Immunotherapy with Dendritic Cells Directed against Tumor Antigens Shared with Normal Host Cells Results in Severe Autoimmune Disease (original) (raw)
Abstract
Vaccination with dendritic cells (DCs) presenting tumor antigens induces primary immune response or amplifies existing cytotoxic antitumor T cell responses. This study documents that antitumor treatment with DCs may cause severe autoimmune disease when the tumor antigens are not tumor-specific but are also expressed in peripheral nonlymphoid organs. Growing tumors with such shared tumor antigens that were, at least initially, strictly located outside of secondary lymphoid organs were successfully controlled by specific DC vaccination. However, antitumor treatment was accompanied by fatal autoimmune disease, i.e., autoimmune diabetes in transgenic mice expressing the tumor antigen also in pancreatic β islet cells or by severe arteritis, myocarditis, and eventually dilated cardiomyopathy when arterial smooth muscle cells and cardiomyocytes expressed the transgenic tumor antigen. These results reveal the delicate balance between tumor immunity and autoimmunity and therefore point out important limitations for the use of not strictly tumor-specific antigens in antitumor vaccination with DCs.
Keywords: dendritic cells, immunotherapy, autoimmunity, tumor immunity, vaccination
Introduction
A large number of tumor antigens has been identified, and antitumor responses against these antigens can be elicited in mice or humans 1 2 3 4. Tumor antigens most suitable for immunotherapy are, ideally, the strictly tumor-specific antigens that are shared by different tumors or mutated proteins expressed uniquely by tumor cells 2. However, many tumor antigens that are shared with other tissues are recognized by tumor-specific CTLs, such as the melanocyte differentiation antigen tyrosinase-related protein 2 (TRP-2; references 5 and 6) or the antigen p53 7, which is specifically overexpressed in tumors. Therefore, for certain tumor antigens, an efficient immune response may potentially be harmful, because immunity against self-antigens may cause autoimmunity. Indeed, efficient tumor control in human patients may be associated with autoimmune phenomena, e.g., antimelanoma immunity that sometimes correlates with vitiligo 8, certain gynecological tumors that appear to induce paraneoplastic cerebellar degeneration 9, or Wegener's granulomatosis, which is often associated with renal cell carcinoma 10. It is therefore important to assess the potential sequelae of immunity against tumor antigens that are also expressed by normal cells of peripheral organs, i.e., antigens that are located strictly outside of the immune system and are shared between tumors and normal host tissues. It has been shown that such antigens are largely ignored by T cells but can be reacted against if sufficient antigen reaches secondary lymphoid organs during a sufficient time period 11.
Dendritic cells (DCs) are potent immunostimulatory cells facilitating antigen transport to lymphoid tissues and efficient stimulation of T cells 12 13. It has thus been suggested that targeting immunogenic tumor antigens to DCs may represent an efficient means for induction of antitumor immunity 14. Indeed, DC vaccination against experimental murine model tumors has given encouraging results 15 16 17 18 19 20. However, to be effective, DC vaccination usually had to be initiated before or within a few days after tumor cell transfer. Although in some of these studies tumor antigens not exclusively expressed by the tumor had been used 18 19 20, autoimmune side-effects had not been observed. This lack of autoimmunity may be due to the fact that only low CTL activities present for a relatively short time were sufficient to control and clear these early and small tumors without or before causing clinically apparent autoimmune disease. In contrast, clinically established tumors seem to require a strong and sustained CTL response to guarantee complete remission of more sizeable tumors 21. Although strong antitumor CTLs directed against a unique and truly tumor-specific antigen may cure the tumor without causing autoimmunity, this may be a serious problem for CTL responses against antigens shared between tumor cells and nonneoplastic cells. In the latter case, it is important to evaluate the optimal conditions for induction of tumor-specific CTLs by antitumor vaccination and whether a therapeutic window exists between induction of tumor immunity and autoimmunity against so far immunologically ignored self-antigens.
Materials and Methods
Mice and Viruses.
Mice were obtained from the Institut für Labortierkunde (University of Zürich, Switzerland). Transgenic mice expressing lymphocytic (L)CMV-GP (glycoprotein) under the control of the rat insulin promotor (RIP-GP mice; reference 22), transgenic mice expressing the LCMV GP33 epitope in all tissues (H8 mice; reference 23), and SM-LacZ mice expressing β-galactosidase under the control of the SM22α promotor (2126nlz; reference 24) have been previously described. Experiments were carried out with age- (8–16 wk) and sex-matched animals. The β-galactosidase–recombinant vaccinia virus (VV-LacZ) was provided by Dr. R. Drillien (Université Louis Pasteur, Strasbourg, France) and grown and plaqued on BSC-40 cells.
Preparation and Peptide Pulse of DCs.
Generation of DCs from bone marrow cultures of H8 and C57BL/6 mice has been described 25. C57BL/6 DCs were pulsed with β-galactosidase peptide β-gal 497–504 (reference 26) at a concentration of 10−6 M for 60 min at 37°C. Cells were washed three times with BSS and injected intravenously in a volume of 0.5 ml of BSS.
Transplantation of Tumor Pieces and Monitoring of Tumor Growth.
The MC-GP cell line 27 and the EL4-LacZ cell line 26 were described previously. Tumor cells in single-cell suspensions were injected subcutaneously into the flanks of T cell–immunodeficient mice (H-2b RAG-1−/−). Growing tumor pieces were dissected into small tumor pieces of 2 × 2 × 2 mm containing ∼2–5 × 106 tumor cells and transplanted into the flanks of naive recipient mice. Tumor size was assessed twice per week, and the animals were killed when the tumor volume reached ∼7 cm3. Tumor volume was calculated by the formula V = π × abc/6, where a, b, and c are the orthogonal diameters. Tumor cells were checked before transplantation and at the end of each experiment for tumor antigen expression.
