Macrophage Inflammatory Protein 3α Is Expressed at Inflamed Epithelial Surfaces and Is the Most Potent Chemokine Known in Attracting Langerhans Cell Precursors (original) (raw)
Abstract
Dendritic cells (DCs) form a network comprising different populations that initiate and differentially regulate immune responses. Langerhans cells (LCs) represent a unique population of DCs colonizing epithelium, and we present here observations suggesting that macrophage inflammatory protein (MIP)-3α plays a central role in LC precursor recruitment into the epithelium during inflammation. (a) Among DC populations, MIP-3α was the most potent chemokine inducing the selective migration of in vitro–generated CD34+ hematopoietic progenitor cell–derived LC precursors and skin LCs in accordance with the restricted MIP-3α receptor (CC chemokine receptor 6) expression to these cells. (b) MIP-3α was mainly produced by epithelial cells, and the migration of LC precursors induced by the supernatant of activated skin keratinocytes was completely blocked with an antibody against MIP-3α. (c) In vivo, MIP-3α was selectively produced at sites of inflammation as illustrated in tonsils and lesional psoriatic skin where MIP-3α upregulation appeared associated with an increase in LC turnover. (d) Finally, the secretion of MIP-3α was strongly upregulated by cells of epithelial origin after inflammatory stimuli (interleukin 1β plus tumor necrosis factor α) or T cell signals. Results of this study suggest a major role of MIP-3α in epithelial colonization by LCs under inflammatory conditions and immune disorders, and might open new ways to control epithelial immunity.
Keywords: dendritic cell, chemokine, migration, regulation, in vivo expression
Introduction
Dendritic cells (DCs) are bone marrow–derived professional APCs with a unique ability to induce primary immune responses. They represent a trace population of cells found in virtually every tissue and biological fluid (for reviews, see references 1 2 3 4 5). DCs exist at different maturation stages interconnected by defined pathways of circulation 1. In the periphery, immature DCs such as Langerhans cells (LCs) capture antigens 6 and, under the influence of inflammatory stimuli, subsequently migrate via the lymphatic system or blood vessels. They reach secondary lymphoid organs and home to the T cell–rich areas, where they present processed antigen to naive T cells and generate antigen-specific primary T cell responses.
Recently, several studies, including ours, have demonstrated that the traffic of DCs from the site of antigen capture to the draining lymphoid organs involves selective chemokines active on maturing DCs through CC chemokine receptor (CCR)7 7 8 9 10. 6Ckine (secondary lymphoid tissue chemokine [SLC], Exodus-2) expressed by lymphatic vessels may direct into the lymph stream antigen-loaded maturing DCs leaving the site of infection 11 12 13 14. Mature DCs entering the draining lymph nodes might then be driven into the paracortical area, in response to the production of MIP-3β (EBV-induced molecule 1 ligand chemokine [ELC], Exodus-3) and/or 6Ckine by cells spread over the T cell zone 7 15 . This role of MIP-3β and 6Ckine is supported by the specific alterations in T cell and DC homing into lymph nodes observed in natural mutant mice deficient for 6Ckine 16 17 18 and in CCR7 genetically deficient mice 19.
In addition to the heterogeneity related to their stage of maturation, DC populations differ in their origin and/or function. In particular, human DC subsets have been reported to exert different functions, with regard to the regulation of B cell proliferation 20 21 and the differentiation of T cell responses towards type I or type II 22. Recently, human circulating blood CD11c− DC precursors 23 24 have been shown to correspond to natural IFN-α–producing cells (NIPCs), suggesting a role of this population during viral infection 25. Finally, LCs represent a population of DCs only found in epithelia, and whose specific function is not yet fully elucidated. Several studies have shown that LCs are regulated independently from other DC populations. In particular, TGF-β has been identified as an essential factor for the development of LCs both in vitro and in vivo 26 27 28 29 30.
The selective recruitment of a specific DC population at the site of infection will likely determine the type of immune response initiated. Indeed, after pathogen invasion, DCs are recruited at the site of inflammation as illustrated in rat, where intratracheal antigen instillation induces accumulation of DCs in the airway epithelium 31 32. Although several chemokines (monocyte chemotactic protein [MCP]-3, MCP-4, MIP-1α, MIP-1β, MIP-3α, regulated upon activation, normal T cell expressed and secreted protein [RANTES], stromal cell–derived factor [SDF]-1, thymus-expressed chemokine [TECK], and macrophage-derived chemokine [MDC]) have been reported to attract immature DCs in vitro 7 9 33 34 35 36, their relative contribution in normal and pathological conditions remains to be understood. In this study, we have investigated the response of LC precursors to chemokines. We show that among all DC-attracting chemokines, MIP-3α is the main chemokine expressed by the epithelium that attracts LC precursors. This specificity may open avenues for the understanding and eventually for the control of the specific function of epithelial LCs.
Materials and Methods
Hematopoietic Factors, Reagents, and Cell Lines.
Recombinant human (rh)GM-CSF (specific activity 2 × 106 U/mg; Schering-Plough) was used at a saturating concentration of 100 ng/ml. rhTNF-α (specific activity 2 × 107 U/mg; Genzyme) was used at an optimal concentration of 2.5 ng/ml 37. Recombinant human stem cell factor (rhSCF) (specific activity 4 × 105 U/mg; R&D Systems) was used at an optimal concentration of 25 ng/ml. rhIL-4 (specific activity 2 × 107 U/mg; Schering-Plough) was used at a saturating concentration of 50 U/ml. Recombinant human chemokines MIP-1α (specific activity 2 × 105 U/mg), MIP-1β (specific activity 104 U/mg), RANTES (specific activity 104 U/mg), MCP-1 (specific activity 104 U/mg), MCP-2 (specific activity 104 U/mg), MCP-3 (specific activity 104 U/mg), MCP-4 (specific activity 104 U/mg), MIP-3α (specific activity 4 × 105 U/mg), and SDF-1 (specific activity 2 × 105 U/mg) were obtained from R&D Systems.
Generation of DCs from Cord Blood CD34_+_ Hematopoietic Progenitor Cells or from Peripheral Blood Monocytes.
Cells bearing CD34+ antigen were isolated from umbilical cord blood mononuclear fractions through positive selection as described 37 38, using anti-CD34+ mAb (Immu-133.3; Immunotech), goat anti–mouse IgG–coated microbeads (Miltenyi Biotec), and Midimacs separation columns (Miltenyi Biotec). After purification, CD34+ cells (80–99% purity) were cryopreserved in 10% DMSO.
Cultures were established in the presence of SCF, GM-CSF, TNF-α, and 2.5% human AB+ serum, as described 37, in endotoxin-free medium consisting of RPMI 1640 (GIBCO BRL) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Flow Laboratories), 10 mM Hepes, 2 mM l-glutamine, 5 × 10−5 M β-mercaptoethanol, 100 μg/ml gentamicin (Schering-Plough). After thawing, CD34+ cells were seeded for expansion in 25–75-cm2 culture vessels (Linbro; Flow Laboratories) at 2 × 104 cells/ml. Cells were used at day 6 where 15–30% of the cells are CD1a+CD14−, representing LC precursors 37.
Monocytes were purified by immunomagnetic depletion (Dynabeads; Dynal) after preparation of PBMCs followed by a 51% Percoll gradient. The depletion was performed with anti-CD3 (OKT3), anti-CD19 (4G1), anti-CD8 (OKT8), anti-CD56 (NKH1; Coulter), and anti-CD16 (ION16; Immunotech) mAbs. Monocyte-derived DCs were produced by culturing purified monocytes for 6–7 d in the presence of GM-CSF and IL-4 39.
Enrichment in CD11c+ DCs and CD11c− DC Precursors from Peripheral Blood, and in LCs from Skin.
CD11c+ DCs and CD11c− cells were enriched as described previously from peripheral blood 23 24. Mononuclear cells were isolated by centrifugation over Ficoll-Hypaque. Then, the depletion was performed with anti-CD3 (OKT3), anti-CD19 (4G7), anti-CD14 (MOP9), anti-NKH1 (Coulter), anti-glycophorine A (Immunotech), anti-CD16 (Immunotech), and anti-CD35 (Immunotech) to remove T and B lymphocytes, monocytes, NK cells, and granulocytes from the resulting low density cells by magnetic beads (anti–mouse Ig–coated Dynabeads; Dynal). All of the depletion procedure was performed in the presence of 0.5 mM EDTA. The enriched population contained between 20 and 35% DCs identified by their expression of HLA-DR (Tricolor; Becton Dickinson) and the lack of FITC-labeled lineage markers CD3, CD14, CD15, CD16, CD20, and CD57.
