fibroblastoid FosER cells were transfected with expression plasmids encoding full-length GFP- or myc-tagged E-cadherin and analyzed for BrdU incorporation and/or double immunofluorescence microscopy 24 h after transfection. (A) Confocal images of cells stained with antibodies to myc and BrdU (top) or to myc and p27KIP1 (bottom) and with the DNA dye Hoechst are shown. (B) Cells expressing E-cadherin–GFP or GFP alone as a control were analyzed for BrdU incorporation, and the percentage of BrdU-positive cells of all GFP-positive cells was determined by statistical analysis (>500 cells). Data represent mean values of at least three independent experiments; SD is shown. White bars, weak expression; black bars, strong expression of E-cadherin or GFP. Bar, 10 μm.
Prolonged expression of ectopic E-cadherin leads to apoptosis
To test whether ectopic E-cadherin–mediated G1 phase arrest is a transient or constitutive effect and to analyze the fate of transfected cells upon prolonged cultivation, we transiently transfected epithelial and fibroblastoid FosER cells with E-cadherin–GFP fusion constructs or GFP alone and followed the number of transfected GFP-positive cells over a period of 4 d after transfection by FACS® analysis. When compared with GFP-expressing cells (set to 100%), the number of GFP–E-cadherin–positive epithelial cells dropped dramatically to a value close to zero after 4 d (Fig. 3A). The number of fibroblastoid cells expressing E-cadherin–GFP was also strongly reduced within 4 d, but as in the BrdU incorporation experiments the reduction was less efficient than in epithelial cells. The decrease in E-cadherin–GFP–expressing cells during prolonged cultivation was consistent with a G1 phase arrest of transfected cells, but the dramatic downregulation within the short time period could not be explained by growth arrest alone. Since the stability of GFP and E-cadherin–GFP fusion proteins were similar (unpublished data), we reasoned that G1 phase–arrested cells might enter apoptosis. Indeed, >50% of cells highly overexpressing E-cadherin showed plasma membrane blebbing and nuclear fragmentation 24–48 h after transfection and were shown by TUNEL staining to undergo apoptosis (Fig. 3 B). After 4 d, only cells expressing low levels of E-cadherin were detected, and <10% of those were apoptotic. Thus, cell cycle arrest by ectopic E-cadherin apparently resulted in cell death upon prolonged cultivation.
Figure 3.

Prolonged expression of ectopic E-cadherin caused apoptosis. Epithelial and fibroblastoid FosER cells were transfected with expression plasmids encoding full-length GFP- or myc-tagged E-cadherin, GFP-cadherin and β-catenin, or GFP as control. (A) GFP and E-cadherin–GFP–expressing cells were analyzed by FACS® within 4 d after transfection. The graph shows the percentage of E-cadherin–GFP– positive cells relative to GFP-positive cells (set to 100%) at each time point. Mean values and SD of at least three independent experiments are shown. (B) Epithelial FosER cells expressing ectopic E-cadherin were analyzed for apoptosis 48 h after transfection using the TUNEL assay and processed for immunofluorescence microscopy using myc antibody to visualize E-cadherin. Confocal images are shown. Bar, 10 μm.
The growth-suppressive effect of E-cadherin is mediated by domains affecting β-catenin transcriptional activity and is cell adhesion independent
To determine the domains of E-cadherin responsible for growth suppression and apoptosis, we generated E-cadherin deletion mutants, missing either the cytoplasmic juxtamembrane p120ctn binding domain (Ecad-βcat) or the COOH-terminal β-catenin binding domain (Ecad-p120), but both constructs contained the complete extracellular NH2-terminal ectodomains and the transmembrane region (Fig. 4A). In addition, we generated nonmembrane-anchored E-cadherin fragments, representing either the p120ctn (BD-p120) or the β-catenin (BD-βcat) binding domains. Upon transient transfection, tagged E-cadherin fragments were expressed at significant levels and demonstrated the expected molecular weights as shown by immunoblotting of total cell lysates using anti-myc or anti-GFP antibodies (Fig. 4 B, asterisks). Unlike the membrane-anchored E-cadherin fragment missing the β-catenin binding site (Fig. 4 C, c, Ecad-p120), the β-catenin–binding E-cadherin fragment (Ecad-βcat) localized to the plasma membrane and led to increased β-catenin levels in both epithelial and fibroblastoid cells and caused a partial translocation of β-catenin from the cytoplasm to the membrane in fibroblastoid cells (Fig. 4 C, a). The overexpressed cytoplasmic E-cadherin domains, BD-βcat and BD-p120, were distributed more diffusely throughout the cytoplasm and nucleus (Fig. 4 C, b and d), and as expected the β-catenin fragment (Fig. 4 C, b, BD-βcat) stabilized endogenous β-catenin.
Figure 4.
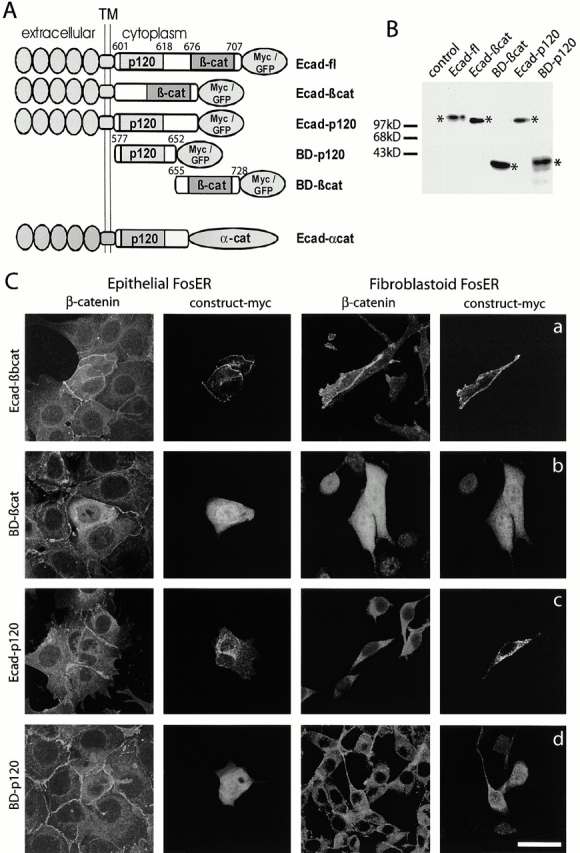
Expression of E-cadherin deletion mutants in epithelial and fibroblastoid FosER cells. (A) Schematic drawing of E-cadherin deletion mutants. p120 and β-cat denote interaction domains of E-cadherin with p120ctn and β-catenin, respectively. TM, transmembrane domain; numbers, position of amino acid in processed E-cadherin (B). Fibroblastoid FosER cells were transfected with plasmids encoding myc-tagged E-cadherin fragments. Cell lysates were prepared 24 h after transfection and analyzed by immunoblotting using antibody to myc. Asterisks denote position of construct. Control, nontransfected cells. (C) Transfected cells were processed for double immunofluorescence microscopy 24 h after transfection using antibodies to myc and to β-catenin. Confocal images are shown. Bar, 10 μm.
