Sonic Hedgehog Opposes Epithelial Cell Cycle Arrest (original) (raw)
Abstract
Stratified epithelium displays an equilibrium between proliferation and cell cycle arrest, a balance that is disrupted in basal cell carcinoma (BCC). Sonic hedgehog (Shh) pathway activation appears sufficient to induce BCC, however, the way it does so is unknown. Shh-induced epidermal hyperplasia is accompanied by continued cell proliferation in normally growth arrested suprabasal cells in vivo. Shh-expressing cells fail to exit S and G2/M phases in response to calcium-induced differentiation and also resist exhaustion of replicative growth capacity. In addition, Shh blocks p21CIP1/WAF1-induced growth arrest. These data indicate that Shh promotes neoplasia by opposing normal stimuli for epithelial cell cycle arrest.
Keywords: Sonic hedgehog, epidermis, cell cycle regulation, basal cell carcinoma, cyclin-dependent kinase inhibitor
Identified as an important regulator of segment polarity and tissue organization in Drosophila 14, the hedgehog signal transduction pathway can play a significant role in human disease 1 15 27. Mutations in hedgehog pathway genes have been associated with human basal cell carcinoma (BCC)1 13 16 29, and hedgehog pathway activation alone is sufficient to induce epidermal neoplasms indistinguishable from BCC in transgenic murine 25 and human skin 11. The basis for these potent neoplastic effects are not fully understood. Specifically, the effects of Shh on cell growth regulation in this context have not been studied.
Here we report that Shh promotes cellular proliferation by opposing triggers for physiologic epithelial growth arrest. In addition, we demonstrate that Shh augments the replicative capacity of normal human epithelial cells. The results of these studies identify a new role for Shh in cell cycle regulation and provide a mechanistic basis for Shh induction of epithelial neoplasia.
Materials and Methods
Genetically Engineered Human Skin
Early passage normal human keratinocytes were transduced with retroviral expression vectors for Shh-IRES-GFP as well as GFP and lacZ controls as noted below and seeded on devitalized human dermis and grafted to SCID mice 5. 4 mice were grafted per vector group; untransduced keratinocytes as well as GFP and lacZ vectors groups were used as additional controls. At 1 and 3 wk, a biopsy was performed and 5-μM-thick tissue cryosections were prepared. The human species origin of regenerated epithelium was confirmed in tissue sections using species-specific antibodies to human involucrin 24 (BTI Inc.). Propidium iodide staining was performed to highlight cellular nuclei in Shh and GFP control human epidermis at 1 and 3 wk using propidium iodide at 40 ng/ml for 1 min. For study of indicators of cellular proliferation in vivo, bromodeoxyuridine (BrdU) (Sigma) 100 mg/kg was administered by intraperitoneal injection to mice bearing genetically engineered human skin grafts. 2 h after injection, human skin was excised and 5-μM cryosections prepared. Antibody staining for BrdU (Becton Dickinson) and Ki-67 (Novocastra) was performed as described 28. Quantitation of fold increase in BrdU[+] and Ki-67[+] cell numbers per 100 μM unit surface length was performed at 1 and 3 wk, with GFP epidermis at 1 wk assigned the standard value of 1.0. Error bars were calculated as SD between four independent grafts in each vector group.
Cell Culture and Gene Transfer
Primary human keratinocytes were freshly isolated from human skin 26 and transduced with retroviral expression vectors for Shh-IRES-GFP as well as GFP and lacZ controls 7 11. Greater than 99% efficiency of both gene transfer and gene expression maintenance was verified by fluorescence microscopy (or X-gal staining at pH 8.0 for E. coli lacZ), both at 48 h after transduction and at serial passaging to passage 15 as previously described 7 11. A similar gene transfer approach was used for the human p21Cip1 retroviral expression vector except that high efficiency gene transfer was confirmed by immunofluorescence staining of cell aliquots using antibodies to p21Cip1 (Santa Cruz Biotechnology). For p21Cip1 studies, transduction with p21Cip1 retroviral expression vectors was performed 48 h after initial transduction with either Shh-IRES-GFP or GFP and lacZ controls. Senescence-associated β-galactosidase (SA-β-gal) expression was determined at pH 6.0 as described 9 before and at day 0, 3, 5, 7, and 9 after transduction with retroviral vector for expression of p21Cip1. Keratinocytes were used from the same donor for each series of experiments to control for possible tissue variations between individuals. All experiments were performed using triplicate independent transductions, with data presented as the average of these independent transductions ± SD.
