CD27 Promotes Survival of Activated T Cells and Complements CD28 in Generation and Establishment of the Effector T Cell Pool (original) (raw)
Abstract
CD27, like CD28, acts in concert with the T cell receptor to support T cell expansion. Using CD27−/− mice, we have shown earlier that CD27 determines the magnitude of primary and memory T cell responses to influenza virus. Here, we have examined the relative contributions of CD27 and CD28 to generation of the virus-specific effector T cell pool and its establishment at the site of infection (the lung), using CD27−/−, CD28−/−, and CD27/CD28−/− mice. We find that primary and memory CD8+ T cell responses to influenza virus are dependent on the collective contribution of both receptors. In the primary response, CD27 and CD28 impact to a similar extent on expansion of virus-specific T cells in draining lymph nodes. CD27 is the principle determinant for accumulation of virus-specific T cells in the lung because it can sustain this response in CD28−/− mice. Unlike CD28, CD27 does not affect cell cycle activity, but promotes survival of activated T cells throughout successive rounds of division at the site of priming and may do so at the site of infection as well. CD27 was found to rescue CD28−/− T cells from death at the onset of division, explaining its capacity to support a T cell response in absence of CD28.
Keywords: costimulation, apoptosis, influenza virus, MHC tetramer, CFSE
Introduction
An effective immune response is based on a dramatic increase in numbers of antigen-specific T cells. It is qualitatively tuned by their differentiation into cells with the appropriate effector functions. Naive T cells generally cannot expand efficiently upon TCR stimulation alone. A second signal is required, which is provided by membrane receptors on the T cell and their ligands on the antigen-presenting cell. CD28 provides such a costimulatory signal. It promotes assembly of the T cell signaling complex and primarily amplifies signals delivered via the TCR (1). This decreases the threshold for T cell recruitment into the dividing pool, resulting in responsiveness at lower doses of antigen (2). CD28 increases IL-2 production and thus enhances cell cycle activity. CD28 also promotes survival of activated T cells through a pathway, which up-regulates the apoptosis inhibitory Bcl-2 family member Bcl-xL (3, 4). These observations support the concept that numerical expansion of activated T cells is not only dependent on cell division, but also on cell survival.
Several members of the TNF receptor family, including CD27 and its close relatives 4-1BB (CD137) and OX40 (CD134), have recently moved to center stage as costimulatory receptors (5). These molecules link to members of the TNF receptor–associated factor (TRAF) family of signaling adaptors via small, ill-conserved motifs in their cytoplasmic tails. Biochemical and genetic studies support a role for TRAFs and TRAF-linked receptors in counteracting apoptotic signals, but there is also evidence for effects on cell proliferation, differentiation, and migration (6, 7).
Expression of these receptors and their transmembrane, TNF-related ligands is selective in time and place and tightly controlled by antigenic stimulation. In man and mouse, CD27 is restricted to lymphoid cells. It is present on the great majority of naive CD4+ and CD8+ T cells, and transiently up-regulated upon priming (8, 9). CD70, the ligand of CD27, is exclusively expressed on activated lymphocytes and mature DCs. On T cells, CD70 is induced by the TCR and modulated by cytokines (10–12). On DCs, it is induced by CD40, or by “danger” signals, such as LPS (12, 13). Therefore, CD27–CD70 interaction can come into play during T–DC and T–T cell communication and is expected to contribute to the T cell response both at the site of priming and at the effector site.
In vitro, CD27 can give a costimulatory signal to purified naive T cells that promotes T cell expansion (14, 15). In CD27−/− mice, as generated in our laboratory, T cell development and homeostasis are normal. However, upon intranasal infection with influenza virus, CD27−/− mice displayed deficits in generation of antigen-specific T cells in lymphoid organs and in establishment of CD4+ and CD8+ T cell pools at the effector site, i.e., the lung. CD27 deletion impacted on the primary as well as the memory response (16). CD70 transgenic mice essentially display the reverse phenotype. Both CD4+ and CD8+ T cells with an effector phenotype accumulate in these mice (17, 18).
Using intranasal infection with influenza virus as a model system, we show that CD27 is as important as CD28 for generation of a virus-specific T cell pool in lung draining lymph nodes (DLNs) and the main determinant for accumulation of these cells at the site of infection (the lung). CD27 makes this contribution not by affecting cell cycle activity, but by stimulating the survival of activated T cells, also in the absence of CD28 signals.
Materials and Methods
Mice.
All mice were bred under pathogen-free conditions and were used for experiments at 6–10 wk of age. Animal experiments were performed according to institutional and national guidelines. CD27−/− mice (16) and CD28−/− mice (19) were backcrossed for 8 and 10 generations to a C57BL/6 background, respectively. Offspring was genotyped by PCR on tail DNA as previously described (16, 19). F5 TCR transgenic mice (20) and H-Y TCR transgenic mice (21) were phenotyped by staining of peripheral blood samples with antibody to Vβ11 or Vβ8, respectively.
Flow Cytometry.
Lung, spleen, and lymph nodes were forced through a nylon mesh in the presence of IMDM with 8% FCS to obtain single cell suspensions. Erythrocytes were lysed by incubation of the harvested cell population in 0.14 M NH4Cl, 0.017 M Tris-HCl, pH 7.2, for 2 min on ice. Cells were preincubated with Fc-Block (mAb to CD16/CD32, 2.4G2; BD Biosciences) and washed in staining buffer (PBS, 0.5% BSA, 0.01% sodium azide). Next, cells were incubated with specific antibodies directly conjugated to FITC, PE, or allophycocyanin (APC). APC-labeled tetramers of the murine MHC class I H2-Db heavy chain, β2 microglobulin, and the influenza virus nucleoprotein (NP)366–374 peptide ASNENMDAM were prepared as previously described (22) and used for immunofluorescence in combination with anti-CD8 mAb. After incubation with respective antibodies and tetramers, cells were washed and analyzed using a FACSCalibur™ and CELLQuest™ software (Becton Dickinson). mAbs used for immunofluorescence were anti-CD3ɛ, 500A2; anti-CD8b.2, 53-5.8; anti-CD27, LG.3A10 (15); anti-CD28, 37.51; anti-CD45R/B220, RA3-6B2; anti-CD45.1, A20; anti-Vβ8, F23.1; and anti-Vβ11, RR3-15. They were purchased from BD Biosciences or donated by investigators.
Preparation of Purified T Cells.
Cell suspensions were prepared as described above, passed over nylon wool (Polysciences) and incubated on ice for 30 min with mAb M5/114.15.2 to MHC class II (BD Biosciences). This was followed by 30 min of incubation on ice with 100 μl goat anti–mouse Ig-coated magnetic beads and 20 μl sheep anti–rat Ig-coated magnetic beads (Advanced Magnetics Inc.) per 107 cells. Beads were removed by magnetic sorting. Purity of the resulting cell population was checked with anti-CD3ɛ and anti-B220 mAbs. Only preparations that contained >98% T cells were used for stimulation or adoptive transfer.
In Vitro T Cell Stimulation.
