HIV-1 Vpr Enhances Viral Burden by Facilitating Infection of Tissue Macrophages but Not Nondividing CD4+ T Cells (original) (raw)
Abstract
Prior experiments in explants of human lymphoid tissue have demonstrated that human immunodeficiency virus type 1 (HIV-1) productively infects diverse cellular targets including T cells and tissue macrophages. We sought to determine the specific contribution of macrophages and T cells to the overall viral burden within lymphoid tissue. To block infection of macrophages selectively while preserving infection of T cells, we used viruses deficient for viral protein R (Vpr) that exhibit profound replication defects in nondividing cells in vitro. We inoculated tonsil histocultures with matched pairs of congenic viruses that differed only by the presence of a wild-type or truncated vpr gene. Although these viruses exhibited no reduction in the infection or depletion of T cells, the ability of the Vpr-deficient R5 virus to infect tissue macrophages was severely impaired compared with matched wild-type R5 virus. Interestingly, the Vpr-deficient R5 virus also exhibited a 50% reduction in overall virus replication compared with its wild-type counterpart despite the fact that macrophages represent a small fraction of the potential targets of HIV-1 infection in these tissues. Collectively, these data highlight the importance of tissue macrophages in local viral burden and further implicate roles for CC chemokine receptor 5, macrophages, and Vpr in the life cycle and pathogenesis of HIV-1.
Keywords: naive T cell, memory T cell, nuclear import, preintegration complex, burst size
Introduction
HIV-1 is a member of the lentivirus subfamily of retroviruses. Among the features that distinguish lentiviruses is their productive infection of cells of the monocyte/macrophage lineage. In addition to the gag, pol, and env genes found in all retroviruses, the HIV-1 genome contains six additional genes: tat, rev, vif, vpr, vpu, and nef. These genes confer upon HIV-1 a number of unique abilities, including the capacity to infect noncycling cells. Viral protein R (Vpr)* in particular is known to play an important role in facilitating infection of nondividing tissue macrophages (1–6) as well as inducing G2 cell-cycle arrest in dividing T cells (7–11). The vpr gene encodes a 96 amino acid, 14-kD nucleophilic protein that is incorporated into mature virions via an interaction with p6, a proteolytic subunit of the p55gag precursor (12–15). Although vpr is frequently deleted during in vitro passaging, Vpr is believed to have numerous functions that contribute to the establishment and pathogenesis of HIV-1 infection in vivo (8, 16).
Unlike simple retroviruses, HIV-1 does not depend on cellular division and the accompanying breakdown of the nuclear envelope for productive infection (17, 18). It is believed that this special property is due to the concerted and perhaps redundant activities of matrix (MA), integrase (IN), the DNA flap, and Vpr. Specifically, both MA (19–21) and IN (22–24) bound to the viral genome contain nuclear localization signals that target the preintegration complex (PIC) to the nucleus via interactions with host nuclear import machinery. The central DNA flap, a triple-stranded helix that is common to retroviruses, may additionally contribute to nuclear targeting through an unknown mechanism (25). Vpr is also highly nucleophilic and utilizes a distinct targeting strategy (1, 3–5, 26, 27). It contains two nonoverlapping and unique nuclear localization signals that likely contribute to the nuclear localization of the PIC (28, 29). Previous work has identified Vpr as a contributing factor in the infection of macrophages in vitro, which presumably is linked to this nuclear localization function (2, 4, 6).
Vpr also causes G2 cell-cycle arrest in infected cells cultured in vitro (7–11, 30). Expression of Vpr in some cell types by transfection, transduction, or productive HIV-1 infection is associated with inactivation of p34Cdc2 kinase, leading to the accumulation of the cells in the G2 phase of the cell-cycle (9, 11). The biologic significance of this arrest during natural infection is not well understood. However, studies have demonstrated that the HIV-1 LTR is most active in the G2 phase, implying that G2 arrest may confer a replicative advantage to viral species encoding a functional Vpr (8, 31, 32). In vitro studies have also revealed that prolonged G2 arrest may induce apoptosis of the infected cell (32–37), although others have not observed this effect (36, 38, 39). Thus, Vpr may variably potentiate or mediate apoptosis, and this function seems to segregate with cell-cycle arrest in mutagenesis studies (36). Based on these studies, it is speculated that Vpr contributes to HIV-mediated immune destruction by promoting depletion of target cells.
To clarify the importance of Vpr to HIV replication and subsequent pathogenesis, we employed a human lymphoid histoculture model. This system is distinguished by its capacity to support the replication of HIV-1, HIV type 2 (HIV-2), or simian immunodeficiency virus (SIV) without the need for exogenous cell activation or growth factors (40–44). Consequently, this ex vivo system preserves the diverse cell types and cellular activation and maturation phenotypes found within lymphoid tissues in vivo. Spleen and tonsil histocultures thus represent a valuable model in which to study the cellular tropism and cytopathic potential of these viruses in a physiologically relevant setting. We therefore employed this model to determine in which cell types Vpr plays a role in infection and to establish the contribution of these cells to the viral burden within lymphoid tissue. These studies reveal that while Vpr augments the infection of macrophages, it does not contribute to the productive infection of proliferating or resting T cells. Furthermore, Vpr-deficient R5 viruses exhibit a significant reduction in the extent of viral replication, emphasizing the importance of tissue macrophages as a permissive reservoir for viral replication in vivo. These findings suggest that other host or viral factors may be responsible for the infection of resting lymphocytes and highlight the importance of Vpr and CC chemokine receptor (CCR)5 coreceptor specificity for HIV-1infection of tissue macrophages.
Materials and Methods
Cloning of Vpr-deficient Viruses
The envelope genes determining CXCR4 (X4) or CCR5 (R5) preference were derived from HIV-1 NL4–3 or HIV-1 BaL. The region extending from the 5′ EcoR1 restriction endonuclease site located within the Vpr gene to the NheI site distal to the V3 region within the envelope gene were cloned from BaL and introduced into the NL4–3 proviral clone thereby creating the 107 strain (45). These sequences are sufficient to confer CCR5-tropism to 107. The Vpr− proviral clones were generated by introducing the AgeI to EcoRI (within vpr) fragment from the pNL4–3-derived partial proviral clone p210–19 (46) (obtained from the National Institutes of Health AIDS Research and Reference Reagent Program) into the AgeI and EcoRI sites of the respective proviruses. This cloning strategy preserved the pol and vif sequence identity between NL4–3 and 107, truncated the Vpr product by introducing a 115-nucleotide deletion, and also introduced two stop codons after the 21st amino acid of Vpr and downstream of the Vif stop codon in an otherwise isogenic virus.
Preparation of Viral Stocks
Plasmids encoding NL4–3 or 107 were transfected into 293T cells using calcium phosphate precipitation and 48 h later the resultant supernatants were centrifuged at 5,000 RPM for 10 min to remove cell debris and then aliquoted for subsequent infection of histocultures. Virus stocks were normalized for infectivity by determining the TCID50 on PHA-stimulated PBMCs obtained from at least two separate donors as described previously (40, 41).
Western Blot of Vpr
For hemagglutinin-Vpr (HA-Vpr) constructs, equal number of 293T cells were transfected using calcium phosphate and then harvested, pelleted, and lysed directly with SDS loading buffer. Similar transfection efficiency was confirmed by cotransfection with pEGFP (CLONTECH Laboratories, Inc.). Western blot was then performed with anti-HA monoclonal antibody (HA.11; Covance). For Vpr within HIV-1 virions, 300 ng of virus in 1 ml of media was concentrated by ultra-centrifugation at 40,000 g at 4°C for 1 h. The precipitate was resuspended in loading buffer containing SDS and β-mercaptoethanol and subjected to standard PAGE and blotted with antibodies to p24 (Coulter) or Vpr (28).
Culture and Infection of Human Lymphoid Tissues Ex Vivo
Human noninflammatory tonsil tissue removed during tonsillectomy (provided by San Francisco General Hospital, Kaiser-San Francisco and San Rafael, CA) were cut into 2–3 mm blocks and placed into culture as described (40). Tissues were inoculated within 24 h of preparation with viruses at ∼50 TCID50/tissue block. After 1- or 2-wk infections, cells were mechanically isolated from infected and uninfected tissue and analyzed by flow cytometry (see below). No significant differences in the ex vivo behavior and HIV-1 or SIV permissivity of spleen or tonsil tissue have been observed.
