Vaccination with Mage-3a1 Peptide–Pulsed Mature, Monocyte-Derived Dendritic Cells Expands Specific Cytotoxic T Cells and Induces Regression of Some Metastases in Advanced Stage IV Melanoma (original) (raw)
Abstract
Dendritic cells (DCs) are considered to be promising adjuvants for inducing immunity to cancer. We used mature, monocyte-derived DCs to elicit resistance to malignant melanoma. The DCs were pulsed with Mage-3A1 tumor peptide and a recall antigen, tetanus toxoid or tuberculin. 11 far advanced stage IV melanoma patients, who were progressive despite standard chemotherapy, received five DC vaccinations at 14-d intervals. The first three vaccinations were administered into the skin, 3 × 106 DCs each subcutaneously and intradermally, followed by two intravenous injections of 6 × 106 and 12 × 106 DCs, respectively. Only minor (less than or equal to grade II) side effects were observed. Immunity to the recall antigen was boosted. Significant expansions of Mage-3A1–specific CD8+ cytotoxic T lymphocyte (CTL) precursors were induced in 8/11 patients. Curiously, these immune responses often declined after the intravenous vaccinations. Regressions of individual metastases (skin, lymph node, lung, and liver) were evident in 6/11 patients. Resolution of skin metastases in two of the patients was accompanied by erythema and CD8+ T cell infiltration, whereas nonregressing lesions lacked CD8+ T cells as well as Mage-3 mRNA expression. This study proves the principle that DC “vaccines” can frequently expand tumor-specific CTLs and elicit regressions even in advanced cancer and, in addition, provides evidence for an active CD8+ CTL–tumor cell interaction in situ as well as escape by lack of tumor antigen expression.
Keywords: dendritic cells, vaccination, active immunotherapy, melanoma, cytotoxic T lymphocytes
It is now established that the immune system has cells, particularly CD8+ CTLs, that can recognize tumor antigens and kill tumors 1 2. Nevertheless, a major problem is that these T cells are either not induced or only weakly induced, i.e., the T cells are not evident in the systemic circulation. One possibility is that there is inadequate tumor antigen presentation by dendritic cells (DCs), “nature's adjuvant” for eliciting T cell immunity 3. Another is that tumor-reactive T cells are tolerized by the tumors 1 4. Melanoma provides a compelling setting in which to pursue a current goal of cancer immunotherapy, the generation of stronger tumor-specific T cell immunity, particularly with CTLs 4. The majority of tumor antigens identified so far are expressed by melanomas 2. Limited antimelanoma CTL responses have been detected 5, and infusions of IL-2 expanded killer cells can lead to rejection of melanoma 6.
Conventional adjuvants promote antibody rather than CTL responses. Therefore, several novel strategies are being explored to induce tumor-specific T cell immunity. DC vaccination is one of these 3. Immature DCs capture antigens but lack full T cell–stimulatory activity 7. In the presence of appropriate stimuli, such as inflammatory cytokines, the DCs mature. DCs upregulate T cell adhesion and costimulatory molecules as well as select chemokine receptors that guide DC migration into lymphoid organs for priming of antigen-specific T cells. The use of DCs as adjuvants is supported by many animal experiments with primarily mature DCs 3 8. These studies have shown that the injection of tumor antigen–loaded DCs reliably induces tumor-specific CTL responses, tumor resistance, and in some cases, regression of metastases 3 8. In the few pilot trials reported so far for humans, immature DCs have been employed 9 10 11. Scattered tumor responses are reported, but evidence for the induction of tumor-specific CTLs by DC vaccination has not been shown.
We have developed a technique to generate large numbers of homogenous populations of mature and stable DCs from monocytes in the absence of nonhuman proteins 12 13. We are now exploring the use of these DCs as vaccine adjuvants in humans. Here we provide the proof of the principle by demonstrating that three intracutaneous injections of Mage-3A1 peptide–pulsed mature DCs reliably enhance Mage-3A1–specific CD8+ and recall CD4+ T cell immunity in heavily pretreated, progressive stage IV melanoma patients with large tumor loads. Expansions of Mage-3A1–specific CTL responses have not been previously detected after Mage-3A1 peptide vaccination in less advanced melanoma patients 14, underscoring the potent adjuvant properties of DCs. As regressions of metastases also occurred upon DC-mediated immunization and were accompanied by CD8+ T cell infiltration, we propose that the induced Mage-3A1–specific CTLs are active in vivo.
Materials and Methods
Patient Eligibility Criteria
Patients were eligible if they suffered from stage IV (i.e., distant metastases) cutaneous malignant melanoma (1988 American Joint Committee on Cancer/Union Internationale Contre Cancer pTNM staging system) that was not curable by resection and was progressive despite chemo(immuno)therapy. Further inclusion criteria were an expected survival ≥4 mo, Karnofsky index ≥60%, age ≥18 yr, measurable disease, HLA-A1 positivity, expression of Mage-3 gene shown by reverse transcriptase (RT)-PCR in at least one excised metastasis, and no systemic chemo-, radio-, or immunotherapy within 4 wk (6 wk in the case of nitrosurea drugs) preceding the first DC vaccination. A positive skin test to recall antigens was not required. Important exclusion criteria were active central nervous system (CNS) metastasis, any significant psychiatric abnormality, severely impaired organ function (hematological, renal, liver), active autoimmune disease (except vitiligo), previous splenectomy or radiation therapy to the spleen, organ allografts, evidence for another active malignant neoplasm, pregnancy, lactation, or participation (or intent to participate) in any other clinical trial. Concomitant treatment (chemo- or immunotherapy, corticosteroids, investigational drugs, paramedical substances) was prohibited. Palliative radiation or surgical therapy of selected metastases and certain medications (acetaminophen/paracetamol, nonsteroidal antiinflammatory drugs, opiates) to control symptoms were allowed.
Clinical Protocol and Study Design
The study was performed at the Departments of Dermatology in Erlangen, Würzburg, and Mainz, Germany according to standards of Good Clinical Practice for Trials on Medicinal Products in the European Community. The protocol was approved by the Protocol Review Committee of the Ludwig Institute for Cancer Research (New York, NY) and performed under supervision of its Office of Clinical Trials Management as study LUD #97-001. The protocol was also approved by the ethics committees of the involved study centers.