For cell cycle analysis, T cells purified from pooled spleen and lymph node cells were labeled with carboxy-fluorescein diacetate succinimidyl ester (CFSE; Molecular Probes Inc.) before stimulation. Cells were incubated at a concentration of 5 × 107 cells/ml in PBS, containing 0.1% BSA and 5 μM CFSE, for 10 min at 37°C. Labeling was quenched with 10 ml cold medium with 10% FCS and cells were washed twice with IMDM with 8% FCS before use. For in vitro T cell stimulation with antibodies, purified T cells were plated at 105 per well in IMDM with 8% FCS in 96-well plates, which were coated with mAb 145-2C11 to CD3ɛ at 2.5 μg/ml, in the presence or absence of 10 μg/ml coated mAb 37.51 to CD28 or 0.1 μg/ml soluble mAb LG.3A10 to CD27. Mouse anti–rat Igκ chain mAb RG7/7.6, which also reacts with hamster Igκ, was added at 25 μg/ml to cross-link anti-CD27 mAb. After culture, live cells were counted on an automated cell counter (CASY®1, model TT; Schärfe-System). Before analysis, cells were incubated with TO-PRO-3 (Molecular Probes) at 1 μM in FACS® buffer for 10 min. Samples were analyzed for CFSE and TO-PRO-3 fluorescence intensity on a FACSCalibur™.
Apoptosis Assays.
To determine nuclear fragmentation (subdiploid DNA content), cells were lysed in 0.1% sodium citrate, 0.1% Triton X-100, and 50 μg/ml propidium iodide (PI). Fluorescence intensity of nuclei and nuclear fragments was measured by flow cytometry as previously described (23). Exposure at the outer plasma membrane leaflet of phosphatidyl serine was determined using FITC-labeled annexin V (APOPTEST™; Nexins Research BV) as previously described (24). 5 μg/ml PI was added before analysis and cells with high PI fluorescence intensity were excluded from analysis.
Virus Infection.
Influenza virus strain A/NT/60/68 was obtained from the Department of Virology, Erasmus University Rotterdam, Netherlands. Mice were anesthetized and infected intranasally with 50 μl Hank's balanced salt solution containing 25 or 100 hemagglutinin units of the same virus for primary and memory responses, respectively.
Adoptive Transfers.
Purified T cells from lymph nodes of TCR transgenic F5 CD27−/−, F5 CD28−/−, and F5 wild-type mice were mixed 1:1 with purified H-Y TCR transgenic nonresponder T cells (5 × 106 of each) and all were labeled with CFSE. The mix was suspended in 200 μl Hank's balanced salt solution and injected intravenously into female C57BL/6 recipients expressing the CD45.1 allotype. Mice were infected with influenza virus 2 d later. At indicated time points, DLNs, lung, and spleen were isolated and prepared for analysis. For the experiment depicted in Fig. 4, purified naive F5 T cells were injected at 15 × 106 into recipient mice, which were infected with influenza virus 2 d later. At day 4 after infection, T cells were purified from DLNs of recipients, mixed at a 1:1 ratio with purified H-Y TCR transgenic T cells, and labeled with CFSE. A mix of 106 T cells of each was injected intravenously into recipient mice, which had been infected with influenza virus on the same day as the donor mice. Cells recovered from DLNs and lung were stained with PE-conjugated anti-Vβ8 and anti-CD45.1 mAbs and APC-conjugated H2-Db NP366–374 tetramers. PI was added just before analysis and PI+ cells were excluded from analysis. Within the window detecting green fluorescence (CFSE), 1,000 nonresponder events (H-Y TCR transgenic T cells, Vβ8+) were collected from DLNs or spleen and 500 from the lung. Analysis of cell division was performed on the simultaneously collected F5 TCR transgenic responder T cells, allowing for quantitative comparison of the CFSE histograms.
Figure 4.



Impact of CD27 deletion on division of influenza virus-specific T cells recovered from the lung. Recipient mice were injected with naive F5 T cells and infected. At day 4 after infection, T cells were purified from DLNs and labeled with CFSE. Together with the H-Y T cell standard, these were injected into recipient mice, which had been infected with influenza virus at the same time point as donor mice. Day 5 after infection corresponds with day 1 after transfer. Donor F5 T cells were discriminated from T cells of the first recipient by means of the CD45.1 marker. (A) Quantitative comparison of CFSE profiles indicating cell divisions of wild-type and CD27−/− F5 T cells in the lung at the indicated days after virus infection. (B) Accumulation of F5 T cells in lung and DLNs of virus-infected mice in the days after adoptive transfer. Data points are means of two mice. Results are representative of at least two independent experiments. (C) Quantitative comparison of CFSE profiles indicating cell divisions of wild-type and CD27−/− F5 T cells in DLNs at the indicated days after virus infection.
Results
Virus-specific T Cell Responses in Mice Lacking CD27, CD28, or Both.
To determine the relative contributions of CD27 and CD28 to the T cell response, intranasal infection of C57BL/6 mice with influenza virus was used. In this model system, influenza virus causes a localized infection in the respiratory tract and is characterized by an influx of both CD4+ and CD8+ effector T cells into the lung (16). The CD8+ T cell response can be monitored with H-2Db MHC class I tetramers, loaded with the immunodominant peptide NP366–374 (22).
Wild-type, CD27−/−, CD28−/−, and CD27/CD28−/− double deficient mice were infected and analyzed 6, 8, 10, or 14 d later for the presence of virus-specific CD8+ T cells. In the lung, accumulation of wild-type virus-specific T cells became detectable at day 6 and peaked at days 8–10 after infection (Fig. 1 A). In CD28−/− mice, the T cell response at this site was partially intact, whereas it was almost completely abrogated upon additional CD27 deletion. Apparently, CD27 can support the virus-specific CD8+ T cell response in the lung in absence of CD28. Accumulation of virus-specific CD8+ T cells in the lung of CD27−/− mice was in fact much lower than in CD28−/− mice, indicating that CD27 is a major determinant for the CD8+ T cell response at the site of infection. The response at the site of priming, the mediastinal lung DLNs, was strongly reduced in both CD27−/− and CD28−/− mice compared with wild-type mice, and virtually absent in CD27/CD28−/− mice, indicating that both receptors make crucial and in part complimentary contributions to generation of the virus-specific CD8+ T cell pool. Strikingly, in the spleen, CD27 deficiency only had a minor effect, whereas CD28 deletion strongly reduced the response. Accumulation of virus-specific CD8+ T cells in the spleen appeared fully dependent on the collective contribution of CD27 and CD28. In the absence of both molecules, increase in tetramer+ T cells was virtually undetectable.
Figure 1.


In vivo primary and memory T cell responses to influenza virus in the presence or absence of CD27 and/or CD28. Wild-type, CD27−/−, CD28−/−, and CD27−/−/CD28−/− mice were infected intranasally with influenza virus. Cells were isolated from the lung, DLNs, and spleen on the indicated days after infection. They were counted, stained with anti-CD8 mAb and NP366–374/H2-Db tetramers, and analyzed by flow cytometry. Each symbol represents an individual mouse (four per group) and dashes represent mean values. (A) Primary response. (B) Memory response. The complete experiment was performed two times with reproducible results.