To test the effects of transiently expressed E-cadherin mutants on cell cycle progression, we determined BrdU incorporation 48 h after transfection. In fibroblastoid cells lacking endogenous E-cadherin, both constructs containing the β-catenin binding site with (Ecad-βcat) or without the transmembrane domain (BD-βcat) clearly reduced the number of BrdU-positive cells compared with GFP-expressing cells in a dose-dependent manner (Fig. 5A, right), whereas E-cadherin fragments missing the β-catenin binding site (Ecad-p120; BD-p120) had no significant effect. Furthermore, analysis of β-catenin transcriptional activity using the TOPFLASH luciferase reporter construct (Korinek et al., 1997) revealed that exclusively the β-catenin–binding E-cadherin fragments causing cell cycle arrest also significantly reduced β-catenin/LEF-1 activity (Fig. 5 B). Thus, the negative effect of E-cadherin fragments on cell proliferation is likely mediated by their interference with the signaling function of endogenous β-catenin and is independent of their function in cell adhesion. To support this hypothesis, we expressed a chimeric E-cadherin–α-catenin protein missing the β-catenin binding site, which is active in mediating cell adhesion but does not interfere with β-catenin signaling in colon carcinoma cells (Fig. 4 A; Gottardi et al., 2001). As expected, the chimeric protein reduced neither BrdU incorporation nor β-catenin activity in fibroblastoid cells.
Figure 5.
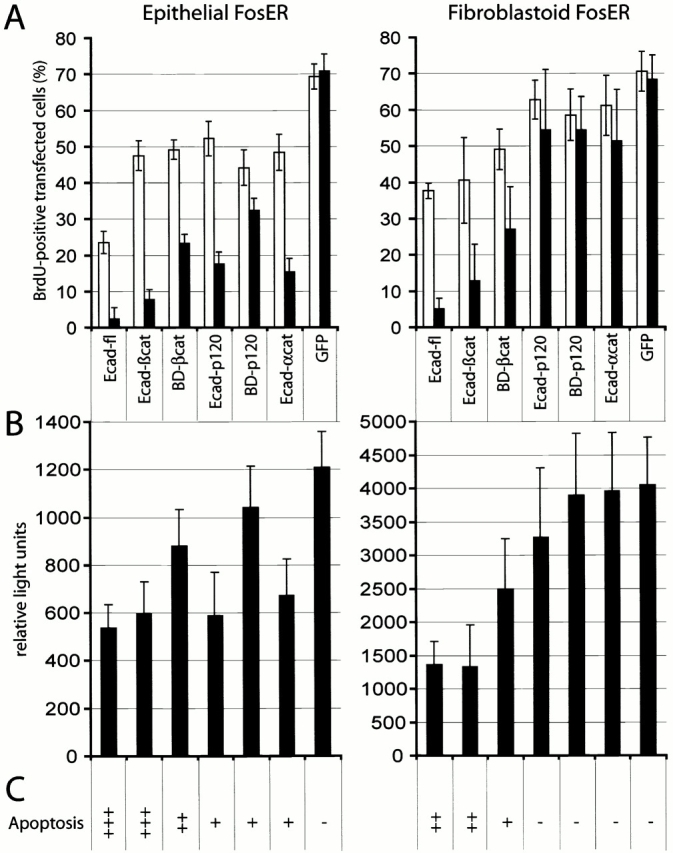
Growth-suppressive effect of E-cadherin fragments is strictly correlated with downregulation of β-catenin/LEF-1 transcriptional activity. Epithelial and fibroblastoid FosER cells were transfected with expression plasmids for GFP-tagged full-length E-cadherin, E-cadherin deletion mutants, E-cadherin–α-catenin chimera, or with GFP alone and analyzed for BrdU incorporation (A) and TOPFLASH activity (B) 24 h after transfection. (A) BrdU incorporation as in Fig. 2, depicting cells with weak (white bar) and strong (black bar) expression of ectopic proteins. (B) Cells were transfected with LEF-1–dependent TOPFLASH luciferase reporter construct together with expression plasmids encoding E-cadherin fragments and cytomegalovirus β-galactosidase reporter plasmid. Luciferase activities were normalized for β-galactosidase control. Mean values and SD of at least three independent experiments are shown. (C) Apoptosis in transfected cells was analyzed by microscopy. +++, >50%; ++, 30–50%; +, <30%; −, <1% of transfected cells were apoptotic.
In epithelial cells containing endogenous E-cadherin–β-catenin complexes, the effects of E-cadherin fragments seemed to be more complex, since all E-cadherin fragments, including those missing the β-catenin binding site, showed a significant reduction of BrdU incorporation and caused reduction in β-catenin transcriptional activity. This can be explained by the fact that these fragments may interfere with endogenous adhesion complexes and indirectly affect transcriptional activity of endogenous β-catenin (see Discussion).
Taken together, these experiments showed that the ability of E-cadherin fragments to arrest cell cycle progression correlated strictly with their effects on β-catenin signaling activity. Furthermore, the ability of E-cadherin fragments to interfere with β-catenin activity correlated nicely with the induction of apoptosis (Fig. 5 C). This raised the interesting possibility that β-catenin transcriptional activity may be important for cell cycle progression.
E-cadherin's growth-inhibiting activity is counteracted by increased β-catenin activity
If β-catenin's transcriptional activity is essential for cell proliferation, an artificial increase of β-catenin activity in E-cadherin–overexpressing cells should release them from cell cycle arrest. As shown in Fig. 6A, E-cadherin overexpression in epithelial FosER cells caused a significant (≤50%) reduction of endogenous β-catenin/LEF-1 activity, but coexpression of ectopic β-catenin or LEF-1 or TCF-3 rescued or even increased β-catenin activity compared with the control.
Figure 6.

Upregulation of β-catenin activity rescues E-cadherin–mediated growth arrest. Epithelial FosER cells were transfected with reporter constructs and with constructs encoding E-cadherin alone or together with constructs expressing β-catenin or LEF-1 or TCF-3. 30 h after transfection, cells were lysed and analysed for β-catenin/LEF-1 reporter gene activity (A) as in Fig. 5 or E2F reporter gene activity (B). Graphs show mean relative light units of reporter gene activities (plus SD) normalized for β-galactosidase activity from at least three independent experiments. Control in B represents activity in asynchronously growing cells.
To test whether rescue of β-catenin/LEF activity can also rescue cell cycle progression, we used a reporter construct containing multiple E2F binding sites in front of the luciferase gene, which was shown to be activated during G1-S phase transition in proliferating cells (Krek et al., 1993). The ectopic E-cadherin–mediated cell cycle arrest is reflected by a reduction of E2F-dependent reporter activity to ∼60% of the control activity in mock-transfected cells (Fig. 6 B). Coexpression of β-catenin or LEF-1 or TCF-3 together with E-cadherin rescued E2F-dependent transcription to values close to the control or approximately twofold higher levels than the control. Thus, an increase in β-catenin–dependent transcriptional activity can overcome E-cadherin–mediated cell cycle arrest. This conclusion was further supported by the observation that coexpression of β-catenin with E-cadherin–GFP partially rescued the steep decrease in the number of GFP-positive cells seen in E-cadherin–GFP–expressing cells within 4 d of cultivation after transfection (Fig. 3 A, broken line), indicating reduced apoptosis in these cells.