The study design is shown in Table . All patients gave written informed consent before undergoing a screening evaluation to determine their eligibility. Extensive clinical and laboratory assessments were conducted at visits 1, 5, and 8 (Table ) and consisted of a complete physical examination, staging procedures, and standard laboratory values as well as special ones (pregnancy test, free testosterone in males, autoantibody profile, and antibodies to HIV-1/2, human T cell lymphotropic virus type I, hepatitis B virus, and hepatitis C virus). Patients were hospitalized and examined the day before each vaccination and were monitored for 48 h after the DC injections. Adverse events and changes in laboratory values were graded on a scale derived from the Common Toxicity Criteria of the National Cancer Institute, National Institutes of Health, Bethesda, MD.
Table 2.
Study Design

Production of the DC Vaccine
During prestudy screening, we tested a small amount of fresh blood to verify that appropriate numbers of mature DCs could be generated from the patient's monocytes 12. Sufficient DC numbers could be successfully generated in all patients, but in some patients the test generation revealed that TNF-α had to be added to assure full maturation. To avoid repetitive blood drawings, we performed a single leukapheresis during visit 2 to generate DCs as described 13. In short, PBMCs from the leukapheresis (≥1010 nucleated cells) were isolated on Lymphoprep™ (Nycomed Pharma) and divided into three fractions. The first fraction of 109 PBMCs was cultured on bacteriological petri dishes (Cat. #1005; Falcon Labware) coated with human Ig (100 μg/ml; Sandoglobin™; Sandoz GmbH) in complete RPMI 1640 medium (BioWhittaker) supplemented with 20 μg/ml gentamicin (Refobacin 10; Merck), 2 mM glutamine (BioWhittaker), and 1% heat-inactivated human plasma for 24 h to generate monocyte-conditioned medium (MCM) for later use as the DC maturation stimulus. The second fraction of 3 × 108 PBMCs was used for the generation of DCs for vaccination 1 and delayed-type hypersensitivity (DTH) test I. Adherent monocytes were cultured in 1,000 U/ml GM-CSF (10 × 107 U/mg; Leukomax™; Novartis) and 800 U/ml IL-4 (purity >98%; 4.1 × 107 U/mg in a bioassay using proliferation of human IL-4R+ CTLL; CellGenix; expressed in Escherichia coli and produced under good laboratory practice conditions but verified for good manufacturing practice [GMP] safety and purity criteria by us) for 6 d, and then MCM was added to mature the DCs. MCM was supplemented in patients 04, 06, 09, 11, and 12 with 10 ng/ml GMP-rhu TNF-α (purity >99%; 5 × 107 U/mg in a bioassay using murine L-M cells; a gift of Dr. G.R. Adolf, Boehringer Ingelheim Austria, Vienna, Austria) to assure full maturation of DCs. Mature DCs were harvested on day 7. The third fraction of PBMCs was frozen in aliquots and stored in the gas phase of liquid nitrogen to generate DCs for later vaccinations and DTH tests.
DCs for vaccinations were pulsed with the Mage-3A1 peptide 15 (EVDPIGHLY, synthesized at GMP quality by Clinalfa) as tumor antigen, and as a recall antigen and positive control, tetanus toxoid (TT) or tuberculin (if at visit 1 the DTH to TT in the Multitest Merieux was >10 mm; both purchased from the Bacterial Vaccines Department of the Statens Serum Institute, Copenhagen, Denmark). The recall antigen was added at 10 μg/ml for the last 24 h, and the Mage-3A1 peptide was added at 10 μM directly to the cultures for the last 8 h (if immunity to recall antigens was strongly boosted, the dose of recall antigen was reduced to 1.0 or 0.1 μg/ml or was omitted for the intravenous DC injections to avoid a cytokine release syndrome). On day 7, mature DCs were harvested, resuspended in complete medium, washed, and pulsed once more with Mage-3A1 peptide (now at 30 μM) for 60 min at 37°C. DCs were finally washed and resuspended in PBS (GMP quality PBS; BioWhittaker) for injection. DCs to be used for Mage-3A1 DTH tests were pulsed with Mage-3A1 (but no recall antigen); DCs that served as negative control in the DTH tests were not pulsed at all. An aliquot of the DCs to be used for vaccinations was analyzed as described 13 to assure that functionally active and mature DCs were generated. The features of the DCs are described in Results. Release criteria were typical morphology (>95% nonadherent veiled cells) and phenotype (>95% HLA-DR+++CD86+++CD40+CD25+CD14− and >65% homogenously CD83++).
Immunization Schedule
A total of five vaccinations (three into the skin followed by two intravenously) with antigen-pulsed DCs were given at 14-d intervals (Table ). This design was chosen to explore the toxicity and efficacy of various routes in this trial. For vaccinations 1–3, 3 × 106 DCs were given subcutaneously at two sites (1.5 × 106 DCs in 500 μl PBS per site) and 3 × 106 intradermally at 10 sites (3 × 105 DCs in 100 μl PBS per site). The injection sites were the ventromedial regions of the upper arms and the thighs close to the regional lymph nodes and were rotated clockwise. Limbs where draining lymph nodes had been removed and/or irradiated were excluded. For intravenous vaccinations 4 and 5, a total of 6 and 12 × 106 antigen-pulsed DCs (resuspended in 25 or 50 ml PBS plus 1% autologous plasma) was administered over 5 and 10 min, respectively. Premedication with an antipyretic (500 mg acetaminophen/paracetamol p.o.) and an antihistamine (2.68 mg clemastinhydrogenfumarat i.v.) was given 30 min before intravenous DC vaccination.
Evaluation of Immune Status
Recall Antigen–specific Proliferation and Cytokine Production.
PBMCs were cultured in triplicate at two dose levels (3 × 104 and 1 × 105 PBMCs/well) plus or minus TT or tuberculin (at 0.1, 1, and 10 μg/ml) and pulsed on day 5 with [3H]thymidine for 12 h. In all cases, the highest cpms were obtained with the highest doses of PBMCs and antigen and are shown in Fig. 2. IL-4 and IFN-γ levels were measured in culture media by ELISA (Endogen, Inc.). In a separate plate, staphylococcal enterotoxin (SEA; Serva) was added at 0.5, 1, and 5 ng/ml, and proliferation was assessed after 3 d to provide a positive control for helper T cell viability and responsiveness.
Figure 2.

Recall antigen–specific immunity (tuberculin in patient 10; TT in all others) as assayed by antigen-specific proliferation. The cpm values determined after therapy (14 d after vaccination 5) are shown as multiples of pretherapy cpm values. Absolute cpm (cpm with recall antigen minus cpm without antigen) after therapy was 68,917 in patient 02, 85,225 in patient 04, 16,759 in patient 05, 7,913 in patient 06, 16,367 in patient 07, 107,923 in patient 09, 22,790 in patient 10, 4,507 in patient 12, and 1,831 in patient 13 (SEM for all measurements was <20%). Patients 08 and 11 could not be evaluated due to shortage of cells after therapy.
