Vav1 Transduces T Cell Receptor Signals to the Activation of Phospholipase C-γ1 via Phosphoinositide 3-Kinase-dependent and -independent Pathways (original) (raw)
Abstract
Vav1 is a signal transducing protein required for T cell receptor (TCR) signals that drive positive and negative selection in the thymus. Furthermore, Vav1-deficient thymocytes show greatly reduced TCR-induced intracellular calcium flux. Using a novel genetic system which allows the study of signaling in highly enriched populations of CD4+CD8+ double positive thymocytes, we have studied the mechanism by which Vav1 regulates TCR-induced calcium flux. We show that in Vav1-deficient double positive thymocytes, phosphorylation, and activation of phospholipase C-γ1 (PLCγ1) is defective. Furthermore, we demonstrate that Vav1 regulates PLCγ1 phosphorylation by at least two distinct pathways. First, in the absence of Vav1 the Tec-family kinases Itk and Tec are no longer activated, most likely as a result of a defect in phosphoinositide 3-kinase (PI3K) activation. Second, Vav1-deficient thymocytes show defective assembly of a signaling complex containing PLCγ1 and the adaptor molecule Src homology 2 domain–containing leukocyte phosphoprotein 76. We show that this latter function is independent of PI3K.
Keywords: thymus, Itk, Tec, Rac1, SLP-76
Introduction
Signals from the TCR play an important role in T cell development in the thymus and in subsequent activation of T cells in response to antigen. Biochemical analysis of TCR signal transduction has shown that receptor stimulation leads to the rapid activation of Src-family tyrosine kinases which phosphorylate tyrosine residues within motifs named immunoreceptor tyrosine-based activation motifs that are found on the ζ and CD3γ, δ, and ɛ subunits of the TCR complex (1). Subsequently, the ZAP-70 tyrosine kinase binds to the phosphorylated immunoreceptor tyrosine-based activation motifs and is activated by further phosphorylation by Src-family kinases. These activated kinases then rapidly phosphorylate multiple intracellular signal transducing proteins, resulting in the generation of an intracellular calcium flux, and activation of the ERK, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase cascades. Ultimately these signaling pathways converge on the nucleus causing activation of a number of transcription factors and induction of gene expression.
Vav1 is a 95-kD protein first isolated as an oncogene in a 3T3 fibroblast transformation assay (2). Subsequently, two related proteins, Vav2 and Vav3, have been identified (3–5). Vav1 was shown to be rapidly tyrosine phosphorylated after stimulation of both the TCR and BCR (6–8). Analysis of its sequence showed that it contained a number of domains characteristic of signal transducing proteins (9, 10). It has a Dbl homology domain common to GDP/GTP exchange factors (GEFs)* for Rho-family GTPases. Vav1 itself was shown to be a specific GEF for Rac1, Rac2, and RhoG, and to be activated by tyrosine phosphorylation (11–13). In addition Vav1 contains an array of SH3-SH2-SH3 domains, suggestive of an adaptor-like function. Its SH2 domain has been proposed to bind to a phosphotyrosine on the adaptor Src homology 2 domain–containing leukocyte phosphoprotein (SLP)-76 which itself is part of a complex including phospholipase C (PLC)γ1 and the adapters linker for activation of T cell (LAT) and Gads which assembles after TCR stimulation (14–17).
The first indications of the importance of Vav1 in TCR signaling came from studies in which we and others showed that TCR-induced proliferation of T cells from Vav1-deficient mice was greatly reduced, in part as a result of decreased IL-2 production (18–20). Subsequently, it was found that overexpression of Vav1 in Jurkat T cells led to hyperactivation of TCR-induced activation of the transcription factor nuclear factor (NF)-AT (21). Further analysis showed that both positive and negative selection was compromised in Vav1 −/− mice, consistent with Vav1 transducing TCR signals required for both of these selective events (22, 23). Biochemical studies demonstrated that in Vav1 −/− CD4+ T cells, TCR-induced calcium flux, ERK activation, and induction of the NF-κB transcription factor were defective (24). In addition it has been shown that Vav1 −/− T cells show reduced TCR capping, as well as TCR clustering at the immunological synapse between a T cell and an antigen-presenting cell (25–27).
In this study we address the mechanism by which Vav1 transduces TCR signals to the activation of an intracellular calcium flux. Using a novel genetic system which allows the study of signaling in highly enriched populations of CD4+CD8+ double positive (DP) thymocytes, we show that Vav1 regulates PLCγ1 phosphorylation by at least two distinct pathways. First, in the absence of Vav1 the Tec-family kinases Itk and Tec are no longer activated, most likely as a result of a defect in phosphoinositide 3-kinase (PI3K) activation. Second, Vav1-deficient thymocytes show defective assembly of a signaling complex containing PLCγ1 and the adaptor molecule SLP-76. We show that this latter function is independent of PI3K.
Materials and Methods
Mice.
The generation of mice carrying a mutation disrupting the Vav1 gene (Vav1 tm1Tyb/tm1Tyb; Vav1 −/−) and F5, Rag-1 −/−, β2m −/− mice have been described previously (22, 28).
Stimulation of Thymocytes for Biochemical Analysis.
For all biochemical analysis, thymi were disaggregated in air-buffered (AB) IMDM. Cells were preincubated with the hamster anti–mouse CD3ɛ mAb (10 μg/ml 2C11; BD PharMingen) on ice for 30 min, washed, and then incubated in AB IMDM for 5 min at 37°C before cross-linking of the antibodies with goat anti-Armenian hamster IgG antiserum (75 μg/ml; Jackson Immunoresearch Laboratories). Where PI3K inhibitors were used, thymocytes were preincubated with 100 nM wortmannin (Sigma-Aldrich) or 10 μM Ly294002 (Sigma-Aldrich) at 37°C for 30 min before standing on ice for 5 min. Preincubation with anti-CD3ɛ on ice then proceeded as above, still in the presence of inhibitor. Subsequent cross-linking of 2C11 also occurred in the presence of inhibitor. For control samples where no inhibitor was used, an equivalent volume of DMSO carrier was added.
Immunoblotting and Immunoprecipitation.
All chemicals were obtained from Sigma-Aldrich unless otherwise indicated. Cells were typically stimulated at 2 × 108 cells per milliliter in AB IMDM for the specified times and stimulation stopped by the addition of an equal volume of 2× lysis buffer (2% n-octyl β-D-glucopyranoside, 50 mM Tris-Cl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 20 mM NaF, 10 mM disodium pyrophosphate, 2 mM sodium orthovanadate, 1:50 [vol/vol] protease inhibitor cocktail [P8340]; Sigma-Aldrich]). Cell lysates were cleared by centrifugation at 15,340 g for 15 min at 4°C and used for immunoprecipitations or if total cytoplasmic lysates were to be analyzed directly, an equal volume of 2× Laemmli sample buffer was added before boiling for 3 min. Immunoprecipitations, immunoblotting, and SDS-PAGE were performed by standard procedures. The following antibodies were used for immunoprecipitation and immunoblotting: anti-PLCγ1 rabbit polyclonal Ab (sc-81; Santa Cruz Biotechnology, Inc.); anti-LAT rabbit polyclonal Ab M41 (gift from M. Turner, Babraham Institute, Babraham, UK); anti–SLP-76 sheep polyclonal Ab (gift from G. Koretzky, University of Massachusetts, Worcester, MA); anti-Tec rabbit polyclonal Ab (Upstate Biotechnology). Anti-Itk rabbit polyclonal Ab (USB) was used for immunoprecipitation. The following antibodies were used for immunoblotting: anti-phosphotyrosine mAb RC20 conjugated directly to HRP (BD Transduction Labs); anti-Itk mAb 2F12 (gift from L. Berg, University of Pennsylvania, Philadelphia, PA); anti-Gads rabbit polyclonal Ab (gift from J. McGlade, Hospital for Sick Children, Toronto, Canada); anti-phosphotyrosine783-PLCγ1 (Biosource International); anti–phosphothreonine308-Akt, anti–phosphoserine473-Akt, anti-Akt, anti-phosphotyrosine319-ZAP-70, and anti-ZAP-70 rabbit polyclonal Ab (Cell Signaling Technology); and anti-Rac1 mAb (USB). For immunoblots, antibody binding was revealed with goat anti–mouse IgG-HRP (Santa Cruz Biotechnology, Inc.) for mouse mAb, goat anti–rabbit IgG-HRP (Cell Signaling Technology) or Protein A-HRP (Amersham Pharmacia Biotech) for rabbit polyclonal Ab, and donkey anti–sheep IgG-HRP (Serotec) for sheep polyclonal Ab. For immunoprecipitations, immunocomplexes were isolated using protein A or protein G Plus agarose (Santa Cruz Biotechnology, Inc.) for rabbit or sheep polyclonal Ab, respectively.