β-Catenin transcriptional activity is upregulated in proliferating versus arrested cells
If β-catenin activity is required for cell cycle progression, the endogenous activity should undergo cell cycle–dependent changes. First, we analyzed endogenous β-catenin transcriptional activity in subconfluent, proliferating versus dense, contact-inhibited cultures. When epithelial cells were grown at low densities (≤40% confluency), they exhibited an increased β-catenin–LEF-1/TCF activity, which was five- to sevenfold reduced when cells reached a confluency of >80% (Fig. 7A). A similar dependence of β-catenin–LEF-1/TCF–dependent reporter activity on cell confluency was detected in fibroblastoid cells except that the overall values of β-catenin transcriptional activity were up to tenfold higher than in epithelial FosER cells. To account for any unspecific changes in basic transcriptional activity during different cell cycle stages, all β-catenin/LEF-1 activities were related to respective reporter activities obtained with a control reporter containing mutated LEF-1 binding sites unable to bind LEF-1 (FOPFLASH).
Figure 7.
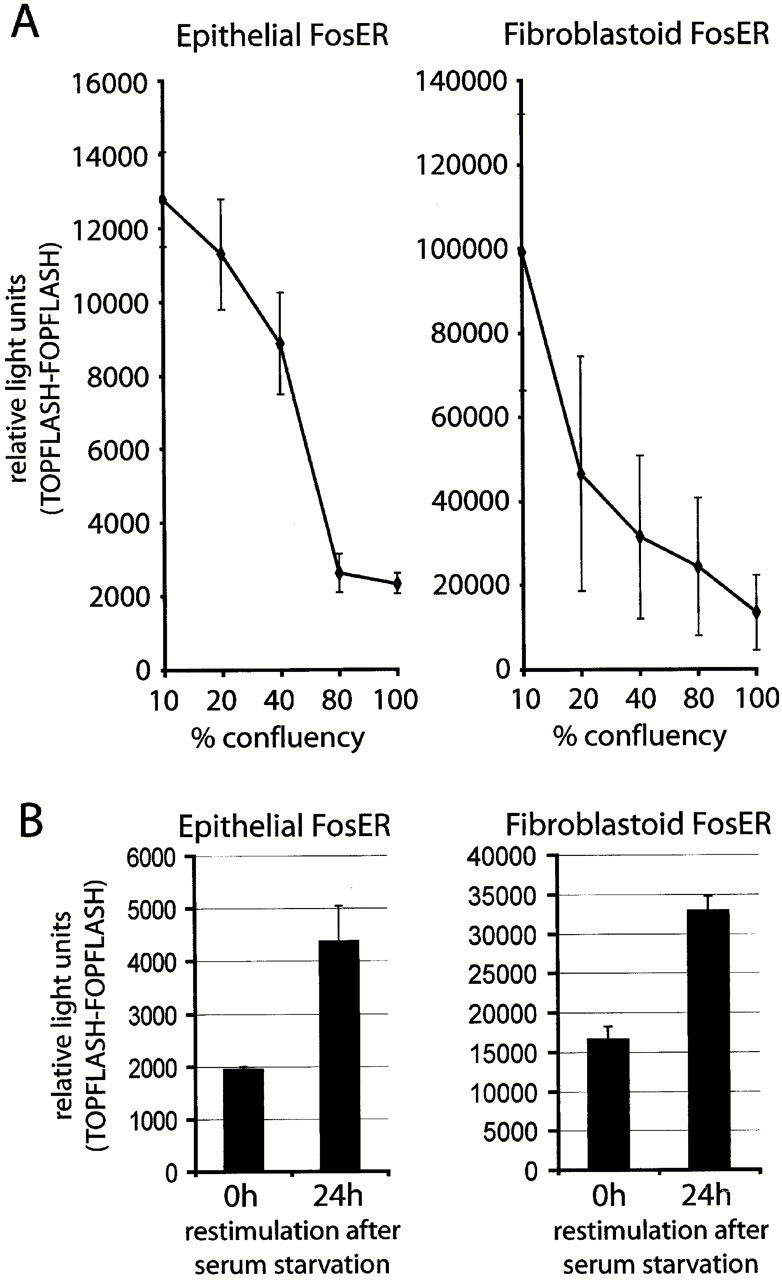
β-Catenin/LEF-1 activity depends on cell proliferation. (A) Epithelial and fibroblastoid FosER cells were transfected with TOPFLASH or FOPFLASH luciferase and β-galactosidase reporter constructs at different cell densities and analyzed after 30 h. (B) FosER cells were transfected with reporter constructs and starved for 48 h in medium without FCS and restimulated for 24 h in complete medium before analysis. Data obtained were normalized for β-galactosidase activity, and mean values of normalized TOPFLASH minus FOPFLASH activities of at least three independent experiments (and SD) are shown.
In a second set of experiments, we arrested cell proliferation by serum starvation of subconfluent epithelial and fibroblastoid FosER cells for 48 h and released them from the cell cycle block for up to 24 h by addition of serum. β-Catenin/LEF-1 activity increased more than twofold upon serum stimulation (Fig. 7 B), confirming a strict correlation of cell proliferation and β-catenin signaling.
Stable expression of ectopic E-cadherin reduced cell proliferation in fibroblastoid cells
Our data suggest that the relative expression levels of E-cadherin versus β-catenin control β-catenin transcriptional activity and cell cycle progression. To address this hypothesis in more detail, we generated fibroblastoid FosER cell clones stably expressing ectopic E-cadherin. Immunofluorescence microscopy revealed that stable E-cadherin–expressing cells reverted to a typical epithelial morphology with a peripheral localization of ectopic E-cadherin at cell contacts (Fig. 8A). Furthermore, endogenous β-catenin was translocated from the cytoplasm and nucleus in fibroblastoid cells to E-cadherin containing cell–cell contacts. Although the expression levels of ectopic E-cadherin in three independent stable clones were found to be <50% of the amount of endogenous E-cadherin in epithelial cells (Fig. 8 B), β-catenin activity was reduced up to fivefold in E-cadherin–expressing clones versus fibroblastoid cells (Fig. 8 C). The analysis of cell proliferation rates (Fig. 8 D) indicated that the three independent E-cadherin–expressing fibroblastoid cell clones grew significantly slower than the E-cadherin–deficient parental fibroblastoid cells, exhibiting generation times of 20.5, 21.2, and 22.9 h versus 16.2 h. Growth retardation may be mediated by a decrease in protein (Fig. 8 E) and mRNA (unpublished data) levels of the positive cell cycle regulator cyclin D1, whose transcription may be directly controlled by β-catenin (Tetsu and McCormick, 1999), and the upregulation of a negative cell cycle regulator, the cdk inhibitor p27KIP1 (St. Croix et al., 1998) (Fig. 8 E). E-cadherin did not increase apoptosis in these clones (unpublished data). The reduction in growth rates upon ectopic expression of E-cadherin in fibroblastoid cells is consistent with the role of β-catenin signaling in cell proliferation and its negative control by E-cadherin.