Cell Cycle Analysis
Cell cycle distribution was determined in proliferating cells grown in a 1:1 mixture of SFM media (GIBCO BRL) and 154 media (Cascade Biologics) growth media as described 28. Terminal differentiation-associated growth inhibition was induced by addition of 1.5 mM calcium to this media for 48 h. Shh and control cells were stained with propidium iodide and analyzed for DNA content via flow cytometry 23.
Cell Growth and Replicative Capacity
Cumulative cell yield was determined as previously described 17, with cells grown in a low calcium 1:1 media mixture of SFM:154 media 28. Cells were passaged at identical densities in triplicate after triplicate independent transductions for each condition. Each independent transduction was passaged separately after initial gene transfer, with the average cell numbers ± SD presented for each condition. Three independent series of these long-term experiments were performed to assess replicative capacity and cumulative cell yield.
Analysis of CDK2 and CDK4
Analysis of CDK2 and CDK4 kinase activity was performed as previously described 20. In brief, cells were rinsed with cold PBS and harvested in lysis buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 2.5 mM EGTA, 1 mM EDTA, and 0.1% Tween-20 with 1 mM DTT, 0.1 mM PMSF, 0.2 U/ml aprotinin, 10 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, and 1 mM sodium fluoride). After a freeze-thaw cycle, lysates were collected after centrifugation at 12,000 g for 10 min and immunoprecipitated using antibodies to human CDK2 and CDK4 (Santa Cruz). Histone H1 (Boehringer-Mannheim) was used as a substrate for kinase assays in 50 mM Hepes, pH 7.0, 10 mM MgCl2, 2.5 mM EGTA, 1 mM DTT, 20 μM ATP, 10 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, and 1 mM sodium fluoride. The amount of CDK2 and CDK4 protein in precipitates was confirmed in parallel by immunoblotting.
Results
Shh Promotes Epithelial Proliferation In Vivo
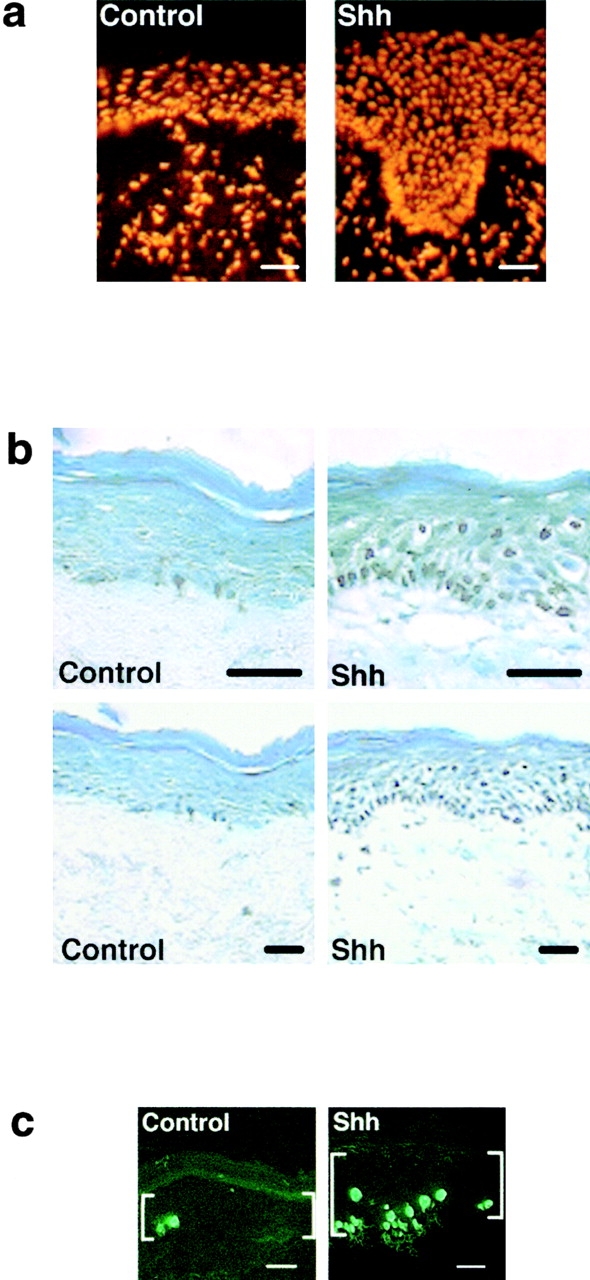
To determine if Shh impacts epithelial cell cycle regulation, we expressed Shh in human epidermis regenerated on immune deficient mice from normal keratinocytes in an approach known to effect hedgehog pathway activation 11. Genetically engineered human epidermal grafts were then examined at 1 and 3 wk after grafting. Compared with normal findings seen in GFP[+] and lacZ[+] controls, Shh[+] epidermis displays hyperplasia and an over sixfold increase in cell numbers per unit surface length by 3 wk (Fig. 1 a). Shh[+] epidermis is hyperproliferative, with >10-fold increases in Ki-67 expression and >8-fold increase