Increased Turnover of T Lymphocytes in HIV-1 Infection and Its Reduction by Antiretroviral Therapy (original) (raw)
Abstract
The mechanism of CD4+ T cell depletion in human immunodeficiency virus (HIV)-1 infection remains controversial. Using deuterated glucose to label the DNA of proliferating cells in vivo, we studied T cell dynamics in four normal subjects and seven HIV-1–infected patients naive to antiretroviral drugs. The results were analyzed using a newly developed mathematical model to determine fractional rates of lymphocyte proliferation and death. In CD4+ T cells, mean proliferation and death rates were elevated by 6.3- and 2.9-fold, respectively, in infected patients compared with normal controls. In CD8+ T cells, the mean proliferation rate was 7.7-fold higher in HIV-1 infection, but the mean death rate was not significantly increased. Five of the infected patients underwent subsequent deuterated glucose labeling studies after initiating antiretroviral therapy. The lymphocyte proliferation and death rates in both CD4+ and CD8+ cell populations were substantially reduced by 5–11 weeks and nearly normal by one year. Taken together, these new findings strongly indicate that CD4+ lymphocyte depletion seen in AIDS is primarily a consequence of increased cellular destruction, not decreased cellular production.
Keywords: deuterated glucose, longitudinal study, mathematical model, apoptosis, mechanisms of CD4+ T cell depletion
Introduction
CD4+ lymphocyte depletion, the immunologic hallmark of AIDS, may be the consequence of HIV-1's ability to increase cellular destruction, decrease cellular production, or both. This important, unresolved issue was initially addressed by quantifying the rebound in CD4+ T cells during effective antiretroviral therapy (1, 2). The observed rapid rise in such cell counts in blood provided indirect evidence that CD4+ lymphocytes were turning over at a high rate during HIV-1 infection. However, this conclusion was called into question by the possibility that the rise could be largely due to lymphocyte redistribution (3). That telomere lengths in CD4+ T cells of infected persons remained relatively stable over time further argued against a discernible increase in lymphocyte turnover (4), a conclusion contradicted by findings of higher frequencies of T cells expressing proliferation antigens in HIV-1 infection (5, 6). Direct measurement of lymphocyte dynamics was then performed using bromodeoxyuridine to label proliferating cells in macaques with and without simian immunodeficiency virus (SIV) infection (7, 8). Conclusive data emerged that show turnover rates for both CD4+ and CD8+ lymphocytes to be several-fold faster in SIV infection. More recently, a labeling technique using deuterated glucose (D-glucose)* was developed to safely quantify T cell dynamics in humans (9). Its initial application demonstrated enhanced CD4+ and CD8+ lymphocyte turnover in HIV-1 infection (10). However, that these rates were even higher in patients successfully treated with antiretroviral therapy (10) was interpreted by some (11, 12) to suggest a block in lymphocyte production as the major dysfunction caused by HIV-1. To resolve these apparent conflicts, we conducted a detailed study using D-glucose labeling in normal subjects as well as in HIV-1–infected patients before and during treatment. The results were then analyzed with a newly developed mathematical model. We now report that both CD4+ and CD8+ lymphocyte turnover rates are elevated several-fold in HIV-1 infection, and these rates begin to normalize within a few months of effective antiretroviral therapy.
Materials and Methods
Study Subjects
Four normal persons and seven HIV-1–infected but untreated patients were recruited into the study (Table I). Each study subject gave informed consent to participating in a clinical protocol, which was approved by the Institutional Review Board of The Rockefeller University Hospital. Five of the infected individuals were given antiretroviral therapy several weeks after the completion of the first D-glucose labeling study. The treatment regimen consisted of nelfinavir (2,500 mg/d), lamivudine (300 mg/d), abacavir (600 mg/d), and efavirenz (600 mg/d). Drug substitutions were permitted for intolerance or adverse events. During treatment, patients P1, P2, and P3 were studied twice, while P4 and P5 were studied once.
Table I.
