Mice Lacking Expression of Secondary Lymphoid Organ Chemokine Have Defects in Lymphocyte Homing and Dendritic Cell Localization (original) (raw)
Abstract
Secondary lymphoid organ chemokine (SLC) is expressed in high endothelial venules and in T cell zones of spleen and lymph nodes (LNs) and strongly attracts naive T cells. In mice homozygous for the paucity of lymph node T cell (plt) mutation, naive T cells fail to home to LNs or the lymphoid regions of spleen. Here we demonstrate that expression of SLC is undetectable in plt mice. In addition to the defect in T cell homing, we demonstrate that dendritic cells (DCs) fail to accumulate in spleen and LN T cell zones of plt mice. DC migration to LNs after contact sensitization is also substantially reduced. The physiologic significance of these abnormalities in plt mice is indicated by a markedly increased sensitivity to infection with murine hepatitis virus. The plt mutation maps to the SLC locus; however, the sequence of SLC introns and exons in plt mice is normal. These findings suggest that the abnormalities in plt mice are due to a genetic defect in the expression of SLC and that SLC mediates the entry of naive T cells and antigen-stimulated DCs into the T cell zones of secondary lymphoid organs.
Keywords: CC chemokines, cellular immunity, leukocyte chemotaxis, T lymphocytes, mutation
Secondary lymphoid organs are the predominant site of lymphocyte sensitization to novel antigens. Their function requires the colocalization of two distinct populations of leukocytes: antigen-presenting dendritic cells (DCs)1 and antigen-responsive naive lymphocytes (1). Chemokines, a rapidly growing family of small chemotactic cytokines, are believed to provide the signals that guide leukocytes to their proper location within lymphoid organs (2, 3). Several recently identified chemokines have been suggested to mediate the constitutive trafficking of leukocytes based on their in vitro characteristics and their expression in lymphoid tissues (4). A role for chemokines in B cell localization was shown by a targeted disruption of the chemokine receptor Burkitt's lymphoma receptor 1 (5–8). However, the involvement of chemokines in the localization of either T cells or DCs within lymphoid organs has not been directly demonstrated.
DCs are distributed throughout the body at sites where they can capture antigens (9). In response to an inflammatory stimulus, these cells migrate into afferent lymphatics, then are carried to draining LNs where they are deposited in the subcapsular space. From the subcapsular space, DCs migrate into T cell zones where they take up residence as interdigitating DCs and present MHC-bound antigens and costimulatory molecules to passing lymphocytes (10–15). To sample this antigen repertoire, T lymphocytes that have never been stimulated by antigen (naive T cells) migrate or “home” specifically to the T cell zones of secondary lymphoid organs. In LNs and Peyer's patches (PPs), lymphocytes leave the blood through specialized high endothelial venules (HEVs) by a series of discrete steps which include selectin-mediated rolling, integrin activation, integrin- mediated firm adhesion, and endothelial extravasation (16, 17). In spleen, which lacks HEVs, lymphocytes exit the blood in the marginal zone and migrate to the T cell zone within the splenic white pulp by a poorly understood route (18). Within the T cell zones of these lymphoid organs, a continuous flow of naive T cells sample the antigens presented by DCs. Most of these lymphocytes eventually return to the circulation. The few that encounter their cognate antigen are retained within the T cell zone, undergo clonal expansion, and differentiate into effector or memory T cells.
SLC (also known as 6Ckine, Exodus-2, and TCA4 [19– 22]) is a recently identified chemokine that is expressed in the HEVs of LNs and PPs, in nondendritic stromal cells within the T cell areas of LNs, spleen, and PPs, in the thymic medulla, and in the lymphatic endothelium of multiple tissues (22, 23). SLC has been hypothesized to mediate the homing of naive lymphocytes to secondary lymphoid tissues based on several findings (23). SLC is the only chemokine known to be constitutively expressed in the endothelial cells of HEVs (23). SLC stimulates the chemotaxis of naive T cells and, to a lesser extent, memory T cells and B cells (23, 24). SLC stimulates both the αLβ2 integrin– mediated adhesion of T cells to intercellular adhesion molecule (ICAM)-1 and the α4β7 integrin–mediated adhesion of these cells to mucosal addressin cell adhesion molecule (MadCAM)-1 (23, 25, 26). Activated αLβ2 function is essential for lymphocyte homing to LNs and PPs, and activated α4β7 is required for homing to PPs. Under physiologic flow conditions, SLC stimulates the arrest of rolling T cells with an efficiency and subset specificity (naive versus memory) similar to that seen in vivo (27). These properties strongly suggest that SLC mediates the homing of naive T cells and perhaps other lymphocytes to secondary lymphoid organs; however, this has not been demonstrated directly.
Recently, Nakano et al. described an autosomal recessive mutation in mice, paucity of lymph node T cells (plt), which leads to a defect in the homing of naive T lymphocytes to secondary lymphoid organs (28, 29). plt mice have greatly decreased numbers of naive T lymphocytes in LNs, PPs, and the white pulp of spleen. When injected into plt mice, T lymphocytes from wild-type (+/+) mice fail to enter LNs and PPs and accumulate only in the red pulp of spleen, whereas lymphocytes from plt mice home normally in +/+ mice. These findings demonstrate that the plt defect affects a gene expressed in the lymphoid organ stroma. Although the exact mutation has not been identified, the plt locus maps to mouse chromosome 4 in a region syntenic to human chromosome 9p13. Two known human chemokine genes map to 9p13: SLC and EBV-induced molecule 1 ligand chemokine (ELC) (19).
In view of the lymphocyte homing defects in plt mice, we hypothesized that the plt mutation involves the murine SLC gene. Here we report that SLC mRNA is not expressed in plt mice. We also explore the possibility that lack of SLC expression in these mice leads to additional leukocyte trafficking abnormalities. Hence, we also find that plt mice have abnormalities in the localization of DCs within lymphoid organs, in the migration of DCs to LNs, and are severely immunocompromised. Our findings suggest that the abnormalities in plt mice are due to a genetic defect in the expression of SLC and that SLC is required for the entry of both naive T lymphocytes and antigen-bearing DCs into the T cell zones of secondary lymphoid organs.
Materials and Methods
Reagents and Antibodies.
FITC (isomer 1) and LPS (serotype 055:B5) were obtained from Sigma Chemical Co. Metrizamide A.G. was obtained from Accurate Chemical and Scientific Co. The following anti–mouse antibodies were used for immunohistochemistry and FACS® analysis: biotinylated anti-CD11c, biotinylated anti-IgMa, biotinylated anti-B220, biotinylated anti-CD3, biotinylated anti–I-Ad, FITC-conjugated anti–I-Ad, PE-conjugated anti–I-Ad, and anti-CD4 (PharMingen); NLDC-145 and MOMA-1 (Bachem). The following secondary reagents were used when applicable: avidin-conjugated FITC (avidin-FITC), streptavidin-conjugated alkaline phosphatase (SA-AP), and SA-conjugated horseradish peroxidase (SA-HRP; Vector Labs); AP-conjugated goat anti–rat Ig (anti–rat Ig-AP; PharMingen); HRP-conjugated goat anti–rat Ig (anti–rat Ig-HRP; Caltag Laboratories); and SA-conjugated peridinine chlorophyll protein (SA-PerCP; Becton Dickinson). Cells were suspended in RPMI/ 25 mM HEPES/5% FCS unless otherwise noted.