Figure 8.
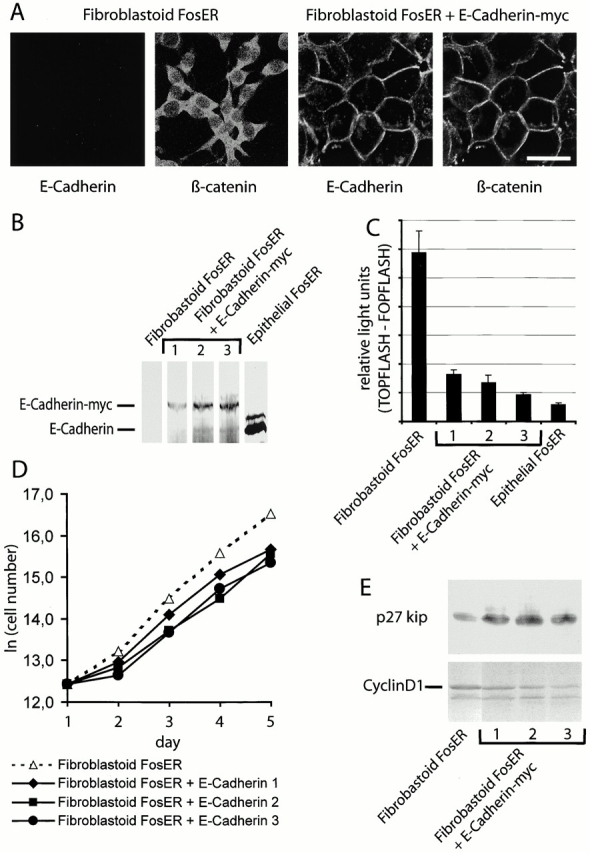
Stable expression of E-cadherin in fibroblastoid FosER cells reduces cell proliferation rate. Fibroblastoid FosER cells were stably transfected with a construct expressing myc-tagged E-cadherin, and three clones were selected by hygromycin resistance. (A) Confocal immunofluorescence images of fibroblastoid cells and stable E-cadherin–expressing cells stained with antibodies to E-cadherin and β-catenin. (B) Cell lysates of stable clones and of fibroblastoid and epithelial FosER cells as control were analyzed by immunoblotting using antibodies to E-cadherin. (C) TOPFLASH minus FOPFLASH reporter activities were measured as described in the legend to Fig. 7. (D) Growth curves of fibroblastoid FosER cells and stable clones were determined by cell counting in a Thoma chamber. Cell numbers were plotted as values against cultivation time to indicate doubling times_._ (E) Immunoblot analysis of total cell lysates using antibodies to cyclin D1 and p27KIP1. Bar, 10 μm.
Discussion
The studies presented here revealed novel aspects on the function and interaction of the well-studied proteins E-cadherin and β-catenin. Our data suggest that β-catenin/LEF-1 transcriptional activity, which has been implicated previously in inducing hyperproliferation in various tumors (Barker and Clevers, 2000), also plays a role in controlled cell proliferation in nontransformed cultured mammary epithelial cells and is regulated by physiological stimuli such as contact inhibition or withdrawal of serum growth factors. Also, we found that E-cadherin downregulated β-catenin/LEF activity and negatively affected cell proliferation in a cell adhesion-independent manner. In previous studies, E-cadherin has been implicated in contact inhibition-induced cell growth arrest in epithelial cells (St. Croix et al., 1998), which is dependent on cell adhesion. Only very recently was a similar cell adhesion-independent effect of E-cadherin on cell proliferation described in SW 480 colon carcinoma cells (Gottardi et al., 2001). These findings have major implications for E-cadherin's role in carcinogenesis. Loss of E-cadherin expression seen in many tumors of epithelial origin (Christofori and Semb, 1999) may have a dual effect, that is, loss of cell adhesion favoring motility and invasion and induction of cell proliferation by upregulation of β-catenin/LEF activity.
Is β-catenin activity essential for cell proliferation?
β-catenin–LEF/TCF signaling has been demonstrated previously to drive cell proliferation during tumor formation by turning on the expression of c-myc (He et al., 1998) and the cell cycle regulator cyclin D1 (Shtutman et al., 1999; Tetsu and McCormick, 1999), whose expression is also regulated in the cell system used in this study. In addition to various tumor models in transgenic mice, where β-catenin/LEF/TCF activity is upregulated in specific tissues (Gat et al., 1998; Harada et al., 1999; Roose et al., 1999) overexpression of β-catenin in cultured epithelial cells (Kolligs et al., 1999; Orford et al., 1999) or the expression of chimeras of LEF-1 fused to VP16-transactivating domain in chicken embryo fibroblasts (Aoki et al., 1999) was found to induce cell proliferation and neoplastic transformation.
Our data that β-catenin activity is increased in proliferating versus arrested cells and that an increase in β-catenin activity can overcome a cell cycle arrest indicate that regulation of β-catenin activity may be a more general mechanism controlling cell proliferation in mammary epithelial cells, not necessarily correlated with neoplastic transformation. This hypothesis is supported by the finding that TCF-4−/− mice exhibited a defect in the proliferation of epithelial precursors in the colon (Korinek et al., 1998). On the other hand, elimination of β-catenin by gene knockout in mice resulted in embryonic lethality due to an inability to form dorsal structures (Huelsken et al., 2000). These defects reflect the importance of β-catenin in wnt-mediated axis formation, but cell proliferation did not seem to be affected at this stage of development. There are two possible explanations for this observation. First, β-catenin–LEF/TCF–mediated cell proliferation might become important only at later stages of development when differentiated cells have to proliferate during homeostasis and reorganization of tissues such as in mammary gland or colon epithelia. Second, plakoglobin, which is upregulated in β-catenin–deficient cells and compensates for the adhesive role of β-catenin in cell junctions (Haegel et al., 1995; Huelsken et al., 2000), might also compensate for β-catenin's role in cell proliferation. Although plakoglobin only inefficiently forms a complex with LEF/TCF and DNA (Zhurinsky et al., 2000a), plakoglobin-dependent transcriptional activity may be favored in the absence of β-catenin. Interestingly, plakoglobin was found to possess transforming activity, which was dependent on LEF/TCF function and unlike β-catenin strongly activated c-Myc expression in a rat kidney epithelial cell line (Kolligs et al., 2000).
In any case, other signaling molecules or pathways are likely to cooperate with β-catenin to induce cell proliferation in different cell types. Since expression of wnt but not the expression of β-catenin in immortalized Rat-1 fibroblasts caused a proliferative response (Young et al., 1998), additional β-catenin–independent wnt-mediated pathways may be important.