Epidermal cell suspensions were obtained from normal skin patients undergoing reconstructive plastic surgery of breast or abdomen, as described elsewhere 40. LC enrichment was achieved by successive density gradient centrifugation steps and depletion of basal keratinocytes. The isolated cells contained 55–75% CD1a+ LCs.
Nonhematopoietic Cell Culture.
Human primary keratinocytes, dermal fibroblasts, and endothelial cells (human umbilical vein endothelial cells) were purchased from Clonetics and cultured in keratinocyte (KGM-2), fibroblast (FGM), or endothelial cell (EGM-2) growth media (Clonetics). For production of supernatants, cells were seeded at 1–2 × 104 cells per ml, and after 3–5 d (80% confluence), fresh medium was added in the presence or absence of activators (IL-1β at 10 ng/ml and TNF-α at 2.5 ng/ml). Supernatants were harvested after 48 h. A similar procedure was used for cell lines of various origins (American Type Culture Collection), including the renal cell carcinoma lines CHA and VER (generated by Léon Bérard Center, Hospital, Lyon, France).
Analysis of CCR6 Expression by FACS®.
Expression of CCR6 was determined using an mAb against CCR6 (R&D Systems). On CD34+-derived DCs, monocyte-derived DCs, and freshly isolated skin LCs, double staining was performed with anti-CD1a–FITC. On monocytes, double stainings were performed with anti-CD14–FITC. On blood DCs, triple stainings were performed with anti–HLA-DR–Tricolor (Becton Dickinson), anti-CD3, -CD14, -CD16, -CD19, -CD20, -CD56–FITC (Becton Dickinson), and anti-CCR6–PE (R&D Systems).
Chemotaxis Assay.
Transwell (5-μm-pore; Costar) experiments were performed to characterized heterogeneous populations. Serial dilutions of chemokines or supernatants were added to 24-well plates. 5 × 105 cells were added to transwell inserts. Plates were incubated for 1.5 h at 37°C. After removal of the transwell inserts, migrated cells were counted and stained for flow cytometry, to either differentiate between CD1a+ and CD14+ DC precursors (from CD34+ hematopoietic progenitor cells [HPCs]) or HLA-DR+Lin−CD11c+ circulating blood DCs and HLA-DR+Lin−CD11c− blood DC precursors.
In Situ Hybridization.
In situ hybridization for MIP-3α was performed as described 7 41. Coupled primers were used for amplifying by reverse transcription PCR the majority of the open reading frame of the MIP-3α gene. Sense and antisense probes for MIP-3α were radiolabeled with 35S-UTP, and then partially degraded by alkaline hydrolysis. 6-μm cryostat sections of human tonsil and normal, lesional, and nonlesional psoriatic skin were prepared on charged electrostatic slides (SuperFrost/Plus; Polylabo) and fixed with cold acetone and 4% paraformaldehyde followed by 0.1 M triethanolamine/0.25% acetic anhydride. The sections were hybridized overnight at 50°C (2–3 × 106 cpm/slide), RNase A treated, and after washing under stringent conditions, were exposed for 40 d. After development, the sections were stained with Harris hematoxylin.
Immunohistochemistry.
Frozen 6-μm tissue sections (human tonsils and skin) were fixed in acetone and in 4% paraformaldehyde before the immunostaining. To block the nonspecific activities, sections were pretreated with avidin D and biotin solutions (Blocking kit; Vector Laboratories) for 10 min each step and with 0.3% hydrogen peroxide (Sigma-Aldrich) for 15 min at room temperature. After a brief washing in PBS, the sections were incubated with blocking serum (2% normal human serum) for at least 30 min before adding both primary antibodies. Sections were immunostained between two (simultaneously) or three (sequentially) of the following antibodies: anti–hMIP-3α goat polyclonal antibody (IgG; R&D Systems), anti-hCytokeratin (IgG1, MNF116; Dako), anti–hE-cadherin (IgG1, HECD-1; Takara), anti-hCD1a (IgG2a, Leu-6; Becton Dickinson), anti-hLangerine (IgG1, DCGM4; Schering-Plough [42]), biotinylated anti–hDC-lysosomal-associated membrane protein (LAMP) (IgG1, 104G4; Schering-Plough [43]), anti-hCollagen type IV (IgG1, CIV22; Dako), anti-hCCR6 (IgG2b, 53103.111; R&D Systems) mouse mAbs for 1 h at room temperature in a humid atmosphere. The binding of goat IgG was detected by biotinylated rabbit anti–goat IgG followed by streptavidin-peroxidase, both included in the Vectastain ABC kit (Goat IgG PK-4005; Vector Laboratories). The binding of mouse IgG2a and IgG2b was revealed by biotinylated sheep anti–mouse IgG2a (AB274; The Binding Site) and IgG2b (AB275; The Binding Site), respectively, followed by ExtrAvidin–Horseradish Peroxidase (E2886; Sigma-Aldrich). The binding of mouse IgG1 was revealed by rabbit alkaline phosphatase–labeled anti–mouse IgG (D0314; Dako) or by biotinylated sheep anti–mouse IgG1 (AB274; The Binding Site) followed by ExtrAvidin–Horseradish Peroxidase (E2886; Sigma-Aldrich) at room temperature in a humid atmosphere. The peroxidase and alkaline phosphatase activities were revealed using 3-amino-9-ethylcarbazole (AEC) substrate (SK-4200; Vector Laboratories) or diamino benzoate (DAB) substrate (Dako) and alkaline phosphatase substrate III (SK-5300; Vector Laboratories) for 1–10 min at room temperature, respectively. Negative controls were established by adding nonspecific isotype controls as primary antibodies or by preincubating the primary antibody with the corresponding recombinant protein (e.g., MIP-3α).
ELISA.
Inbred BALB/c mice were immunized with three successive intraperitoneal injections of complete Freund's adjuvant (Sigma-Aldrich), incomplete Freund's adjuvant, or without Freund's adjuvant, with 50 ng of purified hMIP-3α obtained from supernatants of hMIP-3α transient transfected COP5 cells. Spleens were removed for fusion 3 d after an intravenous injection of hMIP-3α. Hybridization was carried out using the nonsecreting myeloma cell line SP2/0-Ag8 with polyethylene glycol 1000 (Sigma-Aldrich). Human MIP-3α transient transfected COP5 cells were cultured for 2 d in 96-well plates and fixed in acetone. Then, hybridoma supernatants were harvested after 6 d, then incubated for 30 min on fixed hMIP-3α transient transfected COP5 cells. Antibody binding was then revealed with peroxidase-conjugated sheep anti–mouse IgG (Biosys) at a 1:200 dilution in PBS for 30 min at 37°C. Positive hybridomas were cloned by limiting dilution and expanded using a high density culture system (Integra cell line CL1000; Integra Biosciences). After sodium sulfate precipitation, the mAbs were purified by anion exchange chromatography on a Hyper-D column and peroxidase labeled (Sepracor). An ELISA was set up using one of the anti–hMIP-3α mAbs, 319F6, as a capture mAb and a peroxidase-coupled mouse anti–hMIP-3α mAb, 206D9, to reveal the captured hMIP-3α The sensitivity of the ELISA is 0.2 ng/ml of MIP-3α and does not cross-react with all of the CC and CXC chemokines tested (MIP-1α, MIP-1β, MIP-1δ, MIP-3β, Eotaxine, 6Ckine, RANTES, MCP-1, thymus and activation-regulated chemokine [TARC], MDC, TECK, SDF-1, IP-10, monokine induced by IFN-γ (MIG), and Lymphotactine from R&D Systems).
The detection of others chemokines such as hMCP-1, hRANTES, and hMIP-1α was realized by using commercial ELISAs (R&D Systems).
Results
MIP-3α Is the Most Potent Chemokine That Selectively Attracts LC Precursors among DC Populations.
