The Multiple Immune-Evasion Genes of Murine Cytomegalovirus Are Not Redundant: m4 and m152 Inhibit Antigen Presentation in a Complementary and Cooperative Fashion (original) (raw)
Abstract
Both human cytomegaloviruses (HCMVs) and murine cytomegaloviruses (MCMVs) encode multiple genes that interfere with antigen presentation by major histocompatibility complex (MHC) class I, and thus protect infected targets from lysis by virus-specific cytotoxic T lymphocytes (CTLs). HCMV has been shown to encode four such genes and MCMV to encode two. MCMV m152 blocks the export of class I from a pre-Golgi compartment, and MCMV m6 directs class I to the lysosome for degradation. A third MCMV gene, m4, encodes a glycoprotein which is expressed at the cell surface in association with class I. Here we here show that m4 is a CTL-evasion gene which, unlike previously described immune-evasion genes, inhibited CTLs without blocking class I surface expression. m152 was necessary to block antigen presentation to both Kb- and Db-restricted CTL clones, while m4 was necessary to block presentation only to Kb-restricted clones. m152 caused complete retention of Db, but only partial retention of Kb, in a pre-Golgi compartment. Thus, while m152 effectively inhibited Db-restricted CTLs, m4 was required to completely inhibit Kb-restricted CTLs. We propose that cytomegaloviruses encode multiple immune-evasion genes in order to cope with the diversity of class I molecules in outbred host populations.
Keywords: murine cytomegalovirus, cytotoxic T lymphocyte, immune evasion, MHC, class I
Introduction
CMVs, including human CMV (HCMV) and murine CMV (MCMV), belong to the β subfamily of the Herpesviridae, a family of large, double-stranded DNA viruses. CMVs cause little pathology in normal host animals, but cause severe disease when the immune system is compromised. CMVs have developed intimate relationships with the host immune systems which permit the viruses to establish latency and reactivate in the face of primed immune responses. A number of mechanisms have been described by which CMVs modulate host-immune responses; these include chemokine receptor homologues and viral gene products which interfere with the normal functions of T cells and natural killer (NK) cells 1. In particular, both HCMV and MCMV encode a number of gene products which specifically interfere with the ability of infected cells to present antigen to CD8+ CTLs 2 3.
CD8+ T cells recognize a trimolecular complex, consisting of class I heavy chain, β2 microglobulin, and a short antigenic peptide, which is assembled in the endoplasmic reticulum (ER). In cells infected with HCMV, at least four different viral gene products interfere with this assembly 4: US6 blocks the peptide transporter associated with antigen presentation (TAP, references 5 6 7); US3 prevents export to the Golgi 8 9; and both US2 and US11 cause the destruction of class I molecules by retrograde transport into the cytosol 10 11. Similarly, at least three MCMV gene products also alter class I assembly. m152/gp40 blocks transport of class I molecules from the ER to the Golgi 12 13 14; m6/gp48 binds to class I molecules and redirects their transport into the lysosome for destruction 15; and m4/gp34 binds class I in the ER, forming a complex which is transported to the cell surface 16. Only m152 and m6 have been previously shown to affect CTL function. There is no sequence homology between these MCMV genes and any mammalian or HCMV gene 17.
Although HCMV US3 and MCMV m152 both cause class I retention, in HCMV-infected cells the dominant effect on class I is rapid degradation due to the actions of US2 and US11 18 19. In contrast, in MCMV-infected cells, class I is not degraded in the ER, but in fact accumulates in a pre-Golgi compartment 12, and is degraded in the lysosome 15, or reaches the cell surface, sometimes in association with m4/gp34 16. Similarly, TAP function is impaired by HCMV US6, whereas TAP function is apparently normal in MCMV-infected cells (unpublished data). Finally, no molecule analogous to m4/gp34 has been observed to coprecipitate with class I in HCMV-infected cells. Thus, although there is no sequence homology between HCMV and MCMV genes that alter class I assembly, both viruses still effectively inhibit class I antigen presentation through the use of multiple genes.
It seems likely that interference with CTL recognition and the use of multiple genes to do so are both important features of the CMV–host relationship. It is not clear why both these viruses should carry a multiplicity of class I–modulating genes, but a number of possible explanations have been proposed. It has been suggested that individual genes may augment the function of others, or that viruses may require multiple different genes in order to interfere with the function of diverse class I proteins in natural outbred host populations. This report provides evidence in support of the latter hypothesis.
We have described previously the MCMV protein m4/gp34, which binds to MHC class I but whose function was not known. m4/gp34 is a 34-kD type 1 transmembrane glycoprotein, the product of the m4 gene 16. m4/gp34 is expressed abundantly during the early phase of viral gene expression, and accumulates in the ER, where it binds to class I molecules and forms a detergent-stable complex which is exported through the Golgi and to the cell surface. We speculated previously that m4 might serve to oppose the action of m152 by rescuing some class I molecules from retention, thus protecting infected cells from NK cells which might otherwise be activated by the loss of surface class I 16; on the basis of this hypothesis, m4/gp34 has been referred to as an “NK decoy.” However, until now there has been no evidence for an effect of m4 on any immune function.
In this paper, we show that m4 cooperates with m152 to prevent recognition of virus-infected cells by CD8+ T cells. m4 is thus the third MCMV gene demonstrated to interfere with the class I pathway of antigen presentation. We show that m152 has a differential effect on different class I molecules, efficiently retaining Db in a pre-Golgi compartment but only partially retaining Kb. To completely prevent recognition of virus-infected cells by three Kb-restricted CTL clones, both m4 and m152 were necessary. In contrast, m4 was not necessary to prevent recognition of infected cells by two Db-restricted CTL clones. Thus m4 and m152 have complementary effects on different class I molecules.
Materials and Methods
Generation of Mutant MCMVs.
Generation and characterization of recombinants ΔMS94.5 (with a deletion of ORFs m150 to 165), ΔMC96.24 (with a deletion of ORF m152), and rMC96.27 (revertant for ΔMC96.24) were described previously 20 21.