Assessment of CD4+ T Cell Infection by Flow Cytometry
At the indicated time points after inoculation (at peak infection, usually 1 wk after inoculation), dispersed cells from infected and uninfected lymphoid histocultures were immunostained for cell surface markers CD4, CD62L, and CD45RA as described previously (47). Cells were then fixed in 1% paraformaldehyde and subsequently permeabilized and immunostained for intracellular CD4 and p24. 50,000 CD4+ lymphocytes were counted and the data were analyzed with CELLQuest™ software (Becton Dickinson). To identify naive and memory subsets the following mAbs were used from Becton Dickinson: anti-CD4 (clone SK7, allophycocyanin conjugated, 1:20 dilution), and anti-CD62L (phycoerythrin, 1:20 dilution). The following antibody from BD PharMingen was also used: anti-CD45RA (cychrome conjugated, 1:20 dilution). To identify infected cells, anti-p24 (fluorescein isothiocyanate, 1:100 dilution) from Beckman Coulter was also used. Results are reported as the mean with SEM.
Flow Cytometric Analysis of Cellular Proliferation
After culturing for 6 d in standard histoculture media supplemented with 5-bromo-2′-deoxyuridine (BrdU; 50 μM; Sigma-Aldrich), lymphoid histocultures were stained for BrdU incorporation as described previously (47). Briefly, after this incubation, cells were dispersed and fixed and permeabilized overnight. Cells were washed, treated with DNase, and then immunostained with a combination of mAbs recognizing CD4 (Becton Dickinson, clone SK3, allophycocyanin conjugated, 1:40 dilution), CD45RA (BD PharMingen, cychrome conjugated, 1:2.5 dilution), CD62L (Becton Dickinson, phycoerythrin conjugated, 1:20 dilution), and BrdU (BD PharMingen, fluorescein isothiocyanate conjugated, 1:2.5 dilution). 50,000 CD4+ lymphocytes were then collected and analyzed by CELLQuest™.
Assessment of DNA Content by Flow Cytometry
Cell-cycle analysis for HA-Vpr constructs was performed by cotransfecting pEGFP (CLONTECH Laboratories, Inc.) and HA-Vpr DNA constructs into 293T cells in a 1:8 molar ratio to identify plasmid-expressing cells. 36 h later cells were trypsinized, fixed in 2% formaldehyde for 30 min, washed, and treated with 0.1 mg/ml Ribonuclease (RNase) A (Sigma-Aldrich) and 10 mg/ml propidium iodide in PBS for 30 min. Cellular DNA content in the transfected (GFP+) and untransfected (GFP−) cells was assessed using a FACScan™ flow cytometer. For analysis of infected and uninfected lymphoid histocultures, cells were dispersed from the tissue at the indicated time points post-inoculation (at peak infection, usually 1 wk after inoculation), and immunostained for the cell surface marker CD4. Cells were then fixed in 1% paraformaldehyde and subsequently permeabilized and immunostained for intracellular CD4 and p24. Samples were then incubated for 30 min in a solution of 0.01mM To-Pro-3 iodide (Molecular Probes) and 0.1 mg/ml RNase A. 200,000 lymphocytes were counted and the data were analyzed with CELLQuest™ and FlowJo (Tree Star). The following mAb was used from Becton Dickinson: anti-CD4 (clone SK7, phycoerythrin conjugated, 1:20 dilution). To identify infected cells, anti-p24 was also used.
Assessment of CD4+ T Cell Depletion by Flow Cytometry
At the indicated time points (usually 2 wk after inoculation), dispersed cells from infected and uninfected lymphoid histocultures were immunostained for cell surface markers CD3, CD4, CD8, and CCR5 as described previously (40, 41). 5,000–20,000 CD3+ lymphocytes were counted and the data were analyzed with CELLQuest™ and FlowJo. CD4+ T cell depletion was expressed as the ratio of CD4+ to CD8+ T cells in infected relative to uninfected tissues as described previously (40, 41). The following mAbs were used from Becton Dickinson: anti-CD3 (clone SK7, allophycocyanin conjugated, 1:80 dilution), anti-CD4 (clone SK3, fluorescein isothiocyanate conjugated, 1:20 dilution), and anti-CD8 (clone SK1, phycoerythrin conjugated, 1:20 dilution). The following antibody from BD PharMingen was also used: anti-CCR5 (allophycocyanin conjugated, 1:20 dilution). Cell numbers in infected tissues were normalized to those obtained for uninfected control tissues. Results are reported as the mean with SEM.
Assessment of Macrophage Infection by Flow Cytometry
At the indicated time points (usually 2 wk after inoculation), dispersed cells from infected and uninfected lymphoid histocultures were immunostained for cell surface markers CD3 and CD14 as described previously (43). Cells were then fixed in 1% paraformaldehyde and subsequently permeabilized and immunostained for intracellular CD68 and p24. 50,000–100,000 lymphocytes were counted and the data were analyzed with CELLQuest™. Results are reported as the mean with SEM.
Assessment of Viral Replication
At the indicated time points, a sample of culture media was withdrawn from infected tissue, diluted 1:1,000 with diluent, and stored at –20°C. The histoculture media was then replaced. At the end of the experiment, samples were thawed, and the concentration of HIV-1 p24 in the media was measured by ELISA from NEN Life Science Products.
Results
Construction of Vpr-deficient Viruses
To examine the tropism and replication of Vpr-deficient viruses in lymphoid tissue, we studied the matched pair of viruses, NL4–3 and 107. NL4–3 is a molecularly cloned strain of HIV-1 that exhibits strict preference for the coreceptor CXCR4 and does not replicate in macrophages (43, 45). 107 is a recombinant virus derived from NL4–3 that shares the same genetic backbone but incorporates coreceptor-determining envelope sequences cloned from BaL, a CCR5-dependent primary HIV-1 isolate, as well as additional upstream sequences (45). These cloned sequences from BaL extend through the 3′ end of vpr and, as a result of this substitution, some minor differences exist in the amino acid sequences of Vpr derived from NL4–3 and 107 (Fig. 1 A). However, the genomes of NL4–3 and 107 are isogenic upstream of vpr. To assess the functionality of these proteins we transiently transfected 293T cells with expression vectors encoding HA-tagged NL4–3- or 107-derived versions of Vpr. Western blot analysis with anti-HA mAb of whole-cell lysates from these transfectants revealed slightly lower expression of 107 Vpr compared with NL4–3 Vpr (Fig. 1 B). Because mutations or deletions in the 3′ end of vpr can alter expression levels of Vpr (48), we infer that the sequence differences between the NL4–3 and 107 vpr genes may result in stability differences between the two Vpr proteins leading to differential expression. We next transfected 293T cells with empty HA vector, NL4–3-derived, or 107-derived HA-Vpr expression vectors and stained the cells for intracellular DNA content 36 h later. Cell-cycle analysis by flow cytometry revealed that both NL4–3- and 107-Vpr induced significant G2 cell-cycle arrest, thereby establishing the functionality of both NL4–3- and 107-Vpr in vitro (Fig. 1 C).
Figure 1.



NL4–3- and 107-derived Vpr are functional and incorporated into mature virions. (A) HIV-1 Vpr from NL4–3 is identical to the BaL-derived, macrophage-tropic 107 variant except at residues 78 and 83–85. (B and C) DNA encoding HA-tagged NL4–3 Vpr or 107 Vpr and GFP was transfected into 293T cells. (B) Cells were lysed 36 h later, and expression of Vpr was determined by Western blot with anti-HA mAb. (C) Cells were harvested 36 h later, and labeled with propidium iodide. DNA content was measured in GFP+ cells by flow cytometry. (D) Cloning strategy for truncation of vpr within both NL4–3 and 107 proviral constructs. Note that the sequences of vif, pol, and tat are unaffected by these mutations within vpr. The shaded box identifies the deleted region within vpr. (E) DNA encoding each of the HIV-1 recombinants was transfected into 293T cells and supernatants were collected 48 h later. Virus production was normalized for p24 concentration, and equal amounts of virus were concentrated for each strain by ultra-centrifugation. The viral pellet was then subjected to Western blot analysis. Shown are representative experiments.