Enzyme-linked Immunospot Assay for IFN-γ Release from Single Antigen–specific T Cells.
To quantitate antigen-specific, IFN-γ–releasing, Mage-3A1–specific effector T cells, an enzyme-linked immunospot (ELISPOT) assay was used as described 16. PBMCs (105 and 5 × 105/well) or in some cases CD8+ or CD4+ T cells (isolated by MACS™ according to the manufacturer, Miltenyi Biotec) were added in triplicate to nitrocellulose-bottomed 96-well plates (MAHA S4510; Millipore Corp.) precoated with the primary anti–IFN-γ mAb (1-D1K; Mabtech) in 50 μl ELISPOT medium (RPMI 1640 and 5% heat-inactivated human serum) per well. For the detection of Mage-3A1–reactive T cells, the APCs were irradiated T2.A1 cells (provided by P. van der Bruggen, Ludwig Institute of Cancer Research, Brussels, Belgium) pulsed with MHC class I–restricted peptides (Mage-3A1 peptide and the HIV-1 p17-derived negative control peptide GSEELRSLY) added at 7.5 × 104/well (final volume 100 μl/well). After incubation for 20 h, wells were washed six times, incubated with biotinylated second mAb to IFN-γ (7-B6-1; Mabtech) for 2 h, washed, and stained with Vectastain Elite kit (Vector Labs.). For detection of TT-reactive T cells, TT was added at 10 μg/ml directly to the PBMCs (1 or 5 × 105 PBMCs/flat-bottomed 96-well plate). Assays were performed on fresh PBMCs. Spots were evaluated and counted using a special computer-assisted video imaging analysis system (Carl Zeiss Vision) as described 16.
Semiquantitative Assessment of CTL Precursors.
The multiple microculture method developed by Romero et al. 17 was used to obtain a semiquantitative assessment of CTLp (precursors) specific for Mage-3A1 peptide. Aliquots of frozen PMBCs were thawed and assayed together. CD8+ T cells were isolated with magnetic microbeads (MACS™ separation columns; Miltenyi Biotec) and seeded at 104/well in 96-well round-bottomed plates in RPMI 1640 with 10% heat-inactivated human serum. The CD8− PBMCs were pulsed with peptide Mage-3A1 or the influenza PB1 control peptide VSDGGPNLY (10 μg/ml; 30 min at room temperature), irradiated (30 Gy from a cesium source), and added as an APC population at 105/well together with IL-2 (10 IU/ml final) and IL-7 (10 ng/ml final) in a total volume of 200 μl/well. On day 7, 100 μl fresh medium was substituted, and peptide Mage-3A1 or PB1 (1 μg/ml final) and IL-2 (10 U/ml) was added. On day 12, each microwell was divided into three equal samples to test cytolytic activity in a standard 4-h 51Cr-release assay on peptide-pulsed (10 μg/ml for 1 h at 37°C) T2A1 cells, nonpulsed T2A1 cells, and K562 target cells, respectively. All of the assays were performed with an 80-fold excess of nonlabeled K562 to block NK activity. Microwells were scored positive if lysis of T2A1 targets with peptide minus lysis without peptide was ≥12% and this specific lysis was greater than or equal to twice the lysis of T2A1 targets without peptide plus six as described 18. We aimed at testing 30 microwells of 104 CD8+ T cells. Therefore, 1/30 positive wells equals at least one CTLp in 3 × 105 (i.e., 30 wells at 104 CTLp per well) CD8+ T cells (corresponding to ∼3 × 106 PBMCs).
DTH.
DTH to Mage-3A1 peptide was assessed by intradermal injection at two sites of each 3 × 105 Mage-3A1 peptide–loaded DC in 0.1 ml PBS. Negative controls were nonpulsed autologous DCs in 0.1 ml PBS and 0.1 ml PBS. DTH to seven common recall antigens (Multitest Merieux) including TT and tuberculin was performed on visits 1, 5, and 8 (Table ).
Assessment and Analysis of Tumor Tissue
For recruitment into the study, Mage-3 gene expression in at least one metastatic deposit had to be demonstrated by RT-PCR as described 14. Accessible superficial skin metastases were removed whenever possible after the vaccinations and subjected to Mage-3 RT-PCR as well as routine histology and immunohistology (to characterize cellular infiltrates).
Statistical Analysis
For analysis of the immune response, pre- and postimmunization values were compared by paired t test after logarithmic transformation of the data. Significance was set at P < 0.05.
Results
Patient Characteristics
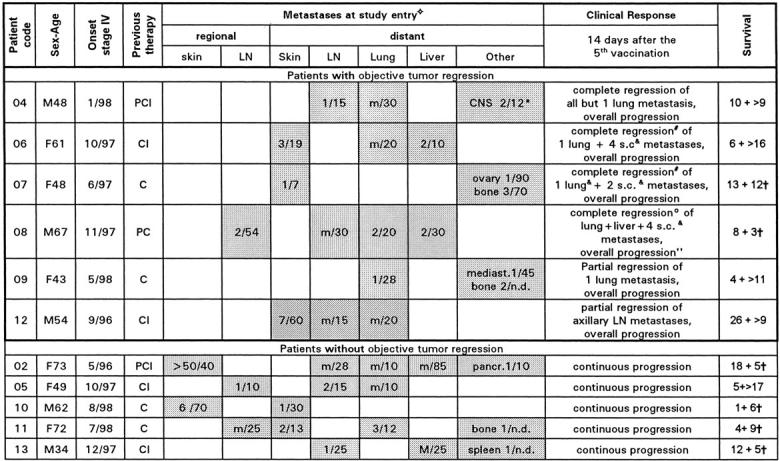
All 13 patients were HLA-A1+, had proven Mage-3 mRNA expression in at least one excised metastasis, and suffered from advanced stage IV melanoma, i.e., distant metastases that were progressive despite chemotherapy and, in some cases, chemoimmunotherapy (Table ). We offered DCs to all patients who fulfilled the inclusion and exclusion criteria, i.e., we did not select for subsets of patients. Two patients (numbers 01 and 03) succumbed to melanoma after two and three vaccinations, respectively. 11 patients received all five planned DC vaccinations in 14-d intervals (Table ) and were thus fully evaluable.
Table 1.