As a way to understand the regulation of LC recruitment, we investigated the response of CD34+-derived LC precursors to various chemokines. CD34+ HPCs were cultured for 6 d in the presence of GM-CSF+TNF-α+SCF, and the responses of LC precursors were assessed by staining for CD1a (specific marker for LCs in this system) after 1.5 h migration in 5-μm transwells. Although the total population migrated in response to several CC chemokines (MIP-1α, MIP-1β, MCP-1–4, MIP-3α, RANTES; Fig. 1 A), the CD1a+ LC precursors responded mainly to MIP-3α and to a lower level to RANTES, MCP-3, and MIP-1α (Fig. 1 B). However, when dose–response analyses were performed, CD1a+ LC precursors migrated only in response to low doses of MIP-3α (1–10 ng/ml; not shown). MIP-3α induced the migration of CD34+-derived CD1a+ LC precursors, but not that of monocytes (not shown) and monocyte-derived DCs or that of circulating blood DCs (Fig. 2 A). These populations responded well to other chemokines such as SDF-1 or MCP-1. In contrast, freshly isolated skin LCs migrated in response to MIP-3α although to a lower level than CD34+-derived LC precursors, probably because they may be in a more differentiated stage than in vitro–generated LC precursors. These chemotactic responses to MIP-3α correlated with the expression of its receptor CCR6, which was detected by FACS® analysis on CD34+-derived CD1a+ LC precursors and freshly isolated LCs but not on other DC populations (Fig. 2 B).
Figure 1.

MIP-3α is the most potent chemokine to induce the selective migration of CD1a+ LC precursors derived from CD34+ HPCs. CD34+ HPCs were cultured in the presence of SCF+GM-CSF+TNF-α for 6 d. Migration assays were performed by seeding 5 × 105 cells in 5-μm-pore transwells for 1.5 h. The migrated cells were analyzed for CD1a and CD14 expression by FACS® after double staining. Results are expressed as the number of migrated cells measured by FACS®, and represent the means of optimal responses for each chemokine obtained in 10 independent experiments. A shows the migration of the total DC precursors (both CD1a+ and CD14+ subpopulations) and B that of CD1a+ LC precursors alone. Chemokines were used at their optimal concentration, from 1 to 500 ng/ml.
Figure 2.

Only LC precursors derived from CD34+ HPCs and isolated skin LCs migrate in response to MIP-3α and express CCR6. CD34+ HPC–derived CD1a+ LC precursors, monocyte-derived DCs, blood CD11c+ DCs, blood CD11c− DC precursors, and epidermal LCs were isolated as described in Materials and Methods. (A) Migration assays were performed by seeding 5 × 105 cells in 5-μm-pore transwells for 1.5 h. Migration in response to MIP-3α was assessed for all populations; the positive controls were RANTES for CD34+ HPC–derived CD1a+ LC precursors and epidermal LCs, MCP-1 for monocyte-derived DCs (Mono-DC), and SDF-1 for blood CD11c+ DCs and blood CD11c− DC precursors. The migrating cells were analyzed for CD1a and CD14 expression for CD34+-derived DCs and monocyte-derived DCs. Blood DCs were analyzed by triple staining for HLA-DR, CD11c, and lineage markers by FACS®. Skin-derived LCs were enumerated and identified based on morphology. Results are expressed as migration index compared with the control (medium alone), and represent the means obtained in 4–10 independent experiments. (B) The expression of CCR6 was determined by double staining with CD1a for CD34+-derived CD1a+ LC precursors, monocyte-derived (Mono-der) DCs, skin LCs, with CD14 for monocytes, and by triple staining with HLA-DR and lineage markers for blood DCs. The results are representative of 5–10 experiments.
These observations show that MIP-3α is the most potent chemokine active on LC precursors and that it recruits only LCs among freshly isolated DC populations.
MIP-3α Is Selectively Expressed at Epithelial Surfaces and Is the Main Keratinocyte Chemoattractant for LC Precursors.
In accordance with our previous study on MIP-3α mRNA expression 7, we confirmed the production of the protein within the epithelial crypts of inflamed tonsils (Fig. 3A–C). The MIP-3α–expressing cells lined the lumen of the crypt, at the interface between the external milieu and the tissue, i.e., the site of pathogen entry. MIP-3α–positive cells represented a subset of cells coexpressing cytokeratin (Fig. 3 A) and E-cadherin (Fig. 3 B), demonstrating their epithelial origin. Moreover, MIP-3α–expressing epithelial cells colocalized with CD1a+ LCs within the inflamed crypts (Fig. 3 C). Double staining on serial sections showed that foci of high MIP-3α expression colocalized with areas of agglomeration of CCR6+ cells (Fig. 3 D).
Figure 3.

In vivo MIP-3α is expressed by epithelial cells lining the crypts of inflamed tonsils and is surrounded by CCR6+ cells. (A–C) Immunohistochemistry of human tonsils performed with anti–hMIP-3α polyclonal antibody (red) reveals that MIP-3α is strongly and exclusively expressed within inflamed epithelial crypts. Double staining with anti–hMIP-3α polyclonal antibody (red) and anticytokeratin (blue) (A) or anti–E-cadherin mAb (blue) (B) shows that MIP-3α is produced by a subset of epithelial cells lining the crypts in tonsils. Moreover, double immunostaining performed with MIP-3α–specific polyclonal antibody (red) and anti-CD1a mAb (blue) (C) reveals that MIP-3α–expressing epithelial cells colocalize with CD1a+ LCs within the inflamed crypts, a site of pathogen entry. (D) Focal accumulation of CCR6+ cells (red) is directly adjacent to MIP-3α–expressing epithelial cells within the areas of E-cadherin+ cells (blue). The specificity of MIP-3α immunostaining was demonstrated either by using isotype controls or by preincubating the anti–MIP-3α polyclonal antibody with the recombinant MIP-3α protein; in both conditions, no staining was detected (data not shown). C, epithelial crypt of tonsils. Original magnifications: (A and B) ×400; (C and D) ×200.
Then, the cellular source of MIP-3α production was determined using an ELISA specific for human MIP-3α (see Materials and Methods). First, cell lines of epithelial origin (renal cell carcinoma and colon carcinoma) constitutively produced detectable amounts of MIP-3α (0.5–9.6 ng/ml), whereas cell lines of other origin (melanoma, sarcoma, neuroblastoma, fibroblasts, and T and B cells) did not (not shown). On primary cultures (Fig. 4 A), MIP-3α was never produced without activation by any cell types, including epithelial cells. Upon activation with IL-1 and TNF-α (see below), MIP-3α was produced by keratinocytes at high level (40–90 ng/ml). Moderate levels of MIP-3α were produced by other cellular constituents of the skin such as IL-1+TNF-α–activated dermal endothelial cells (28–38 ng/ml) and dermal fibroblasts (12–32 ng/ml). Low levels of MIP-3α were secreted by monocyte-derived DCs (3–12 ng/ml), monocytes (4–10 ng/ml), and T cells (≤1 ng/ml) activated by CD40 ligand (CD40L), LPS, or anti-CD3+anti-CD28, respectively. Of note, IL-1β+TNF-α stimulation did not induce the production of MIP-3α by monocyte-derived DCs nor by monocytes (not shown). Finally, B cells activated through CD40 did not secrete detectable levels of MIP-3α.
Figure 4.
MIP-3α is selectively produced by epithelial cells. (A) Nonhematopoietic cells, including endothelial cells (human umbilical vein endothelial cells), keratinocytes, and fibroblasts, were seeded at 1–2 × 104 cells/ml, and after 3–5 d of culture (80% confluence), cells were either activated by IL-1β+TNF-α or left unactivated for 48 h. Monocyte-derived DCs and B lymphocytes were either activated by CD40L-transfected L cells (murine fibroblasts, CD40L L cells) or left unactivated for 24 h. Coculture with untransfected L cells was checked for the absence of MIP-3α production. Peripheral blood monocytes and T lymphocytes were activated for 24 h in the presence of LPS and anti-CD3 plus anti-CD28, respectively. IL-1β+TNF-α did not induce MIP-3α production by monocytes or monocyte-derived DCs (not shown). Supernatants were collected and measured for MIP-3α content using a specific ELISA. (B) Keratinocyte supernatants were collected and measured for MIP-3α, MCP-1, MIP-1α, and RANTES contents using specific ELISAs. Results shown are representative of five independent experiments.