For densitometric analysis the blots were scanned, bands of interest were quantitated and in-lane background was subtracted. To determine specific phosphorylation, the signal from phosphorylated bands was divided by the signal from the appropriate loading control and all values were normalized to the maximum response (set to 100%). Signals below detection were set to 0%.
Rac1 Activation Assay.
For analysis of Rac1 activation, cells were stimulated at 3 × 107 cells per milliliter, lysed by the addition of an equal volume of 2× pulldown buffer (2% Triton X-100, 300 mM NaCl, 20 mM MgCl2, 2 mM sodium orthovanadate, 100 mM NaF, 2 mM PMSF, and 20 μg/ml leupeptin), and cleared by centrifugation at 15,340 g at 4°C for 2 min followed by the addition of 0.03 vol of a slurry containing Glutathione-Sepharose beads (Amersham Pharmacia Biotech) bound to bacterially expressed GST-Pak1-RBD (fusion protein of GST with amino acids 1–125 of rat Pak1, the Rac binding domain of Pak1). Samples were rotated at 4°C for 5 min, washed twice in 1× wash buffer (0.1% Triton X-100, 50 mM Tris, pH 7.5, 500 mM NaCl, 10 mM MgCl2) before elution of Gst-Pak1-RBD-bound protein using Laemmli sample buffer at 95°C.
Intracellular Calcium Analysis.
Intracellular calcium concentrations were analyzed by flow cytometry using Indo-1 loaded thymocytes as described previously, except that the cells were only stained with anti-CD3ɛ (22). CD3ɛ was cross-linked by the addition of goat anti–hamster IgG (75 μg/ml). Where required, cells were preincubated with wortmannin or Ly294002 (concentrations as above) at 37°C for 30 min before the addition of Indo-1AM. Inhibitors were present throughout the anti-CD3ɛ prebinding and cross-linking stages.
Inositol 1,4,5-Trisphosphate Measurement.
Thymocytes were stimulated in AB IMDM (100 μl) by cross-linking of CD3ɛ as described above. The stimulations were terminated by the addition of 15 μl ice-cold 6.1 M TCA followed by 15-min incubation on ice. The samples were centrifuged at 1,400 g, 4°C for 15 min, and the supernatant extracted with 10 vol water-saturated diethyl ether, neutralized with 10 μl 1 M NaHCO3, and the final volume of the aqueous phase was adjusted to 200 μl with water. Inositol 1,4,5-trisphosphate (IP3) was quantitated in duplicate or triplicate 100 μl samples using a competitive [3H]IP3 binding assay (NEN Life Sciences) according to the manufacturer's instructions.
Determination of Phosphatidylinositol 4,5-Bisphosphate Levels.
Pellets obtained after TCA-mediated cell lysis as above were washed with 1 M TCA, 1 mM EDTA, and then water. The pellets were extracted in methanol/chloroform/HCl (0.9 ml; 80:40:1), for 15 min at room temperature with intermittent vortexing. To this, chloroform (0.3 ml) and 0.1 M HCl (0.6 ml) was added and 0.4 ml of the lower phase was removed. After evaporation of solvent under vacuum, 1 M KOH (0.25 ml) was added, and lipids were hydrolyzed at 95°C for 15 min, which converts phosphatidylinositol 4,5-bisphosphate (PIP2) primarily to IP3. Hydrolyzed extracts were passed through Dowex columns (0.4 ml) preequilibrated with 5 ml 0.1 M HCl followed by 25 ml water. IP3 was eluted from the column in 1.25 ml of water. Ether extraction followed by neutralization with NaHCO3 and IP3 estimation was performed as above. PIP2 levels were calculated from the IP3 values as described in the Biotrak IP3 assay kit (Amersham Pharmacia Biotech).
Results
A Genetic System Allowing the Study of the Biochemical Role of Vav1 in DP Thymocytes.
In earlier studies, we and others had shown that Vav1 was required for a normal TCR-induced intracellular calcium flux in both thymocytes and T cells (20, 22–26). Furthermore we showed that in CD4+ T cells this was likely due to defective production of the second messenger IP3 (24). We wished to pursue further the biochemical pathway connecting Vav1 with the production of IP3, however the relatively small number of T cells that could be purified from Vav1 −/− mice was limiting. Thus we switched to thymocytes, which provide a much more abundant source of TCR-expressing cells. Since Vav1 −/− mice have a greatly reduced ratio of single positive to DP cells compared with Vav1 +/+ mice (22, 23), we used mice which were blocked in development so that the only TCR-expressing cells are DP thymocytes poised to receive a positive (or negative) selection signal (28). The system consists of the F5 TCR transgene which expresses a class I–restricted TCR specific for a peptide from influenza nuclear protein presented by H-2Db on a background deficient in recombinase activating gene (Rag)-1 (Rag-1 −/−) and MHC class I (β2m −/−). In the resultant F5, Rag-1−/−, β2m −/− mice, the absence of class I molecules causes a complete block in positive selection and thus all the F5 TCR-expressing thymocytes are blocked at the DP stage. To investigate the function of Vav1 within these cells we bred the Vav1 mutation onto this background, generating Vav1 −/−, F5, Rag-1 −/−, β2m −/− mice. As expected, thymic development in these mice was also completely arrested at the DP stage, which made up >90% of the thymus, and the mutant and control cells were matched in their expression of CD4, CD8, and TCR (Fig. 1 A).
Figure 1.



Vav1 is required for normal TCR-driven calcium flux and IP3 generation. (A) Dot plots showing flow cytometric analysis of CD4 and CD8 expression on thymocytes from F5, Rag-1 −/−, β2m −/−, and Vav1 −/−, F5, Rag-1−/−, β2m−/− mice (Vav1 +/+ and Vav1 −/−) showing percentage of cells falling into a CD4+CD8+ DP gate. Histogram shows levels of TCR-β on DP thymocytes gated as on the dot plots. (B) Vav1 −/− or Vav1 +/+ thymocytes were preloaded with Indo-1, coated with anti-CD3ɛ, and the antibody cross-linked with goat anti–hamster antibody at the time indicated by the break in the calcium trace and the short vertical arrow. The graph shows the mean ratio of Indo-1 violet/blue fluorescence, a measure of intracellular [Ca2+], as a function of time. (C) Graphs showing mean levels of IP3 and PIP2 (±SEM) in DP thymocytes stimulated by cross-linking of CD3ɛ as above. Results in this and all other figures are representative of at least three independent experiments.
Vav1 Is Required for Normal TCR-driven Calcium Flux and IP3 Generation in DP Thymocytes.