The recombinant Δm4-MC95.33, with an insertion of the lacZ gene in place of the m4 ORF, was generated by insertional mutagenesis in eukaryotic cells as described previously 22, using the plasmid construct pm4. The homologous recombining region of pm4 was produced by flanking the lacZ gene with MCMV genomic sequences adjacent to the 5′ (nt 2,739–3,250, left flank) and 3′ (nt 4,041–4,737, right flank) ends of the ORF. Plasmid DNA (pHindIIIA) 23 serving as MCMV genomic template and primer pairs for the left flanking sequence (sense [5′-AACTCGAGCATCACGGTGAACGATACCA], antisense [5′-TTGGATCCTGGAACAACGAATGAGACAGA]) and right flanking sequence [sense (5′-ATGCGGCCGCTCGAACTTCA-AACCGCTTAAGAG), antisense (5′-AACCGCGGACTTAT-CGACGTACAATCCTGT)] were used in separate PCR reactions to produce fragments with convenient restriction sites to ligate to the lacZ gene (XhoI, BamHI and NotI and SacII, respectively in bold). These fragments were inserted into corresponding sites within the plasmid pIC4, which contains the lacZ gene under control of the Rous sarcoma virus (RSV) promoter, SV40 poly(A), and flanking loxP sites 22. 30 fmol of linearized pm4 plasmid DNA was cotransfected with wt MCMV DNA (1.5 μg) into NIH3T3 fibroblasts by calcium phosphate precipitation to generate the recombinant virus Δm4-MC95.33. Recombinant virus was isolated and plaque purified as described previously 22. Correct recombinatorial mutagenesis within the genome of Δm4-MC95.33 was confirmed by restriction enzyme analysis (data not shown).
We have recently cloned the MCMV genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli 24. The MCMV-BAC plasmid pSM3fr contains the complete MCMV genome and was transfected into permissive eukaryotic cells to reconstitute the virus MW97.01 (wild-type; reference 25). MW97.01 (wild-type), which contains the complete MCMV genome without any BAC sequence, has wild-type properties both in vitro and in vivo, indicating that the MCMV genome can be passaged in Escherichia coli without altering the properties of the reconstituted viruses.
Recombinant MCMVs Δm4-MW99.03, Δm152-MW99.05, and Δm4+m152-MW99.04 were generated by transfection of the MCMV BAC plasmids pΔm4, pΔm152, and pΔm4+m152, respectively, into primary mouse embryo fibroblasts (MEFs) by calcium phosphate precipitation technique as described previously 24. The MCMV BAC plasmid pΔm4, which encodes an exact deletion of the m4 ORF (nt 3,270–4,067) by insertion of the prokaryotic kanamycin resistance marker (kanr), was constructed using contiguous m4-kan sequence primer pairs: sense (5′-TAATGATCTAGACGGCAATTTCTGTCTCATTCGTTGTTCCAGAGCGACGGATGGTACAAG) and antisense (5′-TACTCAGAACACCGGAAAATGGTTTACTCAAGGGGATTTTTATTTAGGGGGTTAGTTACT). The plasmid pACYC177 (New England Biolabs) served as template for the kanamycin resistance marker. A linear DNA fragment containing flanking homologies of 55 bp to the m4 gene (nt 3,215–3,269 and nt 4,068–4,123 in the MCMV genome) and the kanr was generated by PCR amplification. This fragment was inserted into the wild-type MCMV BAC plasmid pSM3fr 25 by homologous recombination in Escherichia coli to generate the MCMV BAC plasmid pΔm4. The MCMV BAC plasmid pΔm4+m152 was generated using contiguous _m152_-zeocin primer pair PCR amplification. The fragment containing flanking homologies of 60 bp to the m152 gene (nt 21,0184–21,0243 and nt 21,0378 –21,0437) and the zeocin resistance gene was generated using sense (5′-GCTCGAGCGAGAGCACCCGACGATCTGACATTGTCCAGTGTGCCGGTCGCACGAACATCAGAAGT-TCCTATTCTCTAGAAAGTATAGGAACTTCAACGTTTACAATTTCGCCTGATGCG) and antisense (5′-TCACAA-GCCGTGTCACCGCTCCACGTTTCACCGTCGTCGGT-CTCCCGATCGCTAGCCTGAACAGAAGTTCCTATACT-TTCTAGAGAATAGGAACTTCTGAAGTTTTAGCACGTGTCAGTCCT) primer pairs and the plasmid pZero1 (Invitrogen) as template. This fragment was inserted into the MCMV BAC plasmid pΔm4 by homologous recombination in Escherichia coli, generating plasmid pΔm4+m152.Plasmid pΔm4+m152 thus carries exact deletions of the m4 and m152 ORFs and insertions of the kanamycin resistance marker (in the case of m4) and the zeocin resistance marker (in the case of m152) instead. Plasmid pΔm152 was generated by homologous recombination between pSM3fr and the _m152_-zeocin fragment. Correct mutagenesis was confirmed by restriction enzyme and Southern blot analysis (data not shown).
Recombinant MCMVs m4-Tn3514, m4Tn3516, and m4TnP (with Tn1721 transposon insertions within the m4 gene or putative promoter, at nt 3,514, nt 3,516, and nt 3,099, respectively) were reconstituted from recombinant MCMV-BAC plasmids generated by direct transposon mutagenesis as described previously 26 27. The site of mutagenesis was confirmed by restriction enzyme analysis and sequencing (data not shown).
The genomic organization of all MCMV mutants is shown schematically in Fig. 1 A–C. Loss of m4/gp34 expression in the BAC-derived recombinants was confirmed by Western blot analysis of cell lysates from infected NIH3T3 cells with the antiserum m04-3 that detects m4/gp34 (see Fig. 1 D).
Figure 1.
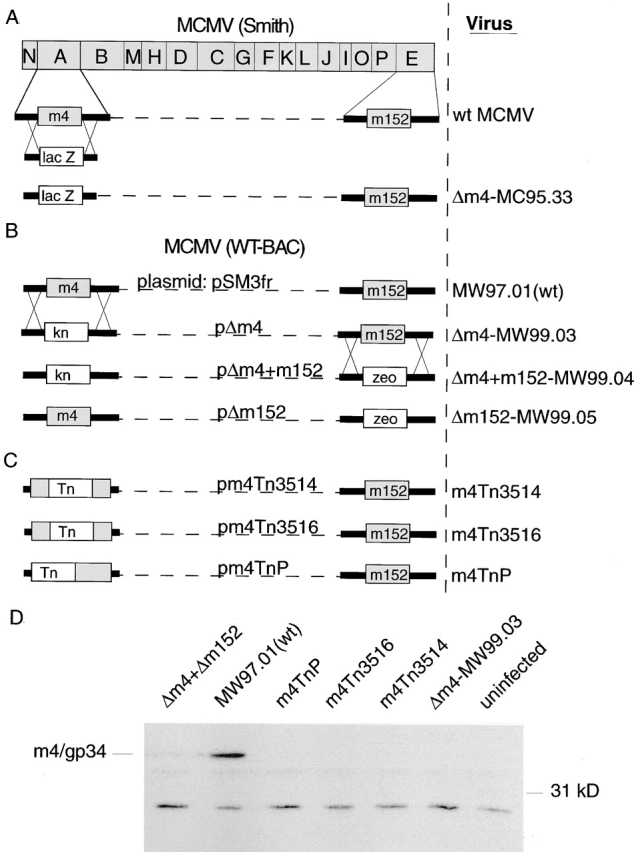
Construction of mutant viruses. (A) Schematic representation of the 230-kb linear MCMV genome. HindIII digestion generates 16 fragments, designated A–P by size, organized in the genome as shown (not to scale). Sequencing of the complete genome (reference 17) revealed 170 potential open reading frames, numbered from the left to the right hand end of the genome, 1–170. ORFs with homology to HCMV genes are given a capitalized M (e.g. M84), and MCMV genes without recognized homology are denoted by a lower case m (e.g. m4). Also shown are the positions of m4 and m152 within the genome, and the strategy for generation of the m4 deletion mutant Δm4-MC95.33 by insertion of the lacZ gene. (B) Generation of BAC mutants by insertional mutagenesis. Kanamycin and zeocin resistance genes were inserted into the pSM3fr BAC plasmid as shown to replace the m4 and m152 ORFs, respectively. Transfection into permissive cells generated the mutant viruses Δm4-MW99.03, Δm4+m152-MW99.04, and Δm152-MW99.05. (C) Generation of BAC mutants by transposon mutagenesis. (D) Western blot analysis using anti-m4/gp34 serum m4-03 of lysates from cells infected with the viruses shown. All viruses were used at the same MOI and all infected cells showed comparable cytopathic effect.