in BrdU incorporation compared with controls (Fig. 1b and Fig. c). Even in Shh[+] tissue sections with comparable thickness to control, Ki-67- and BrdU-positive cells are seen well above the basal layer of Shh[+] epidermis (Fig. 1 b), suggesting a failure to undergo the cell cycle arrest that normally occurs before outward migration in stratified epithelium. Such findings raise the possibility that Shh may oppose stimuli for epithelial cell cycle arrest.
Figure 1.
Shh promotes epithelial proliferation in vivo. Human epidermis genetically engineered to express Shh and GFP control was regenerated on SCID mice. For all studies data presented is the average of three independent grafts each for Shh and control ± SD. (a) Propidium iodide staining to highlight cellular nuclei in Shh and GFP control human epidermis. Quantitation of fold increase in cell numbers per 100 μM unit surface length at 1 and 3 wk is shown at right. (b) Ki-67 proliferation marker expression by immunoperoxidase staining at 3 wk, counterstaining with methyl green; quantitation of fold increase in cell numbers per 100 μM unit surface length is shown at right. Note positive cells (dark nuclei) well above the basement membrane zone in Shh[+] epidermis. (c) BrdU labeling of Shh and control human epidermis at 3 wk; quantitation of fold increase in cell numbers per 100 μM unit surface length is shown at right. Bars: (a) 100 μM; (b) 75 μM; (c) 50 μM.



Shh-expressing Cells Fail to Appropriately Exit S and G2/M Phases in Response to Calcium-induced Differentiation
To determine if Shh confers a resistance to differentiation-associated cell cycle arrest, Shh[+] and control cells were grown to confluence in vitro then media calcium concentration was raised to 1.5 mM in a process that recapitulates features of differentiation seen in vivo, including cell cycle arrest and activation of certain terminal differentiation genes 18. Cell cycle distribution was analyzed both before differentiation stimuli and 48 h after addition of calcium. Shh[+] and control cells are indistinguishable under low calcium conditions promoting cellular proliferation, with the majority of cells in G2/M and S phase (Fig. 2). As anticipated, the addition of calcium induces exit of the majority of normal control cells into G0/G1. Whereas Shh[+] cells alter their cell cycle distribution in response to calcium, instead of redistributing to G0/G1 these cells are disproportionately in G2/M and S phase (Fig. 2). These findings indicate Shh opposes cell cycle inhibitory effects of a primary in vitro stimulus for epithelial differentiation.