Comparison of the F5, Rag-1 −/−, β2m −/− DP thymocytes with wild-type DP cells showed that the peak TCR-induced calcium flux was threefold higher in the monoclonal cells (data not shown). This increase is mainly due to the lack of selecting MHC (data not shown), presumably because in wild-type DP thymocytes engagement of the TCR by peptide/MHC desensitizes TCR signaling, a process which is absent in the F5, Rag1 −/−, β2m −/− thymus. Indeed such increased TCR-induced calcium flux has been previously reported in DP thymocytes and T cells in environments that were MHC-deficient or expressed a limited repertoire of peptide–MHC complexes (29, 30). Nonetheless we found that the Vav1 −/− F5, Rag-1−/−, β2m−/− monoclonal DP cells had a greatly decreased calcium flux compared with F5, Rag-1−/−, β2m−/− cells (Fig. 1 B). This was similar to the defect in calcium flux we and others previously reported in Vav1-deficient thymocytes expressing a polyclonal TCR repertoire, thus validating the use of these cells to study the biochemical role of Vav1 in TCR signaling (19, 22, 23). In all further analysis we compared thymocytes from F5, Rag-1−/−, β2m−/− and Vav1 −/−, F5, Rag-1−/−, β2m−/− mice (hereafter referred to as Vav1 +/+ and Vav1 −/−, respectively).
A key second messenger driving intracellular calcium fluxes in lymphocytes is IP3, which is generated by the action of PLC on PIP2. As shown in Fig. 1 C, the TCR-driven production of IP3 is greatly diminished in Vav1 −/− DP thymocytes relative to Vav1 +/+ cells, which likely accounts for the large decrease in intracellular calcium flux. This result is similar to the reduced TCR-induced IP3 generation that we previously demonstrated in Vav1 −/− CD4+ T cells (24).
The defect in IP3 production could in principle be due to a failure of PLC activation or localization, or to a shortage of its substrate PIP2. It has been proposed that the GTPase Rac1 may activate phosphatidylinositol 4-phosphate 5-kinase (PIP5K), the enzyme that generates PIP2 from its precursor phosphatidylinositol 4-phosphate (31), and thus Vav1, as a GEF for Rac1 could potentially regulate PIP5K and thus the supply of PIP2. Indeed it has been shown that in Vav1-deficient B cells, CD19-induced activation of PIP5K is defective (32). To examine if a shortage of PIP2 might account for the defective IP3 production in Vav1 −/− thymocytes, we directly determined PIP2 levels in cells stimulated through the TCR. Our results show that Vav1 −/− cells had levels of PIP2, at least as high if not higher than those seen in Vav1 +/+ cells (Fig. 1 C). Thus it seems unlikely that the defective TCR-induced IP3 generation is caused by a lack of substrate for PLC. However since our analysis measures total cellular PIP2, we cannot exclude that there is a small pool of PIP2 used by PLC in which there is a selective shortage of the phospholipid.
Vav1 Is Required for Normal Tyrosine Phosphorylation of PLCγ1.
Generation of IP3 by signaling from antigen receptors is mediated by the PLCγ1 and 2 isoforms of the enzyme whose activation is in part mediated by tyrosine phosphorylation (33). Of these isoforms, PLCγ1 is likely to be more important for TCR signaling, since, while PLCγ2 is required for normal antigen receptor signaling in B cells, it appears to be dispensable for TCR signaling (34, 35). Thus we analyzed TCR-induced tyrosine phosphorylation of PLCγ1 in Vav1 +/+ and Vav1 −/− DP thymocytes. Using anti-phosphotyrosine Ab, we saw a clear reduction in TCR-induced PLCγ1 phosphorylation in Vav1 −/− cells (Fig. 2 A). Three distinct sites of phosphorylation have been identified on PLCγ1: tyrosines 771, 783, and 1254 (36). Mutation of tyrosine 783 (Y783) to phenylalanine in PLCγ1 blocked the phosphorylation-induced activation of the enzyme, suggesting that this residue is critical for regulation of PLCγ1 (36). Using antisera specific for phospho-Y783 PLCγ1, we found that in Vav1 −/− DP thymocytes, the TCR-induced phosphorylation of this residue is reduced, consistent with the defective activation of the lipase (Fig. 2 A).
Figure 2.
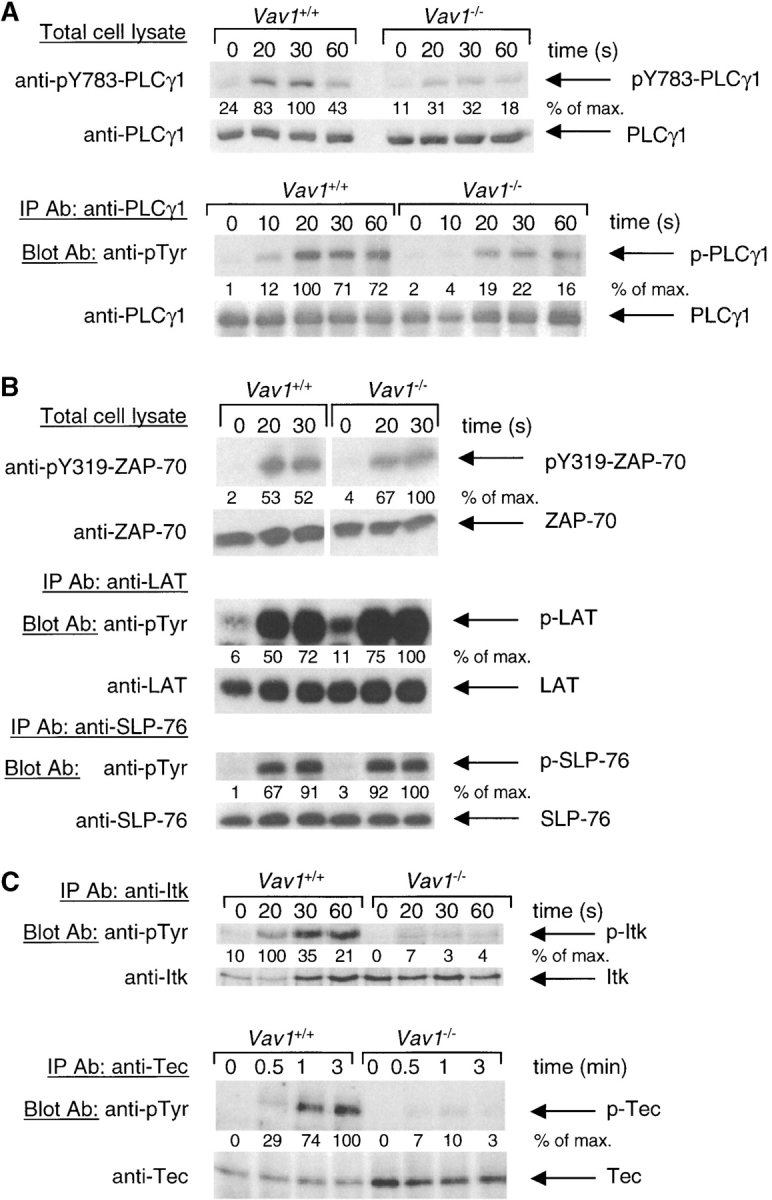
Vav1 is required for normal tyrosine phosphorylation of PLCγ1 and Tec-family kinases. Vav1 +/+ and Vav1 −/− DP thymocytes were coated with anti-CD3ɛ antibody and then stimulated by cross-linking with a secondary antibody for the indicated times, or left unstimulated (0 s). In some cases total cell lysates were analyzed directly by immunoblotting with the indicated antibodies. In other cases the protein of interest was immunoprecipitated with specific antibody (IP Ab) and analyzed by immunoblotting with blotting antibodies (Blot Ab). Equal loading was always evaluated by reprobing immunoblots with antibodies specific for the protein being analyzed. Densitometry was performed as in Materials and Methods, and all signals were normalized to the maximum response (set to 100%). (A) Phosphorylation of PLCγ1 on tyrosine 783 (pY783) was analyzed by immunoblotting total cell lysates directly. Total tyrosine phosphorylation on PLCγ1 (p-PLCγ1) was analyzed by blotting PLCγ1 immunoprecipitates with anti-phosphotyrosine (anti-pTyr) antibodies. (B) Phosphorylation of ZAP-70 on tyrosine 319 (pY319) was analyzed by immunoblotting total cell lysates directly. Tyrosine phosphorylation on LAT and SLP-76 was analyzed by blotting immunoprecipitates with anti-pTyr antibodies. (C) Tyrosine phosphorylation on Itk and Tec was analyzed by blotting immunoprecipitates with anti-pTyr antibodies.