The production of MIP-3α by epithelial cells was compared with that of three other inducible chemokines, MCP-1, RANTES, and MIP-1α, that are also active on DCs and particularly on LC precursors (see Fig. 1). Notably, upon activation, MIP-3α was secreted by skin keratinocytes in much larger amount (40–90 ng/ml) than the other chemokines (RANTES = 2–5 ng/ml, MCP-1 < 1 ng/ml, MIP-1α < 1 ng/ml) (Fig. 4 B). Supernatant from unactivated keratinocytes did not induce the migration of CD1a+ LC precursors (Fig. 5 A), in accordance with the absence of MIP-3α production by using the specific MIP-3α ELISA. In contrast, supernatants from cells activated with IL-1β+TNF-α, at a concentration as low as 12.5% induced a migration comparable to that observed with recombinant MIP-3α (1 μg/ml) (12.5% of the supernatant corresponds to 10 ng/ml detected by the ELISA). Of note, IL-1β and TNF-α, alone or in combination, had no chemotactic effect in those assays. When anti–MIP-3α was added with MIP-3α in the lower well, the migration in response to this recombinant chemokine was abolished whereas the antibody has no effect on the migration induced by other chemokines such as RANTES (Fig. 5 B). The effect of the keratinocyte supernatant on the migratory response of CD1a+ LC precursors was suppressed in the presence of anti–MIP-3α, whereas the control antibody has no effect. In three independent experiments, activated keratinocyte supernatants induced a migration index of CD1a+ LC precursors ranging from 5 to 20, and addition of anti–MIP-3α always resulted in complete block of supernatant activity. These results were confirmed with different cell types of epithelial origin (not shown).
Figure 5.
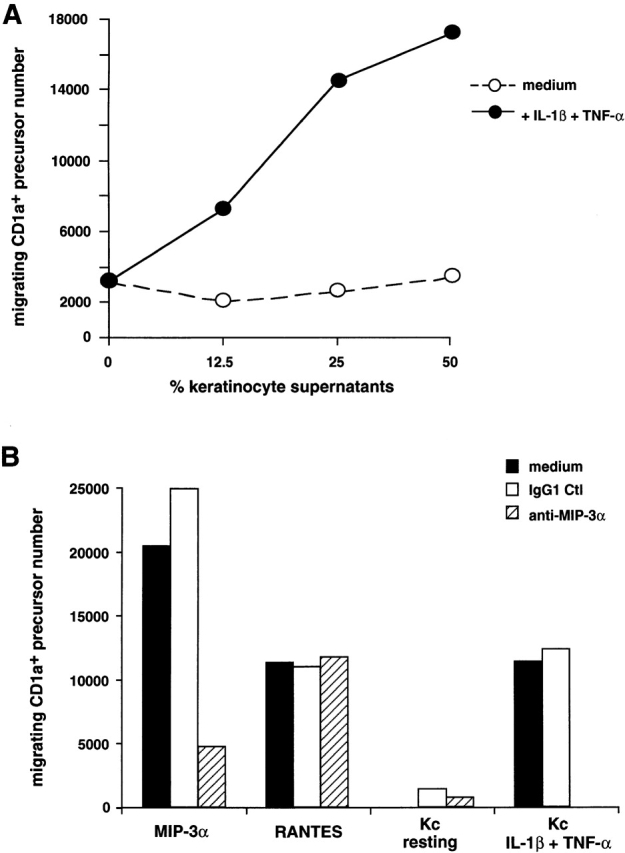
Blocking antibody against MIP-3α abolishes the migration of CD1a+ LC precursors induced by supernatant of activated keratinocyte. Keratinocytes were seeded at 2 × 104 cells/ml, and after 5 d of culture (80% confluence), cells were activated by IL-1β+TNF-α for 48 h. (A) Transwell migration of CD34+-derived CD1a+ LC precursors in response to various concentrations of supernatants from resting or activated keratinocytes is shown. Results are representative of five experiments. (B) Transwell migration of CD1a+ LC precursors, in response to 1 μg/ml of recombinant MIP-3α to 100 ng/ml of recombinant RANTES, or in response to 25% of supernatant from resting or activated keratinocytes (Kc), was assessed in the presence of 10 μg/ml mAb against MIP-3α or isotype-matched control (IgG1 Ctl). Values represent the mean of two independent experimental determinations, and variations between the two measurements were always <10%. Results are expressed as migrating cell numbers and are representative of three independent experiments.
Taken together, these observations show that MIP-3α is expressed by epithelial cells at the site of pathogen entry, and suggest that among the chemokines secreted by those cells, MIP-3α plays a major role in the attraction of LC precursors.
In Lesional Psoriatic Skin, the Upregulation of MIP-3α Expression Appears Associated with an Increased LC Turnover.
With regard to MIP-3α–selective expression in epithelium and activity on LCs, we then investigated its expression in skin, under normal and inflamed conditions. In normal skin, MIP-3α mRNA was not detected by in situ hybridization (Fig. 6 A). However, a very weak expression of the protein was detected by immunohistochemistry within the stratum corneum (Fig. 6 B). As in normal skin, nonlesional keratinocytes of the stratum corneum were the only cells that expressed a very low level of MIP-3α protein (not shown). However, in lesional psoriatic skin, MIP-3α was upregulated in the suprabasal layers of the acanthotic epidermis as shown by in situ hybridization (Fig. 6C and Fig. E) and by immunohistochemistry (Fig. 6F and Fig. G). In all samples (Fig. 6D and Fig. H), the specificity of the MIP-3α staining was demonstrated by the preincubation of the anti–MIP-3α antibody with the recombinant MIP-3α protein (normal and lesional psoriatic skin shown).
Figure 6.
Upregulation of MIP-3α mRNA and protein in lesional psoriatic skin. Detection of MIP-3α by in situ hybridization (A, C, and E) and immunohistochemistry (B, D, F–L). J is a higher magnification of I; B and D, F, and H are serial cryostat sections. Human skin sections were hybridized with antisense (A, C, and E) and sense (not shown) 35S-labeled RNA probes for MIP-3α and were exposed for 40 d. By in situ hybridization, no signal is detected in nonlesional (not shown) and in normal skin (A). In lesional psoriatic skin, MIP-3α mRNA is strongly expressed within the suprabasal layers of the epidermis (C; and E, dark field illumination). The sense riboprobes do not generate any background hybridization (not shown). In normal skin, immunostaining with MIP-3α–specific polyclonal antibody (red) and anti-collagen IV mAb (blue) reveals that MIP-3α protein is very weakly expressed within the stratum corneum (B). The specificity of this MIP-3α immunostaining (red) was demonstrated by preincubating the anti–MIP-3α polyclonal antibody with the recombinant MIP-3α protein (D). Immunohistochemistry of lesional psoriatic skin performed with MIP-3α–specific polyclonal antibody (red) confirms the upregulation of MIP-3α expression in the upper suprabasal layers of the epidermis (F). Double immunostaining performed with MIP-3α–specific polyclonal antibody (red) and anti-Langerin mAb (blue) reveals that MIP-3α–expressing keratinocytes colocalize with Langerin+ LCs near the stratum corneum areas (F) as well as around hair follicles (G) of lesional psoriatic skin. The specificity of MIP-3α immunostaining (red) in lesional psoriatic skin was demonstrated (continues) (continued) either by using isotype controls (H) or by preincubating the anti–MIP-3α polyclonal antibody with the recombinant MIP-3α protein (not shown). In both conditions, no MIP-3α staining is detected. Double staining performed with anti-Langerin mAb (blue) and anti–DC-LAMP (red) indicates the presence of a high number of Langerin+DC-LAMP+ maturing LCs (arrow) and Langerin−DC-LAMP+ mature DCs (arrowhead) in lesional psoriatic skin (I and J), whereas Langerin+DC-LAMP− LCs are detected within the epidermis of normal skin (not shown), lesional (I), and nonlesional psoriatic skin (K). Mature DCs (arrowhead) and most of the maturing DCs (arrow) that are present in the dermis are detected around collagen IV+ vessels (L). E, epidermis; D, dermis; C, stratum corneum. Original magnifications: (A, C, E–H, K, and L) ×200; (I) ×100; (B, D, and J) ×400.