Patients' Characteristics, Status before DC Vaccination, and Response to DC Vaccination
Quality of the Vaccine
All vaccine preparations were highly enriched in mature DCs. More than 95% of the cells were large and veiled in appearance, expressed a characteristic phenotype by flow cytometry (HLA-DR+++CD86+++CD40+CD25+CD14−), and acted as strong stimulators of an MLR at DC/T cell ratios of ≤1:300 13. Most (mean 80%) expressed the CD83 mature DC marker 19. These features were stable upon removal of cytokines and culture for one to two more days 13. The DCs were pulsed with Mage-3A1 peptide as a tumor antigen and TT or tuberculin as a recall antigen. The latter were internal controls for immunization and possibly provided help for CTL responses 20.
Toxicity
No major (above grade II) toxicity or severe side effects were observed in any patient, including the two patients who were not fully evaluable. We noticed, however, local reactions (erythema, induration, pruritus) at the intracutaneous vaccination sites that increased with the number of vaccinations. In 9/11 patients, strong DTH reactions (induration >10 mm in diameter) were noted to DCs carrying a recall antigen (Fig. 1). Elevation of body temperature (grade I and II fever) was observed in most (9/11) patients and was also related to pulsing DCs with recall antigen. The most striking example was patient 02, who progressively developed fever (up to 40°C) upon successive vaccinations but did not show a rise in body temperature when TT was omitted for the final (fifth) vaccination. We observed slight lymph node enlargement, clinically in 63% and by sonography in 83% of patients, after the intracutaneous DC injections. Interestingly, these were delayed, being inapparent during the 2 d of monitoring after vaccinations but detected when patients were investigated again the day before the next vaccination (Table ).
Figure 1.


Local reactions to DCs carrying Mage-3A1 peptide and TT at the intradermal and subcutaneous vaccination sites in patient 09 (24 h after vaccination 2; top panel) and 02 (48 h after vaccination 3; bottom panel). Erythema at the 10 intradermal (left) and 2 subcutaneous (right) vaccination sites was followed by induration >10 mm in diameter (with secondary purpura in patient 02). These local reactions represent strong DTH reactions to DCs carrying TT, as such strong reactions did not occur in response to unpulsed DCs or DCs pulsed with Mage-3A1 peptide alone in DTH tests I–III (Table ; reactions not shown).
Immunological Responses
Boosting of Recall Antigen-specific Immunity.
PBMCs that had been frozen before vaccination and 14 d after vaccination 5 were thawed and assayed together, as specified in the protocol (Table ). In most patients, a significant boost of antigen-specific immunity developed to TT (and tuberculin in patient 10) (P < 0.004; Fig. 2). Supernatants from the proliferative assays contained large amounts of IFN-γ (mean 1,679 pg/ml, range 846–4,325) but little IL-4 (IFN-γ/IL-4, 317:1). In five patients, we studied the kinetics of the immune response to TT by IFN-γ ELISPOT analysis. We found an increase after the intracutaneous vaccinations (P < 0.02) but a peculiar decrease after the intravenous vaccinations (P < 0.008; Fig. 3). Thus, comparing recall immunity before and after all five vaccinations (Fig. 2) as prespecified in the protocol (Table ) obviously underestimated the extent of boosting.
Figure 3.

Kinetic analysis of immunity to recall antigens as assessed by TT-specific IFN-γ ELISPOT (SEM for all measurements was <20%). Blood was drawn (see Table , Study Design) before the first DC vaccination and then every 14 d just before administration of the next DC vaccination (e.g., pre Vacc # 2 means immediately before vaccination 2, i.e., 14 d after vaccination 1), and finally after therapy. Time points at which vaccinations were not performed lack bars. Note the increase after the intracutaneous vaccinations and the decline upon the two vaccinations after intravenous ones. Patient 10, who received tuberculin-pulsed DCs, exhibited no significant change in the TT-specific IFN-γ ELISPOT as expected.
Expansion of Mage-3A1–specific CTLp.
Aliquots of PBMCs, frozen before the first and after the third and fifth vaccinations, were thawed at the same time (Table ) and subjected to a semiquantitative recall assay for CTLp (reference 17; Fig. 4). Before vaccination, CTLp frequencies were low or undetectable. In 8/11 patients, we found a significant expansion of Mage-3A1–specific CTLp as indicated by the increase (mean, eightfold; P < 0.008) of positive microcultures in the multiple microculture procedure employed for the semiquantitative assessment of CTLp 17. Interestingly, in six patients, the CTLp frequencies were maximal after the three intracutaneous vaccinations (P < 0.0013) but then decreased after the two additional intravenous vaccinations in all but one of these patients (P < 0.026). Only in 1/11 patients did we observe an increase of CTLp to an irrelevant PB1 influenza peptide that served as a specificity control (not shown).
Figure 4.

Mage-3A1 CTLp frequency analysis as assessed by semiquantitative recall assay. The y-axis and the numbers above the bars indicate the percentage of positive wells found before vaccination 1, before vaccination 4 (14 d after vaccination 3), and after therapy (usually 14 d after vaccination 5).
ELISPOT Analysis for IFN-γ–releasing, Mage-3A1–specific T Cells.
We also tried to detect Mage-3A1–specific CTL effectors in uncultured fresh, nonfrozen PBMCs by performing ELISPOT analyses at 14-d intervals on all patients. A significant increase of Mage-3A1–reactive IFN-γ spot–forming cells was apparent only in patients 07 and 09 after the first and second vaccinations, respectively, but the frequency of Mage-3A1–specific effectors was very high (∼5,000 and 10,500/107 CD8+ T cells; not shown).
DTH Test to Mage-3A1 Peptide–loaded DCs.
Tests of DTH to Mage-3A1 peptide–loaded DCs yielded erythema and/or induration (>5 mm diameter) in 7/11 patients (not shown). The results were, however, equivocal due to the frequently observed background to nonpulsed DCs (up to 10 mm in diameter) and the variability from test site to test site.
Clinical Responses
At the end of the trial, i.e., ∼2 wk after the fifth vaccination (Table ), we observed temporary growth cessation of some individual metastases, but more intriguingly, in 6/11 patients, complete regression of individual metastases in skin, lymph nodes, lung, and liver (Table and Fig. 5). Resolution of skin metastases was found in three patients (Table , patients 06, 07, and 08) and in two of them (06 and 07), it was preceded by local pain, itching, and slight erythema. The six regressing skin lesions of patients 06 and 07 (Table ) were also excised and examined by immunohistology. Clusters of CD8+ T cells were seen around and in the tumor, the latter often necrotic, suggesting an immune attack (Fig. 6).