To study the impact of CD27 and/or CD28 deletion on the memory T cell response, mice were rechallenged with the same virus 6 wk after primary infection. As shown in Fig. 1 B, the wild-type memory response is characterized by more rapid kinetics and an increased magnitude of virus-specific T cell accumulation, in particular in DLNs and lung. In the lung, the response peaks at day 5 rather than at days 8–10 and peak values of NP-specific CD8+ T cells are about fourfold higher than in the primary response. In mice lacking CD27, CD28, or both, virus-specific T cell accumulation in the lung remained at levels seen in the primary response of these mice, indicating a severe reduction in memory. In DLNs, impact of single CD27 or CD28 deletion was somewhat less severe than in lung. In spleen, memory responses were reduced by either CD27 or CD28 deletion, indicating a stronger impact of CD27 deletion on the memory response in the spleen than on the primary response at this site.
We conclude that during the primary response, generation of the CD8+ virus-specific T cell pool in DLNs and its establishment at the site of infection is greatly dependent on the collective, partially nonredundant contribution of CD27 and CD28. CD27 is able to sustain effector T cell accumulation in the lung in the absence of CD28 during the primary response. The memory response in the lung is more dependent on CD28 than the primary response. Although T cell responses in DLNs and lung show similar requirements for CD27 and CD28, the response in the spleen appears to be regulated differently. In the primary response, CD28 rather than CD27 is required for accumulation of virus-specific CD8+ T cells at this site, whereas the memory response depends similarly, but not fully, on both receptors.
CD27 on Virus-specific T Cells Promotes Their Expansion at the Site of Priming, but Has No Effect on Cell Division.
Subsequent experiments were designed to determine by which mechanisms CD27 and CD28 contributed to generation of the virus-specific T cell pool in DLNs during the primary response. We made use of adoptive transfer of CFSE-labeled T cells to follow successive cycles of cell division. As responder cells, F5 TCR transgenic T cells were used, which are specific for the NP366–374–H-2Db complex (20). To allow for quantitative comparison of the CFSE profiles, a population of nonresponder TCR transgenic T cells was used with specificity for the male antigen H-Y (21). Labeled F5 and H-Y T cells were coinjected in a 1:1 ratio into female mice. 2 d later, mice were challenged with influenza virus. At the indicated time points after infection, cells were harvested and stained with anti-Vβ8 mAb to detect H-Y TCR transgenic T cells and with antibody to CD45.1 to detect cells of the recipient mouse. F5 T cells were characterized by CFSE staining and lack of CD45.1 or Vβ8 expression. MHC class I tetramers proved ineffective to identify primed F5 T cells, due to marked down-regulation of their TCR upon virus recognition (not depicted). Recovery of wild-type and CD27−/− F5 T cells from the spleen of noninfected mice at day 8 after injection was similar, indicating that CD27 deficiency did not alter the ability of T cells to survive in the absence of antigenic stimulation (Fig. 2 A). CFSE staining of F5 T cells, taken from DLNs at day 4 after virus infection, marked successive divisions, whereas H-Y T cells had not divided, corroborating their nonresponsiveness in this system. Recovery of H-Y T cells was similar when coinjected with either wild-type or CD27−/− F5 T cells both in noninfected and infected mice, ensuring the validity of using these cells as a standard. Using this experimental setup, we followed kinetically the response of wild-type, CD27−/−, and CD28−/− F5 T cells in DLNs (Fig. 2 B). Although at day 2 after virus infection, adoptively transferred wild-type F5 T cells had not yet divided, 24 h later a large proportion of these cells had already completed up to five divisions. This finding highlights the capacity of newly activated T cells to cycle very rapidly. At day 4 after infection, wild-type F5 T cells had further progressed through cell cycle. CD27−/− F5 T cells showed response kinetics, which were very similar to those of wild-type T cells. Because the recovery of F5 TCR transgenic responder T cells was calibrated by collecting a fixed number of H-Y nonresponder T cell events during flow cytometry, CFSE profiles of wild-type and CD27−/− F5 T cells could be overlaid (Fig. 2 B). This approach highlighted that both populations were indiscernible with respect to cell cycle entry and activity. However, in all division cycles, fewer cells had accumulated in the absence of CD27. In contrast to wild-type and CD27−/− T cells, CD28−/− T cells had not yet entered cell cycle at day 3 after infection. At day 4, the majority of CD28−/− T cells had responded, but still lagged behind wild-type T cells with respect to cell division. Enumeration of F5 T cells in DLNs at days 2, 3, and 4 after infection emphasized the impact of CD27 or CD28 deletion on the total yield of newly activated T cells (Fig. 2 C). In the absence of either molecule, cell numbers were greatly reduced and to a similar extent.
Figure 2.
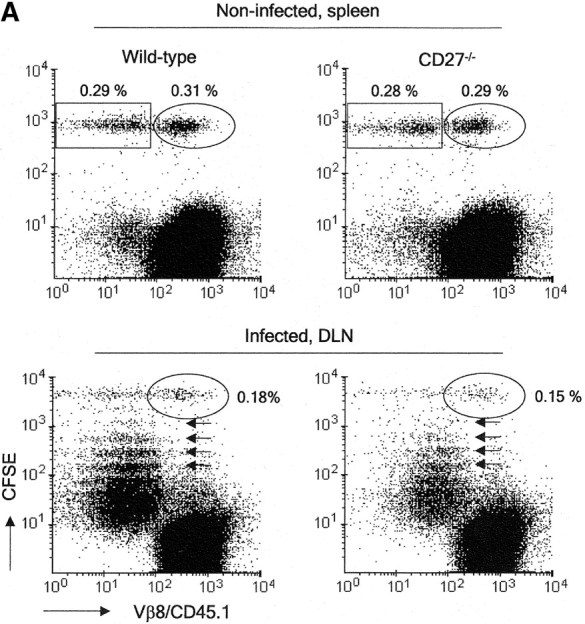


Impact of CD27 or CD28 deletion on division and accumulation of influenza virus-specific T cells in DLNs. CD45.1+ recipient mice were injected with a 1:1 mix of CD45.1− CFSE-labeled F5 and H-Y TCR transgenic naive T cells and infected with influenza virus 2 d later. (A) Recovery of CFSE-labeled wild-type or CD27−/− T cells from the spleen of noninfected recipient mice at day 8 after adoptive transfer, or from DLNs of recipient mice at day 4 after virus infection. The circles indicate nonresponding Vβ8+ H-Y T cells, rectangles Vβ8− F5 T cells, small arrows indicate divisions in the responding F5 population. Percentages of H-Y and F5 T cells were calculated relative to recipient cells. (B) DLNs were harvested at day 2, 3, or 4 after infection. To standardize recovery of F5 T cells, 1,000 H-Y T cells were collected. Wild-type (solid line), CD27−/−, or CD28−/− (dotted lines) F5 T cell responses are quantitatively compared by overlay of CFSE histograms. (C) Kinetics of accumulation of wild-type, CD27−/−, and CD28−/− F5 responder T cells in DLNs of influenza virus-infected mice. The absolute numbers of F5 T cells were calculated from the total number of cells and the percentage of F5 T cells (Vβ8/CD45.1−). Means and standard deviations were derived from four mice per time point out of two independent experiments.