For the present studies, we wished to eliminate Vpr expression in NL4–3 and 107 viral clones for comparisons to their respective parental viruses. We introduced a deletion and two premature stop codons into the nucleotide sequence of vpr in both the NL4–3WT and 107WT proviral contexts, thereby generating NL4–3ΔVpr and 107ΔVpr, respectively (Fig. 1 D). Because NL4–3 and 107 have identical sequences upstream of the EcoR1 site used to delete Vpr, this cloning strategy preserved the isogenicity between NL4–3 and 107 including vif and pol. Neither of these Vpr-deficient viruses produced detectable quantities of the truncated Vpr product (data not shown). Consistent with this observation, immunoblot analysis of wild-type and mutant virions established the presence of Vpr in NL4–3WT and 107WT virions but not in NL4–3ΔVpr and 107ΔVpr virions, confirming that Vpr was not expressed in the deletion mutants (Fig. 1 E). We detected lower levels of Vpr within 107WT virions compared with NL4–3WT virions, consistent with the inferred stability differences between these different versions of the proteins (Fig. 1 B). Comparing wild-type and Vpr-deficient versions of NL4–3 or 107 we subsequently examined the cellular tropism and replication profiles of these viruses within human lymphoid tissues ex vivo.
Vpr Is Not Required for Productive Infection of Resting T Cells
On the basis of morphologic characteristics we have previously identified two distinct lymphocyte populations within lymphoid histocultures (47). We termed one population of cells with small size, low granularity, and low proliferative activity “lymphocytes.” The second population, characterized by cells of greater size, increased granularity, and significant proliferative activity, was termed “blasts.” Although lymphocytes exhibit limited proliferation, our earlier studies have directly demonstrated productive infection of nondividing cells within this subset (47). These results confirm that the virally infected T lymphocytes do not represent a subpopulation of dividing T cells and establish that HIV-1 can productively infect nondividing lymphocytes de novo. To determine if Vpr contributed to the infection of these nondividing cells, we inoculated histoculture tissue with equivalent TCID50 of wild-type or Vpr-deficient strains of NL4–3 or107. After a 7-d incubation, cells were harvested from the tissue and immunostained for intracellular p24, a marker of productive HIV infection. Examination of CD4+ T cells by FACS® revealed significant infection of both lymphocytes and blasts by the wild-type and Vpr-deficient variants of NL4–3 and 107 (Fig. 2). As observed previously, NL4–3 infection resulted in higher levels of productive infection than did 107 due to a broad distribution of CXCR4 on target cells and limited expression of CCR5. Most importantly, within a given X4 (Fig. 2 A) or R5 (Fig. 2 B) proviral backbone, wild-type and Vpr-deficient strains equivalently infected the lymphocyte pool, suggesting that Vpr is not an essential viral factor for the productive infection of nondividing or dividing T cells in such tissue contexts. This finding was unexpected given the putative role of Vpr in the nuclear targeting of the PIC in nondividing cells such as tissue macrophages. Wild-type and Vpr-deficient viruses likewise equivalently infected the blast pool (Fig. 2, A and B). We note that Vpr has been shown to have a greater influence on HIV-1 infectivity at low titers, and only a small impact at high multiplicity of infection (m.o.i.) (22). Importantly, the experiments reported here were performed at a m.o.i. below 2 × 10−4, a very low inoculum, which likely accounts for the 5-d delay in the appearance of detectable intracellular and soluble p24 after inoculation. Experiments performed with one-half the virus inoculum used above also revealed no differences in the infection of CD4+ T lymphocytes and blasts by wild-type or Vpr-deficient viruses under these conditions (data not shown).
Figure 2.

HIV-1 infection of CD4+ T lymphocytes and blasts. (A) Productively infected cell subsets in tonsil tissue following a 7-d infection with NL4–3WT (white bars) or NL4–3ΔVpr (black bars) as measured by intracellular p24 expression detected by FACS®. (B) Productively infected cell subsets in tonsil tissue after a 7-d infection with 107WT (white bars) or 107ΔVpr (black bars). Presented are mean values with SEM (n = 3) from a representative experiment.
We previously demonstrated by pulse-chase analysis that naive and memory T cells in these tissues exhibited significantly different proliferative activity (47). We therefore hypothesized that while Vpr may not be necessary for infection of T cells in general, it may nonetheless be a critical factor for infection of the nonproliferating, naive T cell subset, a population typically accounting for less than 20% of the total CD4+ T lymphocyte population. To measure the cumulative fraction of cycling T cells during a typical HIV infection, we cultured tonsil tissue in medium containing BrdU for 7 d. FACS® analysis of samples harvested after labeling revealed that while greater than 16% of memory CD4+ T lymphocytes incorporated BrdU during this period, only 2.3% of naive CD4+ T lymphocytes did so (Fig. 3 A). This pattern of enhanced labeling of memory cells compared with naive cells was preserved in the blast compartment as well, although the blast subsets exhibited greater proliferative activity overall than did their lymphocyte counterparts.
Figure 3.

BrdU incorporation and HIV-1 infection of naive and memory CD4+ T cell subsets. (A) Tissue samples were cultured continuously in medium containing BrdU for 7 d. Cells were isolated, immunostained for CD4, CD62L, CD45RA, and BrdU, and analyzed by FACS®. Shown are mean values with SEM (n = 3) from a representative experiment. (B and C) Tonsil histocultures were inoculated with equivalent titers of HIV-1, incubated for 7 d, and harvested. Cells were dispersed, immunostained for p24, CD62L, CD45RA, and CD4, and analyzed by FACS®. T cells were also stratified based on forward and side scatter properties defining lymphocyte and blast morphologies. (B) Productively infected subsets in tissue inoculated with NL4–3WT or NL4–3ΔVpr as measured by intracellular p24 expression. (C) Productively infected subsets in tissue inoculated with 107WT or 107ΔVpr. Shown are mean values with SEM (n = 3) from a representative experiment.
We next compared the capacity of wild-type and Vpr-deficient viruses to productively infect each of these subsets. Tonsil histocultures were inoculated with equivalent titers of X4 or R5 wild-type or Vpr-deficient viruses, and after a 7-d incubation, cells were immunostained for maturation markers and intracellular p24. Stratification of T cells into memory and naive subsets revealed no statistically significant differences between cultures infected with wild-type or Vpr-deficient NL4–3 (Fig. 3 B) or 107 (Fig. 3 C) variants. Most importantly, this analysis demonstrated equivalent infection of the nondividing, naive T lymphocyte population by the wild-type and Vpr-deficient viruses. This finding substantiates our earlier conclusion that Vpr does not contribute to lymphocyte infection in general, and specifically indicates that Vpr function is dispensable for productive infection of the nonproliferating subset of T cells.
Vpr Does Not Trigger Cell-Cycle Arrest in Infected Lymphocytes Ex Vivo
We next sought to determine if Vpr induces G2 cell-cycle arrest in these tissues. Tonsil histocultures were inoculated with equivalent titers of NL4–3WT or NL4–3ΔVpr virus, and after a 6-d incubation, the tonsil tissue was harvested and stained for intracellular p24 and DNA content. Samples were then analyzed by flow cytometry; we gated on CD4+ T cells and examined them for intracellular p24 expression. Analysis of the DNA content of p24+ T cells in the samples inoculated with wild-type or Vpr-deficient virus revealed a similar percentage of cells in the G2 phase of the cell-cycle irrespective of the presence or absence of Vpr (Fig. 4, A and C). Interestingly, in samples inoculated with either wild-type or Vpr-deficient virus the percentage of cells in G2 was modestly greater in the fraction of productively-infected T cells (p24+) (Fig. 4, A and C) compared with uninfected T cells (p24−) in the same tissue (Fig. 4, B and D). Similar results were observed over several different infections in lymphoid tissue, suggesting that there is no detectable Vpr-dependent G2 cell-cycle arrest and that other viral factors may play a key role in this phenomenon.