Experimental Animals.
B6 mice were purchased from Simenson, and B10.A5R and B10.A2R from Jackson ImmunoResearch Laboratories. Db−/− mice 28 were a gift from Francois Lemonnier (Institut Pasteur, Paris, France).
Virus Stocks and Cell Culture.
MEFs were grown from Trypsin-digested, day 12–14 mouse embryos and used between passages 3 and 6. Adult mouse fibroblast lines were generated from ears of Db−/− mice and from B6 × 129 backcrossed mice and used between passages 3 and 6. NIH 3T3s (CRL-1658) and Balb3T3s (CCL-163) were obtained from American Type Culture Collection. MEFs and 3T3s were maintained in DMEM supplemented with 10% fetal (for MEFs, adult fibroblast lines, and NIH3T3s) or newborn (for Balb3T3s) calf serum. Virus stocks were generated by infecting subconfluent MEFs with low passage seed stock at an multiplicity of infection (MOI) of 0.001. Cells were then switched to DMEM plus 10% normal calf serum until the monolayer became 100% infected. Stocks were harvested by scraping and sonication of cells. Titer of plaque forming units was determined by serial dilution and agarose overlay on Balb3T3s.
T Cell Line and Clones.
B6 mice were infected intraperitoneally with 5 × 104 PFU MCMV-Smith, ΔMS94.5, or ΔMC96.24. Between 8 and 40 wk later, spleens were harvested. 10% of splenocytes were infected with MCMV (of the same strain with which mice were infected) and returned to culture with the remaining splenocytes. For polyclonal effectors, cultures were used 5 d later in Cr-release assays. To derive CTL clones, the cultures were cloned by limiting dilution on day 3 in the presence of irradiated mixed allogeneic feeder splenocytes and 2 μg/ml concanavalin A (con A; Sigma-Aldrich). Clones were maintained in cloning medium (RPMI medium with 10% FCS, 5 × 10−5 M 2-mercaptoethanol [Sigma-Aldrich], 1% conditioned medium from IL-2–secreting cell line ×63.653 [reference 29], and 10% conditioned medium from conA-stimulated rat splenocytes), and restimulated with conA and irradiated mixed allogeneic feeders each 10 d. Clones have been maintained in culture for >12 mo. Clones were screened for antiviral function based on their ability to specifically kill IFN-γ–boosted ΔMS94.5-infected MEFs compared with uninfected MEFs. Clones 3, 11, and 5 are from ΔMS94.5-infected mice (clone 5 is from a different mouse than clones 3 and 11); clone 96 is from a Smith-infected mouse; and clone 55 is from a ΔMC96.24-infected mouse. Clones 3, 11, and 96 recognize different HPLC fractions of peptides extracted from infected cells (data not shown); clones 5 and 55 have not been tested against HPLC-fractionated extracts.
Cytolytic T Cell Assays.
MEF target cells were plated into 96-well plates at 5,000 cells per well and treated with recombinant mouse IFN-γ (50 U/ml; Sigma-Aldrich) for 24 h, infected with MCMV (at an MOI of 30 for Fig. 3 D and 4, and an MOI of 10 for all other Figures) unless otherwise indicated, and labeled with 51Cr (NEN Life Sciences Products) overnight, in the presence of 0.3 mg/ml phosphonoacetic acid (PAA; Sigma-Aldrich) to prevent expression of viral late genes. CTL clones described here did not kill MEF targets without IFN-γ pretreatment (data not shown). T cells were added at the indicated effector-to-target ratios for 6 h, after which supernatants were harvested and assayed for γ-irradiation with a Topcount scintillation counter (Packard Instrument Co.). Background Cr-release was determined by incubating targets with medium alone, and total Cr release was achieved by lysing targets with medium containing 2% Triton X-100. The percentage of specific lysis was calculated as (experimental cpm background cpm)/(total cpm-background cpm). Each data point represents the mean of triplicate wells.
Figure 3.

m4 is an immune-evasion gene. Polyclonal CTL or CTL clones were tested for their ability to lyse B6 MEFs infected with the viruses shown. Targets were pretreated with IFN-γ and infected overnight in the presence of PAA. (A) Polyclonal CTLs were tested against ΔMS94.5 (lacking ORFs 150–165), Δm4-MC95.33, and wild-type MCMV (Smith). (B–D) MCMV-specific CTL clones were tested against Smith or the wild-type BAC virus MW97.01 and various m4 deletion mutants, as follows. (B) Δm4-MC95.33, ΔMS94.7 (lacking ORFs 1–17), ΔMS94.5, and Smith. (C) Δm4-MW99.03, ΔMS94.5, and Smith. (D) Δm152-99.05, MW97.01 (wild-type), and three m4 deletion viruses: m4Tn3514, m4Tn3516, and m4TnP.
Antibodies.
Serum 8010 (anti-p8) was generated by immunizing rabbits with synthetic peptide corresponding to exon 8 of Kb. Sera 8142 and 8139 (anti-m4/gp34) were both generated as follows. Serum R123 against the cytoplasmic tail of m4/gp34 16 was used to precipitate m4/gp34 from MCMV (Smith)-infected MEFs. After washing, the immune complex was suspended in complete Freund's adjuvant (Sigma-Aldrich) and used to immunize rabbits subcutaneously. Rabbits were boosted first with immune complex suspended in incomplete Freund's adjuvant (IFA; Sigma-Aldrich), and then by infection with recombinant vaccinia virus expressing m4/gp34 (generated by recombination between modified psc11 plasmid expressing the m4 gene and WR strain vaccinia virus), and finally with recombinant m4/gp34 protein purified from baculovirus, (the gift of Pamela Bjorkman, California Institute of Technology, Pasadena, CA) in IFA. Serum m04-3 used for Western blot analysis of m4/gp34 expression was generated by immunizing rabbits with synthetic peptide corresponding to amino acids 34–48 of m4/gp34 (peptide sequence KEYKEKMKYRHSLGC). Monoclonal antibody 28.14.8S (HB-27; American Type Culture Collection) was purified from hybridoma supernatant.