In conclusion, both CD27 and CD28 on activated T cells promote expansion of virus-specific T cells in DLNs, but do so by distinct mechanisms. Although CD28 increments cell cycle entry and activity, CD27 does not, strongly suggesting that it enhances the accumulation of newly activated T cells at the site of priming by improving their survival.
Effects of CD27 Deletion on Virus-specific T Cell Division and Accumulation in Spleen and Lung.
To determine the effect of CD27 deletion on the virus-specific T cell response in the spleen and at the site of infection, we monitored adoptively transferred CFSE-labeled F5 T cells at later time points (days 4–8 after primary infection). Comparison of Figs. 2 C and 3 A shows that the wild-type F5 T cell response in DLNs peaks at day 4 after infection. CD27 deficiency compromised the F5 T cell response in terms of kinetics as well as cell numbers. The peak response of CD27−/− F5 T cells was reached at day 6 and was about twofold lower than for wild-type T cells (Fig. 3 A). In the spleen, the wild-type T cell response peaked later than in DLNs, at day 6 after infection (Fig. 3 A). In accordance with the data obtained in CD27−/− mice (Fig. 1), the accumulation of CD27−/− F5 T cells in the spleen was very similar to that of wild-type F5 T cells, both in terms of kinetics and cell yield. In the lung, wild-type F5 T cells also accumulated subsequent to the peak response in DLNs, reaching maximum levels at day 6 after infection (Fig. 3 A). Accumulation of CD27−/− F5 T cells in the lung was delayed as compared with wild-type and reached a maximum that was almost threefold lower.
Figure 3.


Impact of CD27 deletion on accumulation of influenza virus-specific T cells in the spleen and lung. Adoptive transfer was performed as outlined for Fig. 2. To standardize recovery of F5 responder T cells, 500 nonresponder H-Y T cells were collected from the lung and 1,000 from the spleen and DLNs. (A) Kinetics of accumulation of wild-type and CD27−/− F5 responder T cells. The absolute numbers of F5 T cells were calculated as indicated for Fig. 2 C. Means and standard deviations are derived from four mice per time point out of two independent experiments. (B) Quantitative comparison of CFSE profiles of wild-type and CD27−/− F5 T cells in the spleen and lung at day 6 after infection. Results are representative of at least two independent experiments.
Overlay of CFSE profiles of wild-type and CD27−/− F5 T cells, taken from the spleen at day 6 after infection, indicated that as in DLNs, CD27 deficiency had no effect on the cell cycle profiles. It also emphasized the minor effect of CD27 deletion on T cell accumulation in this organ (Fig. 3 B). Overlay of CFSE profiles of wild-type and CD27−/− F5 T cells recovered from lung at day 6 after infection highlighted the reduced number of CD27-deficient virus-specific T cells at the site of infection (Fig. 3 B). However, because CFSE fluorescence was extinguished in T cells that reached the lung, as has also been observed by others (25), we could not assess a potential effect of CD27 on division of F5 T cells recovered from the lung.
We conclude that during the primary response to influenza virus, CD27 on primed virus-specific T cells strongly promotes their accumulation at the site of infection, both in terms of kinetics and absolute numbers. In the spleen, CD27 has only a marginal effect on virus-specific T cell accumulation and does not affect cell division.
CD27 Promotes Accumulation, but Not Division of Virus-specific T Cells Found in the Lung.
Because we wanted to follow division cycles in virus-specific T cells recovered from the site of infection, we used a different experimental design. First, naive recipients were injected with F5 T cells and subsequently infected. At day 4 after infection, T cells were purified from DLNs and labeled with CFSE. 106 primed wild-type or CD27−/− F5 T cells were then reinjected together with H-Y nonresponder T cells into recipient mice, which had been infected at the same day as the donor mice. This was done to ensure that donor T cells entered in an appropriate environment. In the lung, wild-type and CD27−/− F5 T cells were already observed 1 d after transfer (day 5 after infection; Fig. 4 A). The next day, these cells had not detectably divided. At day 7, a large proportion of F5 T cells in the lung had completed multiple divisions according to CFSE dilution. In the following days, all F5 T cells were seen to participate in further divisions and fully dilute the CFSE marker. CFSE profiles showed that CD27 deletion did not affect cell division in T cells recovered from the lung (Fig. 4 A). Nevertheless, the accumulation of F5 T cells in the lung was greatly reduced upon CD27 deletion (Fig. 4 B). Peak levels were almost threefold lower for CD27−/− compared with wild-type F5 T cells.
The finding that CD27 promotes T cell accumulation in the lung without affecting cell division can be explained in several ways. First, CD27 may exert this effect solely by promoting the generation of the primed T cell pool in DLNs. Second, CD27 may additionally facilitate the migration of primed T cells into the lung. Third, CD27 may additionally promote the survival of primed T cells at the site of infection.
To examine whether CD27 facilitated further expansion of the adoptively transferred primed T cell pool in DLNs, we examined F5 T cell accumulation and CFSE profiles in DLNs (Fig. 4, B and C). Primed F5 T cells were observed in DLNs 1 d after transfer (day 5 after infection; Fig. 4 C). The next day, the majority of F5 T cells had not yet divided. At day 7, a large proportion had divided and by day 8 all cells had participated in division. The response kinetics of DLN T cells were in fact very similar to those in the lung (Fig. 4 A). As expected, no difference in CFSE profiles was observed between wild-type and CD27−/− F5 T cells in DLNs. Examination of F5 T cell accumulation in DLNs revealed two striking features (Fig. 4 B). First, primed F5 T cells were hardly affected by CD27 deletion throughout their further expansion in DLNs. In addition, primed F5 T cells gave rise to only up to 3 × 104 cells on day 7 and then slowly declined, whereas almost 10-fold higher numbers of wild-type F5 T cells and more than fourfold higher numbers of CD27−/− F5 T cells were found at the peak on day 8 in the lung. Possibly, F5 T cells divided after they had left the site of priming. Such additional cell divisions may have occurred in circulation and/or at the site of infection.
We conclude that CD27 does not affect cell division of primed T cells recovered from the lung, but promotes their accumulation at this site. This is achieved by promoting the generation of the primed virus-specific T cell pool in DLNs. In addition, we have evidence to suggest that CD27 supports the migration of primed T cells to the site of infection and/or their survival at this site.
CD27 Promotes Activated T Cell Survival in a CD28-independent Manner.