Figure 4.

Cell-cycle analysis of p24+ or p24− CD4+ T cells. (A) Tonsil specimens were inoculated with equivalent titers of HIV-1 NL4–3WT (A and B) or NL4–3ΔVpr (C and D). After a 7-d incubation, samples were dispersed, stained for intracellular p24, CD4, and To-Pro-3 iodide, and analyzed by FACS®. DNA content is presented for cells stratified into p24+ (A and C) and p24− (B and D) fractions. Shown is a representative experiment.
Vpr Does Not Contribute to HIV-1–mediated Depletion of T Cells in Lymphoid Histoculture
To determine directly if equivalent infectivity correlated with equivalent cytopathicity, we measured virus-induced depletion of CD4+ T cells after infection with HIV-1. We began by subsetting T cells based on expression of the HIV-1 coreceptor CCR5. As expected from previous studies, NL4–3WT extensively depleted both CCR5+ and CCR5− subpopulations of CD4+ T lymphocytes (Fig. 5 A). Likewise, NL4–3ΔVpr also markedly depleted both subpopulations in a manner indistinguishable from its parental virus. In contrast to the extensive depletion by NL4–3, the cytopathic effects of 107WT were limited to the CCR5+ subset of CD4+ T lymphocytes, as has been observed previously (41, 43). Again, the Vpr-deficient 107 strain exhibited equivalent cytopathic activity compared with wild-type 107. These findings demonstrate that Vpr is not essential for the cytopathic effects of HIV-1 on CD4+ T cells within lymphoid tissues.
Figure 5.
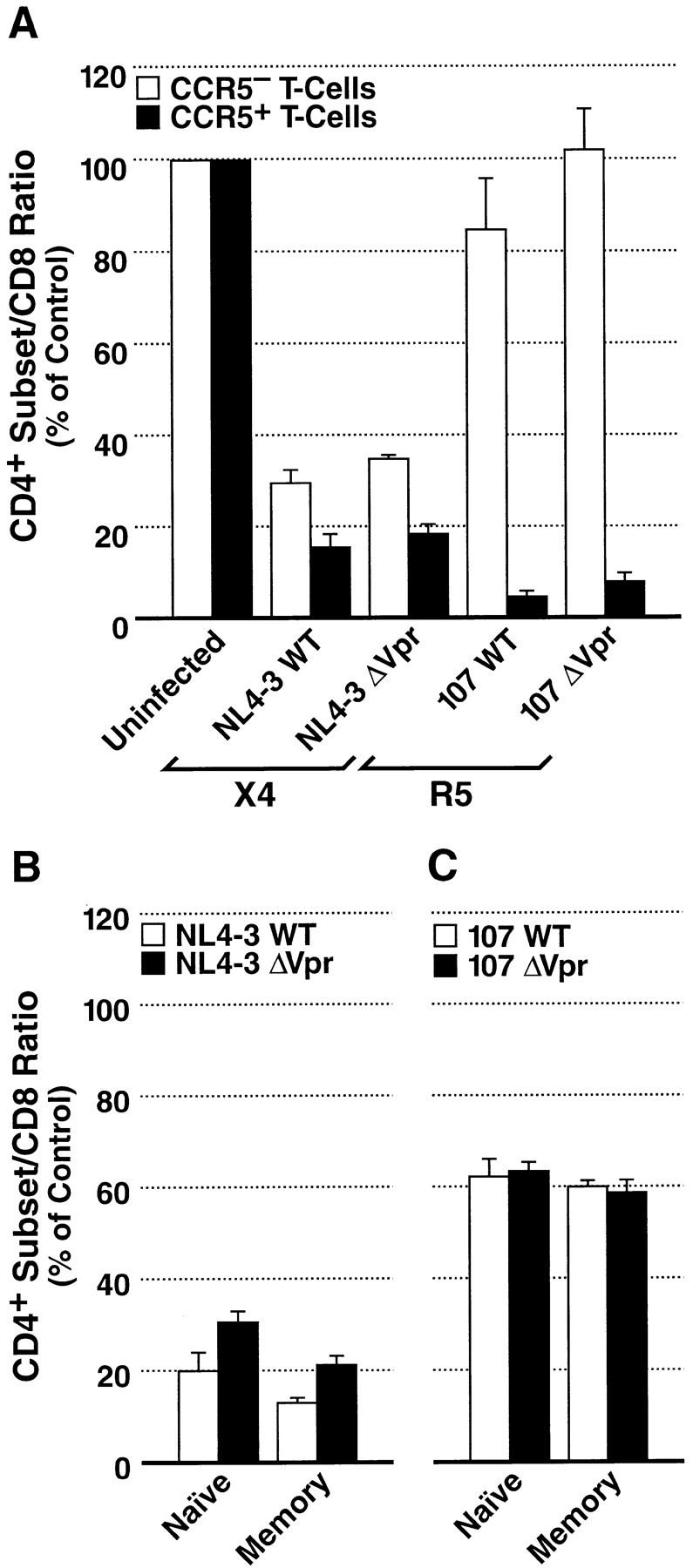
Depletion of CD4+ T cells by HIV-1 in lymphoid histoculture. (A) After a 13-d infection, cells were isolated from histocultures and immunostained for surface expression of CD4, CD3, CD8, and CCR5. The mean CD4+ subset/CD8+ ratio relative to uninfected control tissue for CCR5− T cells (white bars) and CCR5+ T cells (black bars) was then calculated. Shown are mean values with SEM (n = 3) from a representative experiment. (B and C) Depletion of naive and memory CD4+ T cell subsets by HIV-1. After a 12-d infection with NL4–3WT and NL4–3ΔVpr (B) or 107WT and 107ΔVpr (C) cells were dispersed from histocultures and immunostained for CD4, CD62L, CD45RA, and CD8. The mean CD4+ subset/CD8+ ratio relative to uninfected control tissue for memory T cells (white bars) and naive T cells (black bars) was then calculated. Shown are mean values with SEM (n = 3) from a representative experiment.
To determine further whether the state of T cell maturation influences the susceptibility of these cells to depletion by Vpr-deficient viruses, we measured the loss of naive and memory CD4+ lymphocytes after infections with HIV-1. Both NL4–3WT and NL4–3ΔVpr induced severe depletion of naive and memory CD4+ lymphocytes (Fig. 5 B), although the cytopathic effects of NL4–3ΔVpr were slightly reduced compared with that of NL4–3WT, consistent with our earlier finding of slightly reduced infectivity of NL4–3ΔVpr. We also measured depletion in tissue that had been inoculated with 107WT or 107ΔVpr. As is characteristic of all R5 viruses, overall depletion was not severe. Nevertheless, the degree of depletion of the memory and naive populations was equivalent for both 107 variants (Fig. 5 C). Thus, Vpr does not contribute to the cytopathic activity of either HIV-1 R5 or X4 variants in T lymphocytes.
Vpr Contributes Substantively to the Productive Infection of Tissue Macrophages as well as to the Viral Burden in Lymphoid Tissue
In addition to infecting activated T cells, R5 viruses also productively infect cells of the monocyte/macrophage lineage. We therefore examined the putative role of Vpr in mediating the infection of nondividing lymphoid tissue macrophages. We inoculated tonsil histocultures with equivalent titers of 107WT or 107ΔVpr. After incubation, the tissue was dispersed and cells were immunostained for specific macrophage (CD14 and CD68) and T cell (CD3) markers as well as intracellular p24. Because macrophages associate tightly with B- and T cells in lymphoid tissues, macrophages free of complexed T cells (CD14+ or CD68+ and CD3−) were identified by flow cytometry and analyzed for productive infection. In contrast to the behavior observed earlier in T cells, 107ΔVpr exhibited a greater than fourfold reduction in infectivity in lymphoid tissue macrophages compared with 107WT (Fig. 6 A). A similar examination of the same tissue inoculated with NL4–3WT and NL4–3ΔVpr revealed little detectable infection within macrophages by either virus (data not shown). These findings imply that Vpr contributes significantly to the productive infection of tissue macrophages by R5 strains.