Vav1 Is Required for TCR-induced Phosphorylation of Tec-family Kinases.
Genetic experiments had shown that Lck, ZAP-70, and Tec-family kinases as well as the LAT and SLP-76 adapters were essential for normal TCR-induced phosphorylation of PLCγ1 (1). Thus we examined each of these molecules in turn, to identify a cause for the reduced PLCγ1 phosphorylation. The TCR-induced activation of Lck was assessed using an in vitro kinase assay and found to be unaffected by the Vav1 mutation (data not shown), in agreement with previous experiments in Vav1-deficient T cells (24). Using an antiserum specific for phospho-Y319 of ZAP-70, a hallmark of its activation, we could see clear induction of ZAP-70 phosphorylation in both Vav1 +/+ and Vav1 −/− DP thymocytes (Fig. 2 B). Interestingly, while the amount of pY319-ZAP-70 was similar between Vav1 +/+ and Vav1 −/− cells, mutant cells always showed a higher specific phosphorylation of ZAP-70 due to lower amounts of total ZAP-70 in the cells; the reason for this is unclear. Immunoprecipitation of LAT or SLP-76 followed by immunoblotting with anti-phosphotyrosine antibodies showed inducible phosphorylation in Vav1 −/− cells which was similar in magnitude to that observed in Vav1 +/+ cells (Fig. 2 B). Finally, we examined the phosphorylation of Tec-family kinases. Three members of this family are expressed in the T lineage: Tec, Itk, and Rlk. Immunoprecipitation of Itk followed by immunoblotting with anti-phosphotyrosine antibodies showed that its TCR-induced phosphorylation in Vav1 −/− DP thymocytes was greatly reduced compared with Vav1 +/+ cells. Similarly, TCR stimulation caused strong Tec phosphorylation in Vav1 +/+ cells but only very weak phosphorylation in Vav1 −/− cells (Fig. 2 C). We were unable to determine whether phosphorylation of the third Tec-family kinase Rlk was also defective in Vav1 −/− cells due to the low sensitivity of available anti-Rlk antibodies. Thus, in conclusion, we have shown that Vav1 is required for TCR-induced phosphorylation and thus probably activation of Itk and Tec kinases. In view of the demonstrated importance of Tec-family kinases for PLCγ1 phosphorylation (37), it is likely that the defect in PLCγ1 activation in Vav1 −/− cells can at least in part be explained by the defective Itk and Tec phosphorylation.
Vav1 Regulates the Association of PLCγ1 with SLP-76.
While tyrosine phosphorylation is essential for the activation of PLCγ1, it is likely that the enzyme is regulated in other ways as well. In particular it needs to be recruited to the plasma membrane and to associate with a protein complex containing the adaptor proteins linker for activation of T cells (LAT), Gads, and SLP-76 (17). The transmembrane adaptor protein LAT is phosphorylated on multiple tyrosine residues creating docking sites to which several other proteins are recruited via SH2 domain–phosphotyrosine interactions (38). These include the Gads adaptor molecule which binds inducibly to LAT through its SH2 domain and constitutively to the cytoplasmic adaptor SLP-76 via its COOH-terminal SH3 domain (39, 40). PLCγ1 appears to be recruited to this complex through at least two interactions. It binds to phosphorylated LAT in a TCR stimulation-dependent manner through its NH2-terminal SH2 domain and constitutively to SLP-76 via its SH3 domain and both interactions are required for normal TCR-induced phosphorylation (41, 42).
Since Vav1 is also thought to be recruited to this complex (via SLP-76), we investigated whether its formation might be defective in Vav1 −/− DP thymocytes. We immunoprecipitated LAT from Vav1 +/+ and Vav1 −/− cells stimulated through the TCR and immunoblotted the precipitates with anti-PLCγ1, anti-Gads, and anti–SLP-76 antibodies. Our results show that in DP thymocytes, TCR stimulation induces the recruitment of all three of these molecules to LAT, and that the extent of complex formation is similar in Vav1 +/+ and Vav1 −/− cells (Fig. 3 A). We examined the same associations in reverse by immunoprecipitating PLCγ1 or SLP-76 and analyzing the precipitates for the presence of LAT. In both cases we were unable to detect LAT itself, probably due to limitations of antibody sensitivity, however using anti-phosphotyrosine antibodies we could readily see the inducible association of a 36-kD phosphoprotein (pp36) with both PLCγ1 and SLP-76, which comigrated with LAT (data not shown). Our results showed that the extent of TCR-induced association of pp36/LAT with both PLCγ1 and SLP-76 was similar in both Vav1 +/+ and Vav1 −/− cells (data not shown).
Figure 3.



Vav1 regulates association of PLCγ1 and SLP-76. Vav1 +/+ and Vav1 −/− DP thymocytes were stimulated and then analyzed by immunoprecipitation and immunoblotting as described in Fig. 2. (A) Immunoprecipitates of LAT were analyzed for the presence of PLCγ1, SLP-76, and Gads. (B) Immunoprecipitates of SLP-76 were analyzed for the presence of Gads. (C) Immunoprecipitates of PLCγ1 were analyzed for the presence of SLP-76.
Next we analyzed SLP-76 immunoprecipitates for the presence of the Gads adaptor protein. As expected, we saw a constitutive association of SLP-76 with Gads which did not change after TCR stimulation, and was similar in Vav1 +/+ and Vav1 −/− cells (Fig. 3 B). Finally, we immunoprecipitated PLCγ1 and blotted the precipitates for the presence of SLP-76. These experiments showed a weak constitutive association between PLCγ1 and SLP-76 which was strongly enhanced by TCR stimulation in Vav1 +/+ cells (Fig. 3 C). In contrast, in Vav1 −/− cells, the constitutive PLCγ1-SLP-76 association was reduced, and there was very little further TCR-induced association. In view of the demonstrated importance of the SLP-76-PLCγ1 association for PLCγ1 phosphorylation, its large reduction in Vav1 −/− DP thymocytes may well contribute to the observed reduction in TCR-induced PLCγ1 activation.
Vav1 Is Required for TCR-induced PI3K Activation.
The phosphorylation and subsequent activation of Itk and Tec is controlled at several levels. First, the kinases are recruited to the plasma membrane. This is achieved by the binding of phosphatidylinositol 3,4,5-trisphosphate (PIP3), produced by the action of PI3K on PIP2, to the PH domain of Itk or Tec (43). Mutations in the PH domain that block binding of PIP3, or treatment of cells with inhibitors of PI3K inhibit the antigen receptor–induced activation of both kinases (44, 45). Second, it has been proposed for Itk that the inactive kinase exists in a self-inhibited state with intramolecular interactions repressing activity (46). Binding of ligands to various domains of Itk is proposed to promote unfolding of these intramolecular interactions, eventually leading to activation of the kinase after phosphorylation by a Src-family kinase.
To investigate whether the defective phosphorylation of Itk and Tec in Vav1-deficient cells might be due to a failure to activate PI3K, we analyzed the phosphorylation of the serine/threonine kinase Akt as a surrogate for PI3K activity. Akt is activated by at least three steps: membrane recruitment driven by binding of phosphatidylinositol 3,4-bisphosphate to the PH domain, phosphorylation on threonine 308 (T308) by PDK1, a PIP3-activated kinase, and finally phosphorylation on serine 473 (S473) by a kinase whose identity is unclear (47). Since phosphatidylinositol 3,4-bisphosphate is derived from PIP3 by the action of the inositol phosphatase SHIP, the first two steps in the activation of Akt are dependent on PI3K. Analysis of TCR-induced phosphorylation of Akt on T308 showed that it was readily induced in Vav1 +/+ DP thymocytes, but was undetectable in Vav1 −/− cells (Fig. 4 A). Furthermore, analysis of S473 phosphorylation showed that once again Vav1 +/+ cells showed clear TCR-induced phospho-S473-Akt, while Vav1 −/− cells were greatly diminished in this response (Fig. 4 A). Taken together these results strongly suggest that the TCR stimulation–induced activation of PI3K was defective.