Baseline Characteristics of Study Subjects and Their Estimated Values of CD4+ and CD8+ Lymphocyte Proliferation (p), Death (d), and Fractional Input from a Source (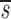 U)
U)
| Baseline values | CD4+ | CD8+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Subjects | Age(sex) | Plasma viral load | CD4+ | CD8+ | p(95% CI) | d(95% CI) | 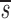 U(95% CI) U(95% CI) |
p(95% CI) | d(95% CI) | 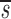 U(95% CI) U(95% CI) |
| (copies/ml) | μl−1 | μl−1 | day−1 | day−1 | day−1 | day−1 | day−1 | day−1 | ||
| Controls: | ||||||||||
| C1 | 43 (M) | - | 880 | 562 | 0.005(0.005-0.006) | 0.057(0.024-0.110) | 0.044(0.016-0.094) | 0.004(0.003-0.005) | 0.076(0.044-0.139) | 0.072(0.041-0.134) |
| C2 | 22 (F) | - | 992 | 576 | 0.004(0.003-0.011) | 0.064(0.024-0.496) | 0.060(0.020-0.485) | 0.002(0.002-0.003) | 0.018(0.013-0.090) | 0.016(0.011-0.087) |
| C3 | 22 (F) | - | 1,366 | 651 | 0.003(0.001-0.131) | 0.013(0.002-10.9) | 0.010(0.000-10.7) | 0.002(0.001-0.006) | 0.008(0.002-1.36) | 0.006(0.000-1.34) |
| C4 | 22 (M) | - | 1,066 | 621 | 0.004(0.003-0.005) | 0.042(0.029-0.120) | 0.020(0.013-0.065) | 0.002(0.002-0.003) | 0.070(0.016-0.196) | 0.067(0.014-0.192) |
| Mean ± SE | 30.0 | - | 1,076 | 603 | 0.004 ± 0.0005 | 0.044 ± 0.011 | 0.033 ± 0.011 | 0.003 ± 0.0004 | 0.043 ± 0.017 | 0.040 ± 0.017 |
| Infected: | ||||||||||
| P1 | 38 (M) | 500,000 | 322 | 806 | 0.033(0.028-0.041) | 0.112(0.071-0.167) | 0.077(0.045-0.123) | 0.036(0.033-0.040) | 0.065(0.038-0.090) | 0.046(0.025-0.078) |
| P2 | 24 (M) | 226,087 | 255 | 494 | 0.034(0.027-0.042) | 0.135(0.086-0.255) | 0.100(0.056-0.214) | 0.018(0.015-0.022) | 0.092(0.064-0.272) | 0.074(0.049-0.249) |
| P3 | 26 (M) | 7,629 | 323 | 617 | 0.010(0.009-0.010) | 0.042(0.025-0.068) | 0.050(0.034-0.081) | 0.010(0.009-0.011) | 0.029(0.018-0.072) | 0.031(0.020-0.077) |
| P4 | 46 (F) | 61,946 | 48 | 323 | 0.039(0.036-0.047) | 0.082(0.053-0.131) | 0.042(0.016-0.086) | 0.024(0.022-0.029) | 0.032(0.024-0.059) | 0.008(0.003-0.032) |
| P5 | 38 (M) | 88,997 | 804 | 1,148 | 0.019(0.015-0.160) | 0.306(0.153-3.8) | 0.285(0.137-3.61) | 0.029(0.026-0.035) | 0.062(0.034-0.157) | 0.022(0.012-0.072) |
| P6 | 21 (M) | 13,367 | 626 | 1,312 | 0.016(0.014-0.019) | 0.151(0.095-0.214) | 0.240(0.115-0.641) | 0.024(0.023-0.026) | 0.049(0.038-0.067) | 0.024(0.015-0.041) |
| P7 | 36 (F) | 22,410 | 337 | 1,011 | 0.022(0.021-0.023) | 0.077(0.060-0.093) | 0.065(0.045-0.079) | 0.017(0.016-0.019) | 0.022(0.015-0.060) | 0.011(0.007-0.050) |
| Mean ± SE | 32.6 | 131,491 | 388 | 816 | 0.025 ± 0.004a | 0.129 ± 0.033a | 0.123 ± 0.037a | 0.023 ± 0.003a | 0.050 ± 0.009 | 0.031 ± 0.009 |
D-Glucose Labeling
All subjects, except one (P5), were hospitalized for a 7-d labeling period, when D-glucose (30 g/d; purchased from Cambridge Isotope Laboratories, Inc.) was administered intravenously while dietary carbohydrate was highly restricted (<46 g/d).