Figure 2.

Shh-expressing cells fail to appropriately exit S and G2/M phases in response to calcium-induced differentiation. Cell cycle distribution was determined in cells in low calcium growth media and in cells undergoing calcium-induced differentiation in vitro via elevation of the media calcium concentration to 1.5 mM. Shh and GFP control cells were analyzed for DNA content via flow cytometry. (a) Control, growth media. (b) Shh[+], growth media. (c) Control, 1.5 mM calcium. (d) Shh[+] 1.5 mM calcium. Representative plots are shown; accompanying percentage figures represent the averages of three independent transductions and flow cytometric analyses for Shh and controls ± SEM
Shh Augments Cell Capacity for Long-Term Growth
These findings suggest that Shh pathway activation may confer resistance to growth inhibitory mechanisms that are of potential importance in epithelial growth control and neoplasia. To test if Shh augments the long-term growth capacity of nonimmortalized human epithelial cells, we performed serial passaging of Shh[+] cells along with GFP- and lacZ-expressing controls. Nonimmortalized keratinocytes exhaust their replicative capacity after multiple passages in vitro, allowing the total cell yield of a defined number of input cells to be determined 17 19. Shh[+] cells display more active sustained proliferation, growing to higher densities and demonstrating evidence of more active DNA replication compared with controls (Fig. 3, a and b). Consistent with this, Shh[+] cells generate a cumulative increased cell yield of 64–150-fold over controls before exhaustion of replicative capacity (Fig. 3 c). Shh[+] cells continue to proliferate until passage 16 whereas controls cease active proliferation after passage 12. Less than 5% of cells were nonviable at each passage for all groups (as judged by trypan blue exclusion), indicating that this increase is not due to alterations in frequency of cell death. These findings indicate that Shh augments the proliferative capacity of normal epithelial cells.
Figure 3.

Shh augments cell capacity for long-term growth. (a) Methylene blue stain of plates of normal human epithelial cells expressing Shh as well as GFP and lacZ controls after passaging at identical densities, shown at 4 d after plating at passage 11. Note the increased cell density and staining in Shh plates compared with controls; duplicate independent transductions passaged separately for the duration of the experiment are shown. (b) BrdU incorporation of Shh[+] cells over a 2-h time period compared with GFP and lacZ (LZRS) controls. Immunofluorescence staining was performed using antibody to BrdU (green) and counterstaining with propidium iodide to visualize all cells in the field (red). BrdU labeling index = number of BrdU[+] cells/total cell number at passage 11 was calculated in three independent transductions with quantitation shown at bottom of each panel. (c) Cumulative cell yield of Shh[+] as well as GFP and lacZ controls was determined after exhaustion of proliferative capacity. The insert shows an expanded scale to more clearly denote the cell yield for controls. Data represents mean total cell yield for three independent experiments ± SEM. (d) Confirmation of retained gene expression in Shh[+] cells as well as controls. mRNA was prepared from GFP[+], lacZ[+] and Shh[+] cells as well as untransduced controls ([−]) at their final passages and subjected to RT-PCR. (Top) Shh cDNA-specific primers. (Bottom) Primers for the ψ+ retroviral packaging sequence common to all vectors.