Cytotoxicity Assays.
Spleen cells (4 × 106 per well) from primed mice were restimulated for 5 d in 24-well tissue culture plates with 2 × 106 β-gal 497–504-pulsed, irradiated (3,000 rads) spleen cells in IMDM supplemented with 10% FCS, penicillin/streptomycin, and 0.001 M 2-ME. Restimulated spleen effector cells from one well were resuspended in 1 ml of MEM/2% FCS, and threefold serial dilutions were made (indicated as dilution of culture). EL-4 cells were pulsed with β-gal 497–504 (10−6 M; 1.5 h at 37°C) and used in a standard 5-h 51Cr-release assay. Unlabeled EL-4 cells served as controls.
Cell Rejection Assay.
To monitor DC-induced CTL activity directly in vivo, naive or SM-LacZ mice primed with 2 × 105 β-gal 497–504-pulsed DCs on days 0 and 2 were adoptively transfused with β-gal 497–504-pulsed splenocytes on day 8. β-gal 497–504-pulsed splenocytes were labeled with a high intensity of CSFE (5- and 6-carboxyfluorescein diacetate succinimidyl ester; Molecular Probes, Inc.) fluorescence, whereas unpulsed control cells were labeled with a low intensity of CSFE fluorescence. 4 × 107 cells from each preparation were transferred intravenously into naive or DC-primed mice, and the percentage of CSFE-positive cells in PBL was determined by FACS® analysis after 24 h. Erythrocytes were lysed with FACS lysis solution (Becton Dickinson), and the cell suspensions were analyzed on a FACScan™ flow cytometer (Becton Dickinson).
Measurement of Blood Glucose.
The glucose concentration in blood obtained from a tail vein was measured using an ELITE hemoglucometer (Bayer AG). Mice were considered diabetic with values >14 mM at two consecutive measurements.
Immunohistology.
Freshly removed organs were immersed in HBSS and snap-frozen in liquid nitrogen or fixed in 4% buffered formalin and subsequently embedded in paraplast. Frozen tissue sections were cut in a cryostat and fixed in acetone for 10 min. Sections were incubated with anti–mouse mAb against CD8+ cells (YTS169.4.2; reference 28), followed by goat anti–rat Ig (Caltag Labs.) and alkaline phosphatase–labeled donkey anti–goat Ig (Jackson ImmunoResearch Labs.). Endothelial cells and smooth muscle cells were stained on deparaffinized sections using rabbit anti–von Willebrand factor (DAKO A/S) or anti–smooth muscle actin (clone 1A4; Sigma Chemical Co.), respectively. Alkaline phosphatase was visualized using AS-BI phosphate/new fuchsin. Sections were counterstained with hemalum.
Results
Development of Autoimmune Diabetes as a Consequence of Curative Tumor Immunity.
We have previously shown that both the strength of the initial activation and the maintenance of CTL activity by repetitive DC immunization are important parameters for the induction of an autoimmune disease via DCs 29. Correspondingly, a single immunization with DCs is usually not sufficient to cure an established and peripherally growing tumor; maintenance of CTL activity by repetitive DC immunization is required to control and to eventually cure such a tumor 21. In a first set of experiments, we evaluated the consequences of an antitumor response against a model tumor antigen that is also expressed on strictly peripheral pancreatic β islet cells. RIP-GP mice express the GP of LCMV exclusively in pancreatic β islet cells, where the neo–self-antigen is immunologically ignored unless mice are infected with LCMV 22 or immunized with GP-expressing DCs 29 to induce an appropriate anti–LCMV-GP–specific immune response in secondary lymphoid organs. LCMV-GP was also transfected into fibrosarcoma cell line MC57 (MC-GP; references 21 and 27). Small pieces of MC-GP tumors (2 × 2 × 2 mm) were implanted subcutaneously into RIP-GP mice, and therapeutic treatment was started when successful tumor growth could be determined by palpation (tumor diameter ≥5 mm, usually around day 14 after transplantation). Normal C57BL/6 mice do not generate an anti–LCMV-GP response after transplantation of MC-GP tumor pieces because tumor cells do not reach lymphoid organs 21. Similarly, in RIP-GP mice, MC-GP tumor pieces grew without inducing a specific immune response, and the mice remained normoglycemic (Fig. 1 A). When treatment of mice was started 14 d after transplantation with repetitive injection of 2 × 105 DCs constitutively expressing the immunodominant CTL epitope of LCMV-GP (H8-DC), the treatment also led to efficient tumor control in RIP-GP mice (Fig. 1 B). However, these recipients rapidly developed diabetes with an early onset and a severe disease outcome (30% of the mice died before day 21; Fig. 1 B), comparable to or slightly more severe than that of RIP-GP mice without MC-GP tumors treated with the same protocol (10% of the mice died before day 21; Fig. 1C).
Figure 1.