Cell Count and Phenotypic Analysis of Peripheral T Cells
CD4+ and CD8+ cell counts were measured using TruCount System (Becton Dickinson). Four-color flow-cytometric analysis was performed for Ki67 expression and TdT-mediated dUTP nick-end labeling (TUNEL) positivity. In brief, freshly isolated PBMCs were stained with anti–CD3-PerCP (Becton Dickinson), anti–CD4-APC (Exalpha), and anti–CD8-PE (Dako). After washing, cells were treated with FACS® Lysing Solution (Becton Dickinson) for 40 min at room temperature, and further stained with anti-Ki67 (MIB-1; Beckman Coulter) in PBS with 0.5% NP40 and 5% FCS at room temperature for 45 min. After two washes, cells were fixed with 1% paraformaldehyde in PBS. For TUNEL positivity, freshly isolated PBMCs were cultured overnight without any stimulation, and then stained for cell surface markers before detecting apoptotic cells using the In Situ Cell Detection Kit (Roche Molecular Biochemicals). The cells were fixed with 1% paraformaldehyde in PBS and analyzed by FACSCaliber™ (Becton Dickinson).
Viral Load Measurement
Plasma viral load was measured by a branched DNA assay (13) with a detection limit of 50 copies/ml (Bayer Diagnostics).
Measurement of Deoxyadenosine Enrichment in DNA of Cell Populations
Monocytes were purified (>95% purity) using an anti-CD14 mAb coupled to Dynabeads (Dynal). Sorting of CD3+ CD4+ and CD3+CD8+ lymphocytes was carried out using MoFlo (Cytomation) after staining with anti–CD3-FITC (Becton Dickinson) together with anti–CD4-PE (Becton Dickinson) and anti-CD8-CyChrome (BD PharMingen). The purity of the sorted cells was >98%. DNA was extracted from each cell preparation using QIAamp DNA Blood Mini Kit (QIAGEN), followed by hydrolysis with nuclease P1 at 45°C overnight, snake venom phosphodiesterase I at 37°C for 2 h, and alkaline phosphatase (Sigma-Aldrich) at 37°C for 1 h. Deoxyadenosine (dA) was separated using Supelco LC-18, SPE tube (Supelco) and converted to triacetylaldonitrile derivative before analysis by gas chromatography and mass spectroscopy as described previously (9, 10). The degree of dA enrichment was calculated using a standard curve for each experiment. Precursor enrichment was assessed based on the amount of deuterium-containing glucose in plasma, and used to convert our data into labeled DNA as described previously (9, 10). Because dA enrichment measured at time t is due to DNA synthesis at earlier times, for samples measured during labeling we used the average precursor enrichment up to time t for this conversion. For samples measured after the completion of labeling, we used the enrichment averaged over the labeling period.