Shh Antagonizes p21Cip1-induced Growth Arrest
Another negative growth regulatory mechanism bypassed in neoplasia are cyclin-dependent kinase inhibitors (CKIs). To determine if Shh could also confer resistance to CKI-induced growth arrest, we studied its impact on the effects of p21Cip1, a CKI of known importance in epithelial growth inhibition 8 23. p21Cip1 is known to be important in the cell growth arrest that occurs before keratinocyte terminal differentiation 8 23. Shh[+] and control cells were transduced at high efficiency with a retroviral expression vector for p21Cip1 and kinetics of cell growth were determined. As anticipated, p21Cip1 profoundly inhibits proliferation of both untransduced and GFP[+] control cells, however, p21Cip1 expression fails to inhibit exponential growth by Shh[+] cells (Fig. 4 a). In addition to resisting p21Cip1 growth arrest, Shh[+] cells fail to induce a biomarker seen in permanent cell cycle arrest, SA-β-gal 9, in response to p21Cip1. Whereas ∼50% of control cells are SA-β-gal[+] by day 5, only 15% of Shh[+] cells express this biomarker (Fig. 4 b). Shh expression in these cells is also associated with augmented levels of CDK2 and CDK4 associated kinase activity, with this activity not fully repressed by p21Cip1 in the case of CDK2 (Fig. 4 c). These data indicate that Shh opposes p21Cip1-induced growth arrest and suggest that Shh leads to activation of key core cell cycle machinery elements CDK2 and CDK4.
Figure 4.
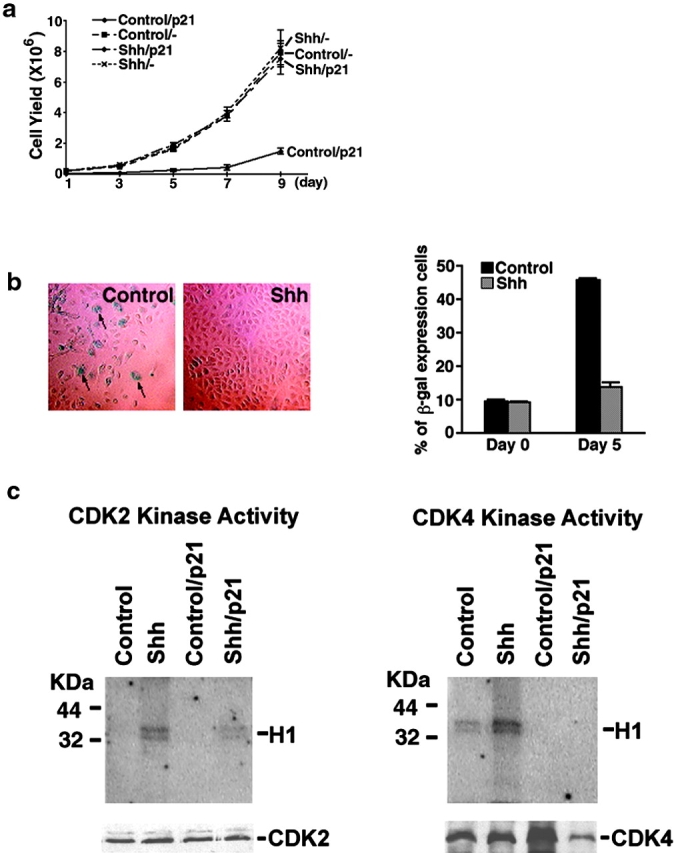
Shh antagonizes p21Cip1-induced growth arrest. (a) Cell growth after p21Cip1 expression. 2 d after high efficiency gene transfer with Shh or GFP retroviral expression vectors, normal human epithelial cells were transduced in triplicate independent transductions at high efficiency with a retroviral expression vector for human p21Cip1 and cell numbers determined in the days following. Data is expressed from triplicate independent transductions ± SEM (b) SA-β-gal induction. Proportion of cells expressing SA-β-gal after p21Cip1 gene transfer. Arrows denote representative SA-β-gal[+] cells. Quantitation of percent SA-β-gal[+] cells ± SEM from three independent transductions is shown at right. (c) Shh effect on CDK2 and CDK4 kinase activity. Extracts from normal human epithelial cells transduced with retroviral expression vectors for Shh, GFP control as well as those cotransduced with vectors for either both Shh and p21Cip1 or GFP and p21Cip1 were immunoprecipitated using antibodies to CDK2 and CDK4. Kinase activity (top panels) was measured using histone H1 as a substrate. Immunoblotting was also performed in parallel to demonstrate the amount of CDK2 and CDK4 protein present in the precipitate (bottom panels below kinase activity).