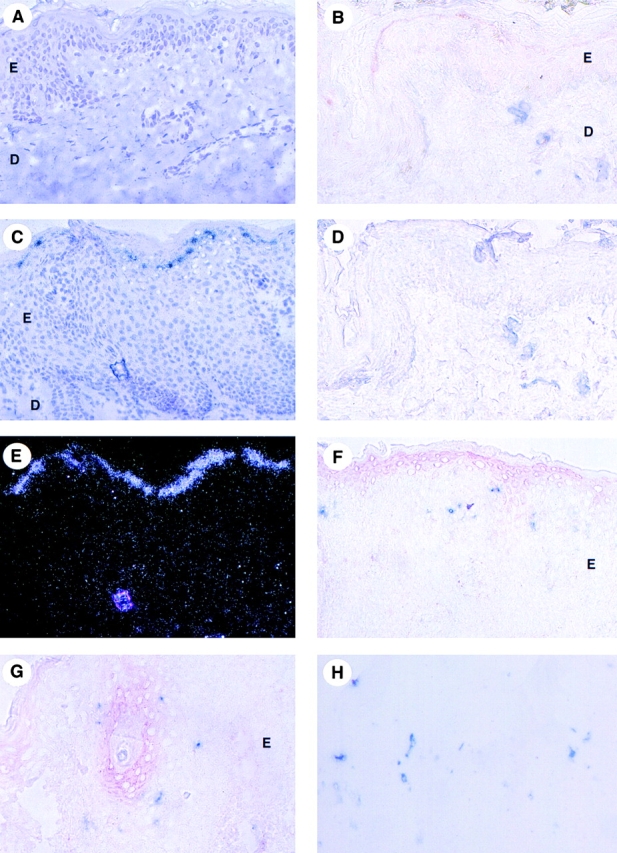

When double staining was performed, accumulation of Langerin+CD1a+ LCs was apparent adjacent to the site of high MIP-3α expression (Fig. 6F and Fig. G). Production of MIP-3α protein was also observed around psoriatic hair follicles, which were surrounded by Langerin+CD1a+ LCs (Fig. 6 G). The sensitivity of the immunohistochemistry technique did not permit the detection of CCR6 expression by inflammatory cells in the skin. We next analyzed the maturation stage of epidermal DCs using mAbs against Langerin and DC-LAMP, two DC-specific markers that characterize immature and mature LCs, respectively 42 43. In normal and in nonlesional psoriatic skin (Fig. 6 K, and Fig. 7), epidermal Langerin+DC-LAMP− cells represent the only LC population detected. In contrast, Langerin+DC-LAMP+ maturing LCs are almost exclusively observed in lesional psoriatic skin, mainly in the dermis but also in the epidermis and the papillary dermis (Fig. 6I and Fig. J, and Fig. 7). Langerin+DC-LAMP+ maturing LCs represent >25% of the LC compartment in lesional psoriatic skin, whereas they represent <2% in nonlesional psoriatic and normal skin (Fig. 7). Furthermore, Langerin− DC-LAMP+ mature DCs were only found in the dermis of lesional skin (20% of total DC population; Fig. 6J–L, and Fig. 7), and they are associated with a veiled morphology (Fig. 6 J). Finally, maturing and mature DCs that are localized in the dermis were always observed around collagen IV+ vessels (Fig. 6 L). Some of these vessels probably represent lymphatic vessels, suggesting that maturing LCs emigrate out of the skin. Thus, the presence of mature LCs in the epidermis and dermis in the local inflammatory psoriatic skin environment is likely to reflect a high LC turnover.
Figure 7.
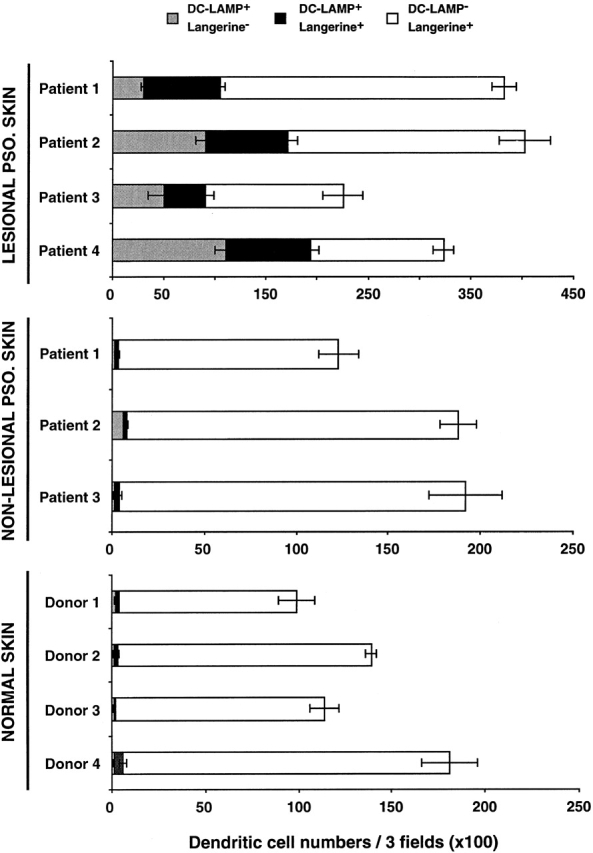
Maturing DC-LAMP+ LCs are strongly increased in lesional psoriatic skin. The stage of DC maturation was determined according to the expression by immunohistochemistry of two DC-specific markers, Langerin and DC-LAMP. Immature LCs (DC-LAMP−Langerin+, present in epidermis only), maturing LCs (DC-LAMP+Langerin+, present in epidermis and dermis), and mature DCs (DC-LAMP+ Langerin−, present in dermis only) were counted in three randomly selected fields (original magnification: ×100) per section. Each count was performed on two or three serial sections, and results were expressed as the mean ± SD of DCs per three fields. Results from three to four independent normal donor and patients are shown. PSO., psoriatic.
Taken together, these observations argue for an active role of MIP-3α in this suggested increase in LC trafficking in lesional psoriatic skin and, thus, in the development and/or chronicity of this disease.
MIP-3α Production Is Induced upon Inflammatory and T Cell Signals.
The regulation of MIP-3α secretion by epithelial cells was further defined by using the kidney carcinoma epithelial cell lines expressing this chemokine at different levels. In vitro, among several inflammatory stimuli (IL-1, IL-6, TNF-α, and GM-CSF), only IL-1α and IL-1β induced a strong production of MIP-3α in all the epithelial cells tested, as shown with CHA and VER (Fig. 8 A). However, TNF-α synergized with IL-17 and in some experiments with IL-1 in inducing MIP-3α production. The optimal production was obtained with 1 ng/ml of IL-1 after 24 h of activation (Fig. 8 B). Because in psoriasis, T cells are involved in the development of the pathology, we next tested the effect of signals delivered by T lymphocytes on MIP-3α production by epithelial cells (Fig. 8 C). On the epithelial cell line CHA, MIP-3α was not detected in absence of activation nor after activation with IFN-γ or CD40L alone. However, CD40L synergized with both IL-1 (from 70 to 234 ng/ml) and IL-17 (from 1 to 19 ng/ml). CD40L and IFN-γ also had a strong synergic effect on MIP-3α production (28 ng/ml) compared with the cytokines alone. We also observed (Fig. 8 C) that coculture of anti-CD3–activated T cells with CHA induced the secretion of MIP-3α at a physiologic and chemotactic level (15.7 ng/ml).
Figure 8.