Mice.
BALB/c-plt/plt mice were produced by backcrossing plt mice 10 generations into a BALB/c genetic background as described previously (29). BALB/c control mice were obtained from The Jackson Laboratory. All experiments were performed on 6–10-wk-old age- and sex-matched mice maintained under specific pathogen–free conditions.
RNA Expression Studies.
For Northern analysis, mRNA from tissues of wild-type and plt mice was subjected to gel electrophoresis, transferred to Hybond-N+ membranes (Amersham Pharmacia Biotech), and probed using randomly primed mouse SLC, ELC, and actin cDNA. For in situ hybridizations, paraffin sections (5 μm) from +/+ and plt mice were deparaffinized, fixed in 4% paraformaldehyde, and treated with proteinase K. After washing in 0.5× SSC, the sections were covered with hybridization solution, prehybridized for 1–3 h at 55°C, and hybridized overnight with sense or antisense 35S-labeled riboprobe transcribed from the mouse SLC or ELC cDNA. After hybridization, sections were washed at high stringency, dehydrated, dipped in photographic emulsion NTB2 (Eastman Kodak Co.), stored at 4°C for 8 wk, developed, and counterstained with hematoxylin and eosin.
Immunohistochemistry.
Tissue samples of spleen and LNs were frozen in OCT, and 10-μm cryostat sections were prepared. Sections were fixed in acetone for 10 min, air dried, blocked with PBS/5% normal goat serum for 30 min, and incubated with the indicated primary antibodies for 1 h. Slides were then washed in PBS, incubated with either SA-AP and anti–rat HRP or anti–rat AP and SA-HRP for 30 min, and developed with 3,3'-diaminobenzidine (DAB) followed by Vector Red (Vector Labs) according to the manufacturer's instructions.
Flow Cytometry.
For DC quantitation, mesenteric, inguinal, axillary, and brachial LNs from four plt and two +/+ mice were pooled, and single cell suspensions were prepared. RBCs were depleted by lysis, and cells at 5 × 106/ml were layered onto metrizamide (14.5 g/100 ml medium) and centrifuged for 10 min at 600 g. Cells at the interface were collected, washed, and resuspended in medium, stained with FITC-conjugated anti–I-Ad, and analyzed by flow cytometry on a FACScan® (Becton Dickinson). Spleen cells were isolated as described previously (30). In brief, spleens were minced, incubated for 1 h at 37°C in RPMI with 130 U/ml collagenase and 0.1 mg/ml DNase, teased through 70-μm nylon mesh, and centrifuged. RBCs were depleted by lysis, then cells were washed once and resuspended. Cells were stained with FITC-conjugated anti–I-Ad, biotinylated anti-B220, and biotinylated anti-CD3 followed by SA-PerCP and analyzed by flow cytometry. For DC quantitation, cells were gated against B220 and CD3 and analyzed for expression of I-Ad.
Contact Sensitization.
The shaved abdomens of mice were painted with 0.4 ml of 5 mg/ml FITC dissolved in a 50:50 (vol/ vol) mixture of acetone and dibutylphthalate. After 24 h, cell suspensions of pooled inguinal, axillary, and brachial LNs were prepared from each mouse and analyzed by flow cytometry. In some experiments, aliquots of LN cells were stained with biotinylated anti-CD11c followed by SA-PerCP. To determine I-A expression on FITC+ cells, aliquots of LN cells from four mice were pooled, partially purified on metrizamide, stained with biotinylated anti–I-Ad followed by SA-PerCP, and analyzed by flow cytometry. For sensitization with a lower dose of antigen, 25 μl of 8 mg/ml FITC was spotted on the shaved right flank of five +/+ and five plt mice. After 24 h, the draining and contralateral inguinal LNs were removed and analyzed individually. Single cell suspensions were quantitated, stained with anti–I-Ad and anti-B220, and analyzed by flow cytometry.
Skin DC Migration and Staining.
Skin explant culture, preparation of dermal and epidermal whole mounts, and staining of DCs were performed as described previously (31). In brief, ears were rinsed with 70% ethanol and split with forceps into dorsal and ventral halves. Dorsal ear halves were floated directly on 2 ml of complete medium in 24-well plates for 3 d. Epidermal and dermal sheets were prepared by floating ear halves (fresh or cultured) or freshly prepared abdominal skin dermal side down on 0.5 M ammonium thiocyanate for 20 min at 37°C. Epidermis was separated from dermis with fine forceps and stained immediately. Epidermal and dermal sheets were cut into 3 × 3–mm sections, fixed in acetone for 30 min, and rehydrated in PBS. They were incubated in 1:20 biotinylated anti–I-Ad overnight at 4°C, washed in PBS three times, and incubated with SA-FITC at 1:100 for 90 min at room temperature. After three washes, sheets were mounted on microscope slides and evaluated by UV microscopy. Nonadherent migratory cells were recovered from the bottom of tissue culture wells after 72 h by gentle rinsing. Aliquots were counted and cells were cytospun onto microscope slides for staining with 1:100 biotinylated anti–I-Ad using a Vectastain ABC kit (Vector Labs) and DAB and counterstained with hematoxylin. The proportion of I-A+ cells was determined by examining 10 fields/slide over 3 slides.
Viral Infection.
Mouse hepatitis virus, strain A59 (MHV-A59), was propagated and plaque assayed on DBT cells as described previously (32, 33). A single pool of virus was divided into aliquots and stored at −80°C until use. Mice were infected by intraperitoneal injection of 0.2–2 × 106 PFU of MHV-A59 in a 0.2 ml vol. Mortality was assessed at 14 d after infection. LD50 were calculated by the PROBIT method.
Sequence Analysis.
DNA fragments for sequencing were generated by PCR amplification of genomic DNA from +/+ and plt mice with the following primer pairs (position of products relative to transcriptional start site is listed in parentheses): GTCAACCTGGTCTATGAATCCCAG and CACGACATCACACTGAACCGATC (−1025 to −428); GCTCAGCACTTATGGAAGGGTG and GCCATGATTGTGGTTGAGTTGAG (−598 to 59); TCTCACCTACAGCTCTGGTCTCATC and GTGAACCACCCAGCTTGAAGTTC (12 to 779); CTGGAAAGAAAGGAAAGGGCTC and ATGGAGAGCAGGTTCAGGTCTTGG (571 to 1046); CTTCAACCATTACATCTGCACGG and TTTACTCCTGCCTGGGGATAGG (873 to 1965). PCR was performed using a Perkin Elmer 9600 thermal cycler in a final volume of 50 μl containing 5 U AmpliTaq enzyme with its 1× buffer, 0.2 mM of each dNTP, and 1 μM of each primer. Samples were heated to 94°C for 4 min, AmpliTaq was added, and samples were cycled for 35 cycles at 94°C for 30 s, 60°C for 1 min, and 72°C for 1 min. PCR products were cloned into PCRII TA (Invitrogen) according to the manufacturer's instructions and sequenced using dye terminator technology.
Results
Lack of SLC Expression in plt Mice.