Discussion
Cell cycle exit before outward migration and terminal differentiation is a cardinal feature of normal stratified epithelium. Here we report that Shh inhibits this process. We observe that Shh induces dramatic increases in the proportion of actively proliferating cells within stratified epithelium, including cells in normally growth-arrested suprabasal layers. This observation suggests that Shh opposes the normal growth arrest necessary for homeostasis in stratified epithelium. Implicated as important in this growth arrest process in vivo are calcium and the CKI p21Cip1. Consistent with this, we found that Shh rendered cells resistant to the exit from S and G2/M that occurs in normal keratinocytes exposed to elevated calcium concentrations in vitro in a process that may recapitulate the local differences in calcium concentration that exist within normal epidermis 10. This resistance was characterized by an enhanced accumulation of cells in G2/M, raising the possibility that Shh could impact regulators acting at later points in the cell cycle. In addition to calcium, another factor implicated in the promotion of normal epithelial growth arrest is the CKI p21Cip1 8. Shh also opposes p21Cip1 growth inhibitory effects, raising the possibility that Shh may exert a dominant positive impact on core cell cycle machinery capable of bypassing negative signals from CKIs.
The possibility that Shh acts dominantly on core cell cycle machinery is supported by the observation that Shh expression is associated with an increase in activity of CDK2 and CDK4 under normal growth conditions. This is most notable in the case of CDK2 whose inhibition by p21Cip1 is an important control point in the G1-S transition 3. Shh[+] cells sustain CDK2 activity even in the presence of p21Cip1, although the undetectable levels of CDK2 kinase activity in controls make conclusions about relative impact of Shh on p21Cip1 inhibition of CDK2 difficult. Detailed knowledge of the impact of loss of Shh pathway function on mammalian epithelial cell growth in settings where it is normally active, is not currently available. The failure of hair follicle morphogenesis in mice lacking Shh 4, however, suggests that such loss may lead to a failure of the epithelial cell expansion required in this process. Consistent with these data, our findings indicate that Shh enhances epithelial proliferation and opposes stimuli for normal cell cycle exit.
Evidence of Shh pathway activation is present in the vast majority of human BCCs, with induction of the Shh target Gli1 a consistent finding even in the absence of abnormal Shh or Ptc expression 6. The fact that misexpression of Gli1 alone can induce tumors in vertebrate embryos in the absence of primary perturbations in Shh expression 6 underscores the fact that Shh pathway downstream effectors may account for the impact of induced Shh expression observed in these studies.
Unlike the classical step-wise model of carcinogenesis requiring multiple genetic lesions, hedgehog pathway activation alone appears sufficient to produce cardinal features of human BCC 11. This observation may account for the fact that BCC arises without precursor lesions, unlike another common cancer of stratified epithelium, cutaneous squamous cell carcinoma 22. In light of the mechanism of Shh pathway induction, it is possible these effects may occur within normal hair-bearing skin and in BCC in a non-cell autonomous manner. This single hit induction of neoplasia by Shh stands in sharp contrast to the step-wise model of carcinogenesis requiring multiple genetic alterations believed for some time to be operative in other neoplasias, such as colorectal carcinoma 12. In addition, this proposed requirement for activation of only one pathway to trigger this cancer may offer an explanation as to why BCC is the most common malignancy in the United States, with an estimated incidence of up to 1,000,000 yearly 21.