Figure 6.

Flow cytometric analysis of HIV-1 and SIV infection of macrophages in lymphoid tissue. After a 9-d incubation with 107WT or 107ΔVpr (A) or PBj6.6WT, PBj6.6ΔVpr or PBj6.6Δvpx (B), cells were dispersed and immunostained for surface expression of CD3 and CD14 as well as intracellular CD68 and p24 or p27. Shown are mean values with SEM (n = 3) from a representative experiment.
Interestingly, within the genomes of HIV-2 and SIV the arrest and nuclear import functions of HIV-1 Vpr are divided between two related gene products, Vpr and viral protein X (Vpx) (49). Cell-cycle arrest is mediated by Vpr in both HIV-2 and SIV, whereas nuclear localization is mediated by Vpx in both species. The significance of this functional division is not well understood; however, the evolutionary conservation of both functions implies their importance in the viral life cycle. To determine if the macrophage-specific defect we observed in 107ΔVpr was specifically attributable to the nuclear entry activity of Vpr, we inoculated tonsil samples with SIV PBj6.6WT, PBj6.6ΔVpx or PBj6.6ΔVpr. Cells were later dispersed and stained for macrophage-lineage markers and intracellular p27. FACS® analysis revealed a greater than sixfold reduction in the number of productively infected macrophages in tissue that had been inoculated with PBj6.6ΔVpx, compared with tissue infected with wild-type virus (Fig. 6 B), consistent with a role for Vpx in nuclear targeting. In contrast, tissue infected with PBj6.6ΔVpr showed no significant decrease in the frequency of infected macrophages (Fig. 6 B). Indeed, across several experiments the frequency was either unchanged or modestly increased compared with PBj6.6WT. Therefore, these results implicate the nuclear targeting function of HIV-1 Vpr in the infection of macrophages by R5 HIV-1.
Finally, we measured the extent and kinetics of viral replication of wild-type and Vpr-deficient viruses in lymphoid histocultures. Tissue was inoculated with equal titers of NL4–3WT or NL4–3ΔVpr (Fig. 7 A) as well as 107WT or 107ΔVpr (Fig. 7 B). Supernatants were collected serially throughout 18-d incubations, and the concentrations of p24 in the media were assessed by ELISA. As expected from their strict T cell tropism and the earlier findings of similar infectivity, NL4–3WT and NL4–3ΔVpr exhibited similar replication profiles (Fig. 7 A). Both the overall growth kinetics as well as the total virus production were similar. In contrast, a substantially different pattern of replication was found for 107WT compared with 107ΔVpr. 107ΔVpr exhibited an ∼50% reduction in viral replication compared with 107WT (Fig. 7 B). Based on the selective effects of Vpr we observed earlier in macrophages rather than T cells, we infer that the differential viral output in tissues infected with 107ΔVpr compared with 107WT is the result of decreased macrophage infection by Vpr-deficient viruses. Thus, these results reveal the significant contribution of tissue macrophages to the viral burden within human lymphoid tissues. Consistent with this conclusion, quantitation of soluble p27 in the culture media of the SIV-infected tissues revealed an eightfold reduction in viral output from tissues inoculated with the Vpx-deficient strain of PBj6.6 compared with the wild-type and Vpr-deficient counterparts (data not shown). Thus, the Vpx-deficient SIV strain behaved similarly to the Vpr-deficient R5 HIV-1 strain. Overall, these results suggest that the major role of Vpr in HIV-1 pathogenesis is to facilitate infection of macrophages, a population that may also contribute substantially to the viral burden in infected individuals.
Figure 7.

Kinetic analysis of HIV-1 replication in lymphoid histoculture. Tissue was inoculated with NL4–3WT or NL4–3ΔVpr (A) or 107WT or 107ΔVpr (B). Culture medium was sampled at the indicated time points and HIV-1 p24 concentration was measured by ELISA. Shown are mean values with SEM (n = 3) from a representative experiment.
Discussion
Peripheral blood mononuclear cells in culture typically require exogenous stimulation in order to support productive infection by either wild-type or Vpr-deficient viruses. This dependency on activation appears to relate to the nuclear membrane breakdown associated with activation-induced cellular division, which evidently abrogates the need for Vpr to facilitate nuclear import of the PIC. In contrast, T cells within the lymphoid tissue microenvironment are permissive for productive HIV-1 infection independent of exogenous stimulation or cytokines. Moreover, our previous studies have revealed no absolute restriction to HIV-1 infection, replication, or depletion of T cells within these cultures on the basis of target cell maturation or proliferation (47). In particular, these studies demonstrated de novo infection and depletion of resting, naive lymphocytes, which may explain the observation of productively infected resting T cells in vivo (50–52). Because these cells divide very infrequently (Fig. 3), infection must proceed independently of nuclear membrane dissolution. Multiple determinants of nuclear import contribute to this unique property of HIV. The apparent redundancy of mechanisms highlights the importance of nuclear entry in the HIV-1 life cycle. However, it is possible that individual import signals may play a more or less dominant role in particular cellular contexts. Thus, the multiple nuclear import signals may not be redundant after all. We therefore examined the importance of one such nuclear import signal, Vpr, in mediating nuclear import in diverse primary cell types including nondividing T cells and macrophages.
Surprisingly, these studies revealed equivalent infection of resting, naive T cells within histocultures inoculated with matched wild-type or Vpr-deficient HIV-1 strains despite differential infection of macrophages. This finding suggests that other host or viral factors contribute to the nuclear import of the PIC in this subset of nondividing T lymphocytes. At least two additional viral gene products may be responsible. We speculate that the weak nuclear localization signals (NLS) within MA (19–21) may function relatively efficiently within T cells, which would reduce the dependence on Vpr-mediated translocation mechanisms. In addition, multiple NLS have also been identified within viral IN (22–24) that may also be relatively effective in T cells. In particular, a novel NLS within IN has recently been shown to contribute significantly to the infection of dividing as well as growth-arrested cells (24). Finally, the nucleophilic properties of the central DNA flap may also play a more significant role in the nuclear import of the PIC within infected T cells (25). Thus, the individual nuclear import signals within the PIC may each be optimized for a specific primary cell type.
The significance of other Vpr functions in viral expansion is less clear. For example, our analysis of infected lymphoid cultures failed to detect specific G2 cell-cycle arrest in samples inoculated with wild-type HIV-1 compared with Vpr-deficient virus, suggesting that Vpr-dependent arrest is not manifest in this tissue context. These observations are also supported by equivalent virus replication of X4 viruses irrespective of the presence of Vpr. Alternatively, it is possible that cells arrested in G2 by Vpr may be rapidly and selectively eliminated within lymphoid histocultures thereby preventing their detection. However, these studies did reveal a modest accumulation of p24+ cells in the G2 phase of the cell cycle in HIV-1–infected cultures independent of Vpr status. This finding implies that selective elimination of Vpr-arrested cells is not a likely feature of this system and suggests that viral factors in addition to Vpr may contribute to arrest in this tissue context. Indeed, a previous study has demonstrated that envelope glycoprotein may induce arrest in cultured cells (53). The individual contributions of these two pathways remain to be determined.
We also detected equivalent depletion of T cells by wild-type and Vpr-deficient HIV-1 variants. Although earlier studies have reported a pro-apoptotic role for Vpr, our findings suggest that Vpr is not required for such effects in lymphoid tissue. Indeed, mutagenesis studies have revealed that the apoptotic function ascribed to Vpr seems to segregate with the cell-cycle arrest activity (36), and we have found that neither activity is evident in these histocultures. We conclude that neither cell-cycle arrest nor apoptosis induced by Vpr contribute significantly to virus-induced T cell death in this context.
The identical replication kinetics of wild-type and Vpr-deficient X4 viruses within lymphoid tissue is consistent with our earlier conclusions regarding X4 HIV-1 infection and depletion. However, we note that a slight trend toward reduced replication by Vpr-deficient strains was evident from these studies, which may correlate with the observed trend toward slightly reduced infectivity and depletion by Vpr-deficient X4 virus. Although few of these differences reached statistical significance, these patterns were observed in multiple tissue specimens, which may indicate a small contribution of Vpr to the fitness and replication of X4 HIV-1 strains in vivo.