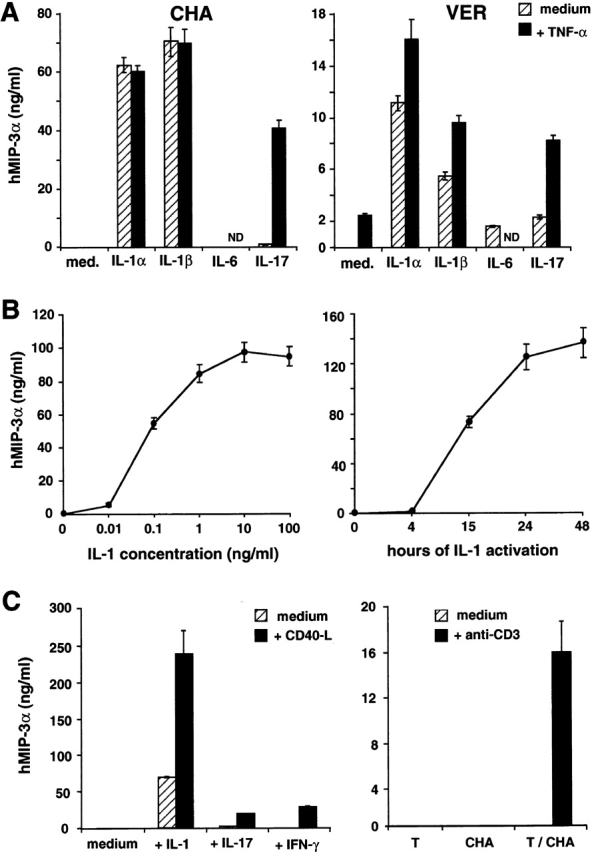
Regulation of MIP-3α production by epithelial cells in response to inflammatory cytokines or T cell signals. (A) Two renal carcinoma cell lines, CHA and VER, were seeded at 1–2 × 104 cells/ml, and after 1–3 d of culture (80% confluence), cells were either activated by IL-1α, IL-1β, IL-6, and IL-17 or left unactivated, with or without stimulation by TNF-α for 48 h. (B) Renal carcinoma cell line, CHA, was seeded at 1–2 × 104 cells/ml, and after 1–3 d of culture (80% confluence), cells were either activated by IL-1β or left unactivated, in a dose dependent on IL-1β (left) or in a time-dependent manner (right). (C) Renal carcinoma cell line, CHA, was seeded at 1–2 × 104 cells/ml, and after 1–3 d of culture (80% confluence), cells were either activated by IL-1β, IL-17, IFN-γ, or left unactivated, with or without stimulation by CD40L L cells for 48 h. Peripheral blood T cells and renal carcinoma cell line (CHA) were independently or cocultivated in the presence or in absence of anti-CD3 antibodies for 48 h. Supernatants were collected and measured for MIP-3α content using a specific ELISA. Results are representative of five independent experiments.
Altogether, these observations show that upon inflammatory or T cell signals, the production of MIP-3α is strongly upregulated by cells of epithelial origin.
Discussion
The capacity of DCs to reach the site of injury/infection and consequently to initiate immunity is determined by their ability to respond to selected chemokines. In this study, we show that MIP-3α is a major chemokine produced by activated epithelial cells, and selectively active on LCs and their precursors. MIP-3α's unique activity suggests that it plays a key role in the control of LC recruitment at inflamed epithelial surfaces and in the regulation of epithelial immunity.
Among all CC chemokines tested, MIP-3α appears to be the most potent chemokine inducing the migration of LC precursors and freshly isolated LCs but not of any other DC population. This result is in line with the specific expression of CCR6 by cells of the LC lineage in contrast with other receptors such as CCR1, CCR2, and CCR5 that are expressed on several other DC populations (9 34; and Caux, C., manuscript in preparation). This observation is in accordance with previous reports showing that both CCR6 expression and MIP-3α responsiveness were detected on CD34+-derived DCs but not on monocyte-derived DCs 36 44. So far, no DC or DC precursors expressing CCR6 or responding to MIP-3α have been identified in the blood from healthy donors. However, it has been recently shown that monocyte-derived DCs can express CCR6 and respond to MIP-3α when cultured with TGF-β 45, a factor previously reported to support LC differentiation from monocytes 46 or from a subset of blood DCs 47. This might suggest that the LC precursors in the bloodstream are not the target of MIP-3α but they acquire MIP-3α responsiveness when they are already mobilized in the tissue and undergo differentiation events in the local cytokine microenvironment 48. This indicates that chemokines play a role not only in the intracellular extravasation across the endothelial barrier, but also in the cellular migration across tissues. Alternatively, in normal condition, LC precursors might not exist in sufficient numbers to be detected in the circulation. But, upon inflammatory conditions, cytokines such as GM-CSF may be released, reach the bone marrow, and induce the margination of LC precursors, which can then be recruited by local MIP-3α secretion.
The selective role of MIP-3α in the recruitment of LC precursors at epithelial surfaces was further supported by (a) its restricted expression by inflamed epithelium at mucosal and nonmucosal sites in vivo; (b) a colocalization of MIP-3α production and the presence of LCs, in situ; and (c) its strong inducible secretion by epithelial cells in response to inflammatory mediators or T cell signals in vitro. In line with the expression of MIP-3α at epithelial surfaces in vivo, the expression of MIP-3α mRNA has been reported in the intestinal mucosa in both mice and humans 49. Experiments with activated keratinocyte supernatants demonstrate that MIP-3α is the main chemokine secreted by epithelial cells under inflammatory conditions that is able to attract LC precursors. Our study is in agreement with the recent publication of Charbonnier and colleagues 50 demonstrating the unique capacity of MIP-3α to induce LC migration. However, in normal skin, they detected the MIP-3α protein in the basal and suprabasal layers of the epidermis, whereas we did not see any expression in those regions by in situ hybridization or by immunohistochemistry. Then, they suggested a role of MIP-3α in the constitutive recruitment of LCs into the skin. As we have detected a very low but specific MIP-3α staining within the stratum corneum, we do not exclude a putative role of this chemokine in LC trafficking under normal conditions. In this context, it would be interesting to determine if MIP-3α secretion can be detected during terminal differentiation of keratinocytes in vitro. However, we observed a strong upregulation of MIP-3α secretion in vitro after inflammatory and T cell signals as well as in vivo_,_ at inflammatory sites such as in tonsils and in lesional psoriatic skin, suggesting a key role of MIP-3α in the recruitment of LC precursors at sites of inflammation. Furthermore, this hypothesis is in agreement with: (a) the upregulation of MIP-3α mRNA observed after in vivo injection of LPS 49; and (b) the presence of LCs in the skin of CCR6-deficient mice in normal situation 51. Thus, it is tempting to consider that MIP-3α may have a more crucial role in the recruitment of epidermal LCs in inflammatory conditions that strongly induce its expression than in their constitutive trafficking.
We report here that MIP-3α is strongly upregulated in lesional psoriatic skin in association with a consistent pool of maturing LCs (DC-LAMP+) concentrated around vessels, arguing for an active role of MIP-3α in an increase of LC trafficking. Furthermore, in this pathology, T cells are responsible for the development of the lesions through the activation of keratinocytes 52 53 54. In this context, MIP-3α expression colocalizes with memory T cells within lesional psoriatic skin and CCR6 expression is upregulated both in lesional biopsies and in peripheral blood cells 55. Furthermore, MIP-3α has been reported to be active on memory T cells with either epithelial or gut-homing properties 56 57, and on intestinal γ/δ T cells 49. Altogether, these observations suggest that MIP-3α through the recruitment of both LCs and T cells of epithelial tropisms may have a unique role in the regulation of epithelial immunity, and may participate in epithelial immune disorders such as psoriasis.
MIP-3α was also expressed by most tumor cell lines of epithelial origin (renal adenocarcinoma and colon adenocarcinoma, among others), and detection of MIP-3α secretion in specimens of tumors such as breast adenocarcinoma (not shown; references 58 59 60) suggests that MIP-3α can be produced by tumor cells in vivo. We and others 61 62 63 have previously shown that adenocarcinomas, in particular renal cell carcinomas, can alter DC development/maturation, preventing the appearance of their T cell stimulatory function. This suggests that tumors of epithelial origin producing MIP-3α can attract LC precursors and divert their function to escape immune surveillance and reach a state of tolerance.
Although, MIP-3α appears to have a single receptor, CCR6, the defensins-β which are also expressed in the epithelium have been demonstrated recently to bind to CCR6 64. The unique role of CCR6 ligands in the recruitment of LC precursors offers novel approaches to assess for the functions of LCs and possibly to manipulate them.
Acknowledgments
We are grateful to Drs. G. Trinchieri and P. Garrone for critical reading of the manuscript. We are also grateful to M. Vatan and C. Alexandre for editorial assistance; to doctors and colleagues from clinics and hospitals in Lyon who provided us with umbilical cord blood samples and tonsils; and to Dr. J.F. Nicolas (Institut National de la Santé, U503, Lyon, France) for providing us with psoriatic skin biopsies and for discussions.
M.C. Dieu-Nosjean is the recipient of a grant from Fondation Marcel Mérieux, Lyon, France. B. Homey is supported by a grant from the Deutsche Forschungsgemeinschaft (DFG Ho 2019/1-1). DNAX Research Institute and the Laboratory for Immunological Research are supported by Schering-Plough Corporation.