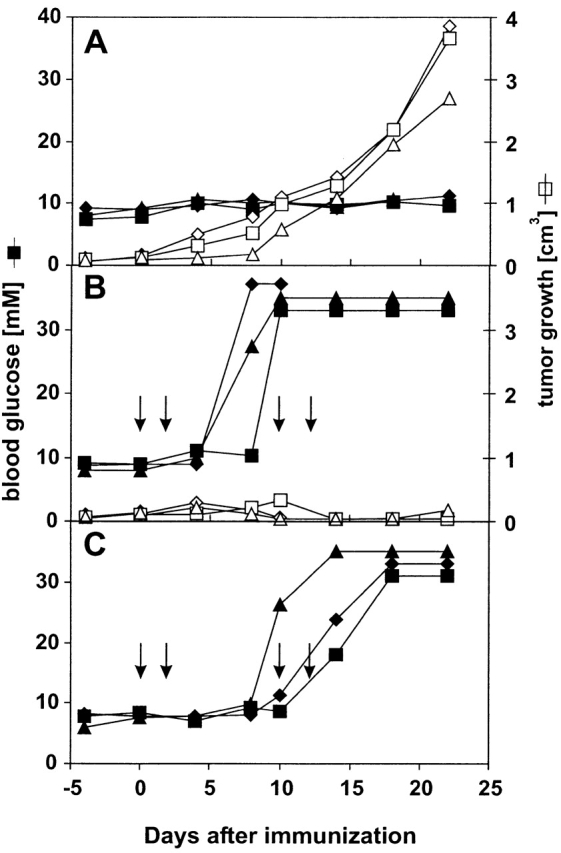
DC treatment of peripherally growing MC-GP tumors leads to diabetes in RIP-GP mice. Mice with established MC-GP tumors (diameter >5 mm on day 14) were used for immunotherapy. Closed symbols indicate blood glucose levels; open symbols represent tumor volume in three individual mice at the indicated time points after beginning of treatment with H8-DC. (A) Tumor growth and blood glucose levels in untreated RIP-GP mice. (B) Successful treatment of MC-GP tumors with GP33-expressing H8-DCs in RIP-GP mice is accompanied by rapidly developing hyperglycemia. Mice were immunized intravenously with 2 × 105 H8-DCs on days 0, 2, 10, and 12. (C) Development of hyperglycemia in RIP-GP mice after repetitive intravenous immunization with 2 × 105 H8-DCs on days 0, 2, 10, and 12. Results from one of three comparable experiments are shown.
Importantly, RIP-GP mice also became diabetic when the tumor treatment was started on day 5 after transplantation, requiring lower doses of H8-DC to achieve tumor control (Table ). Table summarizes all experiments, showing that in RIP-GP mice, antitumor treatment with H8-DC was accompanied by autoimmune diabetes in every case where CTL activity was maintained for a prolonged period of time. Apparently, in this model situation using a tumor antigen with known distribution, there was no window for curative tumor immunity without the side-effect of autoimmune diabetes.
Table 1.
Rejection of Established Fibrosarcomas and Development of Diabetes in RIP-GP and C57BL/6 Mice after Curative Treatment with H8-DCs
| Recipient mice/tumor | Treatment | Tumor control | Incidence of diabetes |
|---|---|---|---|
| Experiment 1: treatment started on day 14 (established, palpable tumors only) | |||
| RIP-GP/MC-GP | 2 × 105 H8-DCs | 10/10 | 10/10 |
| RIP-GP/none | 2 × 105 H8-DCs | — | 10/10 |
| C57BL/6/MC-GP | 2 × 105 H8-DCs | 10/10 | 0/10 |
| RIP-GP/MC-GP | None | 0/6 | 0/6 |
| Experiment 2: treatment started on day 5 (tumors not yet palpable, tumor take 80%) | |||
| RIP-GP/MC-GP | 2 × 104 H8-DCs | 7/10 | 10/10 |
| RIP-GP/none | 2 × 104 H8-DCs | — | 4/4 |
| C57BL/6/MC-GP | 2 × 104 H8-DCs | 6/10 | 0/10 |
| RIP-GP/MC-GP | None | 1/5 | 0/5 |
Induction of Severe Cardiovascular Autoimmune Disease during Antitumor Treatment with DCs.
To evaluate the close linkage between tumor therapy and the possible induction of autoimmunity against so far immunologically ignored peripheral self-antigens by DC treatment, we used a second tumor model. A transgenic mouse line was established that expresses the β-galactosidase antigen in cardiomyocytes of the right ventricle and in arterial smooth muscle cells (SM-LacZ mice; reference 24). Comparable to the RIP-GP mice described above, SM-LacZ mice possess a normal immune reactivity against β-galactosidase. This is shown by the fact that the β-galactosidase–specific CTL response after immunization with recombinant β-galactosidase–expressing vaccinia virus (VV-LacZ) was similar in SM-LacZ and control C57BL/6 mice (Fig. 2 A). Furthermore, SM-LacZ mice rapidly rejected β-gal 497–504-pulsed spleen cells after priming with DCs exogenously loaded with β-gal 497–504 with kinetics similar to those of control C57BL/6 mice (Fig. 2 B). Repetitive priming of SM-LacZ mice with DCs presenting β-galactosidase peptide caused vascular immunopathology with strong lymphocytic infiltration in small and medium-sized arteries and in the right ventricle, whereas repetitive DC treatment of nontransgenic littermates resulted in a mild, nonspecific lymphocyte infiltration in the liver but was not associated with cardiovascular immunopathology. Furthermore, immunization of SM-LacZ mice with DCs pulsed with an irrelevant peptide did not lead to specific anti–β-galactosidase CTL activity and again induced only a mild, nonspecific liver infiltration (Ludewig, B., unpublished results). In naive SM-LacZ mice, specific CTL reactivity was not detectable (Fig. 2 B), and lymphocytes did not infiltrate the right heart muscle or transgene-positive arteries (not shown). Thus, in SM-LacZ mice, the β-galactosidase transgene is immunologically ignored, despite the widespread expression in the vascular system.
Figure 2.