The expression pattern of SLC mRNA and protein in normal mice has been determined (22, 23). To examine the expression of SLC in plt mice, in situ hybridization on tissue sections was performed using 35S-labeled SLC antisense riboprobe. No SLC mRNA was detected in any tissue of plt mice, including LN, spleen, PP, thymus, and lymphatic endothelium, whereas the location and abundance of SLC mRNA was normal in the same tissues of +/+ mice (Fig. 1, A–F, and data not shown). Northern blot analysis of total RNA from peripheral LNs and spleen confirmed the complete absence of SLC transcripts in plt mice (Fig. 1 G).
Figure 1.
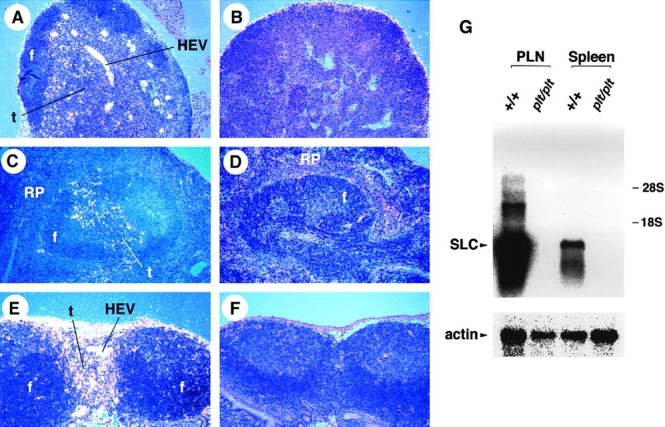
SLC mRNA is not expressed in plt mice. Sections of paraffin-embedded tissues from +/+ (A, C, and E) and plt (B, D, and F) mice were hybridized with 35S-labeled SLC antisense riboprobe and exposed for 8 wk. SLC hybridization signal (white dots) can be seen in LN (A), spleen (C), and PP (E) of +/+ mice. No SLC signal is detected in the LN (B), spleen (D), and PP (F) of plt mice. f, lymphoid follicles; t, T cell zone; RP, red pulp. (G) Total RNA from peripheral LNs (PLN) and spleen of +/+ and plt mice was subjected to Northern blot analysis with 32P-labeled SLC probe and subjected to autoradiography. Blots were stripped and reprobed with actin probe to determine variability in gel loading.
Decreased Accumulation of DCs in the T Cell Zones of plt Mice.
plt mice have a defect in the homing of naive T cells that leads to a marked decrease in the number of these cells in the T cell zones of LNs, PPs, and spleen. The finding that SLC is not expressed in plt mice strongly supports the hypothesis that SLC is required for naive T cell homing. Like naive T cells, activated DCs migrate to the T cell zones of secondary lymphoid organs. Therefore, we examined the distribution of DCs in plt mice. By FACS® analysis, DC numbers were decreased 60% in the LNs of plt mice (Fig. 2 A). The average number of total I-A+ DCs in the pooled LNs of +/+ mice was 24.5 ± 4.7 × 103 per mouse compared with 9.1 ± 2.4 × 103 in plt mice (P < 0.01, n = 3). Immunohistochemistry established that this decrease was most pronounced in the deep cortex. Those DCs that were present in plt mice typically clustered in the internodular cortex (Fig. 3, A and B). In the spleens of plt mice, the total number of DCs was normal (Fig. 2 B), but there was a striking abnormality in cell distribution (Fig. 3). Staining with anti-CD11c revealed a marked decrease in the number of DCs within the white pulp (Fig. 3, C and D). There was a concomitant increase in the number of DCs located outside the white pulp, either in bridging channels or in isolated clusters within the red pulp, such that the total number of splenic DCs remained normal. Staining with NLDC-145, which in the spleen is specific for interdigitating DCs (34), also revealed a decrease in the number of these cells within the white pulp and demonstrated that those DCs outside the white pulp were NLDC-145 negative (Fig. 3, E and F).
Figure 2.
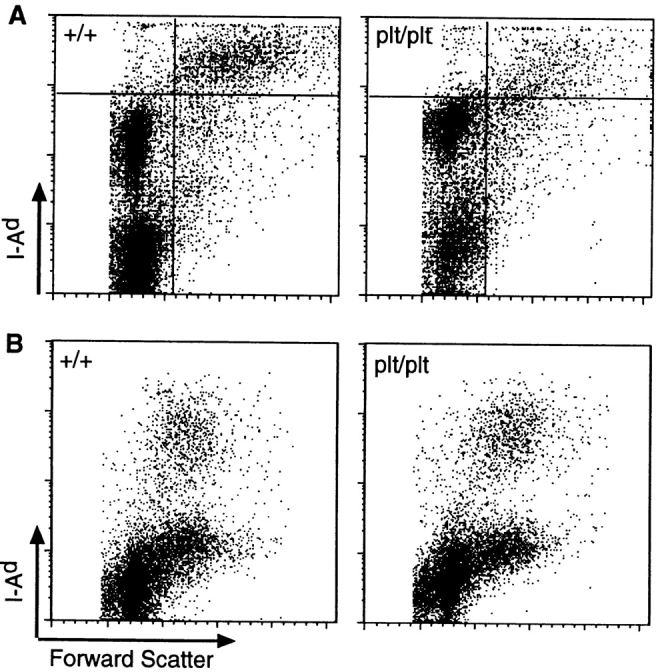
(A) The number of DCs is decreased in LNs of plt mice. LNs of +/+ and plt mice were collected and centrifuged over metrizamide gradients. Cells at the interface were collected, stained with FITC-conjugated anti–mouse I-Ad, normalized to the total number of cells per LN, and analyzed by FACS® to detect I-A+ DCs (top right quadrant). One representative experiment of three is shown. (B) The number of DCs is normal in spleens of plt mice. Spleens of +/+ and plt mice were collected, dissociated with collagenase, and stained with FITC-anti–I-Ad, biotinylated anti-B220, and biotinylated anti-CD3 followed by SA-PerCP, normalized to the total number of cells per spleen, and analyzed by flow cytometry. CD3−, B220− gated cells in one representative experiment of three are shown.
Figure 3.