In contrast to the behavior of cultured peripheral blood T cells (2, 4, 54), monocyte-derived macrophages matured in vitro depend substantially on intact Vpr for productive infection (2, 4, 6, 55). However, this effect is evident only at very low multiplicity of infection and only after weeks of in vitro culture. Indeed, some investigators have observed no effect of Vpr on infection of cultured monocyte-derived macrophages (56). In addition, an important issue that remains unanswered by these studies is the importance of Vpr for infection of macrophages residing within lymphoid tissues. Lymphoid organs are the principal sites of HIV-1 replication in vivo and account for >90% of the viral load during HIV-1 infection (57), particularly during the acute and asymptomatic phases of disease when viruses selectively using CCR5 predominate. CCR5 is expressed on most tissue macrophages, dendritic cells, and a subset of activated T cells. The selection of CCR5-dependent viruses during acute infection suggests that macrophage infection may be a key factor in the establishment of HIV-1 infection as well as the subsequent development of functional immunodeficiency. We therefore examined the role of Vpr in the establishment of HIV-1 infection in tissue-resident macrophages rather than artificially matured blood-derived cells. Our results revealed a substantial defect in the productive infection of macrophages by Vpr-deficient viruses, which stands in sharp contrast to its lack of effect in T cells. This macrophage-restricted phenotype was also apparent in tissues inoculated with Vpx-deficient, but not Vpr-deficient, strains of SIV. These observations confirm the specific importance of nuclear import in the life cycle of HIV-1 and SIV within macrophages. Furthermore, these findings suggest that the nuclear import signals within the viral PIC may be cell type specific and may not be completely redundant in all cellular environments. Indeed, each NLS within the PIC may be conserved so as to preserve the ability of HIV-1 to productively infect distinct cell types. We conclude that Vpr contributes substantially to the replication of R5 HIV-1 within macrophages but does not play a significant role in T cells.
Recent in vitro work has demonstrated that HIV-1 Nef expression within macrophages induces production of the soluble CC-chemokines macrophage inflammatory protein-1α and -1β and other unidentified soluble factors (58). These chemokines may recruit and partially activate neighboring resting lymphocytes and thereby render them permissive for infection by HIV-1, which would assign infection of macrophages an important role in establishing infection in resting lymphocytes. Selective impairment of macrophage infection, as is seen with the Vpr-deficient viruses tested here, would be expected to interrupt this trans-activating function. However, this effect was not evident in the histoculture system. Specifically, the frequency of infected resting, naive T cells by 107ΔVpr or NL4–3ΔVpr was identical to that of their wild-type counterparts. Therefore, experimental conditions or viral factors other than, or in combination with, Nef may play key roles in the ability of HIV-1 to infect resting, naive lymphocytes in a lymphoid tissue microenvironment.
Finally, macrophages may contribute substantially to plasma viral load during late infection, particularly when CD4+ T cells have been extensively depleted (57, 59, 60). However, the contribution of macrophages to viral load during other disease stages has not been well characterized but is proposed to be minimal based on the viral and cellular kinetics observed during acute antiviral treatment (61–64). Based on the assumption that HAART completely suppresses virus replication, such studies suggest that plasma viremia declines with a half-life of ∼1 d, implying that the cells releasing virus into the plasma must have a similar half-life. This conclusion implicates T cells as the principle source of plasma viremia rather than infected tissue macrophages, which are believed to have a half-life of nearly 2 wk (61–64). However, more recent studies have generated evidence of other persistent viral reservoirs that may contribute to plasma viremia (65–68). The studies reported herein address this controversy directly by examining the replication kinetics of viruses that fail to infect macrophages productively while their capacity to infect and replicate within lymphocytes is preserved. Thus, any difference in viral output may be interpreted as the contribution of macrophage infection. The results reveal a significant reduction in the concentration of soluble p24 in cultures infected with 107ΔVpr as compared with wild-type virus. This finding is correlated with a greater than fourfold reduction in the frequency of infected macrophages and preserved numbers of infected T cells in these same samples. We therefore conclude that the differential output between cultures infected with wild-type and Vpr-deficient R5 viruses is attributable to the output of the productively infected macrophage pool.
This conclusion implies that within lymphoid organs infected with R5 viruses the contribution to cell-free virus by infected macrophages is equal to that of all infected CCR5+CD4+ T cells within the particular tissue—a population that significantly outnumbers tissue macrophages by at least 10-fold. Therefore, on a per-cell basis, we infer that productively infected macrophages cumulatively release at least 10-fold more p24 than do productively infected T cells within the same tissue over the 2-wk time interval of these experiments. Thus, it appears that HIV replication within macrophages contributes significantly to tissue viral burden and may account for a substantial fraction of plasma viremia to the extent that plasma virus derives from tissue sources. However, it remains a theoretical possibility that T cells infected by macrophage-derived virus or in the proximity of infected macrophages may produce more p24 than cells infected by T cell–derived or cell-free virus. Nevertheless, our conclusions agree well with recent work demonstrating significant and sustained plasma viremia in SHIV-infected macaques after virus-induced depletion of CD4+ T cells, in which the authors concluded that the viremia was supported by viral replication in productively infected macrophages (60). Our results extend this observation by demonstrating the significant contribution of human lymphoid macrophages to HIV-1 viral burden even when T cells are present.
Given the close physical association between macrophages and T cells within lymphoid organs, it is possible that the majority of virus produced by macrophages may be transmitted to neighboring lymphoid cells by means of cell-to-cell spread. Virus produced by macrophages and transmitted in this manner may not contribute substantially to the cell-free virus detected in the plasma. As a consequence, the dynamics of viral decline within lymphoid tissues may differ from that of the plasma. Although few in number, productively infected lymph node macrophages have in the past been thought to play a large role in viral burden (57, 59). Our findings underscore the potential importance of this relatively small cellular reservoir of virus. Spread of R5 virus directly from macrophages to T cells might contribute substantially to HIV-induced immune pathogenesis during the early and asymptomatic stages of HIV-induced disease, as well as later stages of disease when T cells have been depleted. Tissue macrophages may therefore represent a clinically important cellular reservoir of virus, as macrophage-derived virus would be expected to contribute substantially to the progressive attrition of T cells that characterizes all stages of HIV disease.
Acknowledgments
The authors thank Bruce Chesebro, Malcolm Martin, Ruth Connor, Mario Stevenson, and Beatrice Hahn for kindly providing reagents and Dee J. Holthe, Cecilia Stewart, Claudette Delphis, Ursula Perotti, Mark Weinstein, Nancy Abbey, and the surgical staffs at Kaiser Hospitals (San Rafael, San Francisco) and San Francisco General Hospital for generous assistance in obtaining post-tonsillectomy samples. The authors acknowledge Martin Bigos, Valerie Stepps, and Scott Furlan for technical advice and the assistance of Heather Gravois, John Carroll, and Jack Hull in the preparation of this manuscript. We also thank Mario Stevenson for critically reading this manuscript and providing helpful suggestions.
D.A. Eckstein was supported by the Biomedical Sciences Graduate Program (BMS) and the National Institutes of Health Medical Scientist Training Program (MSTP) at University of California San Francisco (UCSF). M.P. Sherman was supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases grant K08-AI01866. M.L. Penn was supported by BMS, the Universitywide AIDS Research Program, University of California grant D99-GI-015, and the National Institutes of Health MSTP at UCSF. This work was supported in part by National Institutes of Health grant R01-AI43695 (M.A. Goldsmith), the UCSF-California AIDS Research Center (CC99-SF-001, M.A. Goldsmith), the UCSF-GIVI Center for AIDS Research (National Institutes of Health P30-MH59037, W.C. Greene), and the J. David Gladstone Institutes (M.A. Goldsmith and W.C. Greene).
D.A. Eckstein and M.P. Sherman contributed equally to this work.