Immune responses against the β-galactosidase antigen in SM-LacZ mice. (A) SM-LacZ mice or control C57BL/6 mice (B6) were intravenously immunized with 2 × 106 pfu of VV-LacZ. 8 d later, spleen cells were restimulated in vitro for 5 d with peptide-labeled, irradiated spleen cells. Specific lysis was measured on β-gal 497–504-labeled EL4 target cells (closed symbols) or on EL4 cells without peptide (open symbols). Data from two individual mice per group are shown. (B) Rejection of β-gal 497–504-pulsed spleen cells. Naive or primed SM-LacZ mice or C57BL/6 control mice (intravenous injection of 2 × 105 β-gal 497–504-pulsed DCs) were intravenously transfused with 4 × 107 β-gal 497–504-pulsed spleen cells (β-gal+) labeled with a high fluorescence intensity and with 4 × 107 control spleen cells labeled with a low fluorescence intensity (ctrl). 24 h after adoptive transfer, PBLs were analyzed by FACS® analysis. Results are from one of two comparable experiments.
Tumor cells used in this set of experiments were EL-4 thymoma cells expressing β-galactosidase (EL4-LacZ; reference 26). EL4-LacZ tumor cells process and present β-galactosidase peptides on MHC class I antigens; CTLs from C57BL/6 mice primed with VV-LacZ and restimulated with β-gal 497–504 specifically lyse EL4-LacZ cells (Fig. 3 A). Furthermore, EL4-LacZ tumor cells were immunogenic after subcutaneous injection into naive recipients, leading to induction of β-galactosidase–specific CTLs (Fig. 3 B). After subcutaneous injection of up to 2 × 107 tumor cells in suspension, tumor growth was not detectable, indicating the high immunogenicity and efficiency of the CTL response against these tumor cells. However, as for MC-GP tumors, tumors grew rapidly in naive SM-LacZ mice after subcutaneous transplantation of small, solid EL4-LacZ tumor pieces containing 2–5 × 106 cells (Fig. 3 C). Repetitive treatment of SM-LacZ mice with growing EL4-LacZ tumors using DCs pulsed with β-gal 497–504 efficiently controlled tumor growth; it caused tumor eradication in 50% of the mice and significantly delayed tumor growth in the other 50% (Fig. 3 D). However, in mice responding with either complete tumor remission or with reduced tumor growth, this treatment was accompanied by strong lymphocytic infiltrates in the right ventricle (Fig. 4 A) that consisted mainly of CD8+ T cells (Fig. 4 C). In the lungs, DC-induced autoimmune lesions were found associated with pulmonary arteries characterized by strong lymphocyte aggregations in the adventitia and media (Fig. 4 B) and increased lymphocyte adhesion to the inflamed endothelium (Fig. 4 B, inset). Also, in the lesions of the pulmonary arteries, CD8+ T cells dominated the early infiltrate (Fig. 4 D). Strong tumor infiltrates consisting mainly of CD8+ T cells were found in tumors responding to the therapy with complete remission (Fig. 4 E). In most untreated SM-LacZ mice, an anti–β-galactosidase immune response was not induced by the transplanted tumor growing in the periphery; T cells did not infiltrate the right ventricle (Fig. 4 G) or pulmonary arteries (Fig. 4 H). However, in two out of five untreated SM-LacZ mice, tumors were growing considerably more slowly, and some tumor-infiltrating CTLs could be detected (Fig. 4 I). In these mice, few CD8+ T cells infiltrated pulmonary arteries, but none were found in the right ventricle (not shown), indicating that the tumor cells alone had induced both limited tumor immunity and mild autoimmunity.
Figure 3.
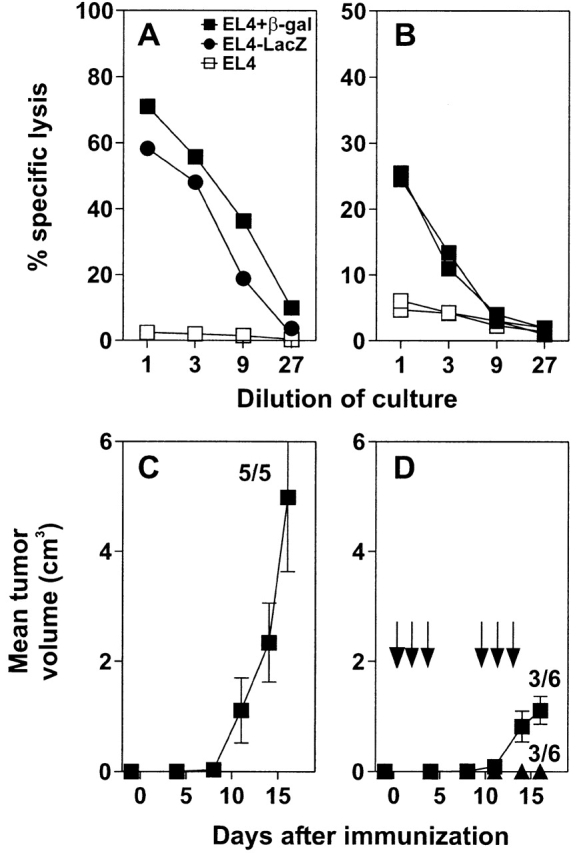
Characterization of EL4-LacZ tumor cells and immunotherapy of EL4-LacZ tumors in SM-LacZ mice. (A) EL4-LacZ (•) and EL4 cells with (▪) or without (□) peptide β-gal 497–504 were used in vitro as targets in a 51Cr-release assay. Effector cells were β-gal 497–504-restimulated spleen cells from C57BL/6 mice infected with 2 × 106 pfu VV-LacZ. (B) 2 × 107 EL4-LacZ tumor cells were injected subcutaneously into the flanks of C57BL/6 mice. 8 d later, splenocytes were restimulated in vitro using irradiated, β-gal 497–504-pulsed spleen cells for 5 d, and the CTL activity was determined in a 51Cr-release assay on β-gal 497–504-labeled EL4 target cells (closed symbols) or on EL4 cells without peptide (open symbols). (C) Small pieces of EL4-LacZ tumors were implanted subcutaneously in the flanks of SM-LacZ mice on day 1, and tumor growth was assessed at the indicated time points. (D) SM-LacZ mice received small EL4-LacZ tumor pieces as in C and were treated from day 0 repetitively by intravenous injection with 3–5 × 105 β-gal 497–504-pulsed DCs. Arrows, day of injection. Numbers indicate the proportion of tumor rejection (or tumor growth) in tumors tested. Results from one of two comparable experiments are shown.