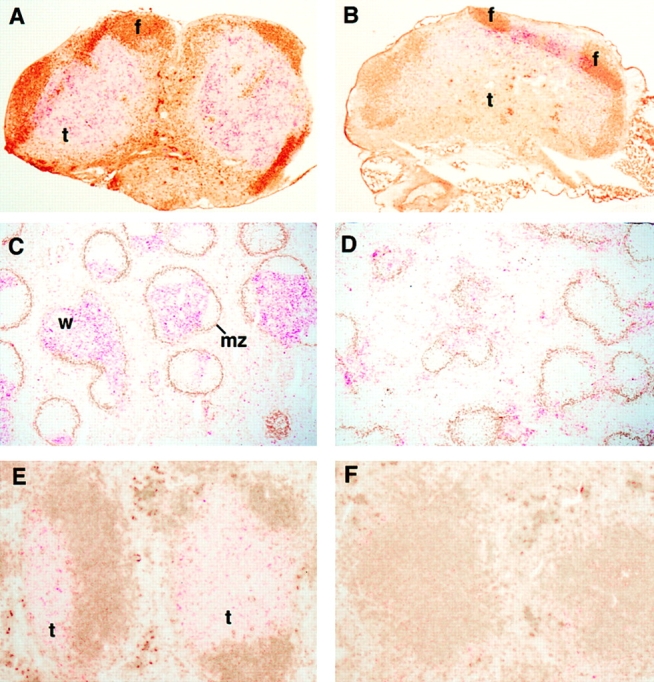
DCs do not accumulate in the T cell zones of plt mice. The distribution of DCs was examined in cryostat sections of +/+ (A, C, and E) and plt (B, D, and F) mice by immunohistochemistry. In LNs from +/+ mice (A), NLDC-145+ DCs (red) are distributed uniformly throughout the T cell zone (t). IgMa + B cells (brown) are shown for orientation. When LNs from plt mice are stained similarly (B), DCs are found only in the outer cortex between B cell follicles (f). In spleens of +/+ mice (C), numerous CD11c+ DCs (red) are seen within the white pulp (w). Marginal zones, which form the outer boundary of the white pulp, are identified by MOMA-1 staining (brown). In spleens of plt mice (D), few DCs appear within the white pulp. In spleens of +/+ mice (E), NLDC-145+ DCs (red) are seen within the T cell zone (t). IgMa + B cells (brown) are shown for orientation. In plt spleen (F), few NLDC-145+ cells are seen.
Decreased Migration of DCs into T Cell Zones of plt Mice.
Interdigitating DCs in the T cell zones of LNs arise from DCs in the periphery which migrate to this area via the lymph after activation. To determine if the paucity of interdigitating cells observed in plt mice was due to a defect in the migration of DCs to LNs, we examined this migration after contact sensitization. 24 h after skin painting with 2 mg FITC, the frequency of FITC+ cells in the draining LNs of plt mice was markedly reduced compared with +/+ mice (Fig. 4 A). The identity of these cells as DCs was confirmed by their characteristic forward and side scatter profiles, their staining with anti-CD11c (Fig. 4 B) and anti–I-A (Fig. 4 C), and their low buoyant density. A comparison of eight FITC-painted plt mice with controls at 24 h revealed a 75% decrease in the number of FITC+ DCs in draining LNs (Fig. 4 D).
Figure 4.
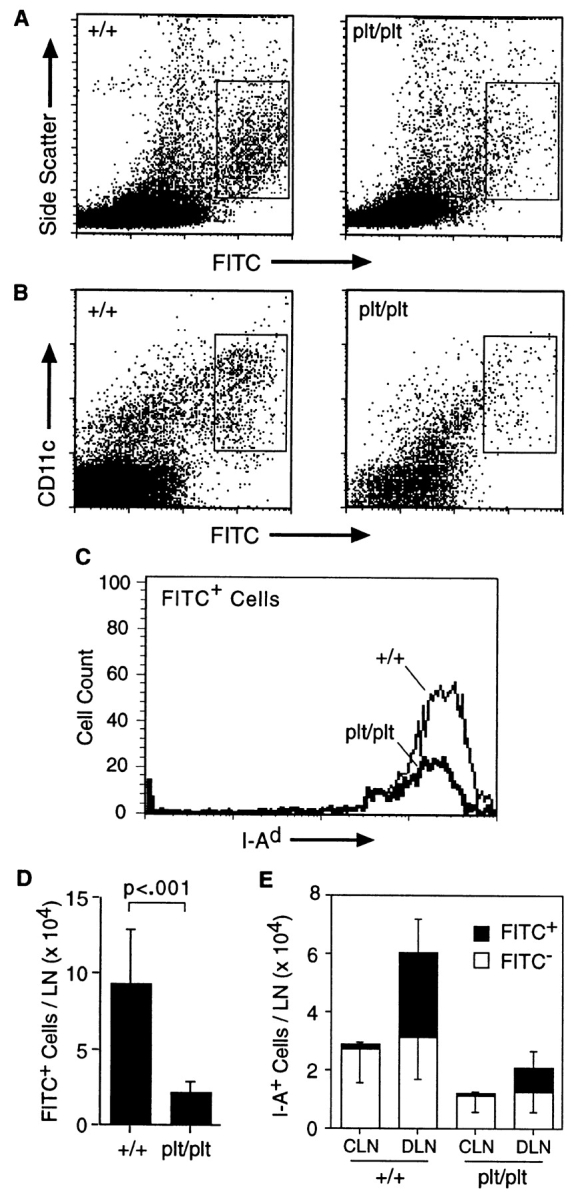
Decreased migration of skin DCs to LNs in plt mice after contact sensitization with FITC. (A) The shaved abdomens of +/+ and plt mice were painted with 2 mg FITC. After 24 h draining LNs were removed, dissociated, normalized to the total number of cells per LN, and analyzed by flow cytometry. A decreased number of large FITC+ cells (boxed area) can be seen in plt mice. One of eight representative experiments is shown. (B) Representative FACS® profile showing a marked decrease of CD11c+ FITC+ cells in plt mice after FITC skin painting. (C) Draining LN cells from FITC-painted mice were partially purified on metrizamide gradients, stained with biotinylated anti–I-Ad followed by SA-PerCP, and analyzed by flow cytometry. Only large FITC+ cells (boxed areas in A) are shown. (D) The number of FITC+ DCs (boxed areas in A) that accumulate in LNs after skin painting with 2 mg FITC is reduced in plt mice. Numbers represent mean ± SD (n = 8). (E) Comparison of DC content in contralateral (CLN) and draining (DLN) inguinal LNs in mice painted on one flank with 0.2 mg FITC. Single cell suspensions were prepared from individual LNs, stained with anti–I-Ad and anti-B220, and analyzed by flow cytometry gated on I-A+ B220− cells.
To ensure that the FITC signal in draining LNs was due to the migration of DCs rather than FITC accumulation within the LNs, we performed unilateral contact sensitization with low dose (0.2 mg) FITC and compared contralateral and draining LNs. In this procedure, resident (FITC−) DCs can be distinguished from newly migrated (FITC+) DCs (35). 24 h after skin painting, contralateral and draining LNs contained similar numbers of FITC− DCs in both +/+ and plt mice (Fig. 4 E). Similar to results shown above, the number of resident DCs was decreased 60% in plt mice. The draining LNs of painted mice demonstrated accumulation of FITC+ I-A+ cells. In plt mice, the number of these newly migrated FITC+ DCs was decreased 73% compared with +/+ mice (29.5 ± 11 × 103 vs. 8.1 ± 5.7 × 103, P = 0.011, n = 5).
To determine if the decreased accumulation of DCs in the LNs of plt mice was secondary to a decreased number of resident skin DCs, abdominal epidermis of +/+ and plt mice was stained with anti–I-A (Fig. 5, A and B). There was no significant difference in the density, morphology, or distribution of I-A+ DCs in the epidermis of untreated plt and +/+ mice. The average number of I-A+ epidermal cells in +/+ mice was 14 ± 2.1 per high power field (HPF) compared with 15.5 ± 2.8 in plt mice.
Figure 5.