Figure 5.
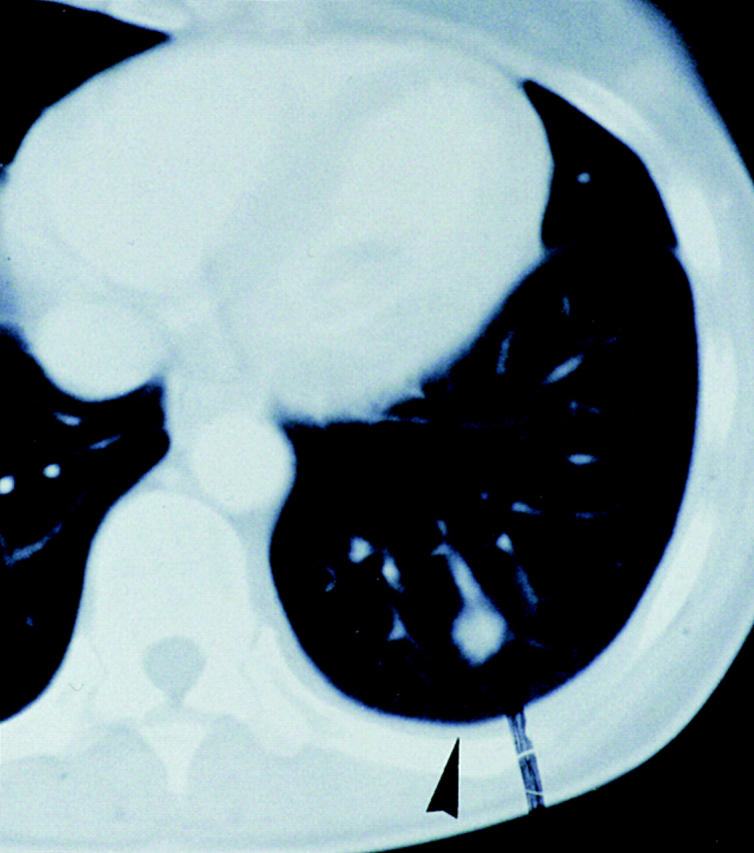
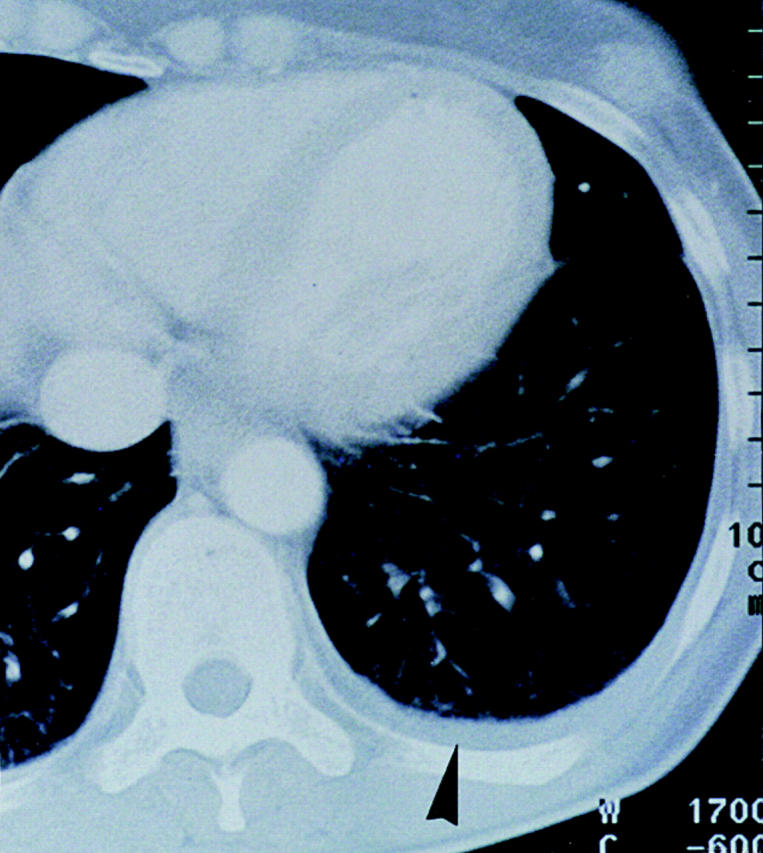
Regression (arrows) of a globular (13 mm in diameter) lung metastasis in patient 07 that was then no longer detectable in serial 6-mm-thick computed tomography scans.
Figure 6.
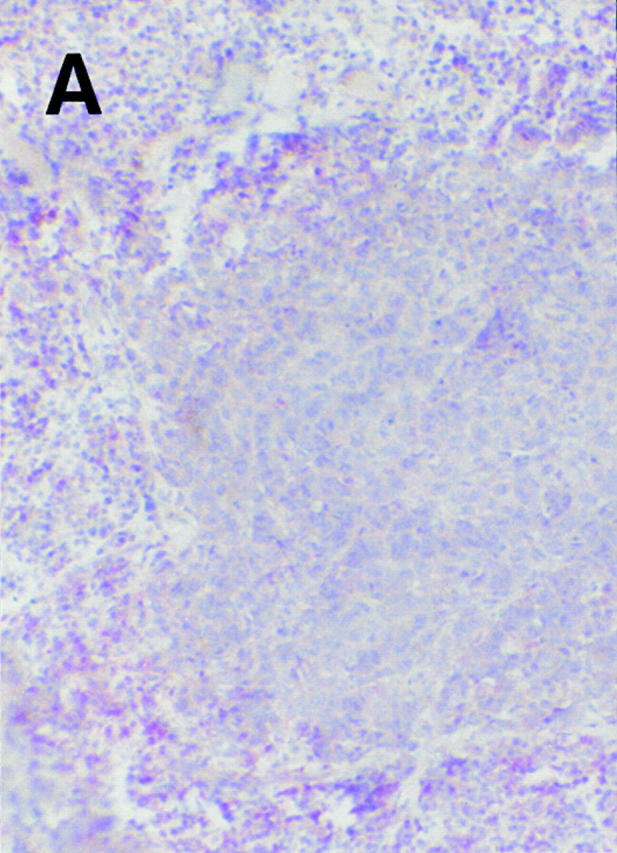
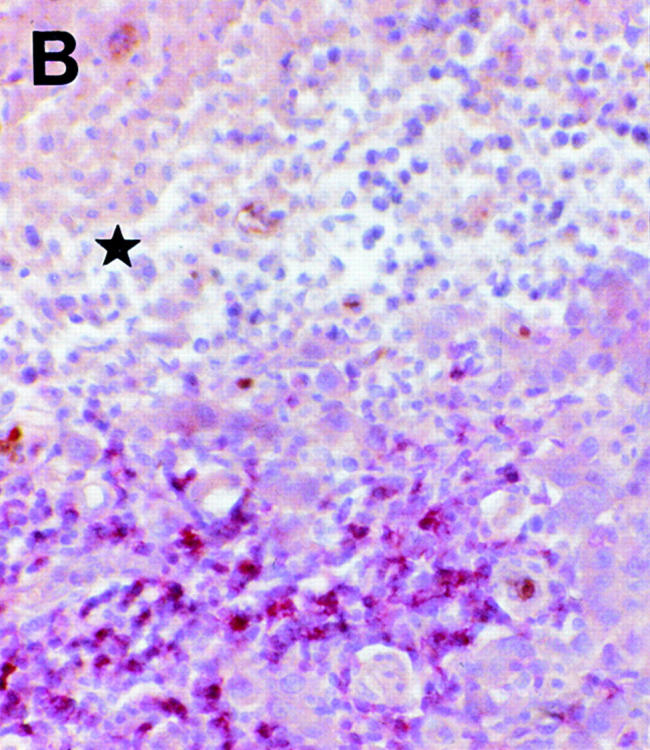
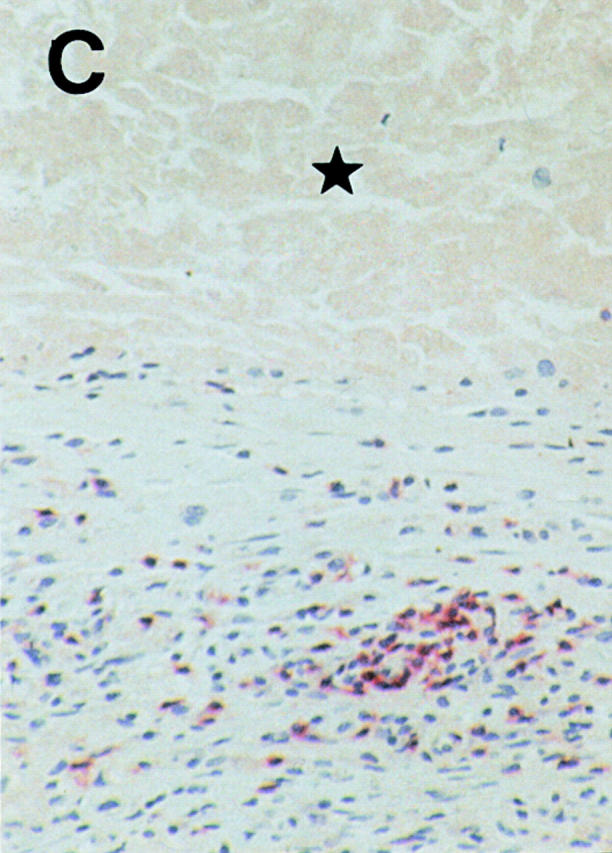
Regressing subcutaneous metastases in patient 06 display a CD8+ lymphocytic infiltrate (alkaline phosphatase/antialkaline phosphatase immunohistochemical staining with anti-CD8 mAb) that surrounds (A) and infiltrates (B) the tumor. Areas of damaged (B, ★) and necrotic (C, ★) melanoma cells are obvious in the vicinity of the CD8+ T cell infiltrate. The metastasis expressed Mage-3, as demonstrated by RT-PCR (data not shown). Magnifications: A, 100; B, 250; C, 160.
In patients 06 and 08, the metastases excised at study entry (four and two, respectively) proved to be Mage-3 mRNA+. However, all of the samples removed at the end (two and six, respectively) were Mage-3 mRNA−, suggesting immunoselection for antigen-negative tumor cells. Remarkably, significant infiltration of CD8+ T cells was not found in any of these lesions.
Discussion
In deciding on the source of DCs for this phase I trial, we selected mature, monocyte-derived DCs for the following reasons. Monocyte-derived DCs currently represent the most homogenous and potent DC populations, with several defining criteria and quality controls 12 13 21. The method for generating production of these DCs is very reproducible and allows the cryopreservation of large numbers of cells at an identical stage of development 12 13. Furthermore, these DCs can be produced in the absence of potentially hazardous FCS 12 13 21. FCS exposure also leads to large syngeneic T cell