In vivo, apoptotic T cells cannot efficiently be quantified due to their efficient removal by phagocytes. Therefore, we performed in vitro experiments to obtain additional evidence for the hypothesis that CD27 promotes the survival of activated T cells. Upon 72 h of stimulation in vitro via the TCR–CD3 complex, purified wild-type T cells from naive mice had expanded significantly as compared with medium controls (Fig. 5 A). Deliberate costimulation with anti-CD27 or anti-CD28 mAb similarly increased the yield of live T cells in such cultures. Consistently, CD27 or CD28 deficiency reduced live cell yield compared with wild-type. Costimulation of CD27−/− cells via CD28 or vice versa revealed that these receptors can give mutually independent signals for the expansion of TCR-activated T cells. Analysis of nuclear fragmentation (23) and the incidence of annexin V+ cells (24) in 72-h cultures showed that stimulation of the TCR–CD3 complex induced apoptosis in a proportion of wild-type–purified T cells (Fig. 5, B and C). The reduction in apoptosis incidence that was reproducibly brought about by stimulation of CD27 in these type of assays correlated with the increased live cell yield (Fig. 5 A).
Figure 5.



CD27 promotes yield of live activated T cells and counteracts apoptosis in vitro. Purified T cells from wild-type, CD27−/−, or CD28−/− mice were stimulated for 72 h with anti-CD3 mAb in the presence or absence of anti-CD27 or anti-CD28 mAb, as indicated. (A) Live cells were counted using an automated cell counter. Means and standard deviations were derived from four cultures of two separate experiments. (B) Incidence of apoptosis as read out by nuclear fragmentation. Representative histograms with the percentage of apoptotic events. The bar diagram shows the mean percentages and standard deviations of apoptotic events determined in three separate experiments. (C) Incidence of apoptosis as read out by annexin V binding. Percentages indicate the proportion of annexin V+ cells (encircled). PIhigh cells were excluded from the analysis. The bar diagram shows the mean percentages and standard deviations of annexin V+ cells determined in three separate experiments.
Because T cell expansion is the net result of cell division and survival, we designed an experiment to discriminate between these two parameters. To study effects on cell division, T cells were labeled with CFSE. To determine effects on cell viability, T cells were stained with TO-PRO-3. This is a blue fluorescent DNA dye that only enters cells in case the plasma membrane has become permeable. This double labeling allowed us to determine the proportion of nonviable T cells in each cycle of cell division by flow cytometry. Fig. 6 A gives an example of the primary data derived from such analysis. At 72 h after the onset of stimulation with anti-CD3 mAb alone, wild-type T cells had divided up to six times according to CFSE staining. Costimulation via CD27 as well as CD28 incremented the overall proportion of live T cells in these cultures.
Figure 6.

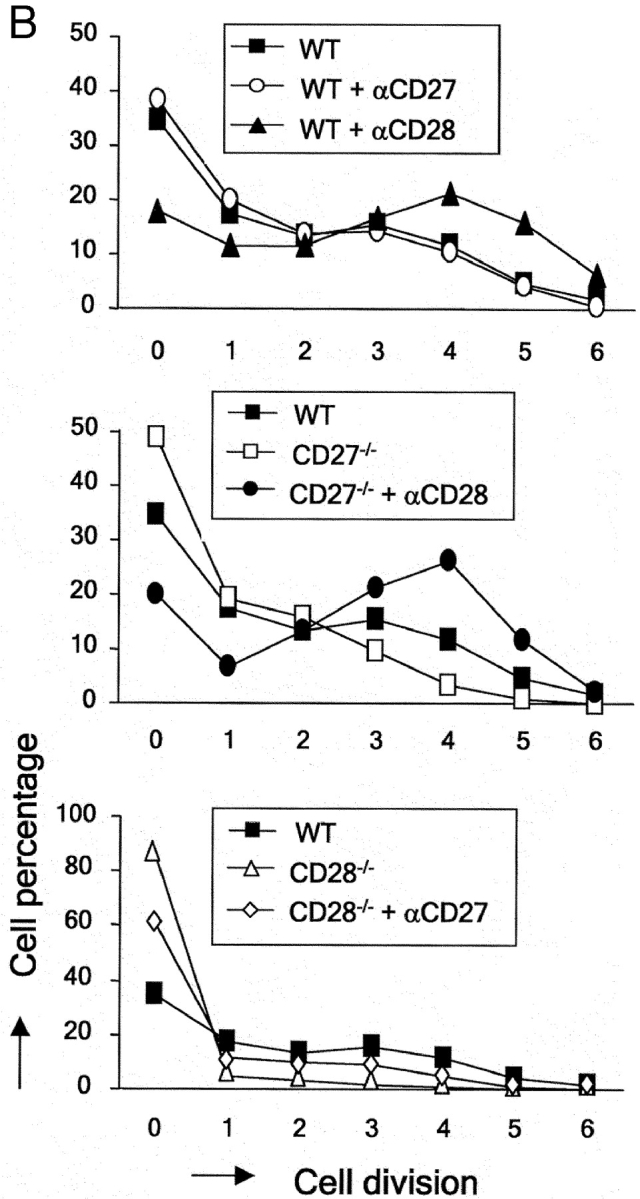

CD27 does not affect cell division but promotes survival and yield of live activated T cells in vitro in the presence and absence of CD28. Purified T cells were stimulated as indicated for Fig. 5. Cells were stained with TO-PRO-3 dye at the end of the culture period and the total cell population was analyzed by flow cytometry. (A) Dot plots showing CFSE and TO-PRO-3 fluorescence intensities and the overall percentage of TO-PRO-3− (viable) and TO-PRO-3+ (nonviable) cells. Lines mark each division round according to CFSE dilution. Division rounds are indicated by numbers 1–6. (B) The percentage of total cells (viable and nonviable) present in each cell division round was determined for wild-type (WT), CD27−/−, or CD28−/− T cells, costimulated with anti-CD27 or anti-CD28 mAb. (C) Live cell yield was calculated per cell division from the total number of cells in the culture and the percentage viable cells in discrete CFSE+ cell populations with successive diminishing fluorescence intensities (indicated by the lines in A). The ratio of nonviable/viable cells per division was calculated from the percentages. Data points represent means of duplicate cultures. Results are representative of four independent experiments.
Quantification of the percentage of cells in each successive cycle of cell division revealed that costimulation of wild-type T cells via CD27 did not affect cell cycle entry or progression (Fig. 6 B). In contrast, costimulation of wild-type T cells via CD28 incremented the proportion of cells participating in cell division, as well as the number of division cycles they had completed in this 72-h period. Consistently, CD27−/− T cells did not display significantly reduced cell cycle entry or activity compared with wild-type. CD28 could clearly stimulate cell cycle entry and progression of CD27−/− T cells, corroborating the independence of these costimulatory events. CD28−/− T cells showed a dramatic reduction in cell cycle entry compared with wild-type T cells. Costimulation via CD27 could rescue this to some extent, but did not bring it back to wild-type levels.
When we plotted the absolute number of live cells present in each division cycle, it became apparent that despite its lack of effect on cell division, costimulation of wild-type T cells via CD27 incremented the live cell yield, from initiation of cycle throughout successive divisions (Fig. 6 C). Plotting the ratio of nonviable/viable cell per division was used as a means to visualize the prosurvival effects of costimulation (Fig. 6 C). Throughout successive division cycles of wild-type T cells, nonviable cells outnumber viable cells. Costimulation via CD27 reverses this ratio in favor of viable cells. A similar effect on cell survival is seen when comparing wild-type and CD27−/− T cells.