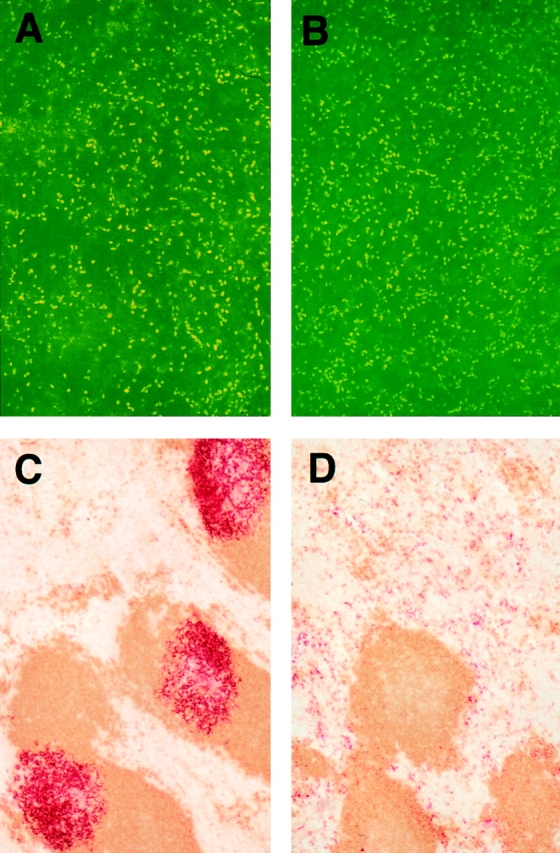
(A and B) Whole mounts of abdominal epidermis from +/+ (A) and plt (B) mice show normal numbers and distribution of DCs. Abdominal epidermis was separated from dermis and stained with anti–I-Ad (reference 31). (C and D) The accumulation of activated DCs in splenic T cell zones is decreased in plt mice. Spleens of +/+ (C) and plt (D) mice were removed 6 h after intraperitoneal injection of LPS. Frozen sections were prepared and stained for CD11c+ DCs (red) and B220+ B cells (brown).
We also examined the migration of splenic DCs upon activation. Mouse spleen normally contains a population of interdigitating DCs located within the T cell zone and a separate population of DCs located in marginal zone bridging channels and red pulp (30, 36, 37). Treatment of mice with LPS has been shown to cause a rapid decrease in the number of DCs located in bridging channels and red pulp and a simultaneous increase in the number of interdigitating DCs (38). This has led to the hypothesis that splenic DCs outside the T cell zone represent an immature population that migrates to the T cell zone upon activation. 6 h after intraperitoneal injection of LPS, spleens of +/+ mice demonstrated intense staining of DCs within T cell zones but few DCs in bridging channels or red pulp (Fig. 5 C). By contrast, the number of interdigitating cells in the splenic T cell zones of plt mice did not increase after LPS treatment. Most DCs remained scattered throughout the red pulp (Fig. 5 D).
Normal DC Migration out of Epidermis in plt Mice.
SLC is expressed (and therefore has the potential to mediate DC migration) at two points along the migration route of DCs from skin to LNs: in lymphatic endothelium and within the T cell zone. To determine if plt mice have a defect in the peripheral mobilization of DCs, we examined the migration of DCs out of cultured skin. Similar to our findings in abdominal epidermis, dorsal ear epidermis from plt mice contains a normal number of DCs (Fig. 6 E). When segments of dorsal ear skin were placed in culture, the number of DCs within the epidermis of +/+ and plt mice decreased to a similar extent over 72 h (Fig. 6, A, B, and E). Over the same time period, the number of DCs within the dermis increased similarly in plt and +/+ mice (not shown). As described previously (31), migrating DCs formed cords within the dermal lymphatics of +/+ mice (Fig. 6 C). A similar formation of cords was seen in plt mice (Fig. 6 D), demonstrating that the plt mutation does not inhibit the entry of DCs into lymphatics. When the cells migrating out of dorsal ear skin were collected over 72 h and counted, the number of DCs released from the skin of plt and +/+ mice was similar (Fig. 6 F).
Figure 6.

Migration of DCs out of skin explants is normal in plt mice. Dorsal ear skin was floated on medium and cultured (reference 31). After 72 h, dermal and epidermal whole mounts were prepared and stained with anti–I-Ad followed by SA-FITC. The density of DCs is similar in the epidermis of +/+ (A) and plt (B) mice after 72 h of culture. DC cords form within dermal lymphatics of both +/+ (C) and plt (D) mice after 72 h of culture. (E) The density of DCs in epidermis decreases similarly in +/+ and plt mice over 72 h of culture. I-A+ cells were counted in 20 fields/slide over 4 slides and calculated as the mean ± SD of cells/ HPF. (F) Emigration of DCs out of cultured skin is normal in plt mice. Nonadherent cells were collected from the bottom of wells in which ear skin had been cultured for 72 h. Total cell number was determined by counting on a hemocytometer. The proportion of DCs was calculated by examining anti–I-Ad–stained cytospins of nonadherent cells. Results represent the mean ± SD of DCs/well over four wells.
Decreased Expression of ELC in plt Mice.
ELC is the chemokine most closely related to SLC. ELC and SLC have similar activities; both activate the CCR7 receptor, and their genes are located within 100 kb of each other in the human genome (24, 39, 40). In contrast to SLC, ELC expression is limited to interdigitating DCs in LNs and spleen (41). To determine if the decreased accumulation of DCs in plt mice leads to a decrease in the production of ELC, we examined ELC expression in plt mice. By Northern analysis, ELC mRNA levels in the LNs and spleen of plt mice were reduced compared with +/+ mice (Fig. 7 E). By in situ hybridization, ELC expression was reduced in the LNs of plt mice compared with +/+ mice (Fig. 7, A and B). ELC antisense probes hybridized most strongly to the outer LN cortex in a region corresponding to the area in which DCs accumulated in plt mice (compare Fig. 7 B and Fig. 3 B). In the spleens of plt mice, ELC expression was also reduced (Fig. 7, C and D). As in LNs, the hybridization signal in plt spleen localized to regions that correspond to the area in which DCs accumulated (compare Fig. 7 D and Fig. 3 D). Similar to results in +/+ mice, no ELC signal was detected in lymphatics or any nonlymphoid tissue of plt mice (data not shown).
Figure 7.

Expression of ELC mRNA is decreased in the LNs and spleens of plt mice. Tissues from +/+ (A and C) and plt (B and D) mice were analyzed as described in the legend to Fig. 1. ELC hybridization signal (white dots) can be seen in LN (A) and spleen (C) of +/+ mice. The intensity of ELC signal is reduced in the LN (B) and spleen (D) of plt mice. f, lymphoid follicles; RP, red pulp. (E) Northern blot analysis. Total RNA from peripheral LNs (PLN) and spleen of +/+ and plt mice was hybridized with 32P-labeled ELC probe and subjected to autoradiography. Blots were stripped and reprobed with actin probe to determine variability in gel loading.
plt Mice Have Decreased Resistance to Viral Infection.
The entry of lymphocytes and DCs into T cell zones is likely to be a critical step in the development of a primary immune response. Because plt mice have defects in lymphocyte and DC migration, they are predicted to be deficient in mounting such a response. To evaluate the response of plt mice to viral infection, these mice were infected with MHV administered by intraperitoneal injection. plt mice demonstrate a markedly enhanced sensitivity to viral infection (Fig. 8). At a dose of 20 PFU, which causes no mortality in wild-type mice, 80% of the infected plt mice died. The LD50 for MHV in wild-type mice was calculated to be 1,840 PFU. In plt mice the LD50 for MHV was 5.67 PFU, a 300-fold decrease.