Figure 4.


Vav1 is required for TCR-induced Akt phosphorylation and Rac1 activation. Vav1 +/+ and Vav1 −/− DP thymocytes were stimulated as described in Fig. 2. (A) Total cell lysates were analyzed for Akt phosphorylation by blotting with antibodies specific for phosphothreonine 308 (pT308) and phosphoserine 473 (pS473). (B) Rac1 activation was evaluated by pulling down active GTP-loaded Rac1 with a GST fusion protein containing the Rac binding domain of Pak1 (GST-Pak1-RBD) and blotting with anti-Rac1 antiserum. Equal quantities of Rac1 in the extracts were confirmed by immunoblotting a fraction of the total cell lysates taken before the GST-Pak1-RBD pulldown.
It has been suggested that Rac1 may activate PI3K (31, 48, 49). Since Vav1 is a GEF for Rac1 and Rac2 GTPases, it is possible to construct a pathway from the TCR leading to Vav1 phosphorylation, and then Rac1 and PI3K activation. To investigate this possibility further, we analyzed TCR-induced Rac1 activation. As shown in Fig. 4 B, we readily detected Rac1 activation after TCR stimulation in Vav1 +/+ cells; in contrast Vav1 −/− cells showed reduced Rac1 activation. Thus, it is plausible that Vav1 regulates PI3K through the activation of the Rac1 GTPase.
Vav1 Regulates PLCγ1 Activation via PI3K-dependent and -independent Pathways.
To investigate whether the role of Vav1 in transducing TCR signals to the activation of PLCγ1 was limited to the activation of PI3K, we compared the effects of the Vav1 mutation with those of inhibitors of PI3K (Ly294002 and wortmannin). Treatment of Vav1 +/+ DP thymocytes with either inhibitor caused a small decrease in the TCR-induced calcium flux relative to untreated cells (Fig. 5 A, and data not shown). However inhibitor-treated Vav1 +/+ cells still showed a much larger TCR-induced calcium flux than Vav1 −/− cells (Fig. 5 A). To control for efficacy of the inhibitors we examined TCR-induced Akt phosphorylation in Vav1 +/+ or Vav1 −/− cells treated with Ly294002 or wortmannin. As expected, both inhibitors completely abolished TCR-induced Akt phosphorylation on S473 (data not shown). Taken together we conclude that in addition to regulating the activation of PI3K, Vav1 must also signal through a PI3K-independent pathway leading to the activation of an intracellular calcium flux.
Figure 5.


Vav1 regulates PLCγ1 activation via PI3K-dependent and -independent pathways. (A) Intracellular calcium concentration in Vav1 +/+ and Vav1 −/− DP thymocytes stimulated by cross-linking CD3ɛ was measured by Indo-1 fluorescence as described in Fig. 1 B. Cells were pretreated with the PI3K inhibitor Ly294002 as indicated. Cells not treated with inhibitor were preincubated with the appropriate concentration of the carrier DMSO. (B) Tyrosine phosphorylation of PLCγ1 and association of PLCγ1 with SLP-76 and Gads was determined using immunoprecipitates of PLCγ1 as described in Fig. 2 A and 3 C, respectively. Cells were pretreated with Ly294002 or with the carrier DMSO as indicated.
To examine this in more detail, we analyzed the effects of inhibiting PI3K on TCR-induced phosphorylation of PLCγ1. Immunoblots with antibodies specific for phosphoY783-PLCγ1 showed that treatment of Vav1 +/+ cells with Ly294002 reproducibly decreased the TCR-induced phosphorylation of this residue on PLCγ1 (Fig. 5 B). However, once again the inhibitor-treated Vav1 +/+ cells showed a larger signal than Vav1 −/− cells. Immunoblotting PLCγ1 immunoprecipitates with anti-phosphotyrosine showed a similar picture; treatment of Vav1 +/+ cells with Ly294002 decreases TCR-induced phosphorylation of PLCγ1, but not as much as disruption of the Vav1 gene (Fig. 5 B). These experiments demonstrate that Vav1 likely regulates the tyrosine phosphorylation of PLCγ1 by at least two distinct pathways, only one of which is dependent on PI3K.
Finally, we examined whether the defect in complex formation between PLCγ1 and SLP-76 is dependent on PI3K. Treatment of Vav1 +/+ DP thymocytes with Ly294002 caused no reproducible change in the TCR-inducible association of PLCγ1 and SLP-76 (Fig. 5 B). In contrast Vav1 −/− cells once again showed a decrease in both the constitutive and inducible associations of these molecules (Fig. 5 B). Reblotting of the same PLCγ1 immunoprecipitates for the Gads adaptor protein, showed that its association with PLCγ1 mirrored that of SLP-76, as would be expected given the constitutive SLP-76-Gads association. Vav1 +/+ cells showed TCR-inducible association of PLCγ1 and Gads which was not affected by Ly294002, whereas in Vav1 −/− cells, the basal and inducible association was greatly decreased (Fig. 5 B). These observations suggest that the PI3K-independent pathway by which Vav1 transduces TCR signals to the induction of PLCγ1 phosphorylation and hence intracellular calcium flux may involve regulation of the association of PLCγ1 with the SLP-76-Gads adaptor molecules (Fig. 6) .
Figure 6.

Cartoon of Vav-regulated pathways leading to PLCγ1 activation. TCR stimulation leads to the activation of the Lck and ZAP-70 kinases and the subsequent phosphorylation and activation of Vav. Vav regulates the activation of PI3K (as readout by Akt phosphorylation) possibly via Rac1, though there is no direct evidence for this. The dashed arrow represents the requirement for PI3K in the activation of Vav (54), leading to a potential positive feedback loop. Vav regulates PLCγ1 activation by at least two pathways, one PI3K-dependent, most likely through the activation of Itk and Tec. Secondly, Vav regulates the association between PLCγ1 and the SLP-76 and Gads adapters independently of PI3K activation. Rlk, another Tec-family kinase involved in PLCγ1 phosphorylation is activated in a PI3K-independent fashion since it does not have a PH domain (reference 61).
Discussion
Using the F5, Rag-1 −/−, β2m −/− system to analyze signaling in DP thymocytes, we showed that in the absence of Vav1, TCR-induced calcium flux, and IP3 production was greatly reduced. This phenotype is similar to the one we and others described for thymocytes and CD4+ T cells expressing a polyclonal TCR repertoire, thus validating the use of this system to study the biochemical role of Vav1 (20, 22–26).
We show here that after TCR stimulation, levels of PIP2 in Vav1 −/− DP cells remain as high if not higher than in Vav1 +/+ cells (Fig. 1 C), strongly suggesting that the defect in IP3 production is due to a failure to activate PLCγ1 rather than a shortage of its substrate. In contrast, it has been proposed that in Vav1 −/− B cells, the defective CD19-induced calcium flux is due to a shortage of PIP2, caused by defective activation of PIP5K (32). An earlier report showed an interaction between Rac1 and PIP5K (31), thus Vav1 could regulate this lipid kinase via its GEF activity toward Rac1. However, a more recent report questions this, showing that PIP5K is regulated by the GTPase ARF6, not Rac1 (50). How can we can reconcile our data with the observed reduction in PIP5K activity in CD19-stimulated Vav1 −/− B cells? One possibility is the difference in cell type and stimulus. Alternatively, we note that rather than measuring PIP2 levels, this study measured the synthesis of PIP2 after CD19 engagement and showed that it is reduced in Vav1 −/− cells. If the levels of PIP2 are controlled by negative feedback, as is common in many biosynthetic pathways, then a failure of PLCγ and PI3K activation would mean that PIP2 is not consumed and would not need to be replenished, thus giving rise to the observed decreased rates of PIP2 synthesis in Vav1 −/− cells.