Because CD28 promotes cell cycle activity, the effects it exerts on the number of live cells in successive divisions cannot be interpreted as an effect on cell survival. However, a striking survival promoting effect of CD28 was revealed in these assays when we compared the survival of TCR-stimulated wild-type and CD28−/− T cells that were still in division round 0. CD28−/− T cells appeared to die dramatically at the onset of stimulation, before completion of the first cell division (Fig. 6 C). Only a very small proportion of cells progressed through cell cycle. Interestingly, costimulation via CD27 could rescue CD28−/− T cells from death at this point: a ratio of ∼20:1 nonviable versus viable cells was changed by costimulation via CD27 to a ratio of 2:1 and thereby the absolute number of live cells in division 0 was restored to wild-type levels. This finding explains the effect of CD27 on cell cycle entry seen in Fig. 6 B. Because CD27 does not promote cell division, live cell yield in the next divisions and overall does not recover to wild-type levels in CD28−/− T cells that are costimulated via CD27.
In conclusion, CD27 rescues activated T cells from death from the onset of TCR/CD3 stimulation, throughout successive divisions. In contrast to CD28, CD27 does not affect cell cycle entry or activity. CD28 also has a survival-promoting effect, which has a dramatic impact at the onset on TCR/CD3 stimulation before completion of the first cell division. CD27 can compensate for the absence of CD28 by promoting activated T cell survival at this point.
Discussion
The total number of T cells present in the antigen-specific pool at the site of priming is determined by three cell-intrinsic parameters: the proportion of T cells entering into the proliferating pool, their cycle activity, and their survival. In accordance with published data (2), we found that CD28 increased cell cycle entry as well as activity. Early studies also report that CD28 promotes the survival of activated T cells (3). From measuring [3H]thymidine incorporation at different time points after TCR stimulation, it was concluded that this prosurvival effect came into play at a late time point and in fact sustained the proliferative response. However, we demonstrate that CD28−/− T cells die in much higher frequency than wild-type T cells when making the transition from G1 to S for the first time. CD28 strongly promotes cell survival at this point, thus greatly increasing the proportion of cells taking part in further divisions. The prosurvival effect of CD28 has been attributed to up-regulation of the antiapoptotic regulator BclxL (4). Retrovirus-mediated expression of Bcl-xL in CD28−/− T cells indeed rescued their survival (26).
In this study, we demonstrate that unlike CD28, CD27 does not affect cell cycle entry or activity of activated T cells. Yet, CD27 incremented the yield of live effector T cells both in vitro and in DLNs in vivo to a similar extent as CD28. Our data demonstrate that CD27 supports the survival of TCR-activated T cells, from the onset of cell division throughout successive cycles. Importantly, we find that CD27 can stimulate survival of in vitro–activated T cells in the absence of CD28. The dramatic cell death of CD28−/− T cells taking place upon TCR stimulation was almost completely rescued by deliberate CD27 stimulation with antibody. However, inasmuch as natural CD27–CD70 interactions took place in this in vitro system with purified T cells alone, they did not suffice to rescue these cells. In vivo, a different picture emerged: adoptively transferred F5 CD28−/− T cells in DLNs showed a 24-h delay in responsiveness as compared with wild-type, but a proportion of these cells did proliferate, in a very efficient manner. In line with published results (27), we found that in CD28−/− mice the antigen-specific CD8+ T cell response to intranasal influenza virus was reduced. However, significant accumulation of virus-specific CD8+ cells in the lung still occurred, which was dependent on CD27. Clearly, in a physiological context, certain virus-specific T cells can enter into and progress through cell cycle efficiently in the absence of a CD28 signal. High level of antigenic stimulation (TCR signals) may allow these cells to cross the threshold for proliferation. Our study emphasizes that such cells must be supported by CD27 for survival. In influenza virus-infected wild-type and CD28−/− mice, CD70 is expressed on T cells and DCs in lymphoid organs and lung, consistent with a role for CD27–CD70 interactions at these sites (12 and unpublished data).
In mice constitutively expressing the CD27 ligand CD70 in B cells, seemingly spontaneous turnover of naive T cells into cycling effector T cells takes place in the absence of deliberate immunization (17, 18). Crossing of CD70 transgenic mice with F5 TCR transgenics has pointed out that this turnover is driven by TCR signals, presumably induced by environmental antigens present in the mouse facility. Based on the data presented here, we postulate that constitutive CD27–CD70 interactions taking place in CD70 transgenics improve survival of T cells induced to divide by low level TCR signals. T cells in CD70 transgenic mice develop into effector cells, which produce increased amounts of IFN-γ, but not IL-2 (17). These findings emphasize that CD27 does not stimulate IL-2 production, unlike CD28. Whether it directs a specific effector cell differentiation program, or is merely permissive for effector cell differentiation to occur, is subject of investigation.
The survival-promoting activity of CD27 is in line with the best-documented effect of TRAF-linked TNF receptor family members, which is activation of nuclear factor κB. Nuclear factor κB signaling has been linked to transcriptional up-regulation of a variety of antiapoptotic proteins, including the Bcl-2 family member Bfl-1 and the death receptor inhibitor c-FlipL (28, 29). At present, we do not know the array of gene products that are controlled by CD27 and have no complete insight into the molecular basis of its survival-promoting effect. However, in human T cells CD27 was found to up-regulate Bcl-xL at the transcriptional level (van Lier, R.A.W., personal communication). Its close homologue, OX40, was recently shown to allow maintenance of Bcl-2 and Bcl-xL expression in the several days after initial T cell activation. The survival defect observed in OX40−/− T cells was rescued by retroviral expression of Bcl-xL or Bcl-2 (30). Interestingly, OX40−/− peripheral T cells were deficient in viability particularly in the later cell divisions after TCR stimulation. CD27 supports cell survival from the onset of TCR stimulation, in line with its expression on naive T cells. This suggests complementarity with OX40, which is induced 1–2 d after T cell activation (31).
4-1BB, another close relative of CD27, which is selectively expressed on activated T cells, also supports activated T cell survival, in a CD28-independent manner (32, 33). Similar to OX40, 4-1BB was recently found to stimulate live cell yield in the later divisions after TCR stimulation (32). Recently, deliberate stimulation of 4-1BB with mAb in vivo was found to enhance the CD8+ T cell response to intranasal influenza virus infection in wild-type mice and to rescue it to a large extent in CD28−/− mice (34). Presumably, in this setting, 4-1BB has a similar effect as we found for CD27, in that it sustained the life of activated T cells that would normally not have survived.