Metabolic Labeling and Immunoprecipitations.
B6 MEFs or adult ear fibroblasts were pretreated with recombinant mouse IFN-γ at 50 U/ ml for 24–48 h before infection. Without IFN-γ, uninfected MEFs do not express detectable amounts of class I. Although infected cells express class I in the absence of IFN-γ, they were also treated with IFN-γ for the sake of consistency. Cells were maintained in the presence of 0.3 mg/ml PAA after infection or mock infection. 1 h before the addition of metabolic label, cells were washed in PBS and placed in cysteine/methionine-free DMEM (GIBCO BRL) supplemented with antibiotics, and 5% FCS. Cells were then labeled with [35S]cysteine/methionine (∼0.2 μCi/ml for long labeling periods and ∼0.5 μCi/ml for pulse labels; NEN Life Sciences Products) for the time periods indicated. For pulse-chase experiments, cells were washed with chase medium (DMEM supplemented with antibiotics, glutamate, 10% FCS, and 1 mM l-cysteine and l-methionine; Sigma-Aldrich) at the end of the labeling period. All lysis and precipitation procedures were carried out at 4°C. Cells were washed in the plates with PBS and lysed in NP-40 lysis buffer (0.5% NP-40, 50 mM Tris-HCl, pH 7.6, 5 mM MgCl2). Just before use, lysis buffer was supplemented with protease inhibitor, either 1 mM PMSF (Sigma-Aldrich) or Complete EDTA-free protease-inhibitor cocktail according to the manufacturer's directions (Boehringer Mannheim). Lysates were precleared by incubation with at least 20 μl of normal rabbit serum and 500 μl of 10% suspension of fixed Staphylococcus aureus for 2 h, and centrifuged for 5 min at 15,000 g. Precleared lysates were then subjected to specific immunoprecipitation as indicated in the Figures. Unless otherwise indicated, each aliquot of lysate received ∼10 μg of antibody plus 150 μl of 5% protein A agarose suspension (Sigma-Aldrich). Immunoprecipitates were washed four times in NET buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 5 mM EDTA, and 0.05% NP40) containing 0.1% SDS. Samples were digested with Endo Hf (New England Biolabs, Inc.) according to manufacturer's protocol, resuspended in reducing sample buffer, and separated by SDS-PAGE on a 12.5% gel. Quantitation of labeled protein was performed using a Molecular Dynamics PhosphorImager.
Results
Construction of MCMV Mutants Lacking m4/gp34 Expression.
MCMV mutants lacking m152/gp40 expression have already been described previously 21. To compare the functions of m4 and m152, we constructed mutant MCMVs with targeted deletions of m4 and/or m152. The process of generating mutant viruses may lead to accidental mutations elsewhere in the genome, so in order to clearly attribute a phenotype to the deleted gene, we have constructed five separate mutant viruses lacking m4, using three different technologies (see Materials and Methods, Fig. 1, and Table ). Δm4-MC95.33 was made by homologous recombination between the viral genome and plasmid in transfected cells (Fig. 1 A). Other mutants were reconstituted from BACs constructed by homologous recombination (Fig. 1 B) or transposon insertion (Fig. 1 C). The correct genomic structure of all BACs was confirmed by restriction analysis and Southern blot analysis (data not shown). Lack of m4/gp34 expression by all Δm4 mutants was confirmed by Western blot analysis (Fig. 1 D).
Table 1.
Viruses Used in This Paper
| Virus | Method | Genotype | Insertion |
|---|---|---|---|
| Smith strain | natural isolate | wild-type | none |
| MW97.01 | BAC derived | wild-type | none |
| ΔMS94.5 | mutagenesis in cells | ΔORFs 150-165 | lacZ |
| ΔMC96.24 | mutagenesis in cells | Δm152 | none |
| rMC96.27 | mutagenesis in cells | wild-type, revertant of ΔMC96.24 | none |
| ΔMS94.7 | spontaneous mutant | ΔORFs 1-17 | none |
| Δm4-MC95.33 | mutagenesis in cells | Δm4 (nt 3250-4041) | lacZ |
| Δm4-MW99.03 | BAC/recombination | Δm4 (nt 3270-4067) | kanr |
| m4Tn3514 | BAC/transposon | m4 disrupted | Tn1721 into m4 at nt 3514 |
| m4Tn3516 | BAC/transposon | m4 disrupted | Tn1721 into m4 at nt 3516 |
| m4TnP | BAC/transposon | putative m4 promoter disrupted | Tn1721 into putative m4 promoter at nt 3099 |
| Δm4+m152-MW99.04 | BAC/recombination | Δm4+m152 (nt 3270-4067 and 210244-211377 | kanr/zeocinr |
| Δm152-MW99.05 | BAC/recombination | Δm152 (nt 210244-211377) | zeocinr |
m4 Does Not Affect the Export of Kb over a 90-min Chase.
NK cells can lyse target cells with low cell surface class I; thus viral functions that reduce cell surface class I in order to protect against CTL recognition might render the infected cells vulnerable to NK attack. We previously hypothesized that m4/gp34 might serve to inhibit NK activity by rescuing some class I molecules from _m152_-induced retention, thus increasing class I expression on the cell surface 16. To test this hypothesis, we infected mouse embryo fibroblast (MEF) cells with either wild-type virus or the mutant virus Δm4-MC95.33, at a range of multiplicities of infection (MOIs), and measured the degree of Kb export as indicated by the acquisition of Endoglycosidase H (Endo H) resistance over a 90-min chase period. Fig. 2 shows that at any given MOI, infection with either Δm4-MC95.33 or wild-type virus caused comparable degrees of Kb retention. In addition, at a fixed MOI of 5, we found no significant difference between the amount of Kb that was exported in wild-type or Δm4-MC95.33-infected cells at a range of timepoints after infection (data not shown). We conclude that m4 does not affect the extent of Kb export in MCMV-infected fibroblasts.
Figure 2.


m4 does not counteract the effects of m152 on Kb. (A) Immunoprecipitation of Kb and m4/gp34 from MEFs infected with increasing doses of wild-type (Smith) or Δm4-MC95.33 MCMV. Cells were infected at the indicated MOI for 5 h, labeled for 30 min with [35S]methionine, and chased for 90 min with unlabeled methionine. Cells were lysed in NP-40 lysis buffer. Kb was immunoprecipitated with anti-p8 and m4/gp34 with serum 8142. All samples were treated with Endo H. The positions of bands corresponding to Endo H-resistant (R) and -sensitive (S) molecules are indicated. (B) Quantitation of Kb export. The amount of Endo H-sensitive (retained) and Endo H-resistant (exported) Kb was determined with phosphorimage analysis of the gel shown in A. The degree of export is calculated as the percentage of export equals (resistant)/(resistant plus sensitive).
m4/gp34 Expression Inhibits CTL Activity.