PLCγ1 activity is regulated at several levels. It is recruited to the plasma membrane through interactions with membrane-associated protein complexes or via binding of PIP3 to its PH domain and is phosphorylated on at least three tyrosine residues (33, 51). Our data shows Vav1 regulates TCR-induced PLCγ1 phosphorylation, and this is likely to account for at least some of the defective PLCγ1 activity in Vav1 −/− DP cells. In contrast, we previously reported that TCR-induced PLCγ1 phosphorylation was normal in Vav1 −/− CD4+ splenic T cells (24). This could be due to a difference in signaling pathways used by the TCR in T cells versus DP thymocytes. However, we note that in our original studies we were hampered by the low number of T cells we could purify and the insensitivity of the antibodies being used, which resulted in a very low signal to background ratio, making it difficult to see partial decreases in PLCγ1 phosphorylation. In contrast, in the current work, we have used a more sensitive anti-PLCγ1 Ab for immunoprecipitation and have had access to a much larger number of cells from the DP thymocyte system, and this may account for why we see a defect in PLCγ1 phosphorylation. Furthermore, the defect was also evident using antiserum specific for phospho-Y783 of PLCγ1 (Fig. 2 A). Mutational analysis has shown that phosphorylation of this residue is critical for PLCγ1 activation (36), so its reduction in Vav1 −/− DP thymocytes is likely to at least partially account for the failure to activate the enzyme. We note, however, that the reduction in PLCγ1 phosphorylation is less pronounced than that in TCR-induced IP 3 production. This may be because Vav1 does something other than just regulate PLCγ1 phosphorylation; as we have shown, Vav1 is required for normal PLCγ1/SLP-76 association. Alternatively, the relationship between PLCγ1 phosphorylation and activity may be nonlinear, if for example several tyrosines need to be phosphorylated to activate the lipase.
To understand the defect in TCR-induced PLCγ1 phosphorylation, we systematically studied most molecules known from genetic studies to be required for this event. While phosphorylation of ZAP-70 on Y319 and total tyrosine phosphorylation of LAT and SLP-76 was normal, the phosphorylation and hence probably activation of Itk and Tec kinases was defective. While mice defective in a single Tec-family kinase show mild defects in thymocyte development and TCR signaling, compound mutations in Itk and Rlk cause a phenotype remarkably similar to that in Vav1 −/− mice (37). As in Vav1 mutants, Itk −/−, Rlk −/− mice show defective positive selection, decreased TCR-driven T cell proliferation and IL-2 production. Furthermore, biochemical analysis showed reduced TCR-induced calcium flux, IP3 production, PLCγ1 phosphorylation, and ERK activation. Unfortunately, we have been unable to determine if Rlk phosphorylation is affected in Vav1 −/− cells, for lack of a sufficiently sensitive antibody and the phenotype of the Itk/Tec double mutant has not yet been reported, so we cannot compare it directly to the Vav1 −/− mice. However, extrapolating from the phenotype of the Itk −/−, Rlk −/− mice, it seems likely that the reduced Itk and Tec activation will contribute significantly to the observed PLCγ1 phosphorylation defect. While the importance of Tec-family kinases for phosphorylation and activation of PLCγ isoforms has been demonstrated in a number of studies, the precise sites of phosphorylation have not been determined. Our data suggests that tyrosine 783 of PLCγ1 may be an important direct target of Tec-family kinases.
The defective activation of PI3K in Vav1 −/− DP thymocytes is the most likely cause for the failure to phosphorylate Itk and Tec. Both kinases contain a PH domain through which they are recruited to the plasma membrane after binding of PIP3, and this binding is a prerequisite for phosphorylation and activation. However, we cannot exclude that Vav1 may regulate these kinases by other mechanisms. Itk has been proposed to exist in an inhibited state maintained by intramolecular interactions (46). Binding of other proteins to domains of Itk involved in these internal associations would open up the kinase allowing its activation by phosphorylation. Vav1 has been reported to bind directly to Itk and Tec, and thus may contribute to their activation by displacing these intramolecular associations (52, 53).
The requirement for Vav1 in TCR-induced activation of PI3K was unexpected, since it has been proposed that PI3K is upstream of Vav1 (54). In these studies it was shown that PIP3 binds to the PH domain of Vav1 and activates its GEF activity. Indeed a recent study has shown that inhibition of PI3K leads to a reduction in TCR-induced Vav phosphorylation (55). Taken together with our results it seems that PI3K is both upstream and downstream of Vav1, potentially creating a positive feedback loop (Fig. 6). In agreement with our conclusions, recent work from other groups has shown that Vav-family proteins may also regulate antigen receptor-induced PI3K activation in mast cells and B cells (56, 57).
It is unclear how Vav1 regulates PI3K. The effect may be direct, in view of a report showing association of Vav1 with p85, a regulatory subunit of class 1 PI3K (58, 59). Alternatively PI3K may be activated by the Rac1 GTPase, since some reports have shown association between these proteins (31, 48, 49). Interestingly, a genetic analysis of PI3K and Rac1 activation in the Jurkat T cell line concluded that in a pathway leading from TCR stimulation to Akt activation, Rac1 was upstream of PI3K whereas in the same cells, cytoskeletal rearrangements were induced by a TCR signaling pathway in which PI3K was upstream of Rac1 (60). Our results are consistent with the idea that in thymocytes, the TCR signaling pathway leading to Akt phosphorylation is mediated by Vav1 via Rac1 and then PI3K activation.
It is most likely that PI3K contributes to PLCγ1 activation and TCR-induced calcium fluxes by regulating Tec-family kinases as described above, however there are other possibilities. PLCγ1 has been shown to bind PIP3 through its PH domain and thus PI3K may regulate PLCγ1 activation directly (51). In addition, we have shown that Vav1 also contributes to PLCγ1 phosphorylation and activation through a PI3K-independent pathway, by demonstrating that the effects of the Vav1 mutation were more severe than PI3K inhibitors (Fig. 5 A and B). Furthermore, we identified a potential route for such a pathway by showing that Vav1 regulates the association of PLCγ1 with the adapters SLP-76 and Gads and that this association was PI3K-independent (Fig. 5 B). This is likely to be significant for the activation of the lipase as mutations in SLP-76 which block its ability to bind PLCγ1 lead to reduced TCR-induced PLCγ1 phosphorylation (42).
The observation that in Vav1 −/− cells, the association of LAT with PLCγ1, Gads, and SLP-76 was normal, whereas that of PLCγ1 with Gads and SLP-76 was affected suggests that there may be two distinct signaling complexes containing PLCγ1. Only one of these includes LAT, whereas the other, Vav1-dependent complex, apparently does not. An interesting possibility is that this complex is nucleated around a different adaptor protein which substitutes for LAT. How Vav1 may regulate the assembly of such a complex is unclear. Since Vav1 binds to SLP-76 after TCR stimulation, it may stabilize the complex by directly binding to PLCγ1, or it might affect the ability of SLP-76 to bind to PLCγ1 by an allosteric mechanism.
In conclusion, we have shown that in primary DP thymocytes Vav1 transduces TCR signals to the phosphorylation and activation of PLCγ1 by at least two independent pathways (Fig. 6). First, Vav1 transduces a signal which leads to the activation of PI3K and phosphorylation of Tec-family kinases, and second, independently of PI3K, Vav1 regulates the formation of a signaling complex between PLCγ1 and the adaptor proteins Gads and SLP-76.
Acknowledgments
We thank M. Turner, G. Koretzky, L. Berg, and J. McGlade for antibodies; Chris Atkins, Keith Williams, and Rosemary Murphy for technical support; and R. Zamoyska, M. Walmsley, and A. Prisco for critical reading of the manuscript. We thank T. Kurosaki for communication of results before publication.