Figure 4.
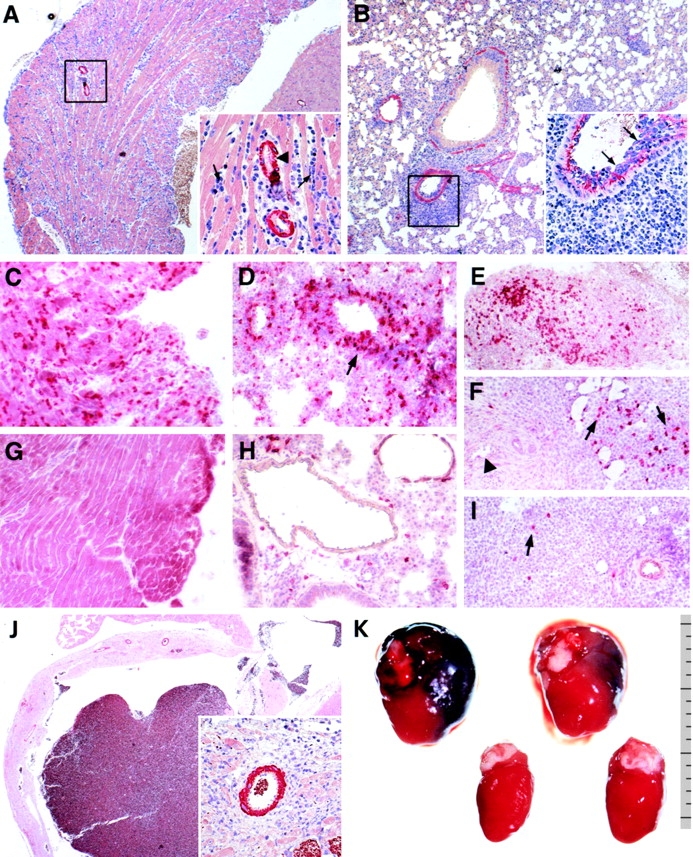
Immunohistological analysis of heart and lung tissue in SM-LacZ mice transplanted with EL4-LacZ tumors and treated repetitively with 3–5 × 105 β-gal 497–504-pulsed DCs (A–F, J, K) or left untreated (G–I). Formalin-fixed and paraffin-embedded sections of heart (A) and lung (B) tissue were stained for smooth muscle actin from a DC-treated SM-LacZ mouse on day 11 after immunization. The inset in A is a magnification of the boxed area in A showing a strong lymphocytic infiltrate between heart muscle cells (arrows) and in the interstitium surrounding coronary arteries (arrowhead). The inset in B is a magnification of the boxed area in B stained for factor VIII showing the strongly increased adhesion of lymphocytes to the inflamed endothelium (arrows) and a strong infiltrate of lymphocytes in the arterial adventitia. Staining for CD8+ lymphocytes in the heart (C), lung (D), and tumor (E and F) of a DC-treated SM-LacZ mouse on day 11. (E) Strong CTL infiltration in a regressing tumor and (F) infiltration of CTL in a growing tumor. Arrows in F indicate tumor infiltrating lymphocytes; the arrowhead indicates an artery not infiltrated by CD8+ lymphocytes. In untreated SM-LacZ mice with transplanted EL4-LacZ tumors, CD8+ lymphocytes were not detected in the right ventricle wall (G) or pulmonary arteries (H) but were occasionally found in small numbers in the tumor tissue (I, arrow). (J) Dilated right ventricle in SM-LacZ mouse 25 d after repetitive immunization with β-gal 497–504-pulsed DCs. Inset is a magnification of the boxed area in J showing infiltration with mononuclear cells, fibrotic tissue, and remaining intact cardiomyocytes. (K) Macroscopic pathology of two hearts from DC-treated SM-LacZ mice on day 25 (top row) in comparison with two control hearts from untreated animals without tumors (bottom row). Ruler indicates the size in millimeters. Original magnifications: A and B, ×50; J, ×20; insets in A, B, and J, ×200; C–I, ×100.
When SM-LacZ mice with tumors were treated with β-galactosidase peptide–pulsed DCs, a dilated cardiomyopathy developed. 3–4 wk after highly repetitive antitumor treatment with DCs, the right ventricle wall was dilated (Fig. 4 J), and cardiomyocytes had been largely destroyed and replaced by fibrotic tissue (Fig. 4 J, inset). Fig. 4 K shows the macroscopic pathology of dilated hearts from SM-LacZ mice after curative tumor treatment with β-galactosidase peptide–pulsed DCs in comparison with hearts from untreated control mice.
Taken together, these results show that previously ignored self-antigens, such as LCMV-GP in RIP-GP mice or β-galactosidase in SM-LacZ mice, may serve as target antigens for vaccination using antigen-expressing DCs. However, potentially harmful autoimmune responses against these self-antigens are induced by DC treatment when a strong and sustained CTL activity is necessary to eliminate the tumor. Depending on the tumor size and the correspondingly needed prolonged maintenance of antitumor/antiself CTL activity, severe and even lethal autoimmunity may develop before tumor growth is controlled.