We next investigated whether m4/gp34 expression could affect recognition of targets by class I–restricted CD8+ CTLs. The first experiments used polyclonal CTLs from spleens of MCMV-infected C57BL/6 (B6) mice restimulated in vitro with virus. Fig. 3 A shows one such experiment. There was minimal specific lysis of targets infected with wild-type MCMV, but strong CTL activity against targets infected with ΔMS94.5, which lacks m152. Thus in wild-type infection the combined effects of the immune-evasion genes were able to completely abrogate recognition; however, a virus lacking m152 was readily detected. There was also significant killing of targets infected with Δm4-MC95.33, demonstrating that m4, in addition to m152, contributes to immune evasion from polyclonal CTLs.
To investigate this phenomenon further, we generated a panel of MCMV-specific CTL clones from mice infected with either wild-type MCMV or two mutant MCMV viruses lacking m152. The antigens recognized by these clones have not yet been identified, but they are all expressed in the early phase of MCMV gene expression (data not shown). Remarkably, none of these clones were able to lyse cells infected with wild-type virus; this included clone 96 which was generated from a mouse infected with wild-type virus. However, all the clones recognized targets infected with viruses lacking m152 (Fig. 3 B–D), confirming the importance of this immune-evasion gene. Next we tested whether the clones could recognize viruses lacking m4 but expressing m152. Fig. 3 B shows an experiment using Δm4-MC95.33, and Fig. 3 C shows an experiment using Δm4-MW99.03. Both m4 deletion mutants, which were independently constructed using different techniques, were recognized, whereas the wild-type virus was not. These results were extended in the assay shown in Fig. 3 D, in which the three m4 deletion mutants generated by transposon insertion, m4Tn3514, m4Tn3516, and m4TnP, were tested for recognition by three different clones. All three mutants were recognized by clones 11 and 96, consistent with the previous results. However, we noted that none of the three mutants was recognized by clone 3.
The results seen with five independent m4 deletion mutants led us to conclude that the observed phenotype is indeed due to the functional deletion of the m4 gene. These results demonstrate for the first time that m4, like m152 and m6, acts as a viral immune-evasion gene. However, the results seen with clone 3 demonstrate that deletion of m4 is not by itself sufficient to permit MCMV recognition by some CTL clones.
Clone 3 Does Not Recognize an Epitope within m4/gp34.
We wondered why only some CTL clones could recognize cells infected with m4 deletion mutants. m4/gp34 provides an epitope recognized by MCMV-specific CTLs from Balb/c mice 30. One possible explanation for inability of clone 3 to recognize m4 deletion viruses was that the epitope recognized by clone 3 could be derived from m4/gp34 itself. Since clone 3 can respond to viruses lacking m152, we constructed a new virus (Δm4+m152-MW99.04) lacking both m152 and m4. Fig. 4 shows that this virus was readily detected by clone 3, indicating that the epitope recognized by clone 3 is not contained within m4/gp34.
Figure 4.

The antigen recognized by clone 3 is not m4/gp34. B6 MEF targets were infected with MCMV MW97.01 (wild-type), m4Tn3516, m4TnP, Δm4+m152-MW99.04, or Δm152-MW99.05. The difference in degree of lysis of targets infected with Δm152 and Δm4+m152 is not reproducible.
m4 Is Necessary for Evasion from Kb- but not Db-restricted CTL Clones.
To further analyze why some clones were able to recognize m4 deletion mutants and others not, we determined the restriction element used by five MCMV-specific CTL clones. MEFs from B10A.2R (KkDb) or B10A.5R (KbLdDd) mice were infected with MCMV-ΔMS94.5 and used as targets in CTL assays. Fig. 5 A shows that clones 3 and 55 are restricted by Db, and clones 5, 11, and 96 are restricted by Kb. These five clones were next tested for their ability to lyse targets infected with m4 deletion mutants. The results are shown in Fig. 5 B. All three Kb-restricted clones were able to lyse targets infected with m4 deletion mutants, indicating that m4 expression was necessary for complete immune evasion from these clones. In contrast, the two Db-restricted clones did not recognize the m4 deletion mutants, indicating that the other immune-evasion genes were sufficient to prevent MCMV-specific killing by these clones.
Figure 5.

m4 is necessary for evasion from Kb- but not Db-restricted CTL clones. (A) Two MCMV-specific clones are Db-restricted and three are Kb-restricted. The class I–restriction elements used by five CTL clones, derived from MCMV-infected B6 mice, were determined by testing the ability of each clone to lyse fibroblasts from B10A.2R (KkDb) or B10A.5R (KbLdDd) mice. Fibroblast targets were either uninfected or infected with MCMV ΔMS94.5. (B) Kb- but not Db-restricted clones respond to MCMV lacking m4. The five CTL clones were tested for ability to lyse of B6 MEFs infected with the indicated viruses.
MCMV Differentially Inhibits Maturation of Different Class I Molecules.
The difference between Kb- and Db-restricted CTL clones suggested that Kb and Db might show different sensitivities to the effects of the immunomodulatory genes. The three Kb-restricted clones can lyse targets infected with virus containing a single deletion of either m152 or m4, indicating that m152 is necessary to prevent antigen presentation by Kb but is not sufficient for this task in the absence of m4. In contrast, the two Db-restricted clones can lyse infected cells if m152 is deleted, but are unable to detect virus in which m4 is deleted while m152 remains. This suggested that Db might be more susceptible to the activity of m152 than Kb. m152 inhibits antigen presentation by retaining class I molecules in the ERGIC. Therefore, we performed a pulse-chase experiment comparing the relative rates of export of Kb and Db in MCMV-infected cells. B6 MEFs were pulsed with [35S]methionine for 15 min and chased for 1, 2, or 4 h. Kb and Db were immunoprecipitated from the same lysates and the respective degree of maturation was determined by Endo H digestion. Fig. 6 A shows that in uninfected cells, both Kb and Db became Endo H-resistant over the chase period, although the maturation of Db was slower than maturation of Kb. In contrast, in infected cells almost all Db was retained in an Endo H-sensitive form over the entire 4 h, while ∼50% of the Kb protein was exported and matured within 2 h. We also noted that the m4/gp34 coprecipitating with the class I molecules displayed a parallel pattern. There was little m4/gp34 associated with Db, and all Db-associated m4/gp34 was Endo H-sensitive; in contrast, there was a significant amount of Kb-associated m4/gp34, which also became 50% Endo H-resistant by 2 h of chase, and nearly 100% Endo H-resistant by 4 h.
Figure 6.