Mathematical Model
Before D-glucose administration, we assume that the number of both CD4+ and CD8+ T cells, T, is determined by equation 1 in Fig. 1 , where s is the rate of T cell entry into the proliferative compartment, p is the proliferation rate per cell, and d is the death rate per cell. During D-glucose administration when cell proliferation occurs, an unlabeled DNA strand, U, is copied and a labeled strand, L, is created while U is preserved. Thus, U→U + L. Similarly, when a labeled strand is copied, L→L + L. These kinetics are described by equations 2 a and b, where the initial amounts of unlabeled and labeled DNA are U(0) and L(0), respectively, p and d are as in equation 1, and the source s now has two components: an unlabled part sU and a labeled one sL, such that s = sU_+ sL. If we assume that the system is at steady state, the amount of DNA in the system will not change over time, and for the first labeling the fraction of labeled DNA is given by fL(t) = L(t)/[U(0)+L(0)]. For the first labeling, L(0) = 0, since initially there is no labeled DNA. From equations 2 a and b we obtain for the fraction of labeled DNA during D-glucose administration, f L(t)=1−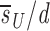 1−_e −dt, where we have defined
1−_e −dt, where we have defined 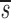 u as sU/U(0); thus, the source is measured relative to the total amount of DNA in the compartment. After D-glucose administration, U→U + U and L→L + U, leading to equations 3 a and b, where we use _sU_′ and _sL_′ for the source terms. The primes indicate that the sources after D-glucose administration may have changed, e.g., more of the source may be labeled, but the total source is still s = _sU_′ + _sL_′. From the solution of equation 3 a, we calculate the fraction of labeled DNA by f L _t_=f L t e_−
u as sU/U(0); thus, the source is measured relative to the total amount of DNA in the compartment. After D-glucose administration, U→U + U and L→L + U, leading to equations 3 a and b, where we use _sU_′ and _sL_′ for the source terms. The primes indicate that the sources after D-glucose administration may have changed, e.g., more of the source may be labeled, but the total source is still s = _sU_′ + _sL_′. From the solution of equation 3 a, we calculate the fraction of labeled DNA by f L _t_=f L t e_−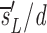 e −_d t_−_t e+
e −_d t_−_t e+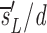 , where
, where 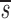 _L_′=
_L_′=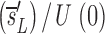 . In this equation, fL(te) represents the fraction of labeled DNA at the time, te, that D-glucose infusion ends. While the proliferation rate does not appear in the equations for fL(t), it can be estimated using the steady state condition p = d − s/T or in the case of DNA, p = d − (_sU_+ sL)/U(0); thus, _p_>_d_−
. In this equation, fL(te) represents the fraction of labeled DNA at the time, te, that D-glucose infusion ends. While the proliferation rate does not appear in the equations for fL(t), it can be estimated using the steady state condition p = d − s/T or in the case of DNA, p = d − (_sU_+ sL)/U(0); thus, _p_>_d_−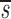 U. Because we expect
U. Because we expect 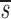 L<
L<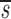 _L_′, p<_d_−
_L_′, p<_d_−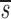 U+
U+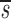 _L_′, these two estimates of p turn out to be close to each other and in Table I we report their mean. The initial slope of the labeling curve is _d_−
_L_′, these two estimates of p turn out to be close to each other and in Table I we report their mean. The initial slope of the labeling curve is _d_−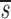 U, which to a good approximation is p. When D-glucose is administered for a second or third time to the same patient, the expressions given above for fL(t) are modified to account for any existing labeled DNA at the start of the infusion. Also, when the measurements of total CD4+ and CD8+ T cells indicate a significant change in the population, we relax the assumption of steady state. In this case, we used the solution of equation 1 to determine the total amount of DNA at time t to be used in the calculation of the fraction of labeled DNA. Finally, because most T cells are in tissues, we assumed that after labeling there would be a delay τ before labeled DNA first appeared in blood. Further, when D-glucose administration ended at time t e, we also assumed labeled DNA would continue to accumulate in the blood until time t e + τ. Thus, on the right hand side of the equations given above for fL(t), t should be replaced by t − τ, and for 0< t < τ, fL(t) = 0. To model monocyte kinetics, we assume no proliferation occurs in the periphery, i.e., p = 0 in equations 1–3, and d is interpreted as a rate of monocyte export out of blood.