Recent studies support a need for cells to surmount several major obstacles in order to progress to neoplasia; apoptosis and irreversible growth arrest have emerged as two of the most important of these obstacles 30. We have found that Shh augments cellular capacity for long-term growth. The fact that Shh opposes p21Cip1-induced growth arrest is consistent with this observation as p21Cip1 loss allows other cell types to resist replicative exhaustion 2. The Shh effects on growth capacity observed here, however, may likely be due to Shh-enhanced resistance to terminal differentiation-associated cell cycle arrest rather than a direct extension of the Hayflick limit on population doublings. Combined with the ability of Shh to induce bcl-2 11, and thus potentially resist apoptosis, the ability to bypass growth arrest stimuli shown has been increasingly recognized as an important milestone in carcinogenesis and may explain the potent effects of the Shh pathway in inducing BCC. In spite of these potent effects, Shh-induced neoplasia appears to lack the aggressiveness of other malignancies characterized by multiple genetic lesions. In general, BCCs are slowly growing tumors that do not exhibit markedly invasive features and characteristically fail to metastasize 22. These biologically mild behavioral characteristics may be due to the fact that BCC lacks the additional genetic lesions necessary for more aggressive behavior.
Our findings indicate that Shh action promotes cellular proliferation by opposing cell cycle arrest. This observation may provide a platform for future studies examining Shh impacts on cell growth during development as well as in efforts aimed at defining the mechanistic basis of normal growth control in epithelial tissues.
Acknowledgments
We thank G. Morrissey, C. Seitz, H. Deng, J. Rheinwald, and Q. Lin for technical advice, reagents, and helpful discussions. We thank A. Oro and M. Scott and for helpful discussions and presubmission review along with J. Swain and P. Jackson.
This work was supported by the Office of Research and Development, Department of Veterans Affairs (V.A.) and National Institutes of Health grants AR43799, AR45192, and AR44012 to P.A. Khavari.
Footnotes
1.used in this paper: Shh, Sonic hedgehog; BCC, basal cell carcinoma; BrdU, bromodeoxyuridine; CDK, cyclin-dependent kinase; CKI, cyclin dependent kinase inhibitor; SA-β-gal, senescence-associated β-galactosidase
References
- Belloni E., Muenke M., Roessler E., Traverso G., Siegel-Bartelt J., Frumkin A., Mitchell H.F., Donis-Keller H., Helms C., Hing A.V. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- Brown J.P., Wei W., Sedivy J.M. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- Cai K., Dynlacht B.D. Activity and nature of p21(WAF1) complexes during the cell cycle. Proc. Natl. Acad. Sci. USA. 1998;95:12254–12259. doi: 10.1073/pnas.95.21.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Swan R.Z., Grachtchouk M., Bolinger M., Litingtung Y., Robertson E.K., Cooper M.K., Gaffield W., Westphal H., Beachy P.A., Dlugosz A.A. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev. Biol. 1999;205:1–9. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- Choate K.A., Medalie D.A., Morgan J.R., Khavari P.A. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat. Med. 1996;2:1263–1267. doi: 10.1038/nm1196-1263. [DOI] [PubMed] [Google Scholar]
- Dahmane N., Lee J., Robins P., Heller P., Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- Deng H., Choate K.A., Lin Q., Khavari P.A. High efficiency gene transfer and pharmacologic selection of genetically engineered human keratinocytes. Biotechniques. 1998;25:274–280. doi: 10.2144/98252gt02. [DOI] [PubMed] [Google Scholar]