Molecular mechanism of E-cadherin–mediated cell cycle control
Our studies demonstrate that E-cadherin can inhibit cell cycle progression and link its growth suppressive effect to the downregulation of β-catenin activity. E-cadherin's proliferation-inhibiting effect has been described in several previous studies in vivo and in vitro (Watabe et al., 1994; Miyaki et al., 1995; Kandikonda et al., 1996; Takahashi and Suzuki, 1996; St. Croix et al., 1998), but the molecular mechanisms of this phenomenon remained mostly elusive.
We show that E-cadherin's growth-repressive effect is clearly dependent on its ability to downregulate β-catenin activity and that this effect is not dependent on the cell adhesion function of E-cadherin. The latter conclusion is based on our findings that cell adhesion inactive E-cadherin fragments, such as the cytoplasmic β-catenin binding domain, clearly reduced β-catenin activity and cell growth, whereas an E-cadherin–α-catenin chimera, which mediates strong cell adhesion but has no effect on β-catenin activity in fibroblastoid cells (Gottardi et al., 2001), did not affect cell proliferation.
Although E-cadherin's cell adhesion activity is not required for cell cycle regulation, it might contribute to this process based on the following observations. First, subconfluent, proliferating and dense, contact-inhibited epithelial cells express similar E-cadherin levels but exhibit different β-catenin–LEF/TCF activities. We speculate that the formation of E-cadherin–mediated cell contacts in dense cultures may stabilize E-cadherin–catenin complexes (Adams et al., 1998) and thereby may reduce the free cytoplasmic pool of β-catenin available for transcriptional activity. This mechanism might cooperate with an E-cadherin– and cell adhesion-independent mechanism, controlling β-catenin activity and allowing a tight control of cell proliferation. In contrast, fibroblastoid FosER cells lacking E-cadherin can only activate E-cadherin–independent pathways, and β-catenin–dependent cell proliferation is less tightly controlled reflected by an overall tenfold higher β-catenin activity than in epithelial cells and the ability of cells to grow in multilayers (Eger et al., 2000).
Second, expression of E-cadherin fragments that increase and/or stabilize cell adhesion in epithelial cells, such as E-cadherin fragments containing the p120ctn binding site (see below) or the E-cadherin–α-catenin chimera (Gottardi et al., 2001), indirectly caused downregulation of β-catenin activity and cell cycle arrest by recruiting β-catenin to cell junctions. Together, these data indicate that β-catenin/LEF–dependent cell proliferation is controlled by a finely tuned balance between free E-cadherin–β-catenin (not engaged in contacts), E-cadherin–β-catenin adhesion complexes, and free β-catenin within a narrow expression window. This balance can be regulated by several means including the change in protein expression, formation of stable E-cadherin–β-catenin cell junctions, and various other signaling pathways in different cell types and tissues. In line with this model, a change in β-catenin activity does not necessarily predict a change in the detectable nuclear localization of β-catenin, since low levels of nuclear β-catenin comprising a small subfraction of the cellular β-catenin pool are sufficient for transcriptional activity (Gottardi et al., 2001).
E-cadherin domains required for growth suppression in different cell types
If the reduction of β-catenin activity is the predominant mechanism by which E-cadherin confers growth arrest, one would expect that ectopic E-cadherin constructs require a functional β-catenin interaction domain to mediate this process. In line with this notion, a similar growth-suppressive effect was observed upon ectopic expression of all β-catenin–binding E-cadherin fragments containing or lacking the transmembrane and extracellular domains. In accordance with the above model, membrane-anchored or cytoplasmic E-cadherin domains lacking the β-catenin binding domain neither mediated downregulation of β-catenin activity nor inhibited cell cycle progression in fibroblastoid FosER cells.
However, interestingly the fragments lacking the β-catenin binding region showed both downregulation of β-catenin activity and growth suppressive effects in epithelial cells. This effect can be explained by the ability of these E-cadherin mutants to interfere with endogenous E-cadherin–β-catenin complexes. Since the β-catenin binding-deficient E-cadherin fragments contained the binding site for p120ctn, it might be argued that their overexpression recruits endogenous p120ctn from endogenous E-cadherin complexes. Since p120ctn has been suggested to negatively effect cell adhesion strength (Ozawa and Kemler, 1998; Aono et al., 1999; for alternative views see Anastasiadis and Reynolds, 2000), depletion of p120ctn from endogenous E-cadherin complexes may lead to increased cell adhesion, stabilization of cell junctions, and recruitment of β-catenin, resulting in significantly lower levels of free signaling-competent β-catenin.
E-cadherin's role during epithelial carcinogenesis
The multiple changes during development of malignant invasive tumors (Hanahan and Weinberg, 2000) include two major events: the initiation of uncontrolled cell proliferation and the loss of cell adhesion, allowing the invasion of surrounding tissues (Birchmeier et al., 1996; Christofori and Semb, 1999). E-cadherin has been viewed as a classical suppressor of invasion because E-cadherin is responsible for stable cell adhesion in epithelial tissues and inhibits cell migration (Larue et al., 1996; Gumbiner, 2000). Observations that E-cadherin can interfere with β-catenin signaling (Sadot et al., 1998; Simcha et al., 1998; Orsulic et al., 1999; Eger et al., 2000) suggested that loss of E-cadherin during carcinogenesis affects not only cell adhesion but causes a switch in gene expression through upregulation of β-catenin–LEF/TCF activity. In line with this hypothesis, the β-catenin–LEF target genes matrilysin and fibronectin were found to be upregulated during EMT (Brabletz et al., 1999; Gradl et al., 1999) and might contribute to cancer progression. Our findings here suggest that loss of E-cadherin and concomitant upregulation of β-catenin–LEF signaling during carcinogenesis might also contribute to increased cell proliferation seen in tumors.
Materials and methods
Cell culture and FACS® analysis
Mouse mammary epithelial cells constitutively expressing FosER (Reichmann et al., 1992) were cultured on plastic dishes as described (Eger et al., 2000). For induction of EMT, cells were cultured in complete medium containing 1 μM β-estradiol (Sigma-Aldrich; Chemie GmbH) for 2–3 wk.
Cells transiently expressing GFP fusion proteins were analyzed by FACS® (FACSCalibur; Becton Dickinson) after trypsinization and suspension in ice-cold PBS/1% FCS.