Footnotes
*
Abbreviations used in this paper: BrdU, 5-bromo-2′-deoxyuridine; CCR, CC chemokine receptor; IN, integrase; MA, matrix; NLS, nuclear localization signal; PIC, preintegration complex; SIV, simian immunodeficiency virus; Vpr, viral protein R.
References
- 1.Bukrinsky, M.I., N. Sharova, M.P. Dempsey, T.L. Stanwick, A.G. Bukrinskaya, S. Haggerty, and M. Stevenson. 1992. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA. 89:6580–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balliet, J.W., D.L. Kolson, G. Eiger, F.M. Kim, K.A. McGann, A. Srinivasan, and R. Collman. 1994. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: mutational analysis of a primary HIV-1 isolate. Virology. 200:623–631. [DOI] [PubMed] [Google Scholar]
- 3.Heinzinger, N.K., M.I. Bukinsky, S.A. Haggerty, A.M. Ragland, V. Kewalramani, M.A. Lee, H.E. Gendelman, L. Ratner, M. Stevenson, and M. Emerman. 1994. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. USA. 91:7311–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connor, R.I., B.K. Chen, S. Choe, and N.R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 206:935–944. [DOI] [PubMed] [Google Scholar]
- 5.Popov, S., M. Rexach, G. Zybarth, N. Reiling, M.A. Lee, L. Ratner, C.M. Lane, M.S. Moore, G. Blobel, and M. Bukrinsky. 1998. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 17:909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vodicka, M.A., D.M. Koepp, P.A. Silver, and M. Emerman. 1998. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 12:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartz, S.R., M.E. Rogel, and M. Emerman. 1996. Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J. Virol. 70:2324–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goh, W.C., M.E. Rogel, C.M. Kinsey, S.F. Michael, P.N. Fultz, M.A. Nowak, B.H. Hahn, and M. Emerman. 1998. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4:65–71. [DOI] [PubMed] [Google Scholar]
- 9.He, J., S. Choe, R. Walker, P. Di Marzio, D.O. Morgan, and N.R. Landau. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jowett, J.B., V. Planelles, B. Poon, N.P. Shah, M.L. Chen, and I.S. Chen. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69:6304–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Re, F., D. Braaten, E.K. Franke, and J. Luban. 1995. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69:6859–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, E.A., E.F. Terwilliger, Y. Jalinoos, J. Proulx, J.G. Sodroski, and W.A. Haseltine. 1990. Identification of HIV-1 vpr product and function. J. Acquir. Immune Defic. Syndr. 3:11–18. [PubMed] [Google Scholar]
- 13.Cohen, E.A., G. Dehni, J.G. Sodroski, and W.A. Haseltine. 1990. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J. Virol. 64:3097–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paxton, W., R.I. Connor, and N.R. Landau. 1993. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J. Virol. 67:7229–7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan, X., Z. Matsuda, M. Matsuda, M. Essex, and T.H. Lee. 1990. Human immunodeficiency virus vpr gene encodes a virion-associated protein. AIDS Res. Hum. Retrovir. 6:1265–1271. [DOI] [PubMed] [Google Scholar]
- 16.Yedavalli, V.R., C. Chappey, and N. Ahmad. 1998. Maintenance of an intact human immunodeficiency virus type 1 vpr gene following mother-to-infant transmission. J. Virol. 72:6937–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis, P.F., and M. Emerman. 1994. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 68:510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphries, E.H., and H.M. Temin. 1974. Requirement for cell division for initiation of transcription of Rous sarcoma virus RNA. J. Virol. 14:531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bukrinsky, M.I., S. Haggerty, M.P. Dempsey, N. Sharova, A. Adzhubel, L. Spitz, P. Lewis, D. Goldfarb, M. Emerman, and M. Stevenson. 1993. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature. 365:666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallay, P., V. Stitt, C. Mundy, M. Oettinger, and D. Trono. 1996. Role of the karyopherin pathway in human immunodeficiency virus type 1 nuclear import. J. Virol. 70:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Schwedler, U., R.S. Kornbluth, and D. Trono. 1994. The nuclear localization signal of the matrix protein of human immunodeficiency virus type 1 allows the establishment of infection in macrophages and quiescent T lymphocytes. Proc. Natl. Acad. Sci. USA. 91:6992–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallay, P., T. Hope, D. Chin, and D. Trono. 1997. HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc. Natl. Acad. Sci. USA. 94:9825–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petit, C., O. Schwartz, and F. Mammano. 2000. The karyophilic properties of human immunodeficiency virus type 1 integrase are not required for nuclear import of proviral DNA. J. Virol. 74:7119–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouyac-Bertoia, M., J.D. Dvorin, R.A. Fouchier, Y. Jenkins, B.E. Meyer, L.I. Wu, M. Emerman, and M.H. Malim. 2001. HIV-1 infection requires a functional integrase NLS. Mol. Cell. 7:1025–1035. [DOI] [PubMed] [Google Scholar]
- 25.Zennou, V., C. Petit, D. Guetard, U. Nerhbass, L. Montagnier, and P. Charneau. 2000. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 101:173–185. [DOI] [PubMed] [Google Scholar]
- 26.Miller, M.D., C.M. Farnet, and F.D. Bushman. 1997. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 71:5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popov, S., M. Rexach, L. Ratner, G. Blobel, and M. Bukrinsky. 1998. Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. J. Biol. Chem. 273:13347–13352. [DOI] [PubMed] [Google Scholar]
- 28.Jenkins, Y., M. McEntee, K. Weis, and W.C. Greene. 1998. Characterization of HIV-1 vpr nuclear import: analysis of signals and pathways. J. Cell Biol. 143:875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman, M.P., C.M. de Noronha, M.I. Heusch, S. Greene, and W.C. Greene. 2001. Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J. Virol. 75:1522–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hrimech, M., X.J. Yao, F. Bachand, N. Rougeau, and E.A. Cohen. 1999. Human immunodeficiency virus type 1 (HIV-1) Vpr functions as an immediate-early protein during HIV-1 infection. J. Virol. 73:4101–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forget, J., X.J. Yao, J. Mercier, and E.A. Cohen. 1998. Human immunodeficiency virus type 1 vpr protein transactivation function: mechanism and identification of domains involved. J. Mol. Biol. 284:915–923. [DOI] [PubMed] [Google Scholar]
- 32.Yao, X.J., A.J. Mouland, R.A. Subbramanian, J. Forget, N. Rougeau, D. Bergeron, and E.A. Cohen. 1998. Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1-infected dividing Jurkat T cells. J. Virol. 72:4686–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart, S.A., B. Poon, J.B. Jowett, and I.S. Chen. 1997. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71:5579–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart, S.A., B. Poon, J.B. Jowett, Y. Xie, and I.S. Chen. 1999. Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc. Natl. Acad. Sci. USA. 96:12039–12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart, S.A., B. Poon, J.Y. Song, and I.S. Chen. 2000. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J. Virol. 74:3105–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conti, L., P. Matarrese, B. Varano, M.C. Gauzzi, A. Sato, W. Malorni, F. Belardelli, and S. Gessani. 2000. Dual role of the HIV-1 vpr protein in the modulation of the apoptotic response of T cells. J. Immunol. 165:3293–3300. [DOI] [PubMed] [Google Scholar]
- 37.Poon, B., K. Grovit-Ferbas, S.A. Stewart, and I.S.Y. Chen. 1998. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science. 281:266–269. [DOI] [PubMed] [Google Scholar]
- 38.Ayyavoo, V., A. Mahboubi, S. Mahalingam, R. Ramalingam, S. Kudchodkar, W.V. Williams, D.R. Green, and D.B. Weiner. 1997. HIV-1 Vpr suppresses immune activation and apoptosis through regulation of nuclear factor kappa B. Nat. Med. 3:1117–1123. [DOI] [PubMed] [Google Scholar]