Footnotes
Abbreviations used in this paper: CCR, CC chemokine receptor; DC, dendritic cell; HPC, hematopoietic progenitor cell; LAMP, lysosomal-associated membrane protein; LC, Langerhans cell; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; RANTES, regulated upon activation, normal T cell expressed and secreted protein; SCF, stem cell factor; SDF, stromal cell–derived factor.
References
- Steinman R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Caux C., Dezutter-Dambuyant C., Liu Y.J., Banchereau J. Isolation and propagation of human dendritic cells. In: Kabelitz D., Ziegler K., editors. Methods in MicrobiologyImmunological Methods. Vol. 25. Academic Press, Ltd.; New York, NY: 1998. pp. 505–538. [Google Scholar]
- Shortman K., Caux C. Dendritic cell developmentmultiple pathways to nature's adjuvants. Stem Cells. 1997;15:409–419. doi: 10.1002/stem.150409. [DOI] [PubMed] [Google Scholar]
- Hart D.N.J. Dendritic cellsunique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–3287. [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Austyn J.D. New insights into the mobilization and phagocytic activity of dendritic cells. J. Exp. Med. 1996;183:1287–1292. doi: 10.1084/jem.183.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieu M.C., Vanbervliet B., Vicari A., Bridon J.M., Oldham E., Aït-Yahia S., Brière F., Zlotnik A., Lebecque S., Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998;188:1–14. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani S., Allavena P., D'Amico G., Luini W., Bianchi G., Kataura M., Imai T., Yoshie O., Bonecchi R., Mantovani A. Differential regulation of chemokine receptors during dendritic cell maturationa model for their trafficking properties. J. Immunol. 1998;161:1083–1086. [PubMed] [Google Scholar]
- Sallusto F., Schaerli P., Loetscher P., Schaniel C., Lenig D., Mackay C.R., Qin S., Lanzavecchia A. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 1998;28:2760–2769. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Yanagihara S., Komura E., Nagafune J., Watarai H., Yamaguchi Y. EBI1/CCR7 is a new member of dendritic cell chemokine receptor that is up-regulated upon maturation. J. Immunol. 1998;161:3096–3102. [PubMed] [Google Scholar]
- Gunn M.D., Tangemann K., Tam C., Cyster J.G., Rosen S.D., Williams L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA. 1998;95:258–263. doi: 10.1073/pnas.95.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki H., Moore A.M., Brown M.J., Hwang S.T. Cutting edgesecondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J. Immunol. 1999;162:2472–2475. [PubMed] [Google Scholar]
- Kellermann S.A., Hudak S., Oldham E.R., Liu Y.J., McEvoy L.M. The CC chemokine receptor-7 ligands 6Ckine and macrophage inflammatory protein-3 beta are potent chemoattractants for in vitro- and in vivo-derived dendritic cells. J. Immunol. 1999;162:3859–3864. [PubMed] [Google Scholar]
- Chan V.W., Kothakota S., Rohan M.C., Panganiban-Lustan L., Gardner J.P., Wachowicz M.S., Winter J.A., Williams L.T. Secondary lymphoid-tissue chemokine (SLC) is chemotactic for mature dendritic cells. Blood. 1999;93:3610–3616. [PubMed] [Google Scholar]
- Ngo V.N., Tang H.L., Cyster J.G. Epstein-Barr virus–induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J. Exp. Med. 1998;188:181–191. doi: 10.1084/jem.188.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H., Tamura T., Yoshimoto T., Yagita H., Miyasaka M., Butcher E.C., Nariuchi H., Kakiuchi T., Matsuzawa A. Genetic defect in T lymphocyte-specific homing into peripheral lymph nodes. Eur. J. Immunol. 1997;27:215–221. doi: 10.1002/eji.1830270132. [DOI] [PubMed] [Google Scholar]
- Nakano H., Mori S., Yonekawa H., Nariuchi H., Matsuzawa A., Kakiuchi T. A novel mutant gene involved in T-lymphocyte-specific homing into peripheral lymphoid organs on mouse chromosome 4. Blood. 1998;91:2886–2895. [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R., Schubel A., Breitfeld D., Kremmer E., Renner-Muller I., Wolf E., Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Durand I., Cella M., Lanzavecchia A., Banchereau J. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF-αII. Functional analysis. Blood. 1997;90:1458–1470. [PubMed] [Google Scholar]
- Dubois B., Barthélémy C., Durand I., Liu Y.J., Caux C., Brière F. Toward a role of dendritic cells in the germinal center reactiontriggering of B cell proliferation and isotype switching. J. Immunol. 1999;162:3428–3436. [PubMed] [Google Scholar]
- Rissoan M.C., Soumelis V., Kadowaki N., Grouard G., Brière F., de Waal Malefyt R., Liu Y.J. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- O'Doherty U., Peng M., Gezelter S., Swiggard W.J., Betjes M., Bhardwaj N., Steinman R.M. Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology. 1994;82:487–493. [PMC free article] [PubMed] [Google Scholar]
- Grouard G., Rissoan M.C., Filgueira L., Durand I., Banchereau J., Liu Y.J. The enigmatic plasmacytoid T cells develop into dendritic cells with IL-3 and CD40-ligand. J. Exp. Med. 1997;185:1101–1111. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal F.P., Kadowaki N., Shodell M., Fitzgerald-Bocarsly P.A., Shah K., Ho S., Antonenko S., Liu Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- Borkowski T.A., Letterio J.J., Farr A.G., Udey M.C. A role for endogenous transforming growth factor β1 in Langerhans cell biologythe skin of transforming growth factor β1 null mice is devoid of epidermal Langerhans cells. J. Exp. Med. 1996;184:2417–2422. doi: 10.1084/jem.184.6.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl E., Strobl H., Majdic O., Knapp W. TGF-beta 1 promotes in vitro generation of dendritic cells by protecting progenitor cells from apoptosis. J. Immunol. 1997;158:1591–1597. [PubMed] [Google Scholar]
- Strobl H., Riedl E., Scheinecker C., Bello-Fernandez C., Pickl W.F., Rappersberger K., Majdic O., Knapp W. TGF-β1 promotes in vitro development of dendritic cells from CD34+ hemopoietic progenitors. J. Immunol. 1996;157:1499–1507. [PubMed] [Google Scholar]
- Strobl H., Bello-Fernandez C., Riedl E., Pickl W.F., Majdic O., Lyman S.D., Knapp W. Flt3 ligand in cooperation with transforming growth factor-beta1 potentiates in vitro development of Langerhans-type dendritic cells and allows single-cell dendritic cell cluster formation under serum-free conditions. Blood. 1997;90:1425–1434. [PubMed] [Google Scholar]
- Caux C., Massacrier C., Dubois B., Valladeau J., Dezutter-Dambuyant C., Schmitt D., Saeland S. Respective involvement of TGFβ and IL-4 in the development of Langerhans cells and non Langerhans dendritic cells from CD34+ progenitors. J. Leukoc. Biol. 1999;66:781–791. doi: 10.1002/jlb.66.5.781. [DOI] [PubMed] [Google Scholar]
- McWilliam A.S., Nelson D., Thomas J.A., Holt P.G. Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J. Exp. Med. 1994;179:1331–1336. doi: 10.1084/jem.179.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam A.S., Nelson D.J., Holt P.G. The biology of airway dendritic cells. Immunol. Cell. Biol. 1995;73:405–413. doi: 10.1038/icb.1995.63. [DOI] [PubMed] [Google Scholar]
- Sozzani S., Sallusto F., Luini W., Zhou D., Piemonti L., Allavena P., van Damme J., Valitutti S., Lanzavecchia A., Mantovani A. Migration of dendritic cells in response to formyl peptides, C5a, and a distinct set of chemokines. J. Immunol. 1995;155:3292–3295. [PubMed] [Google Scholar]
- Sozzani S., Luini W., Borsatti A., Polentarutti N., Zhou D., Piemonti L., D'Amico G., Power C.A., Wells T.N., Gobbi M. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997;159:1993–2000. [PubMed] [Google Scholar]
- Xu L.L., Warren M.K., Rose W.L., Gong W., Wang J.M. Human recombinant monocyte chemotactic protein and other CC chemokines bind and induce directional migration of dendritic cells in vitro. J. Leukoc. Biol. 1996;60:365–371. doi: 10.1002/jlb.60.3.365. [DOI] [PubMed] [Google Scholar]