Constructs
To add a COOH-terminal myc tag to full-length E-cadherin, the plasmid pSK+UM (provided by I. Fialka, Institute of Molecular Pathology) containing the complete cDNA of murine E-cadherin was cut at an internal NheI site and a KpnI site downstream of the stop codon. A PCR fragment containing the internal NheI site of E-cadherin and an external ClaI site instead of the stop codon and a PCR fragment encoding a 6× myc tag and ClaI/KpnI sites at its 5′ and 3′ ends, respectively (derived from pCS2) (Aberle et al., 1997) were inserted into pSK+UM cut with NheI/KpnI. The cDNA encoding complete E-cadherin and the 3′ myc tag were excised from this construct via SpeI/StuI and inserted blunt end into pCS2BC6myc cut with BamHI/StuI or into the GFP tag expression vector pEGFP-N3 (CLONTECH Laboratories, Inc.) blunt end via EcoRI. E-cadherin/myc cDNA cut with HindIII/NotI was cloned into pcDNA3.1/hygro (Invitrogen). cDNAs encoding cytoplasmic E-cadherin fragments were generated by PCR using the following oligonucleotide primers: 5′-CGGGATCCATGGATGAAAACCTGAAGGCAGCCGAC-3′ and 5′-CCATCGATTGTCGTCCTCGCCACCGCCG-3′ for BD-βcat and 5′-CGGGATCCATGCGGAGGAGAACGGTGGTCAAAGAG-3′ and 5′-CCATCGATCAATTTCATCAGGATTGGCAGGACG-3′ for BD-p120. PCR products were cloned into pCS2 in front of the 6× myc tag via external BamHI/ClaI sites. For the E-cadherin constructs lacking the β-catenin (Ecad-p120) or the p120ctn (Ecad-βcat) interaction domains, cDNAs encoding NH2-terminal fragments were generated via PCR using upper primer 5′-CGCGGATAACCAGAACAAAGACC-3′ and lower primers 5′-GCTCTAGACAGAGAGTCGTAAGGGGGTGC-3′ and 5′-GCTCTAGAATAGTAATACACATTGTCCCGGG-3′, respectively, containing external 3′ XbaI sites. The COOH terminal fragments were generated by PCR using upper primers 5′-GCTCTAGACTGAACGAGTGGGGCAACCGATTC-3′ for E-cad120 and 5′-GCTCTAGACACAGGGGCCTGGATGCCCGACCGG-3′ for Ecad-βcat, both containing an external 5′XbaI site, and the lower primer 5′-CCATCGATTGTCGTCCTCGCCACCGCCG-3′ containing an external ClaI site instead of the stop codon. NH2- and COOH-terminal fragments were cloned into pCS2-Ecad in one step using internal BstEII/ClaI. The connection of 5′ and 3′ cDNA fragments by XbaI generated two additional amino acids (ser arg) in the E-cadherin sequence at position 601/602 for processed Ecad-βcat and at position 676/677 for Ecad-p120. For GFP-tagged constructs, the respective pCS2 constructs were cut with HindIII/StuI and put into pEGFP-N3 blunt end via EcoRI.
The plasmid encoding the E-cadherin–α-catenin fusion protein was provided by B. Gumbiner (Memorial Sloan-Kettering Cancer Center, New York, NY) (Gottardi et al., 2001). LEF/TCF-dependent reporter constructs pTOPFLASH or pFOPFLASH, containing the minimal thymidine kinase (TK) promoter, and the LEF-1 and TCF-3 expression vectors were gifts from H. Clevers (University Hospital, Utrecht, Netherlands) (Korinek et al., 1997), and the E2F-dependent reporter construct (Krek et al., 1993) was provided by Hans Rotheneder (Institute of Medical Biochemistry, Vienna Biocenter). The β-catenin expression vector (pCS2+mtMMBC 6× myc) was obtained from R. Kemler (Max Planck Institute for Immunology, Freiburg, Germany) (Aberle et al., 1997).
Transfections and reporter gene assays
Subconfluent epithelial and fibroblastoid FosER cells in a 60-mm plastic dish were transfected with 4 μg plasmid DNA using calcium phosphate or lipofectamine (Life Technologies). Cells were cultivated for 24–96 h after transfection in normal medium before analyses. For stable transfections, fibroblastoid cells transfected with pcDNA3.1/hygro containing full-length myc-tagged E-cadherin were selected in complete medium containing 200 μg/ml hygromycin B.
For reporter gene assays, cells were cotransfected with the luciferase reporter construct, the β-galactosidase control reporter plasmid (pAD-CMV1:βgal), and expression plasmids for E-cadherin, β-catenin, LEF-1, or TCF-3 as described (Eger et al., 2000). Luciferase activity was normalized to β-galactosidase activity as an internal control for transfection efficiency.
Antibodies and immunofluorescence microscopy
The following immunoreagents were used for immunofluorescence microscopy and for immunoblot analyses: mouse monoclonal antibodies to β-catenin, p120ctn, E-cadherin, and p27KIP1 (Transduction Laboratories), antiserum to β-catenin (Sigma-Aldrich), and mouse monoclonal and polyclonal antibodies to c-Myc (CRL-1729; American Type Culture Collection and Santa Cruz Biotechnology, Inc.). Secondary antibodies coupled to peroxidase or alkaline phosphatase were obtained from Promega or Bio-Rad Laboratories, secondary antibodies conjugated to Alexa Fluor 488 were from Molecular Probes, and antibodies coupled to Texas red were from Accurate Chemical & Scientific Corp. or Jackson ImmunoResearch Laboratories.
For immunofluorescence microscopy, cells were fixed with methanol-acetone, stained with primary and secondary antibodies and 0.1 μg/ml Hoechst dye (Eger et al., 2000). Samples were viewed on a ZEISS Axiovert 100M equipped with a LSM 510 confocal microscope.
For analyses of BrdU incorporation, cells were pulsed with 10 μmol/BrdU for 2 h, rinsed with PBS, and fixed with 70% ethanol/50 mM glycine, pH 2.0, for 30 min at −20°C before immunostaining (Boehringer). Promega's apoptosis detection system was used to identify apoptotic cells.
Other procedures
SDS-PAGE and electrotransfer of proteins was done as described previously (Eger et al., 2000). For the immunological detection of the proteins, the Protoblot Immunoscreening system (Promega) or the Super Signal ECL system (Pierce Chemical Co.) was used.
Acknowledgments
We wish to thank Hans Clevers, University Hospital, Utrecht, Netherlands, for providing the TCF-dependent luciferase reporter constructs and the LEF-1 and TCF-3 expression plasmids; Barry Gumbiner, Memorial Sloan-Kettering Cancer Center, New York, NY, for the E-cadherin–α-catenin fusion construct; Hans Rotheneder, Institute of Medical Biochemistry, Vienna Biocenter, for the E2F-dependent reporter gene; Rolf Kemler, Max Planck Institute for Immunology, Freiburg, Germany, for pCS2 β-catenin expression constructs, and Irene Fialka, Institute of Molecular Pathology, for the pSK+UM E-cadherin construct.
This study was supported by grants from the Austrian Science Research Fund (SFB 006) to R. Foisner and H. Beug and from the Hochschuljubiläumsstiftung, City of Vienna, to A. Eger. J. Wolf is a fellow in the International Ph.D. Programme at the Vienna Biocenter supported by the Austrian Science Research Fund (WK001).
Footnotes
*
Abbreviations used in this paper: EMT, epithelial-mesenchymal transition; FosER, cFos estrogen receptor fusion protein; LEF, lymphoid enhancer binding factor; TCF, T cell factor.