U, which to a good approximation is p. When D-glucose is administered for a second or third time to the same patient, the expressions given above for fL(t) are modified to account for any existing labeled DNA at the start of the infusion. Also, when the measurements of total CD4+ and CD8+ T cells indicate a significant change in the population, we relax the assumption of steady state. In this case, we used the solution of equation 1 to determine the total amount of DNA at time t to be used in the calculation of the fraction of labeled DNA. Finally, because most T cells are in tissues, we assumed that after labeling there would be a delay τ before labeled DNA first appeared in blood. Further, when D-glucose administration ended at time t e, we also assumed labeled DNA would continue to accumulate in the blood until time t e + τ. Thus, on the right hand side of the equations given above for fL(t), t should be replaced by t − τ, and for 0< t < τ, fL(t) = 0. To model monocyte kinetics, we assume no proliferation occurs in the periphery, i.e., p = 0 in equations 1–3, and d is interpreted as a rate of monocyte export out of blood.
Figure 1.

A newly developed mathematical model to track labeled and unlabeled strands of DNA before, during and after D-glucose administration. A full explanation is given in Materials and Methods.
Data Fitting
We fitted the appropriate equations for fL(t) to the experimental data using a nonlinear least-square regression method described previously (14). We calculated 95% confidence intervals by bootstrapping (15). At each time point, we also measured the total number of CD4+ and CD8+ T cells, and used a threepoint running average to smooth the data. When there was a significant variation in cell numbers, instead of assuming steady state we fitted simultaneously the smoothed number of T cells and the fraction of labeled DNA. In addition, we used a jackknife method in which zero weight was given to one outlying data point with a disproportionate influence in the data fitting from the labeled DNA measurements of CD4+ T cells in P5 for the first and second labeling, and the CD8+ T cells in C4 and P2 for the first labeling; however, all data points are shown below. The average initial labeling delay, τ, computed using a trimmed mean, was 0.5 d. In a few individuals who exhibited poor initial labeling, the data were fitted with a fixed delay of 0.5 d.
Statistical Analyses
Except where otherwise indicated, comparisons between groups were done using the two-tailed Wilcoxon rank sum test, which is more appropriate for the small sample sizes involved. Correlations, involving all the data (between 15 and 19 data points), were done using standard linear regression calculations. Significance was assessed at the P = 0.05 level.
Results
Four normal subjects and seven HIV-1-infected, treatment-naive patients enrolled in our study of lymphocyte dynamics using D-glucose as the label in what might be considered an in-vivo pulse-chase experiment. Both groups were of similar age (Table I). The infected patients had baseline CD4+ counts of 48–804 cells/μl and plasma viral loads of 7,629–500,000 copies/ml. All but one subject were hospitalized for the first 7 d of the study, when D-glucose was administered intravenously. In P5, for personal reasons, D-glucose was given for 4.3 d only. Blood was obtained from each subject on multiple occasions, including before, during (nearly daily), and after (every 7–10 d) the infusion. No adverse reactions related to D-glucose administration were noted.
Turnover of Blood Monocytes Is Very Rapid in both Healthy Controls and HIV-1–infected Patients
To ensure that the new labeling technique would yield reliable results, we first examined the incorporation of deuterium into the DNA of monocytes since their half-life (t1/2) or transit time in blood had been defined (16). Monocytes were purified from three normal subjects and two patients using an anti-CD14 mAb coupled to Dynabeads. DNA was then extracted from each cell preparation, processed, and tested for the degree of deuterium incorporation into the dA fraction using gas chromatography and mass spectroscopy. As shown in Fig. 2 , labeled DNA became detectable in blood after a delay of ∼2 d. Thereafter, the fraction of labeled DNA rose rapidly, reaching a peak of 0.60–0.85 before falling promptly upon the cessation of D-glucose infusion. These results were then analyzed using a simple mathematical model that assumes monocytes label in the bone marrow and then appear in blood with some time delay before moving into tissues as macrophages. Fitting the data to the model (Fig. 2) yielded a t1/2 of monocytes in blood of ∼2 d, regardless of HIV-1 infection status, in close agreement with published results (16) and thereby validating the D-glucose labeling method (9, 10, 17).
Figure 2.

Sequential changes in the fraction of labeled DNA in blood monocytes. The data points are represented by symbols, and the lines show the best fit of the data to a mathematical model. The calculated half-lives (t1/2) for monocytes in blood are indicated.