Figure 8.

Increased sensitivity of plt mice to MHV infection. Mice were injected intraperitoneally with the indicated doses of MHV and scored for mortality over the next 2 wk. Each data point represents at least eight infected mice. The calculated LD50 for MHV infection of +/+ and plt mice are indicated.
SLC Introns and Exons Are Intact in plt Mice.
Although the plt mutation (29) and the SLC gene (Gunn, M.D., unpublished data) map to the same genetic locus on mouse chromosome 4, the molecular basis of the plt mutation has not been identified. To identify the genetic abnormality that leads to a loss of SLC expression in plt mice, we have initiated a search for the plt mutation at the DNA level. The murine SLC gene contains four exons spanning a total of 1 kb (Gunn, M.D., unpublished data). To determine if the plt mutation occurs within SLC introns or exons, overlapping PCR fragments were generated from genomic DNA of both plt and +/+ mice. Comparison of plt and +/+ sequence over a 3-kb region extending from 1 kb upstream of the transcriptional start site to 1 kb downstream of the polyadenylation signal revealed several single base changes but no mutation that would account for a loss of SLC expression (data not shown). Thus, while the plt mutation is located in the proximity of the SLC gene, it is not within SLC introns or exons.
Discussion
Chemokines are implicated in two distinct steps in lymphocyte extravasation. First, they can stimulate integrin activation and the firm adhesion of rolling lymphocytes. Second, they can provide a chemotactic signal for lymphocyte migration through the endothelium and into the underlying tissue. We believe that SLC is the chemokine that mediates the first of these steps during the homing of naive T cells to LNs and PPs. Evidence in support of this conclusion includes the expression of SLC on HEVs, the ability of SLC to activate both αLβ2 and α4β7 integrins on T cells, the ability of SLC to stimulate the rapid arrest of rolling T cells with an efficiency similar to that seen in vivo, and the absence of any other known chemokine with similar characteristics. We now add to this body of evidence by demonstrating that mice homozygous for the plt mutation do not express SLC (Fig. 1). The lymphocyte homing defect in plt mice is similar to that seen when naive T cells are treated with pertussis toxin—failure of naive T cells to enter LNs, PPs, or the white pulp of spleen, suggesting that the defect in plt mice occurs in the initial steps of lymphocyte extravasation (29, 42, 43). Like pertussis toxin treatment, the lymphocyte homing defect in plt mice probably represents a loss of the signal for the integrin-mediated firm adhesion of naive T cells.
Although the molecular mechanisms have not been elucidated, naive lymphocytes are believed to enter the white pulp of spleen by a series of adhesive and chemotactic steps similar to those that occur in LNs. The lack of T cells in the white pulp of spleen in plt mice suggests that SLC is also required for the entry of naive T cells into the T cell zones of spleen. By analogy with the defect seen in LNs, it is plausible that SLC provides the stimulus for an integrin activation step that is required for movement of naive T cells into the white pulp of spleen. SLC may also provide a chemotactic stimulus that guides naive T cells into the T cell zones of LNs and spleen. However, this function is unlikely to be unique, as other lymphocyte-specific chemokines such as ELC and DCCK1 are expressed in T cell zones (41, 44). ELC has been shown to be more potent than SLC against naive T cells, and therefore may predominate in providing a chemotactic stimulus within the T cell zone (39).
The involvement of SLC in naive lymphocyte homing was initially suggested by its expression on HEVs (23). Similarly, the expression of SLC at sites such as lymphatic endothelium suggested to us that SLC may be involved in other leukocyte trafficking events and that plt mice may have other leukocyte trafficking defects. We now find that plt mice have a defect in the migration of DCs into the T cell zones of lymphoid organs. Four lines of evidence support this finding. First, the number of DCs in the T cell zones of LNs and spleen in plt mice is decreased (Fig. 3). In the spleen, this decrease occurs despite a normal total number of splenic DCs (Fig. 2). Second, DCs appear to accumulate at the periphery of T cell zones in both LNs and spleen of plt mice (Fig. 3). The pattern of this accumulation is consistent with a block in the entry of DCs into T cell zones. Third, we directly demonstrate a decrease in the migration of FITC-activated DCs to draining LNs of plt mice (Fig. 4). It is possible that this defect in DC migration is secondary to the paucity of T cells in the LNs of plt mice. However, in both L-selectin knockout mice, which have a defect in the homing of naive lymphocytes to LNs similar to that of plt mice, and nude mice, which lack mature T cells, FITC-stimulated migration of DCs to LNs is normal (12, 35, 45, 46). This suggests that DC migration to LNs is not lymphocyte dependent. Fourth, splenic DCs in the bridging channels and red pulp of plt mice fail to redistribute to the T cell zone upon activation with LPS (Fig. 5, C and D). Taken together, these results suggest that plt mice lack a factor that acts directly on DCs to stimulate their migration. Due to its lack of expression, the probable identity of this factor is SLC.
Because SLC is expressed in lymphatic endothelium, we examined the possibility that the defect in DC migration from skin to LNs in plt mice is due to an inability of activated DCs to enter afferent lymphatics. Our findings argue against this possibility, as DCs in skin cultured from plt mice were able to migrate normally out of the epidermis, collect in lymphatics, and move out of the skin in normal numbers (Fig. 6). Although these results do not rule out the existence of a subtle defect in the mobilization of peripheral DCs, they suggest that the DC homing defect in plt mice occurs at the level of DC entry into T cell zones. At present, the function of SLC in lymphatic endothelium remains unknown.
Additional support for our conclusion that SLC is involved in DC homing to T cell zones comes from recent studies of chemokine receptor expression by DCs in vitro (47–49). DCs derived in culture from monocytes or CD34+ precursors do not express CCR7, the receptor for SLC and ELC. CCR7 expression is induced in these cells by activation with LPS, CD40 ligand, or TNF. Similarly, activated DCs increase intracellular calcium and undergo chemotaxis in response to ELC. These results suggest that immature DCs become responsive to SLC and ELC upon activation in vivo and that one or both of these chemokines is involved in the migration of activated DCs. It has been suggested that it is the activation-induced downregulation of chemokine receptors such as CCR1, CCR5, and CCR6 that allows DCs to leave sites of inflammation, whereas the more gradual induction of CCR7 on these cells allows them to enter T cell zones in response to ELC (47, 48). Our results support this hypothesis, though we would suggest that SLC, rather than ELC, may play the predominant role.
Finally, we demonstrate that plt mice have a severe immune deficiency. The LD50 of MHV in plt mice is reduced >300-fold compared with +/+ mice (Fig. 8). In comparison, the LD50 of Sendai virus is reduced 10-fold in mice lacking cytotoxic T cell function and 300-fold in nude mice compared with +/+ controls (50). In our view, the most likely cause of this immune deficiency is a defect in the presentation of viral antigen due to the failure of naive T cells and antigen-bearing DCs to enter the T cell zones of plt mice. It is also possible that SLC, like some other chemokines, provides a signal that enhances the antigen- dependent activation of lymphocytes (51, 52). A third possibility is that SLC, expressed in the thymus and in lymphatics, has a role in lymphocyte development or in the efferent limb of immune response that has not yet been characterized.