References
- Aberle, H., A. Bauer, J. Stappert, A. Kispert, and R. Kemler. 1997. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16:3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, C.L., Y.T. Chen, S.J. Smith, and W.J. Nelson. 1998. Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 142:1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiadis, P.Z., and A.B. Reynolds. 2000. The p120 catenin family: complex roles in adhesion, signaling and cancer. J. Cell Sci. 113:1319–1334. [DOI] [PubMed] [Google Scholar]
- Aoki, M., A. Hecht, U. Kruse, R. Kemler, and P.K. Vogt. 1999. Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid-enhancing factor 1. Proc. Natl. Acad. Sci. USA. 96:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono, S., S. Nakagawa, A.B. Reynolds, and M. Takeichi. 1999. p120(ctn) acts as an inhibitory regulator of cadherin function in colon carcinoma cells. J. Cell Biol. 145:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, N., and H. Clevers. 2000. Catenins, wnt signaling and cancer. Bioessays. 22:961–965. [DOI] [PubMed] [Google Scholar]
- Behrens, J., J.P. von Kries, M. Kuhl, L. Bruhn, D. Wedlich, R. Grosschedl, and W. Birchmeier. 1996. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 382:638–642. [DOI] [PubMed] [Google Scholar]
- Behrens, J., B.A. Jerchow, M. Wurtele, J. Grimm, C. Asbrand, R. Wirtz, M. Kuhl, D. Wedlich, and W. Birchmeier. 1998. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 280:596–599. [DOI] [PubMed] [Google Scholar]
- Bienz, M., and H. Clevers. 2000. Linking colorectal cancer to Wnt signaling. Cell. 103:311–320. [DOI] [PubMed] [Google Scholar]
- Birchmeier, C., W. Birchmeier, and B. Brand-Saberi. 1996. Epithelial-mesenchymal transitions in cancer progression. Acta Anat. 156:217–226. [DOI] [PubMed] [Google Scholar]
- Brabletz, T., A. Jung, S. Dag, F. Hlubek, and T. Kirchner. 1999. Beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol. 155:1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori, G., and H. Semb. 1999. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 24:73–76. [DOI] [PubMed] [Google Scholar]
- Cox, R.T., C. Kirkpatrick, and M. Peifer. 1996. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J. Cell Biol. 134:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger, A., A. Stockinger, B. Schaffhauser, H. Beug, and R. Foisner. 2000. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J. Cell Biol. 148:173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto, F., N. Funayama, U. Gluck, and B.M. Gumbiner. 1996. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J. Cell Biol. 132:1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gat, U., R. DasGupta, L. Degenstein, and E. Fuchs. 1998. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 95:605–614. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., E. Wong, and B.M. Gumbiner. 2001. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradl, D., M. Kuhl, and D. Wedlich. 1999. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol. Cell. Biol. 19:5576–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B.M. 2000. Regulation of cadherin adhesive activity. J. Cell Biol. 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegel, H., L. Larue, M. Ohsugi, L. Fedorov, K. Herrenknecht, and R. Kemler. 1995. Lack of beta-catenin affects mouse development at gastrulation. Development. 121:3529–3537. [DOI] [PubMed] [Google Scholar]
- Hanahan, D., and R.A. Weinberg. 2000. The hallmark of cancer. Cell. 10:57–70. [DOI] [PubMed] [Google Scholar]
- Harada, N., Y. Tamai, T. Ishikawa, B. Sauer, K. Takaku, M. Oshima, and M.M. Taketo. 1999. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18:5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, T.C., A.B. Sparks, C. Rago, H. Hermeking, L. Zawel, L.T. da Costa, P.J. Morin, B. Vogelstein, and K.W. Kinzler. 1998. Identification of c-MYC as a target of the APC pathway. Science. 281:1509–1512. [DOI] [PubMed] [Google Scholar]
- Heasman, J., A. Crawford, K. Goldstone, P. Garner-Hamrick, B. Gumbiner, P. McCrea, C. Kintner, C.Y. Noro, and C. Wylie. 1994. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 79:791–803. [DOI] [PubMed] [Google Scholar]
- Hecht, A., and R. Kemler. 2000. Curbing the nuclear activities of β-catenin. EMBO Reports. 1:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht, A., C.M. Litterst, O. Huber, and R. Kemler. 1999. Functional characterization of multiple transactivating elements in beta-catenin, some of which interact with the TATA-binding protein in vitro. J. Biol. Chem. 274:18017–18025. [DOI] [PubMed] [Google Scholar]
- Hecht, A., K. Vleminckx, M.P. Stemmler, F. van Roy, and R. Kemler. 2000. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19:1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, O., R. Korn, J. McLaughlin, M. Ohsugi, B.G. Herrmann, and R. Kemler. 1996. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59:3–10. [DOI] [PubMed] [Google Scholar]
- Huelsken, J., R. Vogel, V. Brinkmann, B. Erdmann, C. Birchmeier, and W. Birchmeier. 2000. Requirement for beta-catenin in anterior-posterior axis formation in mice. J. Cell Biol. 148:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, S., S. Kishida, H. Yamamoto, H. Murai, S. Koyama, and A. Kikuchi. 1998. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17:1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandikonda, S., D. Oda, R. Niederman, and B.C. Sorkin. 1996. Cadherin-mediated adhesion is required for normal growth regulation of human gingival epithelial cells. Cell Adhes. Commun. 4:13–24. [DOI] [PubMed] [Google Scholar]
- Kelly, G.M., D.F. Erezyilmaz, and R.T. Moon. 1995. Induction of a secondary embryonic axis in zebrafish occurs following the overexpression of beta-catenin. Mech. Dev. 53:261–273. [DOI] [PubMed] [Google Scholar]
- Kolligs, F.T., G. Hu, C.V. Dang, and E.R. Fearon. 1999. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol. Cell. Biol. 19:5696–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolligs, F.T., B. Kolligs, K.M. Hajra, G. Hu, M. Tani, K.R. Cho, and E.R. Fearon. 2000. Gamma-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of beta-catenin. Genes Dev. 14:1319–1331. [PMC free article] [PubMed] [Google Scholar]
- Korinek, V., N. Barker, P.J. Morin, D. van Wichen, R. de Weger, K.W. Kinzler, B. Vogelstein, and H. Clevers. 1997. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 275:1784–1787. [DOI] [PubMed] [Google Scholar]
- Korinek, V., N. Barker, P. Moerer, E. Van Donselaar, G. Huls, P.J. Peters, and H. Clevers. 1998. Depletion of epithelial stem-cell compartments the small intestine of mice lacking Tcf-4. Nat. Genet. 19:379–383. [DOI] [PubMed] [Google Scholar]
- Krek, W., D.M. Livingston, and S. Shirodkar. 1993. Binding to DNA and the retinoblastoma gene product promoted by complex formation of different E2F family members. Science. 262:1557–1560. [DOI] [PubMed] [Google Scholar]