The contribution of CD28 to T cell memory has not yet been studied extensively. In a model of infection with lymphocytic choriomeningitis virus, viral clearance during a recall response was as effective in CD28−/− mice as in wild-type mice (35). CD8+ T cell memory was reduced only twofold compared with wild-type, probably reflecting the similarly reduced clonal burst size of the primary response. In our model of local infection with influenza virus, the memory response in CD28−/− animals was in the same order of magnitude as the primary response, indicating a much more profound effect of CD28 deficiency. CD27 deletion had a similar effect on the CD8+ memory T cell response to influenza virus as CD28 deletion. It is currently under investigation whether CD27 promotes CD8+ T cell memory in this influenza virus system by increasing the number of memory T cells and/or their capacity to respond to secondary challenge. Preliminary data indicate that the memory pool is reduced in size in the absence of CD27. Most likely, CD27 also promotes survival of memory T cells during secondary expansion. Primary and secondary infections were performed with the same virus. Because CD27 deletion does not affect Ig responses to this virus (unpublished data), the memory T cell response in CD27−/− mice will not be altered via effects on neutralizing antibody levels. In CD28−/− and CD27/CD28−/− mice, antiviral Ig production is severely reduced. Therefore, we have avoided quantitative comparisons between the memory T cell responses of the different recombinant mice.
Adoptive transfer of CFSE-labeled primed F5 T cells, taken from DLNs at day 4 after infection, allowed us to demonstrate that these cells continue to divide extensively. At least a proportion of these cells still had the potential to home to DLNs. Their relative lack of dependency on CD27 for further accumulation in DLNs suggests that this population represents a selection of the total pool of primed F5 T cells recovered from DLNs of donor mice. Numbers of primed F5 T cells in DLNs only increased from ∼1.5 × 104 to maximally 3 × 104, whereas peak values of F5 T cells found in the lung were 30 × 104. Possibly, a proportion of adoptively transferred primed F5 T cells directly migrated to the lung or migrated from DLNs into circulation before completing cell division. Because CD27 did not affect cell division of primed T cells recovered from the lung, the decrease in F5 T cell accumulation in the lung must be explained by effects on cell migration into the lung and/or cell survival in the lung. Our accumulated evidence strongly argues that a key function of CD27 is to prevent death in activated T cells. Therefore, we postulate that it promotes accumulation of effector T cells at the site of infection not only by improving the survival of activated T cells at the site of priming, but also by doing so at the site of infection. Additional experiments are needed to test this hypothesis. Whether this possible survival-promoting effect of CD27 in the lung is exerted in bronchus-associated lymphoid tissue and/or in lung tissue remains to be established as well.
In contrast to the virus-specific CD8+ T cells in DLNs and the lung, those in the spleen were only affected to a minor extent by CD27 deletion. We suggest that in the spleen, other costimulatory receptors compensate for the absence of CD27. Possibly, expression of such receptors and/or their ligands is more pronounced in the splenic microenvironment than in DLNs or the lung. Our data suggest that the spleen, in contrast to DLNs, makes only a minor contribution to the response at the site of infection. Alternatively, the lack of effect of CD27 deletion on activated T cells in the spleen might be masked by its profound effect once these T cells have reached the lung.
Because the immunodominant epitopes are undefined, the CD4+ antigen-specific T cell response to influenza virus can presently not be followed in C57BL/6 mice with MHC tetramers. However, analysis of the lung infiltrate showed that CD27 deletion similarly decreased the size of both CD8+ and CD4+ effector pools (16). CD27 is expressed on both naive subsets and collective in vitro experiments unambiguously show that it impacts on both (10). Also, in CD70 transgenic mice, CD4+ and CD8+ T cells respond in a similar fashion (17, 18). Reported work on the immune response in mice lacking 4-1BB or OX40 signals suggest that these receptors have a more selective impact on either CD8+ or CD4+ T cells (36–38). However, side by side comparison of mice lacking CD27, 4-1BB, or OX40 signals in the same antigenic model system, as ongoing in our laboratory, should clarify the relative impact of these molecules and their potential complementarity.
Acknowledgments
We thank the members of the Animal Facility, E. Noteboom and A. Pfauth, for expert technical assistance. R.A.W. van Lier, T.N.M. Schumacher, M. Oosterwegel, M. van Stipdonk, and K. Schepers are acknowledged for experimental advice and critical reading of the manuscript.
This work was supported by The Netherlands Organization for Scientific Research.
Abbreviations used in this paper: APC, allophycocyanin; CFSE, carboxy-fluorescein diacetate succinimidyl ester; DLN, draining lymph node; NP, nucleoprotein; PI, propidium iodide; TRAF, TNF receptor–associated factor.
References
- 1.Viola, A., S. Schroeder, Y. Sakakibara, and A. Lanzavecchia. 1999. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 283:680–682. [DOI] [PubMed] [Google Scholar]
- 2.Wells, A.D., H. Gudmundsdottir, and L.A. Turka. 1997. Following the fate of individual T cells throughout activation and clonal expansion. J. Clin. Invest. 100:3137–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noel, P.J., L.H. Boise, J.M. Green, and C.B. Thompson. 1996. CD28 costimulation prevents cell death during primary T cell activation. J. Immunol. 157:636–642. [PubMed] [Google Scholar]
- 4.Okkenhaug, K., L. Wu, K.M. Garza, J. La Rose, W. Khoo, N. Odermatt, T.W. Mak, P. Ohashi, and R. Rottapel. 2001. A point mutation in CD28 distinghuishes proliferative signals from survival signals. Nat. Immunol. 2:325–332. [DOI] [PubMed] [Google Scholar]
- 5.Watts, T.H., and M.A. DeBenedette. 1999. T cell costimulatory molecules other than CD28. Curr. Opin. Immunol. 11:286–293. [DOI] [PubMed] [Google Scholar]
- 6.Gravestein, L.A., and J. Borst. 1998. Tumor necrosis factor receptor family members in the immune system. Semin. Immunol. 10:423–434. [DOI] [PubMed] [Google Scholar]
- 7.Locksley, R.M., N. Killeen, and M.J. Lenardo. 2001. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 104:487–501. [DOI] [PubMed] [Google Scholar]
- 8.Van Lier, R.A.W., J. Borst, T.M. Vroom, H. Klein, P. van Mourik, W.P. Zeijlemaker, and C.J. Melief. 1987. Tissue distribution and biochemical and functional properties of Tp55 (CD27), a novel T cell differentiation antigen. J. Immunol. 139:1589–1596. [PubMed] [Google Scholar]