Kinetics of both CD4+ and CD8+ T Cells Are Faster in HIV-1–infected Patients than in Healthy Controls
We then turned our attention to CD3+CD4+ and CD3+CD8+ lymphocytes that have been purified to >98% homogeneity by a cell sorter from each blood specimen. Again, every cellular DNA sample was examined for deuterium incorporation into the dA fraction (9, 10). From the results summarized in Fig. 3 , it is readily apparent that the labeling and delabeling profiles in the four normal subjects were rather uniform, with CD4+ T cells showing slightly higher levels of deuterium incorporation than CD8+ T cells. Labeled DNA became detectable with a mean time lag of 0.5 d, and its level increased slowly during D-glucose administration followed by a gradual decrease thereafter. The peak fraction of labeled DNA did not exceed 0.04 in either cell population. In contrast, the results are substantially different in seven HIV-1–infected patients who were not on antiretroviral therapy. Their rates of rise and fall of labeled DNA were obviously greater than those observed in normal controls, even in P5 who was given D-glucose for 4.3 d. Likewise, the peak labeling was consistently higher in infected individuals, with some reaching a labeled fraction of ∼0.20. Interestingly, the lowest deuterium incorporation in infected subjects was seen in P3, the one with the lowest viral load (Table I). There was no consistent relationship noted when comparing labeling profiles in CD4+ versus CD8+ lymphocytes. Overall, the findings in Fig. 3 provide a qualitative impression that lymphocyte turnover is substantially more rapid in infected patients than in normal persons.
Figure 3.

Sequential changes in the fraction of labeled DNA in blood T lymphocytes. The data of healthy controls (C1–C4) versus HIV-1–infected patients (P1–P7) are shown in each graph. The period of D-glucose administration is indicated by a box (top left corner). The data points are represented by symbols, and the lines show the best fit of the data to a mathematical model.
Proliferation and Death Rates of CD4+ and CD8+ T Cells Are Elevated Several-fold in HIV-1 Infection
To obtain quantitative estimates of CD4+ and CD8+ T cell turnover from the data, we developed a new mathematical model of lymphocyte dynamics that tracks the number of deuterium-labeled and unlabeled strands of cellular DNA before, during and after D-glucose administration (Fig. 1 and Materials and Methods). Then we used the model to fit the labeling results generated from CD4+ and CD8+ T cells from each study subject as described in Materials and Methods. As shown in Fig. 3, a good fit of the theory to the data was obtained in every case. In so doing, rate estimates of cellular proliferation (p) and death (d) were derived for both lymphocyte populations in each subject (Table I). By comparing the mean value of p for CD4+ T cells of normal individuals (0.004/d) to that of infected patients (0.025/d), it was clear that proliferation rates were significantly increased (∼6.3-fold) in HIV-1 infection (P value <0.01). A similar comparison of mean values of d (0.044/d vs. 0.129/d) showed CD4+ lymphocyte death rates to be significantly elevated (approximately threefold) in infected individuals (P value <0.04). Likewise, the mean value of p for CD8+ T cells was significantly higher (∼7.7-fold) in patients (0.023/d) than in normal controls (0.003/d) (P value <0.01), although mean values of d for the two groups were not significantly different (0.050/d vs. 0.043/d). These quantitative estimates, therefore, validate the qualitative impression above that HIV-1 infection results in a faster turnover of both CD4+ and CD8+ T cells.
Source of CD4+ T Cells May Increase in HIV-1 Infection
Note that in Table I the data fitting also provided estimates for 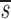 U, which is, to a good approximation, the fraction of T cells in the proliferating compartment input from a source per day (Fig. 1, references 18 and 19). The mean value of
U, which is, to a good approximation, the fraction of T cells in the proliferating compartment input from a source per day (Fig. 1, references 18 and 19). The mean value of 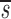 U for CD4+ lymphocytes in infected patients (0.123/d) was significantly higher (P value <0.05) than in normal persons (0.033/d); however, the mean values of
U for CD4+ lymphocytes in infected patients (0.123/d) was significantly higher (P value <0.05) than in normal persons (0.033/d); however, the mean values of 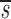 U for CD8+ lymphocytes were not very different (0.031/d vs. 0.040/d) between the two groups. There was no evidence to suggest that HIV-1 infection results in a decrease of CD4+ T cells coming from a source. As we have discussed previously (7, 18, 19), this source could be the thymus or merely a population of resting or slowly dividing lymphocytes that upon activation would undergo rapid clonal expansion and enter the pool of cells acquiring label during the experiment. A model that includes separate populations of resting and proliferating T cells can also fit the data and supports the concept that the source corresponds to activation of resting cells (unpublished data).