Discussion
The above results indicate an important if not crucial problem of immunotherapeutic treatment of tumors: if tumors can be controlled and even eliminated by CTLs and if tumor epitopes are also expressed by other peripheral tissues exclusively outside the immune system, is there a window in which specifically activated CTLs reject the tumor but do not induce autoimmune disease? In our two examples of shared tumor antigens, effective immunization against the tumor is paralleled by induction of autoimmunity to a set of self-antigens that so far had been immunologically ignored in the periphery. These results indicate that a window for the delicate balance between induction of an effective antitumor immunity and a possible simultaneous autoimmune pathogenesis is either very small or nonexistent when the antitumor response is directed against such antigens that are not strictly tumor specific.
Immunological Ignorance as an Avenue for Therapeutic Tumor Immunotherapy.
It is conceivable that therapeutic tumor immunotherapy is most promising when tumor antigen–specific T cells of high avidity have not been centrally or peripherally deleted, i.e., the antigen has stayed outside of lymphatic tissues and therefore has been immunologically ignored. It appears that immunological ignorance of antigens strictly expressed in peripheral tissues is a function of antigen distribution and expression level. For example, in transgenic model situations with low levels of antigen expression exclusively in pancreatic β islets 22 30 or solely in cells of the cardiovascular system, this report shows that the antigens are immunologically ignored. In contrast, high levels of self-antigen expression leads either to central 31 32 or peripheral tolerance 33 34. Similarly, tumor antigens can be either immunologically ignored when expressed at low levels in the periphery 21 35 36 or induce a CTL immune response when they are expressed abundantly and reach local secondary lymphoid organs 37. It has been suggested that presentation of such tumor antigens by bone marrow–derived APCs in secondary lymphoid organs may lead to tumor-specific T cell unresponsiveness, particularly in the CD4+ T cell compartment 38 39. In a study with patients with metastatic melanoma, Lee et al. 40 found that in 1 out of 11 patients, tumor-specific CD8+ T cells might also become unresponsive. From these examples, it is not clear whether such tumor-specific CD8+ and/or CD4+ T cell unresponsiveness is the general rule that leads to a failure of tumor immunity or rather the exception.
We favor the view that central and/or peripheral tolerance mechanisms usually do not exist against peripheral tumor antigens of sarcomas and carcinomas and that the majority of tumor and self-antigens are immunologically ignored. This is supported by the findings that high frequencies of tumor-specific CTLs exist in both tumor patients and healthy individuals 41 42 and that antitumor CTLs may be of high avidity 43. Most importantly, the potent (unimpaired) function of antitumor T cells can be observed in melanoma patients suffering from vitiligo, where tumor-specific T cells mediate autoimmune depigmentation 8. It is therefore important for tumor immunotherapy to use as targets antigens that have been previously immunologically ignored. However, immunization with, for example, DCs efficiently presenting so far immunologically ignored shared tumor antigens will cause rejection of tumors but will also lead to autoimmunity. The two experimental systems used in this report model such a shared natural tumor situation: immune surveillance against potentially immunogenic tumor antigens fails because the tumor antigens are immunologically ignored for too long. Consequently, neither autoimmunity nor an antitumor response was induced. DC priming controlled the tumor and elicited a severe autoimmune response against pancreatic β-islet cells or cells of the cardiovascular system that was life threatening. Of course, immunotherapy with antigens derived from less essential organs may cause transient and less severe disease.
Implications for the use of DCs in Antitumor Immunotherapy.
Some experimental murine tumors express target antigens restricted solely to the tumor and can therefore be efficiently cured without inducing autoimmune phenomena using, for example, DCs 15 16 17. In cases in which non–tumor-restricted antigens are used, autoimmune sequelae are probably not observed when the requirements for immune activity are rather low and tumor elimination is achieved before autoimmune symptoms appear 18 19 20. This probably depends largely on the relative size of the tumor versus the size and importance of the autoimmune target, as well as on the relative precursor and effector T cell frequencies. Furthermore, it is possible that tumor cells may provide certain factors compensating for the lack of healthy tissue that is destroyed during curative antitumor treatment (e.g., in the model study of Speiser et al. [36], where insulin-producing insulinoma cells compensate for the destruction of pancreatic β-islet cells during antitumor treatment).
Under certain experimental transgenic conditions, high-avidity CTLs reactive for both tumor and self-antigens may be deleted. The remaining appropriately activated low-avidity CTLs may provide protection against subsequent tumor challenges but may not suffice to provoke autoimmunity 44. Alternatively, it is possible that CD4+ T cells alone mediate tumor immunity. For example, it has been shown in a transgenic mouse model that CD4+ T cell–mediated antitumor immune responses do not lead to obvious autoimmune sequelae when a retroviral tumor antigen is expressed in normal lymphoid tissues 45. However, it is questionable whether CD4+ T cell–mediated antitumor immune responses alone may be sufficient to cure rapidly growing tumors in the periphery via adoptive immunotherapy.