- 9.Gravestein, L.A., B. Blom, L.A. Nolten, E. de Vries, G. van der Horst, F. Ossendorp, J. Borst, and W.A.M. Loenen. 1993. Cloning and expression of murine CD27: comparison with 4-1BB, another lymphocyte-specific member of the nerve growth factor receptor family. Eur. J. Immunol. 23:943–950. [DOI] [PubMed] [Google Scholar]
- 10.Lens, S.M., K. Tesselaar, M.H.J. van Oers, and R.A.W. van Lier. 1998. Control of lymphocyte function through CD27-CD70 interactions. Semin. Immunol. 10:491–499. [DOI] [PubMed] [Google Scholar]
- 11.Oshima, H., H. Nakano, C. Nohara, T. Kobata, A. Nakajima, N.A. Jenkins, D.J. Gilbert, N.G. Copeland, T. Muto, H. Yagita, et al. 1998. Characterization of murine CD70 by molecular cloning and mAb. Int. Immunol. 10:517–526. [DOI] [PubMed] [Google Scholar]
- 12.Tesselaar, K., Y. Xiao, R. Arens, G.M.W. van Schijndel, D.H. Schuurhuis, R. Mebius, J. Borst, and R.A.W. van Lier. 2002. Expression of the murine CD27 ligand CD70 in vitro and _in vivo_J. Immunol. 169:33–40. [DOI] [PubMed] [Google Scholar]
- 13.Futagawa, T., H. Akiba, T. Kodama, K. Takeda, Y. Hosoda, H. Yagita, and K. Okumura. 2002. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 14:275–286. [DOI] [PubMed] [Google Scholar]
- 14.Hintzen, R.Q., S.M. Lens, K. Lammers, H. Kuiper, M.P. Beckmann, and R.A.W. van Lier. 1995. Engagement of CD27 with its ligand CD70 provides a second signal for T cell activation. J. Immunol. 154:2612–2623. [PubMed] [Google Scholar]
- 15.Gravestein, L.A., J.D. Nieland, A.M. Kruisbeek, and J. Borst. 1995. Novel monoclonal antibodies reveal potent costimulatory activity of murine CD27. Int. Immunol. 7:551–557. [DOI] [PubMed] [Google Scholar]
- 16.Hendriks, J., L.A. Gravestein, K. Tesselaar, R.A.W. van Lier, T.N.M. Schumacher, and J. Borst. 2000. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 1:433–440. [DOI] [PubMed] [Google Scholar]
- 17.Arens, R., K. Tesselaar, P.A. Baars, G.M.W. van Schijndel, J. Hendriks, S.T. Pals, P. Krimpenfort, J. Borst, M.H.J. van Oers, and R.A.W. van Lier. 2001. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNγ-mediated B cell depletion. Immunity. 15:801–812. [DOI] [PubMed] [Google Scholar]
- 18.Tesselaar, K., R. Arens, G.M.W. van Schijndel, P.A. Baars, M.A. van der Valk, J. Borst, M.H.J. van Oers, and R.A.W. van Lier. 2003. Lethal T cell immunodeficiency induced by chronic costimulation via CD27/CD70 interactions. Nat. Immunol. 4:49–54. [DOI] [PubMed] [Google Scholar]
- 19.Shahinian, A., K. Pfeffer, K.P. Lee, T.M. Kündig, K. Kishihara, A. Wakeham, K. Kawai, P.S. Ohashi, C.B. Thompson, and T.W. Mak. 1993. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 261:609–612. [DOI] [PubMed] [Google Scholar]
- 20.Mamalaki, C., J. Elliott, T. Norton, N. Yannoutsos, A.R. Townsend, P. Chandler, E. Simpson, and D. Kioussis. 1993. Positive and negative selection in transgenic mice expressing a T-cell receptor specific for influenza nucleoprotein and endogenous superantigen. Dev. Immunol. 3:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teh, H.S., P. Kisielow, B. Scott, H. Kishi, Y. Uematsu, H. Blüthmann, and H. von Boehmer. 1988. Thymic major histocompatibility complex antigens and the αβ T-cell receptor determine the CD4/CD8 phenotype of T cells. Nature. 335:229–233. [DOI] [PubMed] [Google Scholar]
- 22.Haanen, J.B.A., M. Wolkers, A.M. Kruisbeek, and T.N.M. Schumacher. 1999. Selective expansion of cross-reactive CD8+ memory T cells by viral variants. J. Exp. Med. 190:1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicoletti, I., G. Migliorati, M.C. Pagliacci, F. Grignani, and C.A. Riccardi. 1991. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 139:271–279. [DOI] [PubMed] [Google Scholar]
- 24.Koopman, G., C.P.M. Reutelingsperger, G.A.M. Kuijten, R.M.J. Keehnen, S.T. Pals, and M.H.J. van Oers. 1994. Annexin V for flow cytometric detection of phosphatidyl serine expression on B cells undergoing apoptosis. Blood. 84:1415–1420. [PubMed] [Google Scholar]
- 25.Roman, E., E. Miller, A. Harmsen, J. Wiley, U.H. van Andrian, G. Huston, and S.L. Swain. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J. Exp. Med. 196:957–968. [DOI] [PMC free article] [PubMed]
- 26.Dahl, A.M., C. Klein, P.G. Andres, C.A. London, M.P. Lodge, R.C. Mulligan, and A.K. Abbas. 2000. Expression of Bcl-xL restores cell survival, but not proliferation and effector differentiation, in CD28-deficient T lymphocytes. J. Exp. Med. 191:2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lumsden, J.M., J.M. Roberts, N.L. Harris, R.J. Peach, and F. Ronchese. 2000. Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J. Immunol. 164:79–85. [DOI] [PubMed] [Google Scholar]
- 28.Wang, C.-Y., D.C. Guttridge, M.W. Mayo, and A.S. Baldwin. 1999. NF-κB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell. Biol. 19:5923–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Micheau, O., S. Lens, O. Gaide, K. Alevizopoulos, and J. Tschopp. 2001. NF-κB signals induce the expression of c-FLIP. Mol. Cell. Biol. 21:5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogers, P.R., J. Song, I. Gramaglia, N. Killeen, and M. Croft. 2001. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 15:445–455. [DOI] [PubMed] [Google Scholar]
- 31.Weinberg, A.D., A.T. Vella, and M. Croft. 1998. OX-40: life beyond the effector T cell stage. Semin. Immunol. 10:471–480. [DOI] [PubMed] [Google Scholar]
- 32.Cooper, D., P. Bansal-Pakala, and M. Croft. 2002. 4-1BB (CD137) controls the clonal expansion and survival of CD8 T cells in vivo but does not contribute to the development of cytotoxicity. Eur. J. Immunol. 32:521–529. [DOI] [PubMed] [Google Scholar]
- 33.Sauilli, K., S.Y. Lee, J.L. Cannons, W.C. Yeh, A. Santana, M.D. Goldstein, N. Bangia, M.A. DeBenedette, T.W. Mak, Y. Choi, et al. 1998. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J. Exp. Med. 187:1849–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halstead, E.S., Y.M. Mueller, J.D. Altman, and P.D. Katsikis. 2002. In vivo stimulation of CD137 broadens primary antiviral CD8+ T cell responses. Nat. Immunol. 3:536–541. [DOI] [PubMed] [Google Scholar]
- 35.Suresh, M., J.K. Whitmire, L.E. Harrington, C.P. Larsen, T.C. Pearson, J.D. Altman, and R. Ahmed. 2001. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J. Immunol. 167:5565–5573. [DOI] [PubMed] [Google Scholar]
- 36.Kopf, M., C. Ruedl, N. Schmitz, A. Gallimore, K. Lefrang, B. Ecabert, B. Odermatt, and M.F. Bachmann. 1999. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL responses after virus infection. Immunity. 11:699–708. [DOI] [PubMed] [Google Scholar]
- 37.Tan, J.T., J.K. Whitmire, R. Ahmed, T.C. Pearson, and C.P. Larsen. 1999. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J. Immunol. 163:4859–4868. [PubMed] [Google Scholar]
- 38.Bertram, E.M., P. Lau, and T.H. Watts. 2002. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J. Immunol. 168:3777–3785. [DOI] [PubMed] [Google Scholar]