- Power C.A., Church D.J., Meyer A., Alouani S., Proudfoot A.E., Clark-Lewis I., Sozzani S., Mantovani A., Wells T.N. Cloning and characterization of a specific receptor for the novel CC chemokine MIP-3α from lung dendritic cells. J. Exp. Med. 1997;186:825–835. doi: 10.1084/jem.186.6.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caux C., Vanbervliet B., Massacrier C., Dezutter-Dambuyant C., de Saint-Vis B., Jacquet C., Yoneda K., Imamura S., Schmitt D., Banchereau J. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF-α. J. Exp. Med. 1996;184:695–706. doi: 10.1084/jem.184.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caux C., Saeland S., Favre C., Duvert V., Mannoni P., Banchereau J. Tumor necrosis factor-alpha strongly potentiates interleukin-3 and granulocyte-macrophage colony-stimulating factor-induced proliferation of human CD34+ hematopoietic progenitor cells. Blood. 1990;75:2292–2298. [PubMed] [Google Scholar]
- Sallusto F., Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J. Exp. Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A., Schmitt D., Dezutter-Dambuyant C., Fargier M.C., Thivolet J. Reappearance of CD1a antigenic sites after endocytosis on human Langerhans cells evidenced by immunogoldrelabeling. J. Investig. Dermatol. 1989;92:217–224. doi: 10.1111/1523-1747.ep12276745. [DOI] [PubMed] [Google Scholar]
- Peuchmaur M., Emilie D., Crevon M.C., Solal-Celigny P., Maillot M.C., Lemaigre G., Galanaud P. IL-2 mRNA expression in Tac-positive malignant lymphomas. Am. J. Pathol. 1990;136:383–390. [PMC free article] [PubMed] [Google Scholar]
- Valladeau J., Duvert-Frances V., Pin J.-J., Dezutter-Dambuyant C., Vincent C., Massacrier C., Vincent J., Yoneda K., Banchereau J., Caux C. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell-surface. Eur. J. Immunol. 1999;29:2695–2704. doi: 10.1002/(SICI)1521-4141(199909)29:09<2695::AID-IMMU2695>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- de Saint-Vis B., Vincent J., Vandenabeele S., Vanbervliet B., Pin J.J., Aït-Yahia S., Patel S., Mattei M.G., Banchereau J., Zurawski S. A novel lysosome associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity. 1998;9:325–336. doi: 10.1016/s1074-7613(00)80615-9. [DOI] [PubMed] [Google Scholar]
- Greaves D.R., Wang W., Dairaghi D.J., Dieu M.C., de Saint-Vis B., Franz-Bacon K., Rossi D., Caux C., McClanahan T., Gordon S. CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3α and is highly expressed in human dendritic cells. J. Exp. Med. 1997;186:837–844. doi: 10.1084/jem.186.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Howard O.M., Chen Q., Oppenheim J.J. Cutting edgeimmature dendritic cells generated from monocytes in the presence of TGF-beta1 express functional C-C chemokine receptor 6. J. Immunol. 1999;163:1737–1741. [PubMed] [Google Scholar]
- Geissmann F., Prost C., Monnet J.P., Dy M., Brousse N., Hermine O. Transforming growth factor β1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J. Exp. Med. 1998;187:961–966. doi: 10.1084/jem.187.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T., Inaba M., Inaba K., Toki J., Sogo S., Iguchi T., Adachi Y., Yamaguchi K., Ryuichi A., Valladeau J. A CD1a+/CD11c+ subset of human blood dendritic cells is a direct precursor of Langerhans cells. J. Immunol. 1999;163:1409–1419. [PubMed] [Google Scholar]
- Randolph G.J., Inaba K., Robbiani D.F., Steinman R.M., Muller W.A. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Imai T., Baba M., Ishikawa I., Ueshira M., Nomiyama H., Yoshie O. Selective expression of liver and activation-regulated chemokine (LARC) in intestinal epithelium in mice and humans. Eur. J. Immunol. 1999;29:633–642. doi: 10.1002/(SICI)1521-4141(199902)29:02<633::AID-IMMU633>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Charbonnier A.S., Kohrgruber N., Kriehuber E., Stingl G., Rot A., Maurer D. Macrophage inflammatory protein 3α is involved in the constitutive trafficking of epidermal langerhans cells. J. Exp. Med. 1999;190:1755–1768. doi: 10.1084/jem.190.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D.N., Prosser D.M., Forster R., Zhang J., Kuklin N.A., Abbondanzo S.J., Niu X.D., Chen S.C., Manfra D.J., Wiekowski M.T. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- Gottlieb S.L., Gilleaudeau P., Johnson R., Estes L., Woodworth T.G., Gottlieb A.B., Krueger J.G. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat. Med. 1995;5:442–447. doi: 10.1038/nm0595-442. [DOI] [PubMed] [Google Scholar]
- Nicolas J.F., Chamchick N., Thivolet J., Wijdenes J., Morel P., Revillard J.P. CD4 antibody treatment of severe psoriasis. Lancet. 1991;8762:321. doi: 10.1016/0140-6736(91)90465-2. [DOI] [PubMed] [Google Scholar]
- Krueger J.G., Wolfe J.T., Nabeya R.T., Vallat V.P., Gilleaudeau P., Heftler N.S., Austin L.M., Gottlieb A.B. Successful ultraviolet B treatment of psoriasis is accompanied by a reversal of keratinocyte pathology and by selective depletion of intraepidermal T cells. J. Exp. Med. 1995;6:2057–2068. doi: 10.1084/jem.182.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homey B., Dieu-Nosjean M.C., Wiesenborn A., Massacrier C., Pin J.J., Oldham E., Catron D., Buchanan M.E., Muller A., de Waal Malefyt R. Up-regulation of macrophage inflammatory protein-3alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J. Immunol. 2000;164:6621–6632. doi: 10.4049/jimmunol.164.12.6621. [DOI] [PubMed] [Google Scholar]
- Campbell J.J., Hedrick J., Zlotnik A., Siani M.A., Thompson D.A., Butcher E.C. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- Liao F., Rabin R.L., Smith C.S., Sharma G., Nutman T.B., Farber J.M. CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J. Immunol. 1999;162:186–194. [PubMed] [Google Scholar]
- Bell D., Chomarat P., Broyles D., Netto G., Harb G.M., Lebecque S., Valladeau J., Davoust J., Palucka K.A., Banchereau J. In breast carcinoma tissue, immature dendritic cells reside with tumor, whereas mature dendritic cells are located in peritumoral areas. J. Exp. Med. 1999;190:1417–1426. doi: 10.1084/jem.190.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J., Kusama T., Rossi D.L., Ishiwata T., Maruyama H., Friess H., Buchler M.W., Zlotnik A., Korc M. Detection and localization of Mip-3alpha/LARC/Exodus, a macrophage proinflammatory chemokine, and its CCR6 receptor in human pancreatic cancer. Int. J. Cancer. 1999;81:650–657. doi: 10.1002/(sici)1097-0215(19990517)81:4<650::aid-ijc23>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Ménétrier-Caux C., Montmain G., Dieu M.C., Bain C., Favrot M.C., Caux C., Blay J.Y. Inhibition of the differentiation of dendritic cells from CD34+ progenitors by tumor cellsrole of interleukin-6 and macrophage colony-stimulating factor. Blood. 1998;92:1–15. [PubMed] [Google Scholar]
- Gabrilovich D.I., Chen H.L., Girgis K.R., Cunningham H.T., Meny G.M., Nadaf S., Kavanaugh D., Carbone D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- Enk A.H., Jonuleit H., Saloga J., Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int. J. Cancer. 1997;73:309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Chaux P., Favre N., Martin M., Martin F. Tumor-infiltrating dendritic cells are defective in their antigen-presenting function and inducible B7 expression in rats. Int. J. Cancer. 1997;72:619–624. doi: 10.1002/(sici)1097-0215(19970807)72:4<619::aid-ijc12>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Yang D., Chertov O., Bykovskaia S.N., Chen Q., Buffo M.J., Shogan J., Anderson M., Schroder J.M., Wang J.M., Howard O.M., Oppenheim J.J. Beta-defensinslinking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]