In this study, we cannot fully distinguish the biological effects of SLC from those of ELC. Because SLC and ELC signal through the same receptor, some of their functions may overlap. plt mice express no detectable SLC mRNA while ELC mRNA expression is only partially reduced (Figs. 1 and 7), and the abnormalities observed in plt mice are likely to be due to the sum of these defects. Still, some conclusions can be drawn from our results. The near-total loss of naive T cell homing in plt mice suggests an absolute requirement for SLC and the existence of at least one function that cannot be performed by ELC. In contrast, plt mice appear to exhibit only a partial defect in DC migration, suggesting that SLC and ELC may both contribute to this process and that some DCs reach the T cell zone of plt mice in response to low levels of ELC.
While we suggest that SLC is required for the normal homing of naive T cells and DCs to secondary lymphoid organs, we would emphasize that our findings do not constitute proof of this hypothesis. It is possible that the plt mutation directly affects the expression of a gene other than SLC or ELC which is responsible for some of the phenotypic abnormalities observed in plt mice. Although the plt mutation maps to the same genetic locus as SLC and plt mice express no detectable SLC, no DNA abnormality has been identified in these mice. Based on mapping data, the distance between the plt mutation and the SLC gene is between 1 and 1,000 kb. Given their normal SLC intron and exon sequences, we believe that the most likely cause of the lack of SLC expression in plt mice is a mutation involving a _cis_-acting regulatory region of the SLC gene. Such regions do not have to be in the immediate vicinity of the genes they regulate. For example, the deletion of a region 50 kb upstream of the human β-globin gene leads to a loss of its expression (53). Because the plt mutation may involve a large deletion in the vicinity of the SLC gene, we cannot rule out the possibility that this defect involves more than one gene.
Despite this uncertainty, our working hypothesis continues to be that the genetic defect in plt mice represents a single-gene null mutation of SLC. Thus far, all of the recognized abnormalities in plt mice occur in areas of SLC expression and involve cell types known to be responsive to SLC. This may change, as we are currently examining the phenotype of plt mice in more detail while attempting to identify the DNA abnormality responsible for the plt mutation. At present, however, our findings strongly suggest that SLC mediates the homing of both naive T cells and DCs to secondary lymphoid organs and that the abnormalities in plt mice are due to a genetic defect in the expression of SLC.
Acknowledgments
We thank Linda Prentice and Dale Milfay for expert technical support, Dr. K. Machii (Institute of Public Health, Tokyo, Japan) for his cooperation in MHV infection, and Jason Cyster and Steve Rosen for critical review of this manuscript.
This work was supported by an unrestricted award from the Howard Hughes Medical Institute, by the Uchida grant from the Japan Foundation for Cardiovascular Research, and by a Grant-in-Aid for Scientific Research (09770332) from the Ministry of Education, Science, Sports and Culture to H. Nakano.
Abbreviations used in this paper
AP
alkaline phosphatase
DC
dendritic cell
ELC
EBV-induced molecule 1 ligand chemokine
HEV
high endothelial venule
HPF
high power field
HRP
horseradish peroxidase
MHV
murine hepatitis virus
PerCP
peridinine chlorophyll protein
plt
paucity of lymph node T cells
PP
Peyer's patch
SA
streptavidin
SLC
secondary lymphoid organ chemokine
References
- 1.Gretz JE, Kaldjian EP, Anderson AO, Shaw S. Sophisticated strategies for information encounter in the lymph node: the reticular network as a conduit of soluble information and a highway for cell traffic. J Immunol. 1996;157:495–499. [PubMed] [Google Scholar]
- 2.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 3.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 4.Yoshie O, Imai T, Nomiyama H. Novel lymphocyte-specific CC chemokines and their receptors. J Leukocyte Biol. 1997;62:634–644. doi: 10.1002/jlb.62.5.634. [DOI] [PubMed] [Google Scholar]
- 5.Goodnow CC, Cyster JG. Lymphocyte homing: the scent of a follicle. Curr Biol. 1997;7:R219–R222. doi: 10.1016/s0960-9822(06)00105-9. [DOI] [PubMed] [Google Scholar]
- 6.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1–20. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 7.Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature. 1998;391:799–803. doi: 10.1038/35876. [DOI] [PubMed] [Google Scholar]
- 8.Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B. B cell–attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hart DN, McKenzie JL. Interstitial dendritic cells. Int Rev Immunol. 1990;6:127–138. doi: 10.3109/08830189009056624. [DOI] [PubMed] [Google Scholar]
- 10.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 11.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 12.Kupiec-Weglinski JW, Austyn JM, Morris PJ. Migration patterns of dendritic cells in the mouse. Traffic from the blood, and T cell–dependent and –independent entry to lymphoid tissues. J Exp Med. 1988;167:632–645. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Austyn JM, Kupiec-Weglinski JW, Hankins DF, Morris PJ. Migration patterns of dendritic cells in the mouse. Homing to T cell–dependent areas of spleen, and binding within marginal zone. J Exp Med. 1988;167:646–651. doi: 10.1084/jem.167.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med. 1987;166:1654–1667. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knight SC, Krejci J, Malkovsky M, Colizzi V, Gautam A, Asherson GL. The role of dendritic cells in the initiation of immune responses to contact sensitizers. I. In vivo exposure to antigen. Cell Immunol. 1985;94:427–434. doi: 10.1016/0008-8749(85)90266-7. [DOI] [PubMed] [Google Scholar]
- 16.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 17.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 18.van Ewijk W, Nieuwenhuis P. Compartments, domains and migration pathways of lymphoid cells in the splenic pulp. Experientia. 1985;41:199–208. doi: 10.1007/BF02002614. [DOI] [PubMed] [Google Scholar]
- 19.Nagira M, Imai T, Hieshima K, Kusuda J, Ridanpaa M, Takagi S, Nishimura M, Kakizaki M, Nomiyama H, Yoshie O. Molecular cloning of a novel human CC chemokine secondary lymphoid-tissue chemokine that is a potent chemoattractant for lymphocytes and mapped to chromosome 9p13. J Biol Chem. 1997;272:19518–19524. doi: 10.1074/jbc.272.31.19518. [DOI] [PubMed] [Google Scholar]
- 20.Hedrick J, Zlotnik A. Identification and characterization of a novel beta chemokine containing six conserved cysteines. J Immunol. 1997;159:1589–1593. [PubMed] [Google Scholar]
- 21.Hromas R, Kim C, Klemsz M, Krathwohl M, Fife K, Cooper S, Schnizlein-Bick C, Broxmeyer H. Isolation and characterization of Exodus-2, a novel C-C chemokine with a unique 37 amino acid carboxyl-terminal extension. J Immunol. 1997;159:2554–2558. [PubMed] [Google Scholar]