As stated, in situations where self- and tumor-reactive, high-avidity CTLs are not centrally or peripherally deleted and the antigens are immunologically ignored, such as in RIP-GP or SM-LacZ mice, strong antitumor responses that are also directed against self-antigens will eventually cause autoimmunity. This has also been observed in nontransgenic situations when mice were immunized with the melanoma antigen gp75/tyrosinase-related protein 1, leading to mainly T helper–mediated protection against tumor challenge and also to mild depigmentation 46 47. Similarly, in clinical studies, induction of autoimmune depigmentation has been associated with a good prognosis for melanoma patients, indicative of successful control of the tumor by the immune system 8. Recently, it has been shown that paraneoplastic cerebellar degeneration (PCD) is probably mediated by CTLs specific for tumor antigens present on gynecological tumors and in neuronal tissue 48. Interestingly, PCD-associated gynecological tumors have been documented to regress with the onset of autoimmune neurological disease 9. The clinical consequences of autoimmune reactions against normal melanocytes causing vitiligo during melanoma treatment or a strong autoimmune reaction against organs where the loss of function can be substituted, e.g., with insulin for damage of pancreatic β islet cells, are acceptable during immunotherapeutic antitumor treatment. In contrast, other autoimmune diseases, such as reactions against cells in the cardiovascular system or neuronal tissue, may impose limitations on antitumor therapy with DCs using antigens also expressed by cells in vital organs.
Acknowledgments
We thank Kevin Maloy for helpful discussions and critical reading of the manuscript, Lenka Vlk and Anne Henzelin for expert technical assistance, and Ida Schmieder and Norbert Wey for excellent photography.
This work was supported by the Swiss National Science Foundation, the Deutsche Forschungsgemeinschaft (to B. Ludewig), and the Kanton Zürich.
Footnotes
B. Ludewig and A.F. Ochsenbein contributed equally to this work.
Abbreviations used in this paper: DCs, dendritic cells; GP, glycoprotein; RIP, rat insulin promotor.
References
- Houghton A.N. Cancer antigensimmune recognition of self and altered self. J. Exp. Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Eynde B.J., van der Bruggen P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- Old L.J., Chen Y.T. New paths in human cancer serology. J. Exp. Med. 1998;187:1163–1167. doi: 10.1084/jem.187.8.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S.A. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- Wang R.F., Appella E., Kawakami Y., Kang X., Rosenberg S.A. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J. Exp. Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom M.B., Perry-Lalley D., Robbins P.F., Li Y., el-Gamil M., Rosenberg S.A., Yang J.C. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J. Exp. Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropke M., Hald J., Guldberg P., Zeuthen J., Norgaard L., Fugger L., Svejgaard A., Van der Burg S., Nijman H.W., Melief C.J. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc. Natl. Acad. Sci. USA. 1996;93:14704–14707. doi: 10.1073/pnas.93.25.14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S.A., White D.E. Vitiligo in patients with melanomanormal tissue antigens can be targets for cancer immunotherapy. J. Immunother. Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- Darnell R.B., DeAngelis L.M. Regression of small-cell lung carcinoma in patients with paraneoplastic neuronal antibodies. Lancet. 1993;341:21–22. doi: 10.1016/0140-6736(93)92485-c. [DOI] [PubMed] [Google Scholar]
- Tatsis E., Reinhold-Keller E., Steindorf K., Feller A.C., Gross W.L. Wegener's granulomatosis associated with renal cell carcinoma. Arthritis Rheum. 1999;42:751–756. doi: 10.1002/1529-0131(199904)42:4<751::AID-ANR19>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Zinkernagel R.M., Ehl S., Aichele P., Oehen S., Kundig T., Hengartner H. Antigen localisation regulates immune responses in a dose- and time-dependent fashiona geographical view of immune reactivity. Immunol. Rev. 1997;156:199–209. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- Steinman R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Schuler G., Steinman R.M. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J. Exp. Med. 1997;186:1183–1187. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayordomo J.I., Zorina T., Storkus W.J., Zitvogel L., Celluzzi C., Falo L.D., Melief C.J., Ildstad S.T., Kast W.M., DeLeo A.B. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat. Med. 1995;1:1297–1302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- Song W., Kong H.L., Carpenter H., Torii H., Granstein R., Rafii S., Moore M.A., Crystal R.G. Dendritic cells genetically modified with an adenovirus vector encoding the cDNA for a model antigen induce protective and therapeutic antitumor immunity. J. Exp. Med. 1997;186:1247–1256. doi: 10.1084/jem.186.8.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht J.M., Wang G., Do M.T., Lam J.S., Royal R.E., Reeves M.E., Rosenberg S.A., Hwu P. Dendritic cells retrovirally transduced with a model antigen gene are therapeutically effective against established pulmonary metastases. J. Exp. Med. 1997;186:1213–1221. doi: 10.1084/jem.186.8.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Chen D., Kashiwaba M., Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat. Med. 1997;3:558–561. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- Nair S.K., Boczkowski D., Morse M., Cumming R.I., Lyerly H.K., Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat. Biotechnol. 1998;16:364–369. doi: 10.1038/nbt0498-364. [DOI] [PubMed] [Google Scholar]