- Di Cunto F., Topley G., Calautti E., Hsiao J., Ong L., Seth P.K., Dotto G.P. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science. 1998;280:1069–1072. doi: 10.1126/science.280.5366.1069. [DOI] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias P.M., Nau P., Hanley K., Cullander C., Crumrine D., Bench G., Sideras-Haddad E., Mauro T., Williams M.L., Feingold K.R. Formation of the epidermal calcium gradient coincides with key milestones of barrier ontogenesis. J. Invest. Dermatol. 1998;110:399–404. doi: 10.1046/j.1523-1747.1998.00151.x. [DOI] [PubMed] [Google Scholar]
- Fan H., Oro A.E., Scott M.P., Khavari P.A. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat. Med. 1997;3:788–792. doi: 10.1038/nm0797-788. [DOI] [PubMed] [Google Scholar]
- Fearon E.R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Hahn H., Wicking C., Zaphiropoulous P.G., Gailani M.R., Shanley S., Chidambaram A., Vorechovsky I., Holmberg E., Unden A.B., Gillies S. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M., Brook A., McMahon A.P. The world according to hedgehog. Trends Genet. 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- Ingham P.W. The patched gene in development and cancer. Curr. Opin. Genet. Dev. 1998;8:88–94. doi: 10.1016/s0959-437x(98)80067-1. [DOI] [PubMed] [Google Scholar]
- Johnson R.L., Rothman A.L., Xie J., Goodrich L.V., Bare J.W., Bonifas J.M., Quinn A.G., Myers R.M., Cox D.R., Epstein E.H., Jr., Scott M.P. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Li A., Simmons P.J., Kaur P. Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc. Natl. Acad. Sci. USA. 1998;95:3902–3907. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Tucker R.W., Hennings H., Yuspa S.H. Inhibitors of the intracellular Ca(2+)-ATPase in cultured mouse keratinocytes reveal components of terminal differentiation that are regulated by distinct intracellular Ca2+ compartments. Cell Growth Differ. 1995;6:1171–1184. [PubMed] [Google Scholar]
- Mathor M.B., Ferrari G., Dellambra E., Cilli M., Mavilio F., Cancedda R., De Luca M. Clonal analysis of stably transduced human epidermal stem cells in culture. Proc. Natl. Acad. Sci. USA. 1996;93:10371–10376. doi: 10.1073/pnas.93.19.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H., Quelle D.E., Shurtleff S.A., Shibuya M., Sherr C.J., Kato J.Y. D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.L., Weinstock M.A. Nonmelanoma skin cancer in the United Statesincidence. J. Am. Acad. Dermatol. 1994;30:774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- Miller S.J. Biology of basal cell carcinoma (Part I) J. Am. Acad. Dermatol. 1991;24:1–13. doi: 10.1016/0190-9622(91)70001-i. [DOI] [PubMed] [Google Scholar]
- Missero C., Di Cunto F., Kiyokawa H., Koff A., Dotto G.P. The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev. 1996;10:3065–3075. doi: 10.1101/gad.10.23.3065. [DOI] [PubMed] [Google Scholar]
- Murphy G.F., Flynn T.C., Rice R.H., Pinkus G.S. Involucrin expression in normal and neoplastic human skina marker for keratinocyte differentiation. J. Invest. Dermatol. 1984;82:453–457. doi: 10.1111/1523-1747.ep12260945. [DOI] [PubMed] [Google Scholar]
- Oro A.E., Higgins K.M., Hu Z., Bonifas J.M., Epstein E.H., Scott M.P. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- Rheinwald J.G., Green H. Serial cultivation of strains of human epidermal keratinocytesthe formation of keratinizing colonies from single cells. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Roessler E., Belloni E., Gaudenz K., Jay P., Berta P., Scherer S.W., Tsui L.C., Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Seitz C.S., Lin Q., Deng H., Khavari P.A. Alterations in NF-kB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kB. Proc. Natl. Acad. Sci. USA. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Murone M., Luoh S.M., Ryan A., Gu Q., Zhang C., Bonifas J.M., Lam C.W., Hynes M., Goddard A. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- Yeager T.R., DeVries S., Jarrard D.F., Kao C., Nakada S.Y., Moon T.D., Bruskewitz R., Stadler W.M., Meisner L.F., Gilchrist K.W. Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998;12:163–174. doi: 10.1101/gad.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]