responses in culture, so their clinical use 11 might produce nonspecific immunostimulatory effects. Unlike other investigators 9 10 11, we chose to use mature rather than immature DCs for our first melanoma trial. The DCs that have been used with efficacy in animal experiments were primarily mature 3 8. Mature DCs are much more potent in inducing CTL and Th1 responses in vitro (reference 22 and Jonuleit, H., A. Gieseke, A. Kandemir, L. Paragnik, J. Knop, and A.H. Enk, manuscript in preparation), and the DCs are also resistant to the immunosuppressive effects of IL-10 23 that can be produced by tumors 24 25 26. Mature DCs also display an extended half-life of antigen-presenting MHC class I 26a and class II molecules 27. Finally, mature DCs have a high migratory activity 21 and express CCR7 28, a receptor for chemokines produced constitutively in lymphoid tissues 28. Mature DCs, as used in this cancer therapy trial, have recently also been shown to rapidly generate broad T cell immunity in healthy subjects 28.
Mature DCs were loaded with only one melanoma peptide, Mage-3A1, to avoid uncertainties regarding loading of DCs with multiple peptides 11 of varying affinity and off rate. Successful loading was verified with a Mage-3A1–specific CTL clone and ELISPOT analysis (not shown). The Mage-3A1 peptide 15 was selected for several reasons. It is essentially tumor specific 2 and expressed in tumors other than melanoma 2, and the Mage-3A1 epitope is likely a rejection antigen 14. Moreover, the Mage-3A1 CTLp frequency is exceedingly low in noncancer patients (reference 18; 0.4–3 per 107 CD8+ T cells) as well as in cancer patients, even after peptide vaccination 14. Thus, any induction or boost of Mage-3A1 CD8+ T cell responses would indicate a significant superiority in the adjuvant capacities of DCs.