This work was supported by the Medical Research Council (United Kingdom).
Footnotes
*
Abbreviations used in this paper: AB, air-buffered; DP, double positive; GEF, GDP/GTP exchange factor; IP3, inositol 1,4,5-trisphosphate; ITAM, immunoreceptor tyrosine-based activation motif; LAT, linker for activation of T cell; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PIP5K, phosphatidylinositol 4-phosphate 5-kinase; PLC, phospholipase C; SLP, Src homology 2 domain–containing leukocyte phosphoprotein.
References
- 1.Kane, L.P., J. Lin, and A. Weiss. 2000. Signal transduction by the TCR for antigen. Curr. Opin. Immunol. 12:242–249. [DOI] [PubMed] [Google Scholar]
- 2.Katzav, S., D. Martin-Zanca, and M. Barbacid. 1989. Vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 8:2283–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henske, E.P., M.P. Short, S. Jozwiak, C.M. Bovey, S. Ramlakhan, J.L. Haines, and D.J. Kwiatkowski. 1995. Identification of VAV2 on 9q34 and its exclusion as the tuberous sclerosis gene TSC1. Ann. Hum. Genet. 59:25–37. [DOI] [PubMed] [Google Scholar]
- 4.Schuebel, K.E., X.R. Bustelo, D.A. Nielsen, B.J. Song, M. Barbacid, D. Goldman, and I.J. Lee. 1996. Isolation and characterization of murine vav2, a member of the vav family of proto-oncogenes. Oncogene. 13:363–371. [PubMed] [Google Scholar]
- 5.Movilla, N., and X.R. Bustelo. 1999. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell. Biol. 19:7870–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bustelo, X.R., and M. Barbacid. 1992. Tyrosine phosphorylation of the vav proto-oncogene product in activated B cells. Science. 256:1196–1199. [DOI] [PubMed] [Google Scholar]
- 7.Bustelo, X.R., J.A. Ledbetter, and M. Barbacid. 1992. Product of vav proto-oncogene defines a new class of tyrosine protein kinase substrates. Nature. 356:68–71. [DOI] [PubMed] [Google Scholar]
- 8.Margolis, B., P. Hu, S. Katzav, W. Li, J.M. Oliver, A. Ullrich, A. Weiss, and J. Schlessinger. 1992. Tyrosine phosphorylation of vav proto-oncogene product containing SH2 domain and transcription factor motifs. Nature. 356:71–74. [DOI] [PubMed] [Google Scholar]
- 9.Boguski, M.S., A. Bairoch, T.K. Attwood, and G.S. Michaels. 1992. Proto-vav and gene expression. Nature. 358:113. [DOI] [PubMed] [Google Scholar]
- 10.Bustelo, X.R. 2000. Regulatory and signaling properties of the Vav family. Mol. Cell. Biol. 20:1461–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crespo, P., K.E. Schuebel, A.A. Ostrom, J.S. Gutkind, and X.R. Bustelo. 1997. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature. 385:169–172. [DOI] [PubMed] [Google Scholar]
- 12.Han, J., B. Das, W. Wei, L. Van Aelst, R.D. Mosteller, R. Khosravi Far, J.K. Westwick, C.J. Der, and D. Broek. 1997. Lck regulates Vav activation of members of the Rho family of GTPases. Mol. Cell. Biol. 17:1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schuebel, K.E., N. Movilla, J.L. Rosa, and X.R. Bustelo. 1998. Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 17:6608–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu, J., D.G. Motto, G.A. Koretzky, and A. Weiss. 1996. Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity. 4:593–602. [DOI] [PubMed] [Google Scholar]
- 15.Tuosto, L., F. Michel, and O. Acuto. 1996. p95vav associates with tyrosine-phosphorylated SLP-76 in antigen-stimulated T cells. J. Exp. Med. 184:1161–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onodera, H., D.G. Motto, G.A. Koretzky, and D.M. Rothstein. 1996. Differential regulation of activation-induced tyrosine phosphorylation and recruitment of SLP-76 to Vav by distinct isoforms of the CD45 protein-tyrosine phosphatase. J. Biol. Chem. 271:22225–22230. [DOI] [PubMed] [Google Scholar]
- 17.Leo, A., and B. Schraven. 2001. Adapters in lymphocyte signalling. Curr. Opin. Immunol. 13:307–316. [DOI] [PubMed] [Google Scholar]
- 18.Tarakhovsky, A., M. Turner, S. Schaal, P.J. Mee, L.P. Duddy, K. Rajewsky, and V.L.J. Tybulewicz. 1995. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature. 374:467–470. [DOI] [PubMed] [Google Scholar]
- 19.Zhang, R., F.W. Alt, L. Davidson, S.H. Orkin, and W. Swat. 1995. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature. 374:470–473. [DOI] [PubMed] [Google Scholar]
- 20.Fischer, K.D., A. Zmuidzinas, S. Gardner, M. Barbacid, A. Bernstein, and C. Guidos. 1995. Defective T-cell receptor signalling and positive selection of Vav-deficient CD4+ CD8+ thymocytes. Nature. 374:474–477. [DOI] [PubMed] [Google Scholar]
- 21.Wu, J., S. Katzav, and A. Weiss. 1995. A functional T-cell receptor signaling pathway is required for p95vav activity. Mol. Cell. Biol. 15:4337–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turner, M., P.J. Mee, A. Walters, M.E. Quinn, A.L. Mellor, R. Zamoyska, and V.L.J. Tybulewicz. 1997. A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity. 7:451–460. [DOI] [PubMed] [Google Scholar]
- 23.Kong, Y.Y., K.D. Fischer, M.F. Bachmann, S. Mariathasan, I. Kozieradzki, M.P. Nghiem, D. Bouchard, A. Bernstein, P.S. Ohashi, and J.M. Penninger. 1998. Vav regulates peptide-specific apoptosis in thymocytes. J. Exp. Med. 188:2099–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costello, P.S., A.E. Walters, P.J. Mee, M. Turner, L.F. Reynolds, A. Prisco, N. Sarner, R. Zamoyska, and V.L.J. Tybulewicz. 1999. The Rho-family GTP exchange factor Vav is a critical transducer of TCR signals to the calcium, ERK and NF-κB pathways. Proc. Natl. Acad. Sci. USA. 96:3035–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer, K.-D., Y.-Y. Kong, H. Nishina, K. Tedford, L.E.M. Marengère, I. Kozieradzki, T. Sasaki, M. Starr, G. Chan, S. Gardener, et al. 1998. Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol. 8:554–562. [DOI] [PubMed] [Google Scholar]
- 26.Holsinger, L.J., I. Graef, W. Swat, T. Chi, D.M. Bautista, L. Davidson, R.S. Lewis, F.W. Alt, and G.R. Crabtree. 1998. Defects in actin cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol. 8:563–572. [DOI] [PubMed] [Google Scholar]
- 27.Wülfing, C., A. Bauch, G.R. Crabtree, and M.M. Davis. 2000. The vav exchange factor is an essential regulator in actin-dependent receptor translocation to the lymphocyte-antigen-presenting cell interface. Proc. Natl. Acad. Sci. USA. 97:10150–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth, L.A., O. Williams, R.D. Huby, T. Norton, O. Acuto, S.C. Ley, and D. Kioussis. 1998. Altered peptide ligands induce quantitatively but not qualitatively different intracellular signals in primary thymocytes. Proc. Natl. Acad. Sci. USA. 95:8193–8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong, P., G.M. Barton, K.A. Forbush, and A.Y. Rudensky. 2001. Dynamic tuning of T cell reactivity by self-peptide-major histocompatibility complex ligands. J. Exp. Med. 193:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, K., B. Seddon, M.A. Purbhoo, R. Zamoyska, A.G. Fisher, and M. Merkenschlager. 2001. Sensory adaptation in naive peripheral CD4 T cells. J. Exp. Med. 194:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tolias, K.F., L.C. Cantley, and C.L. Carpenter. 1995. Rho family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 270:17656–17659. [DOI] [PubMed] [Google Scholar]