Differential effects of MCMV infection on maturation of different class I proteins. (A) In a 4-h pulse chase, Kb partially escapes from MCMV-mediated retention, but Db does not. B6 MEFs were infected with MCMV (Smtih) for 5 h, pulsed with [35S]methionine/ cysteine for 15 min, and chased for the indicated time period. Kb and Db were sequentially immunoprecipitated from the same lysates. (B) The differential effect is also apparent over a 14-h labeling period. Fibroblast lines from adult H-2b (B6 × 129) mice were infected with MCMV (Smith). Cells were continuously labeled with [35S]methionine/cysteine from 2–16 h after infection. Kb and Db were sequentially immunoprecipitated from the same lysates. (C) Escape of Kb from the effects of m152 is not due to competition between Kb and Db. Fibroblasts from Db−/− mice were infected with the indicated virus, pulsed for 30 min, and lysed immediately or chased for 3 h. The positions of bands corresponding to Endo H-resistant (R) and -sensitive (S) molecules are indicated.
Because of the slower rate of maturation of Db in uninfected cells, we wondered whether the effect of m152 might simply be a general retardation of the maturation of both molecules with no eventual effect on the relative steady-state degree of export. To address this we labeled cells continuously from 2 to 16 h after infection, and sequentially immunoprecipitated first Kb and then Db from the same lysates. Fig. 6 B shows that whereas all the Db from infected cells remained Endo H sensitive, the majority of Kb acquired Endo H resistance. Again, a parallel maturation pattern of class I–associated m4/gp34 was observed. The results shown in Fig. 6 A and B are typical of a series of similar experiments in which Db retention was always nearly complete, while Kb retention, although variable, was always less. Thus, in infected fibroblasts relatively little Db is available to reach the cell surface, but a large portion of Kb, some of which is m4/gp34-associated, eventually passes through the Golgi to the cell surface.
The observed differential effects of MCMV infection on Kb and Db indicate that these molecules are differently affected by m152. Although a sustained interaction between m152/gp40 and class I has not been demonstrated, we reasoned that Kb might be able to escape retention because of competition by Db (which is fully retained) for a limiting amount of m152/gp40. To test this possibility we determined the extent of export of Kb molecules in infected fibroblasts from mice with a targeted deletion of Db. If competition for m152/gp40 were the cause of the differential retention of Kb and Db, then in the absence of Db, Kb should be fully retained during the 3-h chase period. However, as shown in Fig. 6 C, even in the absence of Db, a significant amount of Kb escaped _m152_-mediated retention. We conclude that the difference in susceptibility to m152 is intrinsic to the individual class I proteins and not due to intermolecular competition.
Discussion
In this paper we have provided the first evidence of a function for the MCMV gene m4. Previous discussion of the function of m4 has been limited to speculation, based on its biochemical association with MHC class I molecules. We have demonstrated here that expression of m4 by MCMV-infected target cells protects those targets from killing by some class I–restricted CTL clones. Thus m4 joins m152 and m6 as the third MCMV CTL-evasion gene. Furthermore, the mechanism of action of m4 is likely to be entirely novel, since all previously described viral CTL-evasion genes have had the effect of reducing class I surface expression, by inhibition of class I transport, removal of class I from the cell surface, or TAP blockade. In contrast, we show here that while m4 does not inhibit Kb export from the ER, (Fig. 2), it significantly inhibits killing of MCMV-infected targets by Kb-restricted clones. This inhibition is demonstrated in Fig. 3, where we show that MEF targets infected with wild-type MCMV were not recognized by CTLs, while targets infected with mutant viruses lacking m4 were recognized to a significant extent. Thus m4 inhibits CTL recognition of infected targets even though they express significant amounts of mature Kb (Fig. 2 and Fig. 6, and reference 16).
The mechanism by which m4 inhibits CTL recognition is not yet known. We have found that between 50 and 70% of mature Kb synthesized over the course of MCMV infection coprecipitates with m4/gp34 in the presence of 0.5% NP40. In addition, immature Kb forms complexes with m4/gp34 which are observed in lysates made with the weaker detergent digitonin 1a. Thus we imagine two mechanisms by which m4 may inhibit CTL activity, either or both of which may be operative: ER-localized m4/gp34 may alter peptide-loading of Kb, and/ or surface-exposed m4/gp34 may alter class I recognition by the TCR or CD8. We are currently in the process of identifying peptide epitopes recognized by MCMV-specific CTLs, which will facilitate the investigation of these possibilities.
In addition to demonstrating the immune-evasive function of m4, our results describe, for the first time, the functional interaction of multiple immune-evasion genes in cells infected with a herpes virus. It has been a longstanding puzzle why CMVs should encode multiple genes (at least four in HCMV and at least three in MCMV) which all have the general effect of reducing class I–restricted antigen presentation. Multiple genes could interact in any of several ways, ranging from complete redundancy to cooperation or synergy. Many previous papers describing viral immune-evasion genes have relied on transfected cells overexpressing single viral genes, and thus can shed no light on this question; however, some possibilities have been discussed in the case of HCMV. Ahn et al_._ raised the hypothesis of synergy 8. They observed that the HCMV gene US3 is expressed earlier in the viral cycle than US2 and US11, and thus might augment the function of the latter genes by retaining class I. Machold et al_._ proposed another reason for HCMV to encode both US2 and US11, which both have the effect of targeting class I for degradation by the proteasome. They suggested these genes might preferentially target different class I molecules 31. Using cell lines transfected with either US2 or US11, and infected with vaccinia viruses encoding various alleles of murine class I genes, they noted that US2 degraded only a subset of the class I molecules that were degraded by US11. However, since no functional assays were done, and only murine class I was tested (while HCMV infects only humans), the biological relevance of the finding was unclear.
Here we have employed a biologically relevant system, using MCMV-infected primary cells to assess the effect of m4 and m152 on antigen presentation to MCMV-specific CTLs. The first clear conclusion from the results reported here is that the genes are not redundant. Deletion of either m152 or m4 allows detection of infected cells by Kb-restricted CTL clones. Thus a contribution from both of these genes (and perhaps also from m6 which was not tested here) is necessary for complete abrogation of antigen presentation in this experimental system. At present we have no data to indicate whether the effects of m4 and m152 are synergistic or merely additive. We also report a differential effect of the immune-evasion genes on antigen presentation by two different class I molecules, Kb and Db. We found that while expression of both m152 and m4 was necessary for complete abrogation of antigen presentation to three Kb-restricted clones, expression of m152, but not of m4, was required to completely block antigen presentation to two Db-restricted clones (Fig. 5).