DTH assays with peptide-pulsed DCs were carried out as described by Nestle et al. 11 to detect Mage-3A1 immunity (not shown). However, we did not detect unequivocal DTH. This was due to the frequently observed background to nonpulsed DCs (possibly due to cytokine production by DCs) and the noteworthy variability from test site to test site. As Mage-3A1–specific T cells are CD8+ T cells and DTH assays typically detect primed CD4+ T cells, we suspect that DTH to MHC class I peptide–pulsed DCs may also for this reason prove not to be a sensitive or reliable way to monitor specific CD8+ T cell–mediated immunity.
In contrast, we found sizable expansions of Mage-3A1–specific CTL precursors in PBMCs from a majority (8/11) of patients (P < 0.008; Fig. 4). This is an important proof of the principle of DC-based immunization, and it is also significant from the point of view that tumors can induce tolerance or anergy. It is very promising that CTLp expansions can be induced in far advanced and heavily pretreated stage IV melanoma patients. However, active Mage-3A1–specific effectors were generally not observed in ELISPOT assays, except for in two patients with high frequencies (>5,000/107 CD8+ T cells). Perhaps active CD8+ effectors were rapidly sequestered in the numerous metastases, as suggested by the biopsy studies illustrated in Fig. 6. An alternative explanation is that looking for effectors in peripheral blood 14 d after a preceding vaccination might simply be too late.
Interestingly, in six patients, CTLp had increased to their highest levels after the three intracutaneous vaccinations (P < 0.0013) and then decreased (P < 0.026) with subsequent intravenous immunizations (Fig. 4). The decrease in CTLp might be due to emigration of activated Mage-3–reactive CTLs into tissues, tolerance induction, or clonal exhaustion via the intravenous route. We also observed decreased responses to recall antigens in the five patients that we studied before and after intravenous vaccination (Fig. 3). The effect of the intravenous route requires additional study, as it may be counterproductive. In contrast, our results clearly demonstrate that the intracutaneous route is effective, so that the less practical intranodal injection propagated by other investigators 11 does not seem essential. It will, however, be necessary to compare subcutaneous and intradermal routes to find out if one is superior.
We found regression of individual metastases in 6/11 patients when patients were staged 14 d after the fifth vaccination (Table ). This percentage of responses was unexpected in far advanced stage IV melanoma patients who were all progressive despite standard chemotherapy and even chemoimmunotherapy. In the study by Nestle et al. 11, chemotherapy was only given to 4/16 melanoma patients, and objective tumor responses were observed in 5/16. Therefore, we attribute the regressions to DC-mediated induction of Mage-3A1–specific CTLs. This interpretation is supported by the heavy infiltration with CD8+ T cells of regressing but not nonregressing (skin) metastases. The observation that all of the metastases in patients 06 and 08 that were excised at the end of the study were Mage-3 mRNA− (whereas those removed at the onset were uniformly positive) suggests immune escape of and selection for Mage-3 antigen–negative tumors. Immune escape might also have been responsible for the lack of tumor response in those nonresponders that had mounted a Mage-3A1–specific CTL response.
After the end of the trial, surviving patients received further vaccinations with DCs and several tumor peptides (Mage-1, tyrosinase, and Mage-3) that were no longer part of the protocol. It is encouraging that 5/11 patients are still alive (Table ) 9–17 mo after study entry, as the expected median survival in patients progressive after chemo(immuno)therapy is only 4 mo 29 30. One of the initial responders (patient 06) has recently experienced a complete response and has now been disease free for 2 mo. It is interesting that Marchand et al. 14 have also observed that regressions, once they have started, proceed slowly and may take months to complete.
In conclusion, the use of a defined DC vaccine combined with detailed immunomonitoring provides proof that vaccination with mature DCs expands tumor-specific T cells in advanced melanoma patients. In addition, we have found some evidence for the direct interaction between CD8+ CTLs and tumor cells as well as for escape of antigen-negative metastases. We are convinced that DC-mediated immunization can be intensified further to reveal the presence of expanded populations of effector cells. Efficacy might be increased at the level of the DC, e.g., by optimizing variables such as DC maturational state, route, dose, and schedule or by improving the short life span of DCs in vivo 31 32; at the level of the T cell, e.g., by providing melanoma-specific CD4+ T cell help 33 34 or IL-2 35; and by treating patients earlier in their disease course.
Acknowledgments
We are grateful to all patients for their confidence and cooperation. We appreciate the support of J. Knop, Head of the Department of Dermatology in Mainz. We thank our colleagues H.B.-R. Balda, H. Hintner, F.S.M. Meurer, and C.R. Neumann for referring patients and T.L. Diepgen for statistical analysis. We are particularly grateful to T. Boon and P. van der Bruggen, who helped us in many ways, and to A. Knuth and T. Woelfel for help in establishing the semiquantitative CTL and ELISPOT assays, respectively. We are obliged to the Protocol Review Committee and Office of Clinical Trials Management of the Ludwig Institute for Cancer Research, in particular H.F. Oettgen and E. Hoffman, for their suggestions on improving the protocol and for supervising the trial.
This work was supported by a grant from the Deutsche Krebshilfe (project #70-2291). B. Thurner was supported by the Training and Mobility of Researchers (TMR) programme of the European Union (EUNIDI).
Footnotes
Abbreviations used in this paper: CNS, central nervous system; DCs, dendritic cells; DTH, delayed-type hypersensitivity; MCM, monocyte-conditioned medium; RT, reverse transcriptase; TT, tetanus toxoid.