- 39.Conti, L., G. Rainaldi, P. Matarrese, B. Varano, R. Rivabene, S. Columba, A. Sato, F. Belardelli, W. Malorni, and S. Gessani. 1998. The HIV-1 vpr protein acts as a negative regulator of apoptosis in a human lymphoblastoid T cell line: possible implications for the pathogenesis of AIDS. J. Exp. Med. 187:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glushakova, S., B. Baibakov, J. Zimmerberg, and L.B. Margolis. 1997. Experimental HIV infection of human lymphoid tissue: correlation of CD4+ T cell depletion and virus syncytium-inducing/non-syncytium-inducing phenotype in histocultures inoculated with laboratory strains and patient isolates of HIV type 1. AIDS Res. Hum. Retrovir. 13:461–471. [DOI] [PubMed] [Google Scholar]
- 41.Penn, M.L., J.C. Grivel, B. Schramm, M.A. Goldsmith, and L. Margolis. 1999. CXCR4 utilization is sufficient to trigger CD4+ T cell depletion in HIV-1-infected human lymphoid tissue. Proc. Natl. Acad. Sci. USA. 96:663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schramm, B., M.L. Penn, E.H. Palacios, R.M. Grant, F. Kirchhoff, and M.A. Goldsmith. 2000. Cytopathicity of human immunodeficiency virus type 2 (HIV-2) in human lymphoid tissue is coreceptor dependent and comparable to that of HIV-1. J. Virol. 74:9594–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grivel, J.C., M.L. Penn, D.A. Eckstein, B. Schramm, R.F. Speck, N.W. Abbey, B. Herndier, L. Margolis, and M.A. Goldsmith. 2000. Human immunodeficiency virus type 1 coreceptor preferences determine target T-cell depletion and cellular tropism in human lymphoid tissue. J. Virol. 74:5347–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schramm, B., M.L. Penn, R.F. Speck, S.Y. Chan, E. De Clercq, D. Schols, R.I. Connor, and M.A. Goldsmith. 2000. Viral entry through CXCR4 is a pathogenic factor and therapeutic target in human immunodeficiency virus type 1 disease. J. Virol. 74:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toohey, K., K. Wehrly, J. Nishio, S. Perryman, and B. Chesebro. 1995. Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology. 213:70–79. [DOI] [PubMed] [Google Scholar]
- 46.Gibbs, J.S., D.A. Regier, and R.C. Desrosiers. 1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retrovir. 10:343–350. [DOI] [PubMed] [Google Scholar]
- 47.Eckstein, D.A., M.L. Penn, Y.D. Korin, D.D. Scripture-Adams, J.A. Zack, J.F. Kreisberg, M. Roederer, M.P. Sherman, P.S. Chin, and M.A. Goldsmith. 2001. HIV-1 actively replicates in naive CD4+ T-cells residing within human lymphoid tissue. Immunity. 15:671-682. [DOI] [PubMed] [Google Scholar]
- 48.Yao, X.J., R.A. Subbramanian, N. Rougeau, F. Boisvert, D. Bergeron, and E.A. Cohen. 1995. Mutagenic analysis of human immunodeficiency virus type 1 Vpr: role of a predicted N-terminal alpha-helical structure in Vpr nuclear localization and virion incorporation. J. Virol. 69:7032–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fletcher, T.M., III, B. Brichacek, N. Sharova, M.A. Newman, G. Stivahtis, P.M. Sharp, M. Emerman, B.H. Hahn, and M. Stevenson. 1996. Nuclear import and cell cycle arrest functions of the HIV-1 Vpr protein are encoded by two separate genes in HIV-2/SIV(SM). EMBO J. 15:6155–6165. [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang, Z., T. Schuler, M. Zupancic, S. Wietgrefe, K.A. Staskus, K.A. Reimann, T.A. Reinhart, M. Rogan, W. Cavert, C.J. Miller, et al. 1999. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science. 286:1353–1357. [DOI] [PubMed] [Google Scholar]
- 51.Ostrowski, M.A., T.W. Chun, S.J. Justement, I. Motola, M.A. Spinelli, J. Adelsberger, L.A. Ehler, S.B. Mizell, C.W. Hallahan, and A.S. Fauci. 1999. Both memory and CD45RA+/CD62L+ naive CD4+ T cells are infected in human immunodeficiency virus type 1-infected individuals. J. Virol. 73:6430–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaak, H., A.B. van't Wout, M. Brouwer, B. Hooibrink, E. Hovenkamp, and H. Schuitemaker. 2000. In vivo HIV-1 infection of CD45RA(+)CD4(+) T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4(+) T cell decline. Proc. Natl. Acad. Sci. USA. 97:1269-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kolesnitchenko, V., L.M. Wahl, H. Tian, I. Sunila, Y. Tani, D.P. Hartmann, J. Cossman, M. Raffeld, J. Orenstein, L.E. Samelson, et al. 1995. Human immunodeficiency virus 1 envelope-initiated G2-phase programmed cell death. Proc. Natl. Acad. Sci. USA. 92:11889–11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dedera, D., W. Hu, N. Vander Heyden, and L. Ratner. 1989. Viral protein R of human immunodeficiency virus types 1 and 2 is dispensable for replication and cytopathogenicity in lymphoid cells. J. Virol. 63:3205–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hattori, N., F. Michaels, K. Fargnoli, L. Marcon, R.C. Gallo, and G. Franchini. 1990. The human immunodeficiency virus type 2 vpr gene is essential for productive infection of human macrophages. Proc. Natl. Acad. Sci. USA. 87:8080–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kootstra, N.A., and H. Schuitemaker. 1999. Phenotype of HIV-1 lacking a functional nuclear localization signal in matrix protein of gag and Vpr is comparable to wild-type HIV-1 in primary macrophages. Virology. 253:170–180. [DOI] [PubMed] [Google Scholar]
- 57.Pantaleo, G., C. Graziosi, L. Butini, P.A. Pizzo, S.M. Schnittman, D.P. Kotler, and A.S. Fauci. 1991. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. USA. 88:9838–9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swingler, S., A. Mann, J. Jacqué, B. Brichacek, V.G. Sasseville, K. Williams, A.A. Lackner, E.N. Janoff, R. Wang, D. Fisher, and M. Stevenson. 1999. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat. Med. 5:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hockett, R.D., J.M. Kilby, C.A. Derdeyn, M.S. Saag, M. Sillers, K. Squires, S. Chiz, M.A. Nowak, G.M. Shaw, and R.P. Bucy. 1999. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J. Exp. Med. 189:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Igarashi, T., C.R. Brown, Y. Endo, A. Buckler-White, R. Plishka, N. Bischogberger, V. Hirsch, and M.A. Martin. 2001. Macrophages are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4+ T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of humans. Proc. Natl. Acad. Sci. USA. 98:658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ho, D.D., A.U. Neumann, A.S. Perelson, W. Chen, J.M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 373:123–126. [DOI] [PubMed] [Google Scholar]
- 62.Wei, X., S.K. Ghosh, M.E. Taylor, V.A. Johnson, E.A. Emini, P. Deutsch, J.D. Lifson, S. Bonhoeffer, M.A. Nowak, B.H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 373:117–122. [DOI] [PubMed] [Google Scholar]
- 63.Perelson, A.S., A.U. Neumann, M. Markowitz, J.M. Leonard, and D.D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 271:1582–1586. [DOI] [PubMed] [Google Scholar]
- 64.Perelson, A.S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D.D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 387:188–191. [DOI] [PubMed] [Google Scholar]
- 65.Wong, J.K., M. Hezareh, H.F. Günthard, D.V. Havlir, C.C. Ignacio, C.A. Spina, and D.D. Richman. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 278:1291–1295. [DOI] [PubMed] [Google Scholar]
- 66.Finzi, D., M. Hermankova, T. Pierson, L.M. Carruth, C. Buck, R.E. Chaisson, T.C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, et al. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 278:1295–1300. [DOI] [PubMed] [Google Scholar]
- 67.Chun, T.W., L. Stuyver, S.B. Mizell, L.A. Ehler, J.A. Mican, M. Baseler, A.L. Lloyd, M.A. Nowak, and A.S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA. 94:13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chun, T.W., R.T. Davey, Jr., M. Ostrowski, J. Shawn Justement, D. Engel, J.I. Mullins, and A.S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757–761. [DOI] [PubMed] [Google Scholar]