These observations, using a limited number of CTL clones, suggested that Db would be more affected by m152 than Kb. This prediction was confirmed by our biochemical analysis of class I assembly in MCMV-infected fibroblasts. Fig. 6 demonstrates that the combined effects of m152 and m6 were insufficient to completely prevent maturation of Kb. During a 120-min chase, ∼50% of newly synthesized Kb molecules became mature (i.e., were exported past the medial Golgi). In contrast, almost no Db became Endo H-resistant >4-h chase. Furthermore, we note that the mature (Endo H-resistant) Kb molecules had significant amounts of m4/gp34 associated with them, while there was relatively little m4/gp34 associated with Db. Thus, the class I molecule which escapes from the effects of m152 and m6, Kb, is preferentially targeted by m4. The difference in retention of Kb and Db is even more strikingly evident over the course of a 16-h labeling period, as shown in Fig. 6 B. The CTL assays monitored antigen presentation by a small subset of total class I, that which was loaded with cognate peptides. The biochemical experiments, on the other hand, monitor the potential for antigen presentation of all the class I synthesized during infection. The almost complete retention of Db due to m152 contrasts with the significant export of Kb. This fully supports the prediction, based on the CTL assays, that Kb would need m4 as a “backup” mechanism for m152 in order to fully inhibit antigen presentation, whereas Db may not. We conclude that m4 complements the function of other MCMV immune-evasion genes.
These observations raise some interesting questions regarding the coevolution of viruses and the immune system. Class Ia loci are both polygenic and highly polymorphic, and it is generally accepted that this diversity reflects evolutionary selection for the ability to present a broad array of different peptides. In addition to differences in peptide binding, however, different class Ia molecules also assemble at different intrinsic rates (Fig. 6 and references 32 33 34) and with different dependence on various chaperones 35 36 37; the evolutionary implications of these differences are less clear. We have now shown that Kb and Db have differential susceptibility to the effects of MCMV m152, and that the virus requires a “backup gene”, m4, in order to achieve complete protection against CTL lysis in vitro. This raises the possibility that intrinsic differences in the assembly behavior of Kb and Db may reflect evolutionary pressure to avoid the effects of viral genes such as m152. Such a tit-for-tat evolutionary model is already widely accepted in the case of NK cells, in which the “missing self” response is believed to have evolved to counteract virally induced class I downregulation; in turn, CMVs encode genes (the signal sequence of HCMV UL40 [references 38 and 39], MCMV m144 [reference 40], and perhaps HCMV UL18 [reference 41]) which inhibit NK activity.
We have provided evidence suggesting that one function of the multiplicity of immune-evasion genes of MCMV is to provide more effective coverage of the diverse class I molecules present in natural outbred host populations. This does not preclude the possibility that some of the other hypothetical advantages discussed previously may also be operative. It is interesting to note that the CTL evasion genes of both MCMV and HCMV are encoded within families of related membrane glycoproteins which are not essential for virus replication in vitro, and which contain many genes whose functions have not yet been identified. There is much still to be learned about the ways that CMVs manipulate the cellular immune response, and the ways that the multiple genes interact to provide selective advantage for the virus.
Acknowledgments
We thank Sabine Linke for her excellent technical assistance, Irena Crnkovic-Mertens for generating the virus ΔMC95.33, and Maurits Kleijnen for VV-m4. We thank Pamela Bjorkman for the gift of recombinant m4/gp34 and Francois Lemonier for the gift of Db−/− mice. We thank David Parker, Dan Mourich, and Klaus Früh for helpful discussions.
This work was supported by grants from the American Heart Association (grant-in-aid 9650521N), the Pew Scholars Program in Biomedical Sciences (95-002844), the Medical Research Foundation of Oregon to A.B. Hill, and by the Deutsche Forschungsgemeinschaft through SFB455, project A7, and Ko571/15-1 to U.H. Koszinowski. D.G. Kavanagh is supported by the National Institutes of Health training grant EY07123-09.
Footnotes
Abbreviations used in this paper: BAC, bacterial artificial chromosome; ER, endoplasmic reticulum; MEF, mouse embryo fibroblast; MOI, multiplicities of infection; NK, natural killer; PAA, phosphoroacetic acid; TAP, transporter associated with antigen presentation.
References
- Alcami A., Koszinowski U.H. Viral mechanisms of immune evasion. Trends Microbiol. 2000;8:410–418. doi: 10.1016/S0966-842X(00)01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh D.G., Koszinowsi U.H., Hill A.B. Themurine cytomegalovirus immune evasion protein m41gp34engages class I MHC in biochemically distinct complexes atthe cell surface and in a pre-Golgi compartment. J. Immunol. 2001;In press doi: 10.4049/jimmunol.167.7.3894. [DOI] [PubMed] [Google Scholar]
- Hengel H., Brune W., Koszinowski U.H. Immune evasion by cytomegalovirus-survival strategies of a highly adapted opportunist. Trends Microbiol. 1998;6:190–197. doi: 10.1016/s0966-842x(98)01255-4. [DOI] [PubMed] [Google Scholar]
- Kavanagh D.G., Hill A.B. Evasion of cytotoxic T lymphocytes by murine cytomegalovirus. Semin. Immunol. 2001;13:19–26. doi: 10.1006/smim.2001.0292. [DOI] [PubMed] [Google Scholar]
- Jones T.R., Hanson L.K., Sun L., Slater J.S., Stenberg R.M., Campbell A.E. Multiple independent loci within the human cytomegalovirus unique short region down-regulate expression of major histocompatibility complex class I heavy chains. J. Virol. 1995;69:4830–4841. doi: 10.1128/jvi.69.8.4830-4841.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K., Gruhler A., Galocha B., Jones T.R., Wiertz E.J., Ploegh H.L., Peterson P.A., Yang Y., Fruh K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity. 1997;6:613–621. doi: 10.1016/s1074-7613(00)80349-0. [DOI] [PubMed] [Google Scholar]
- Hengel H., Koopmann J.O., Flohr T., Muranyi W., Goulmy E., Hammerling G.J., Koszinowski U.H., Momburg F. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity. 1997;6:623–632. doi: 10.1016/s1074-7613(00)80350-7. [DOI] [PubMed] [Google Scholar]