- 32.O'Rourke, L., R. Tooze, M. Turner, D.M. Sandoval, R.H. Carter, V.L.J. Tybulewicz, and D.T. Fearon. 1998. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 8:635–645. [DOI] [PubMed] [Google Scholar]
- 33.Rhee, S.G. 2001. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70:281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, D., J. Feng, R. Wen, J.C. Marine, M.Y. Sangster, E. Parganas, A. Hoffmeyer, C.W. Jackson, J.L. Cleveland, P.J. Murray, and J.N. Ihle. 2000. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity. 13:25–35. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto, A., K. Takeda, M. Inaba, M. Sekimata, T. Kaisho, S. Ikehara, Y. Homma, S. Akira, and T. Kurosaki. 2000. Cutting edge: essential role of phospholipase C-γ2 in B cell development and function. J. Immunol. 165:1738–1742. [DOI] [PubMed] [Google Scholar]
- 36.Kim, H.K., J.W. Kim, A. Zilberstein, B. Margolis, J.G. Kim, J. Schlessinger, and S.G. Rhee. 1991. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-γ 1 phosphorylation on tyrosine residues 783 and 1254. Cell. 65:435–441. [DOI] [PubMed] [Google Scholar]
- 37.Schaeffer, E.M., J. Debnath, G. Yap, D. McVicar, X.C. Liao, D.R. Littman, A. Sher, H.E. Varmus, M.J. Lenardo, and P.L. Schwartzberg. 1999. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 284:638–641. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, W., R.P. Trible, M. Zhu, S.K. Liu, C.J. McGlade, and L.E. Samelson. 2000. Association of Grb2, Gads, and phospholipase C-γ 1 with phosphorylated LAT tyrosine residues. J. Biol. Chem. 275:23355–23361. [DOI] [PubMed] [Google Scholar]
- 39.Liu, S.K., N. Fang, G.A. Koretzky, and C.J. McGlade. 1999. The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT adaptors. Curr. Biol. 9:67–75. [DOI] [PubMed] [Google Scholar]
- 40.Asada, H., N. Ishii, Y. Sasaki, K. Endo, H. Kasai, N. Tanaka, T. Takeshita, S. Tsuchiya, T. Konno, and K. Sugamura. 1999. Grf40, A novel Grb2 family member, is involved in T cell signaling through interaction with SLP-76 and LAT. J. Exp. Med. 189:1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoica, B., K.E. DeBell, L. Graham, B.L. Rellahan, M.A. Alava, J. Laborda, and E. Bonvini. 1998. The amino-terminal Src homology 2 domain of phospholipase Cγ1 is essential for TCR-induced tyrosine phosphorylation of phospholipase Cγ1. J. Immunol. 160:1059–1066. [PubMed] [Google Scholar]
- 42.Yablonski, D., T. Kadlecek, and A. Weiss. 2001. Identification of a phospholipase C-γ1 (PLC-γ1) SH3 domain-binding site in SLP-76 required for T-cell receptor-mediated activation of PLC-γ1 and NFAT. Mol. Cell. Biol. 21:4208–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis, C.M., C. Broussard, M.J. Czar, and P.L. Schwartzberg. 2001. Tec kinases: modulators of lymphocyte signaling and development. Curr. Opin. Immunol. 13:317–325. [DOI] [PubMed] [Google Scholar]
- 44.Ching, K.A., Y. Kawakami, T. Kawakami, and C.D. Tsoukas. 1999. Emt/Itk associates with activated TCR complexes: role of the pleckstrin homology domain. J. Immunol. 163:6006–6013. [PubMed] [Google Scholar]
- 45.Yang, W.C., K.A. Ching, C.D. Tsoukas, and L.J. Berg. 2001. Tec kinase signaling in T cells is regulated by phosphatidylinositol 3-kinase and the Tec pleckstrin homology domain. J. Immunol. 166:387–395. [DOI] [PubMed] [Google Scholar]
- 46.Andreotti, A.H., S.C. Bunnell, S. Feng, L.J. Berg, and S.L. Schreiber. 1997. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 385:93–97. [DOI] [PubMed] [Google Scholar]
- 47.Kandel, E.S., and N. Hay. 1999. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 253:210–229. [DOI] [PubMed] [Google Scholar]
- 48.Zheng, Y., S. Bagrodia, and R.A. Cerione. 1994. Activation of phosphoinositide 3-kinase activity by Cdc42Hs binding to p85. J. Biol. Chem. 269:18727–18730. [PubMed] [Google Scholar]
- 49.Bokoch, G.M., C.J. Vlahos, Y. Wang, U.G. Knaus, and A.E. Traynor-Kaplan. 1996. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem. J. 315:775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Honda, A., M. Nogami, T. Yokozeki, M. Yamazaki, H. Nakamura, H. Watanabe, K. Kawamoto, K. Nakayama, A.J. Morris, M.A. Frohman, and Y. Kanaho. 1999. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell. 99:521–532. [DOI] [PubMed] [Google Scholar]
- 51.Falasca, M., S.K. Logan, V.P. Lehto, G. Baccante, M.A. Lemmon, and J. Schlessinger. 1998. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 17:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Machide, M., H. Mano, and K. Todokoro. 1995. Interleukin 3 and erythropoietin induce association of Vav with Tec kinase through Tec homology domain. Oncogene. 11:619–625. [PubMed] [Google Scholar]
- 53.Bunnell, S.C., M. Diehn, M.B. Yaffe, P.R. Findell, L.C. Cantley, and L.J. Berg. 2000. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 275:2219–2230. [DOI] [PubMed] [Google Scholar]
- 54.Han, J., K. Luby-Phelps, B. Das, X. Shu, Y. Xia, R.D. Mosteller, U.M. Krishna, J.R. Falck, M.A. White, and D. Broek. 1998. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 279:558–560. [DOI] [PubMed] [Google Scholar]
- 55.Fang, D., and Y.C. Liu. 2001. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat. Immunol. 2:870–875. [DOI] [PubMed] [Google Scholar]
- 56.Manetz, T.S., C. Gonzalez-Espinosa, R. Arudchandran, S. Xirasagar, V. Tybulewicz, and J. Rivera. 2001. Vav1 regulates phospholipase cγ activation and calcium responses in mast cells. Mol. Cell. Biol. 21:3763–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inabe, K., M. Ishiai, A.M. Scharenberg, N. Freshney, J. Downward, and T. Kurosaki. 2002. Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J. Exp. Med. 195:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramos-Morales, F., B.J. Druker, and S. Fischer. 1994. Vav binds to several SH2/SH3 containing proteins in activated lymphocytes. Oncogene. 9:1917–1923. [PubMed] [Google Scholar]
- 59.Shigematsu, H., H. Iwasaki, T. Otsuka, Y. Ohno, F. Arima, and Y. Niho. 1997. Role of the vav proto-oncogene product (Vav) in erythropoietin-mediated cell proliferation and phosphatidylinositol 3-kinase activity. J. Biol. Chem. 272:14334–14340. [DOI] [PubMed] [Google Scholar]
- 60.Genot, E.M., C. Arrieumerlou, G. Ku, B.M. Burgering, A. Weiss, and I.M. KrAm. 2000. The T-cell receptor regulates Akt (protein kinase B) via a pathway involving Rac1 and phosphatidylinositide 3-kinase. Mol. Cell. Biol. 20:5469–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chamorro, M., M.J. Czar, J. Debnath, G. Cheng, M.J. Lenardo, H.E. Varmus, and P.L. Schwartzberg. 2001. Requirements for activation and RAFT localization of the T-lymphocyte kinase Rlk/Txk. BMC Immunol. 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]