- Zitvogel L., Mayordomo J.I., Tjandrawan T., DeLeo A.B., Clarke M.R., Lotze M.T., Storkus W.J. Therapy of murine tumors with tumor peptide-pulsed dendritic cellsdependence on T cells, B7 costimulation, and T helper cell 1–associated cytokines. J. Exp. Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenbein A.F., Klenerman P., Karrer U., Ludewig B., Pericin M., Hengartner H., Zinkernagel R.M. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl. Acad. Sci. USA. 1999;96:2233–2238. doi: 10.1073/pnas.96.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi P.S., Oehen S., Buerki K., Pircher H., Ohashi C.T., Odermatt B., Malissen B., Zinkernagel R.M., Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- Ehl S., Hombach J., Aichele P., Rulicke T., Odermatt B., Hengartner H., Zinkernagel R., Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998;187:763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessler H., Mericskay M., Li Z., Nagl S., Paulin D., Small J.V. The SM 22 promotor directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development. 1996;122:2415–2425. doi: 10.1242/dev.122.8.2415. [DOI] [PubMed] [Google Scholar]
- Ludewig B., Ehl S., Karrer U., Odermatt B., Hengartner H., Zinkernagel R.M. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 1998;72:3812–3818. doi: 10.1128/jvi.72.5.3812-3818.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oukka M., Cohen-Tannoudji M., Tanaka Y., Babinet C., Kosmatopoulos K. Medullary thymic epithelial cells induce tolerance to intracellular proteins. J. Immunol. 1996;156:968–975. [PubMed] [Google Scholar]
- Kundig T.M., Bachmann M.F., DiPaolo C., Simard J.J., Battegay M., Lother H., Gessner A., Kuhlcke K., Ohashi P.S., Hengartner H. Fibroblasts as efficient antigen-presenting cells in lymphoid organs. Science. 1995;268:1343–1347. doi: 10.1126/science.7761853. [DOI] [PubMed] [Google Scholar]
- Cobbold S.P., Jayasuriya A., Nash A., Prospero T.D., Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 1984;312:548–551. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- Ludewig B., Odermatt B., Landmann S., Hengartner H., Zinkernagel R.M. Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J. Exp. Med. 1998;188:1493–1501. doi: 10.1084/jem.188.8.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Kosaka H., Miller J.F., Carbone F.R. The peripheral deletion of autoreactive CD8+ T cells induced by cross-presentation of self-antigens involves signaling through CD95 (Fas, Apo-1) J. Exp. Med. 1998;188:415–420. doi: 10.1084/jem.188.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehen S.U., Ohashi P.S., Burki K., Hengartner H., Zinkernagel R.M., Aichele P. Escape of thymocytes and mature T cells from clonal deletion due to limiting tolerogen expression levels. Cell. Immunol. 1994;158:342–352. doi: 10.1006/cimm.1994.1281. [DOI] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Carbone F.R., Allison J., Miller J.F., Kosaka H. Constitutive class I–restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Carbone F.R., Barnden M., Blanas E., Allison J., Heath W.R., Miller J.F. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D.J., Kreuwel H.T., Sherman L.A. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 1999;163:723–727. [PubMed] [Google Scholar]
- Prehn R.T., Main J.M. Immunity to methyl-collanthrene-induced sarcomas. J. Natl. Cancer Inst. 1957;18:769–782. [PubMed] [Google Scholar]
- Speiser D.E., Miranda R., Zakarian A., Bachmann M.F., McKall-Faienza K., Odermatt B., Hanahan D., Zinkernagel R.M., Ohashi P.S. Self-antigens expressed by solid tumors do not efficiently stimulate naive or activated T cellsimplications for immunotherapy. J. Exp. Med. 1997;186:645–653. doi: 10.1084/jem.186.5.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo A.L., Lake R.A., Lo D., Sherman L., McWilliam A., Nelson D., Robinson B.W., Scott B. Tumor antigens are constitutively presented in the draining lymph nodes. J. Immunol. 1999;162:5838–5845. [PubMed] [Google Scholar]
- Staveley-O'Carroll K., Sotomayor E., Montgomery J., Borrello I., Hwang L., Fein S., Pardoll D., Levitsky H. Induction of antigen-specific T cell anergyan early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA. 1998;95:1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor E.M., Borrello I., Tubb E., Rattis F.M., Bien H., Lu Z., Fein S., Schoenberger S., Levitsky H.I. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- Lee P.P., Yee C., Savage P.A., Fong L., Brockstedt D., Weber J.S., Johnson D., Swetter S., Thompson J., Greenberg P.D. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- Romero P., Dunbar P.R., Valmori D., Pittet M., Ogg G.S., Rimoldi D., Chen J.L., Lienard D., Cerottini J.C., Cerundolo V. Ex vivo staining of metastatic lymph nodes by class I major histocompatibility complex tetramers reveals high numbers of antigen-experienced tumor-specific cytolytic T lymphocytes. J. Exp. Med. 1998;188:1641–1650. doi: 10.1084/jem.188.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet M.J., Valmori D., Dunbar P.R., Speiser D.E., Lienard D., Lejeune F., Fleischhauer K., Cerundolo V., Cerottini J.C., Romero P. High frequencies of naive melan-A/MART-1–specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999;190:705–716. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C., Savage P.A., Lee P.P., Davis M.M., Greenberg P.D. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J. Immunol. 1999;162:2227–2234. [PubMed] [Google Scholar]
- Morgan D.J., Kreuwel H.T., Fleck S., Levitsky H.I., Pardoll D.M., Sherman L.A. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 1998;160:643–651. [PubMed] [Google Scholar]
- Hu J., Kindsvogel W., Busby S., Bailey M.C., Shi Y.Y., Greenberg P.D. An evaluation of the potential to use tumor-associated antigens as targets for antitumor T cell therapy using transgenic mice expressing a retroviral tumor antigen in normal lymphoid tissues. J. Exp. Med. 1993;177:1681–1690. doi: 10.1084/jem.177.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L.W., Bowne W.B., Wolchok J.D., Srinivasan R., Qin J., Moroi Y., Clynes R., Song P., Lewis J.J., Houghton A.N. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J. Clin. Invest. 1998;102:1258–1264. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk W.W., Lee D.S., Surman D.R., Irvine K.R., Touloukian C.E., Chan C.C., Carroll M.W., Moss B., Rosenberg S.A., Restifo N.P. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in micerequirement for CD4(+) T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:2982–2987. doi: 10.1073/pnas.96.6.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M.L., Darnell J.C., Bender A., Francisco L.M., Bhardwaj N., Darnell R.B. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat. Med. 1998;4:1321–1324. doi: 10.1038/3315. [DOI] [PubMed] [Google Scholar]