References
- Schreiber H. Tumor Immunology. In: Paul W.E., editor. Fundamental Immunology. Lippincott-Raven Publishers; Philadelphia: 1999. pp. 1237–1270. [Google Scholar]
- Van den Eynde B.J., van der Bruggen P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- Schuler G., Steinman R.M. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J. Exp. Med. 1997;8:1183–1187. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D.M. Cancer vaccines Nat. Med. 4Suppl.1998. 525 531 [DOI] [PubMed] [Google Scholar]
- Romero P., Dunbar P.R., Valmori D., Pittet M., Ogg G.S., Rimoldi D., Chen J.L., Lienard D., Cerottini J.C., Cerundolo V. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J. Exp. Med. 1998;188:1641–1650. doi: 10.1084/jem.188.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S.A. New opportunities for the development of cancer immunotherapies Cancer J. Sci. Am. 4Suppl.1998. S1 4 [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;393:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Lotze M.T., Farhood H., Wilson C.W., Storkus W.J. Dendritic cell therapy of cancer and HIV infection. In: Lotze M.T., Thomson A., editors. Dendritic CellsBiology and Clinical Applications. Academic Press; San Diego, CA: 1999. pp. 459–485. [Google Scholar]
- Hsu F.J., Benike C., Fagnoni F., Liles T.M., Czerwinski D., Taidi B., Engleman E.G., Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- Murphy G., Tjoa B., Ragde H., Kenny G., Boynton A. Phase I clinical trialT-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate. 1996;29:371–380. doi: 10.1002/(SICI)1097-0045(199612)29:6<371::AID-PROS5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Nestle F.O., Alijagic S., Gilliet M., Sun Y., Grabbe S., Dummer R., Burg G., Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- Romani N., Reider D., Heuer M., Ebner S., Kämpgen E., Eibl B., Niederwieser D., Schuler G. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J. Immunol. Methods. 1996;196:137–151. doi: 10.1016/0022-1759(96)00078-6. [DOI] [PubMed] [Google Scholar]
- Thurner B., Röder C., Dieckmann D., Heuer M., Kruse M., Glaser A., Keikavoussi P., Kämpgen E., Bender A., Schuler G. Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J. Immunol. Methods. 1999;223:1–15. doi: 10.1016/s0022-1759(98)00208-7. [DOI] [PubMed] [Google Scholar]
- Marchand M., van Baren N., Weynants P., Brichard V., Dreno B., Tessier M.H., Rankin E., Parmiani G., Arienti F., Humblet Y. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int. J. Cancer. 1999;80:219–230. doi: 10.1002/(sici)1097-0215(19990118)80:2<219::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Gaugler B., Van den Eynde B., van der Bruggen P., Romero P., Gaforio J.J., De Plaen E., Lethe B., Brasseur F., Boon T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 1994;179:921–930. doi: 10.1084/jem.179.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr W., Linn B., Leister N., Wandel E., Meyer zum Buschenfelde K.H., Wölfel T. The use of computer-assisted video image analysis for the quantification of CD8+ T lymphocytes producing tumor necrosis factor alpha spots in response to peptide antigens. J. Immunol. Methods. 1997;203:141–152. doi: 10.1016/s0022-1759(97)00019-7. [DOI] [PubMed] [Google Scholar]
- Romero P., Cerottini J.C., Waanders G.A. Novel methods to monitor antigen-specific cytotoxic T-cell responses in cancer immunotherapy. Mol. Med. Today. 1998;4:305–312. doi: 10.1016/s1357-4310(98)01280-5. [DOI] [PubMed] [Google Scholar]
- Chaux P., Vantomme V., Coulie P., Boon T., van der Bruggen P. Estimation of the frequencies of anti-MAGE-3 cytolytic T-lymphocyte precursors in blood from individuals without cancer. Int. J. Cancer. 1998;77:538–542. doi: 10.1002/(sici)1097-0215(19980812)77:4<538::aid-ijc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Zhou L.J., Tedder T.F. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J. Immunol. 1995;154:3821–3835. [PubMed] [Google Scholar]
- Lanzavecchia A. Immunology. Licence to kill. Nature. 1998;393:413–414. doi: 10.1038/30845. [DOI] [PubMed] [Google Scholar]
- Jonuleit H., Kuhn U., Muller G., Steinbrink K., Paragnik L., Schmitt E., Knop J., Enk A.H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- Dhodapkar M.V., Steinman R.M., Sapp M., Desai H., Fossella C., Krasovsky J., Donahoe S.M., Dunbar P.R., Cerundolo V., Nixon D.F. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J. Clin. Invest. 1999;104:173–180. doi: 10.1172/JCI6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrink K., Jonuleit H., Müller G., Schuler G., Knop J., Enk A.H. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8+ T cells resulting in a failure to lyse tumor cells. Blood. 1999;93:1634–1642. [PubMed] [Google Scholar]
- Enk A.H., Jonuleit H., Saloga J., Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int. J. Cancer. 1997;73:309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Sato T., McCue P., Masuoka K., Salwen S., Lattime E.C., Mastrangelo M.J., Berd D. Interleukin 10 production by human melanoma. Clin. Cancer Res. 1996;2:1383–1390. [PubMed] [Google Scholar]
- Dummer W., Becker J.C., Schwaaf A., Leverkus M., Moll T., Bröcker E.B. Elevated serum levels of interleukin-10 in patients with metastatic malignant melanoma. Melanoma Res. 1995;5:67–68. doi: 10.1097/00008390-199502000-00008. [DOI] [PubMed] [Google Scholar]
- Keikavoussi P., Carstens C., Scheicher C., Koch F., Fries W., Brocker E.B., Koch N., Kämpgen E. Full maturation of human monocyte derived dendritic cells results in stable expression of MHC class I molecules Arch. Dermatol. Res. 291 1999. 110(Abstr.) [Google Scholar]
- Cella M., Engering A., Pinet V., Pieters J., Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- Dieu M.C., Vanbervliet B., Vicari A., Bridon J.M., Oldham E., Ait-Yahia S., Briere F., Zlotnik A., Lebecque S., Caux C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch C.M., Houghton A.N., Sober A.J., Soong S. Cutaneous Melanoma 3rd ed 1998. Quality Medical Publishing Corporation; St. Louis, MO: pp. 596 [Google Scholar]
- Kamanabrou D., Straub C., Heinsch M., Lee C., Lippold A. Sequential biochemotherapy of INF-α/IL-2, cisplatin (CDDP), dacarbacine (DTIC) and carmustine (BCNU). Result of a monocenter phase II study in 109 patients with advanced metastatic malignant melanoma (MMM) Proc. Am. Soc. Clin. Oncol 18 1999. 2044a(Abstr.) [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois B., Van Kooten C., Durand I., Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong B.R., Josien R., Lee S.Y., Sauter B., Li H.L., Steinman R.M., Choi Y. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell–specific survival factor. J. Exp. Med. 1997;186:2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S.L., Gonzales M.I., Parkhurst M., Li Y.F., Southwood S., Sette A., Rosenberg S.A., Robbins P.F. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR–restricted tyrosinase epitopes. J. Exp. Med. 1996;183:1965–1971. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaux P., Vantomme V., Stroobant V., Thielemans K., Corthals J., Luiten R., Eggermont A.M., Boon T., van der Bruggen P. Identification of MAGE-3 epitopes presented by HLA-DR molecules to CD4(+) T lymphocytes. J. Exp. Med. 1999;189:767–778. doi: 10.1084/jem.189.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K., Fields R.C., Giedlin M., Mule J.J. Systemic administration of interleukin 2 enhances the therapeutic efficacy of dendritic cell-based tumor vaccines. Proc. Natl. Acad. Sci. USA. 1999;96:2268–2273. doi: 10.1073/pnas.96.5.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]