- Larue, L., C. Antos, S. Butz, O. Huber, V. Delmas, M. Dominis, and R. Kemler. 1996. A role for cadherins in tissue formation. Development. 122:3185–3194. [DOI] [PubMed] [Google Scholar]
- Miyaki, M., K. Tanaka, R. Kikuchi-Yanoshita, M. Muraoka, M. Konishi, and M. Takeichi. 1995. Increased cell-substratum adhesion, and decreased gelatinase secretion and cell growth, induced by E-cadherin transfection of human colon carcinoma cells. Oncogene. 11:2547–2552. [PubMed] [Google Scholar]
- Molenaar, M., M. van de Wetering, M. Oosterwegel, J. Peterson-Maduro, S. Godsave, V. Korinek, J. Roose, O. Destree, and H. Clevers. 1996. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 86:391–399. [DOI] [PubMed] [Google Scholar]
- Morin, P.J., A.B. Sparks, V. Korinek, N. Barker, H. Clevers, B. Vogelstein, and K.W. Kinzler. 1997. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 275:1787–1790. [DOI] [PubMed] [Google Scholar]
- Nathke, I.S., L. Hinck, J.R. Swedlow, J. Papkoff, and W.J. Nelson. 1994. Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J. Cell Biol. 125:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo, T., and M. Ozawa. 1999. p120(ctn) binds to the membrane-proximal region of the E-cadherin cytoplasmic domain and is involved in modulation of adhesion activity. J. Biol. Chem. 274:21409–21415. [DOI] [PubMed] [Google Scholar]
- Orford, K., C. Crockett, J.P. Jensen, A.M. Weissman, and S.W. Byers. 1997. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J. Biol. Chem. 272:24735–24738. [DOI] [PubMed] [Google Scholar]
- Orford, K., C.C. Orford, and S.W. Byers. 1999. Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J. Cell Biol. 146:855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic, S., O. Huber, H. Aberle, S. Arnold, and R. Kemler. 1999. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112:1237–1245. [DOI] [PubMed] [Google Scholar]
- Ozawa, M., and R. Kemler. 1998. The membrane-proximal region of the E-cadherin cytoplasmic domain prevents dimerization and negatively regulates adhesion activity. J. Cell Biol. 142:1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa, M., H. Baribault, and R. Kemler. 1989. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 8:1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, A.K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 392:190–193. [DOI] [PubMed] [Google Scholar]
- Polakis, P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837–1851. [PubMed] [Google Scholar]
- Reichmann, E., H. Schwarz, E.M. Deiner, I. Leitner, M. Eilers, J. Berger, M. Busslinger, and H. Beug. 1992. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 71:1103–1116. [DOI] [PubMed] [Google Scholar]
- Roose, J., and H. Clevers. 1999. TCF transcription factors: molecular switches in carcinogenesis. Biochim. Biophys. Acta. 1424:M23–M37. [DOI] [PubMed] [Google Scholar]
- Roose, J., M. Molenaar, J. Peterson, J. Hurenkamp, H. Brantjes, P. Moerer, M. van de Wetering, O. Destree, and H. Clevers. 1998. The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature. 395:608–612. [DOI] [PubMed] [Google Scholar]
- Roose, J., G. Huls, M. van Beest, P. Moerer, K. van der Horn, R. Goldschmeding, T. Logtenberg, and H. Clevers. 1999. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science. 285:1923–1926. [DOI] [PubMed] [Google Scholar]
- Rubinfeld, B., I. Albert, E. Porfiri, C. Fiol, S. Munemitsu, and P. Polakis. 1996. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 272:1023–1026. [DOI] [PubMed] [Google Scholar]
- Rubinfeld, B., P. Robbins, M. El-Gamil, I. Albert, E. Porfiri, and P. Polakis. 1997. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 275:1790–1792. [DOI] [PubMed] [Google Scholar]
- Sadot, E., I. Simcha, M. Shtutman, A. Ben-Ze'ev, and B. Geiger. 1998. Inhibition of beta-catenin-mediated transactivation by cadherin derivatives. Proc. Natl. Acad. Sci. USA. 95:15339–15344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, L., A.M. Fannon, P.D. Kwong, A. Thompson, M.S. Lehmann, G. Grubel, J.F. Legrand, J. Als-Nielsen, D.R. Colman, and W.A. Hendrickson. 1995. Structural basis of cell-cell adhesion by cadherins. Nature. 374:327–337. [DOI] [PubMed] [Google Scholar]
- Shtutman, M., J. Zhurinsky, I. Simcha, C. Albanese, M. D'Amico, R. Pestell, and A. Ben-Ze'ev. 1999. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA. 96:5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcha, I., M. Shtutman, D. Salomon, J. Zhurinsky, E. Sadot, B. Geiger, and A. Ben-Ze'ev. 1998. Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J. Cell Biol. 141:1433–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Croix, B., C. Sheehan, J.W. Rak, V.A. Florenes, J.M. Slingerland, and R.S. Kerbel. 1998. E-cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol. 142:557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y., F.T. Kolligs, M.O. Hottiger, R. Mosavin, E.R. Fearon, and G.J. Nabel. 2000. Regulation of beta-catenin transformation by the p300 transcriptional coactivator. Proc. Natl. Acad. Sci. USA. 97:12613–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K., and K. Suzuki. 1996. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp. Cell Res. 226:214–222. [DOI] [PubMed] [Google Scholar]
- Takemaru, K.I., and R.T. Moon. 2000. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 149:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu, O., and F. McCormick. 1999. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 398:422–426. [DOI] [PubMed] [Google Scholar]
- Thoreson, M.A., P.Z. Anastasiadis, J.M. Daniel, R.C. Ireton, M.J. Wheelock, K.R. Johnson, D.K. Hummingbird, and A.B. Reynolds. 2000. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 148:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe, M., A. Nagafuchi, S. Tsukita, and M. Takeichi. 1994. Induction of polarized cell–cell association and retardation of growth by activation of the E-cadherin–catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 127:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert, K., and R. Nusse. 1998. Beta-catenin: a key mediator of Wnt signaling. Curr. Opin. Genet. Dev. 8:95–102. [DOI] [PubMed] [Google Scholar]
- Yap, A.S., C.M. Niessen, and B.M. Gumbiner. 1998. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost, C., M. Torres, J.R. Miller, E. Huang, D. Kimelman, and R.T. Moon. 1996. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 10:1443–1454. [DOI] [PubMed] [Google Scholar]
- Young, C.S., M. Kitamura, S. Hardy, and J. Kitajewski. 1998. Wnt-1 induces growth, cytosolic beta-catenin, and Tcf/Lef transcriptional activation in Rat-1 fibroblasts. Mol. Cell. Biol. 18:2474–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. a. Differential mechanisms of LEF/TCF family-dependent transcriptional activation by beta-catenin and plakoglobin. Mol. Cell. Biol. 20:4238–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. b. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J. Cell Sci. 113:3127–3139. [DOI] [PubMed] [Google Scholar]