- 22.Tanabe S, Lu Z, Luo Y, Quackenbush EJ, Berman MA, Collins-Racie LA, Mi S, Reilly C, Lo D, Jacobs KA, Dorf ME. Identification of a new mouse beta-chemokine, thymus-derived chemotactic agent 4, with activity on T lymphocytes and mesangial cells. J Immunol. 1997;159:5671–5679. [PubMed] [Google Scholar]
- 23.Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci USA. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell JJ, Bowman EP, Murphy K, Youngman KR, Siani MA, Thompson DA, Wu L, Zlotnik A, Butcher EC. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3beta receptor CCR7. J Cell Biol. 1998;141:1053–1059. doi: 10.1083/jcb.141.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 26.Pachynski R, Wu SW, Gunn MD, Erle DJ. Secondary lymphoid-tissue chemokine (SLC) stimulates integrin α4β7-mediated adhesion of lymphocytes to mucosal addressin cell adhesion molecule-1 (MadCAM-1) under flow. J Immunol. 1998;161:952–956. [PubMed] [Google Scholar]
- 27.Tangemann K, Giblin P, Gunn MD, Rosen SD. A high endothelial cell-derived chemokine induces rapid, efficient, and subset-selective arrest of rolling T lymphocytes on a reconstituted endothelial substrate. J Immunol. 1998;161:6330–6337. [PubMed] [Google Scholar]
- 28.Nakano H, Tamura T, Yoshimoto T, Yagita H, Miyasaka M, Butcher EC, Nariuchi H, Kakiuchi T, Matsuzawa A. Genetic defect in T lymphocyte-specific homing into peripheral lymph nodes. Eur J Immunol. 1997;27:215–221. doi: 10.1002/eji.1830270132. [DOI] [PubMed] [Google Scholar]
- 29.Nakano H, Mori S, Yonekawa H, Nariuchi H, Matsuzawa A, Kakiuchi T. A novel mutant gene involved in T-lymphocyte-specific homing into peripheral lymphoid organs on mouse chromosome 4. Blood. 1998;91:2886–2895. [PubMed] [Google Scholar]
- 30.Leenen PJ, Radosevic K, Voerman JS, Salomon B, van Rooijen N, Klatzmann D, van Ewijk W. Heterogeneity of mouse spleen dendritic cells: in vivo phagocytic activity, expression of macrophage markers, and subpopulation turnover. J Immunol. 1998;160:2166–2173. [PubMed] [Google Scholar]
- 31.Larsen CP, Steinman RM, Witmer-Pack M, Hankins DF, Morris PJ, Austyn JM. Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med. 1990;172:1483–1493. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyuwa S. Replication of murine coronaviruses in mouse embryonic stem cell lines in vitro. Exp Anim. 1997;46:311–313. doi: 10.1538/expanim.46.311. [DOI] [PubMed] [Google Scholar]
- 33.Okumura A, Machii K, Azuma S, Toyoda Y, Kyuwa S. Maintenance of pluripotency in mouse embryonic stem cells persistently infected with murine coronavirus. J Virol. 1996;70:4146–4149. doi: 10.1128/jvi.70.6.4146-4149.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witmer-Pack MD, Swiggard WJ, Mirza A, Inaba K, Steinman RM. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol. 1995;163:157–162. doi: 10.1006/cimm.1995.1110. [DOI] [PubMed] [Google Scholar]
- 35.Hill S, Edwards AJ, Kimber I, Knight SC. Systemic migration of dendritic cells during contact sensitization. Immunology. 1990;71:277–281. [PMC free article] [PubMed] [Google Scholar]
- 36.Agger R, Witmer-Pack M, Romani N, Stossel H, Swiggard WJ, Metlay JP, Storozynsky E, Freimuth P, Steinman RM. Two populations of splenic dendritic cells detected with M342, a new monoclonal to an intracellular antigen of interdigitating dendritic cells and some B lymphocytes. J Leukocyte Biol. 1992;52:34–42. doi: 10.1002/jlb.52.1.34. [DOI] [PubMed] [Google Scholar]
- 37.Vremec D, Shortman K. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J Immunol. 1997;159:565–573. [PubMed] [Google Scholar]
- 38.De Smedt T, Pajak B, Muraille E, Lespagnard L, Heinen E, De Baetselier P, Urbain J, Leo O, Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida R, Nagira M, Kitaura M, Imagawa N, Imai T, Yoshie O. Secondary lymphoid-tissue chemokine is a functional ligand for the CC chemokine receptor CCR7. J Biol Chem. 1998;273:7118–7122. doi: 10.1074/jbc.273.12.7118. [DOI] [PubMed] [Google Scholar]
- 40.Kim CH, Pelus LM, White JR, Applebaum E, Johanson K, Broxmeyer HE. CK beta-11/macrophage inflammatory protein-3 beta/EBI1-ligand chemokine is an efficacious chemoattractant for T and B cells. J Immunol. 1998;160:2418–2424. [PubMed] [Google Scholar]
- 41.Ngo VN, Tang HL, Cyster JG. Epstein-Barr virus–induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J Exp Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spangrude GJ, Braaten BA, Daynes RA. Molecular mechanisms of lymphocyte extravasation. I. Studies of two selective inhibitors of lymphocyte recirculation. J Immunol. 1984;132:354–362. [PubMed] [Google Scholar]
- 43.Cyster JG, Goodnow CC. Pertussis toxin inhibits migration of B and T lymphocytes into splenic white pulp cords. J Exp Med. 1995;182:581–586. doi: 10.1084/jem.182.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adema GJ, Hartgers F, Verstraten R, de Vries E, Marland G, Menon S, Foster J, Xu Y, Nooyen P, McClanahan T, et al. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature. 1997;387:713–717. doi: 10.1038/42716. [DOI] [PubMed] [Google Scholar]
- 45.Catalina MD, Carroll MC, Arizpe H, Takashima A, Estess P, Siegelman MH. The route of antigen entry determines the requirement for L-selectin during immune responses. J Exp Med. 1996;184:2341–2351. doi: 10.1084/jem.184.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knight SC, Bedford P, Hunt R. The role of dendritic cells in the initiation of immune responses to contact sensitizers. II. Studies in nude mice. Cell Immunol. 1985;94:435–439. doi: 10.1016/0008-8749(85)90267-9. [DOI] [PubMed] [Google Scholar]
- 47.Sozzani S, Allavena P, D'Amico G, Luini W, Bianchi G, Kataura M, Imai T, Yoshie O, Bonecchi R, Mantovani A. Differential regulation of chemokine receptors during dendritic cell maturation: a model for their trafficking properties. J Immunol. 1998;161:1083–1086. [PubMed] [Google Scholar]
- 48.Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, Qin S, Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1988;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 50.Kast W, Bronkhorst A, de Waal L, Melief C. Cooperation between cytotoxic and helper T lymphocytes in protection against lethal Sendai virus infection. Protection by T cells is MHC-restricted and MHC-regulated; a model for MHC–disease associations. J Exp Med. 1986;164:723–738. doi: 10.1084/jem.164.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science. 1995;269:1727–1730. doi: 10.1126/science.7569902. [DOI] [PubMed] [Google Scholar]
- 52.Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation. I. Beta chemokines costimulate human T lymphocyte activation in vitro. J Immunol. 1996;156:2095–2103. [PubMed] [Google Scholar]
- 53.Driscoll MC, Dobkin CS, Alter BP. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5′ beta-globin gene activation-region hypersensitive sites. Proc Natl Acad Sci USA. 1989;86:7470–7474. doi: 10.1073/pnas.86.19.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]