- Lehner P.J., Karttunen J.T., Wilkinson G.W., Cresswell P. The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. PNAS. 1997;94:6904–6909. doi: 10.1073/pnas.94.13.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K., Angulo A., Ghazal P., Peterson P.A., Yang Y., Fruh K. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. PNAS. 1996;93:10990–10995. doi: 10.1073/pnas.93.20.10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.R., Wiertz E.J., Sun L., Fish K.N., Nelson J.A., Ploegh H.L. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. PNAS. 1996;93:11327–11333. doi: 10.1073/pnas.93.21.11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz E.J., Jones T.R., Sun L., Bogyo M., Geuze H.J., Ploegh H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Wiertz E.J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T.R., Rapoport T.A., Ploegh H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- del Val M., Hengel H., Hacker H., Hartlaub U., Ruppert T., Lucin P., Koszinowski U.H. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J. Exp. Med. 1992;176:729–738. doi: 10.1084/jem.176.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H., Thale R., Lucin P., Muranyi W., Flohr T., Hengel H., Farrell H., Rawlinson W., Koszinowski U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- Ziegler H., Muranyi W., Burgert H.G., Kremmer E., Koszinowski U.H. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 2000;19:870–881. doi: 10.1093/emboj/19.5.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusch U., Muranyi W., Lucin P., Burgert H.G., Hengel H., Koszinowski U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999;18:1081–1091. doi: 10.1093/emboj/18.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleijnen M.F., Huppa J.B., Lucin P., Mukherjee S., Farrell H., Campbell A.E., Koszinowski U.H., Hill A.B., Ploegh H.L. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 1997;16:685–694. doi: 10.1093/emboj/16.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlinson W.D., Farrell H.E., Barrell B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996;70:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beersma M.F., Bijlmakers M.J., Ploegh H.L. Human cytomegalovirus down-regulates HLA class I expression by reducing the stability of class I H chains. J. Immunol. 1993;151:4455–4464. [PubMed] [Google Scholar]
- Warren A.P., Ducroq D.H., Lehner P.J., Borysiewicz L.K. Human cytomegalovirus-infected cells have unstable assembly of major histocompatibility complex class I complexes and are resistant to lysis by cytotoxic T lymphocytes. J. Virol. 1994;68:2822–2829. doi: 10.1128/jvi.68.5.2822-2829.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thale R., Szepan U., Hengel H., Geginat G., Lucin P., Koszinowski U.H. Identification of the mouse cytomegalovirus genomic region affecting major histocompatibility complex class I molecule transport. J. Virol. 1995;69:6098–6105. doi: 10.1128/jvi.69.10.6098-6105.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krmpotic A., Messerle M., Crnkovic-Mertens I., Polic B., Jonjic S., Koszinowski U.H. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 1999;90:1285–1296. doi: 10.1084/jem.190.9.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crnkovic-Mertens I., Messerle M., Milotic I., Szepan U., Kucic N., Krmpotic A., Jonjic S., Koszinowski U.H. Virus attenuation after deletion of the cytomegalovirus Fc receptor gene is not due to antibody control. J. Virol. 1998;72:1377–1382. doi: 10.1128/jvi.72.2.1377-1382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebeling A., Keil G.M., Knust E., Koszinowski U.H. Molecular cloning and physical mapping of murine cytomegalovirus DNA. J. Virol. 1983;47:421–433. doi: 10.1128/jvi.47.3.421-433.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerle M., Crnkovic I., Hammerschmidt W., Ziegler H., Koszinowski U.H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. PNAS. 1997;94:14759–14763. doi: 10.1073/pnas.94.26.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M., Jonjic S., Koszinowski U.H., Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 1999;73:1056–1060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune W., Menard C., Hobom U., Odenbreit S., Messerle M., Koszinowski U.H. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nat. Biotech. 1999;17:360–364. doi: 10.1038/7914. [DOI] [PubMed] [Google Scholar]
- Hobom U., Brune W., Messerle M., Hahn G., Koszinowski U.H. Fast screening procedures for random transposon libraries of cloned herpes virus genomesmutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 2000;74:7720–7729. doi: 10.1128/jvi.74.17.7720-7729.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund P., Glas R., Menard C., Kase A., Johansson M.H., Franksson L., Lemmonier F., Karre K. β2-microglobulin-deficient NK cells show increased sensitivity to MHC class I-mediated inhibition, but self tolerance does not depend upon target cell expression of H-2 Kb and Db heavy chains. Eur. J. Immunol. 1998;28:370–378. doi: 10.1002/(SICI)1521-4141(199801)28:01<370::AID-IMMU370>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Karasuyama H., Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur. J. Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- Holtappels R., Thomas D., Podlech J., Geginat G., Steffens H.P., Reddehase M.J. The putative natural killer decoy early gene m04 (gp34) of murine cytomegalovirus encodes an antigenic peptide recognized by protective antiviral CD8 T cells. J. Virol. 2000;74:1871–1884. doi: 10.1128/jvi.74.4.1871-1884.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R.P., Wiertz E.J., Jones T.R., Ploegh H.L. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J. Exp. Med. 1997;185:363–366. doi: 10.1084/jem.185.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill A., Takiguchi M., McMichael A. Different rates of HLA class I molecule assembly which are determined by amino acid sequence in the α2 domain. Immunogenetics. 1993;37:95–101. doi: 10.1007/BF00216831. [DOI] [PubMed] [Google Scholar]
- Neefjes J.J., Ploegh H.L. Allele and locus-specific differences in cell surface expression and the association of HLA class I heavy chain with β2-microglobulindifferential effects of inhibition of glycosylation on class I subunit association. Eur. J. Immunol. 1988;18:801–810. doi: 10.1002/eji.1830180522. [DOI] [PubMed] [Google Scholar]
- Emerson S.G., Murphy D.B., Cone R.E. Selective turnover and shedding of H-2K and H-2D antigens is controlled by the major histocompatibility complex. J. Exp. Med. 1980;152:783–795. doi: 10.1084/jem.152.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neisig A., Wubbolts R., Zang X., Melief C., Neefjes J. Allele-specific differences in the interaction of MHC class I molecules with transporters associated with antigen processing. J. Immunol. 1996;156:3196–3206. [PubMed] [Google Scholar]
- Peh C.A., Burrows S.R., Barnden M., Khanna R., Cresswell P., Moss D.J., McCluskey J. HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity. 1998;8:531–542. doi: 10.1016/s1074-7613(00)80558-0. [DOI] [PubMed] [Google Scholar]
- Peh C.A., Laham N., Burrows S.R., Zhu Y., McCluskey J. Distinct functions of tapasin revealed by polymorphism in MHC class I peptide loading. J. Immunol. 2000;164:292–299. doi: 10.4049/jimmunol.164.1.292. [DOI] [PubMed] [Google Scholar]
- Ulbrecht M., Martinozzi S., Grzeschik M., Hengel H., Ellwart J.W., Pla M., Weiss E.H. Cutting edgethe human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 2000;164:5019–5022. doi: 10.4049/jimmunol.164.10.5019. [DOI] [PubMed] [Google Scholar]
- Tomasec P., Braud V.M., Rickards C., Powell M.B., McSharry B.P., Gadola S., Cerundolo V., Borysiewicz L.K., McMichael A.J., Wilkinson G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287:1031–1033. doi: 10.1126/science.287.5455.1031. [DOI] [PubMed] [Google Scholar]
- Farrell H.E., Vally H., Lynch D.M., Fleming P., Shellam G.R., Scalzo A.A., Davis-Poynter N.J. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- Reyburn H.T., Mandelboim O., Vales-Gomez M., Davis D.M., Pazmany L., Strominger J.L. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature. 1997;386:514–517. doi: 10.1038/386514a0. [DOI] [PubMed] [Google Scholar]