U for CD8+ lymphocytes were not very different (0.031/d vs. 0.040/d) between the two groups. There was no evidence to suggest that HIV-1 infection results in a decrease of CD4+ T cells coming from a source. As we have discussed previously (7, 18, 19), this source could be the thymus or merely a population of resting or slowly dividing lymphocytes that upon activation would undergo rapid clonal expansion and enter the pool of cells acquiring label during the experiment. A model that includes separate populations of resting and proliferating T cells can also fit the data and supports the concept that the source corresponds to activation of resting cells (unpublished data).
Enhanced Turnover of both CD4+ and CD8+ T Cells in HIV-1 Infection Decreases upon Antiretroviral Therapy
Five of the infected patients were reevaluated to assess the impact of successful antiretroviral therapy on lymphocyte dynamics. A potent four-drug regimen was initiated in P1 through P5 several weeks after the completion of their first labeling study. As shown in Fig. 4 A, plasma viremia dropped precipitously in each case, eventually reaching undetectable levels in those patients treated beyond 150 d. At the same time, the CD4+ T cell numbers of these patients showed an increasing trend (Fig. 4 A). The second episode of D-glucose labeling was carried out in these five patients beginning on days 35–77 of treatment. P4 and P5 left the study after finishing the second study, whereas P1, P2, and P3 went on to have the third episode of D-glucose labeling starting on days 245–375 of treatment when plasma viral load was persistently undetectable save for one transient “blip” in P2.
Figure 4.

Comparison of T cell kinetics before and during antiretroviral therapy. (A) Changes in viral load as well as CD4+ and CD8+ lymphocyte counts during treatment. The data from each patient are represented by a unique symbol. The periods of repeat D-glucose labeling studies are indicated by rectangular boxes marked by each patient's identification number followed by the episode of labeling. (B) Fraction of labeled DNA in CD4+ and CD8+ lymphocytes from P1–P5 during the first (pretreatment) (○), second (gray square), and third (▴) episodes of labeling. The duration of D-glucose administration is indicated by a box in each graph. Again, the data points are represented by symbols, and the lines show the best fit of the data to the mathematical model. Note that the fraction of labeled DNA on day 0 in the second or third episode is not zero because of residual label from the previous labeling episode. (C) Changes in the estimated values of p (proliferation rate) and d (death rate) with antiretroviral therapy.
The results of the sequential D-glucose labeling studies are presented case by case in Fig. 4 B. In all but one patient (P3), the second labeling yielded slower rates of deuterium incorporation and loss in CD4+ and CD8+ lymphocytes as compared with the first labeling. Similarly, the peak values for the fraction of labeled DNA were consistently lower for the second episode, despite having higher, non-zero baseline values (0.005–0.05) that were attributable to the low-level persistence of label from the first study. In P1 and P2, even slower rates of rise and fall of labeled DNA were evident for the third labeling episode, as were the peak values. In P3, the labeling profiles in CD4+ T cells were indistinguishable for the three longitudinal studies, although a slight reduction in labeling was noted in CD8+ T cells for the second and third episodes. It should be pointed out, however, that this patient had the lowest plasma viral load before treatment (Table I) and his first labeling profile was only modestly higher than those of normal individuals (Fig. 3). Thus, in this case, only minimal reductions in labeling could be expected for antiretroviral therapy. The results of P3 notwithstanding, the overall changes in labeling profiles during effective treatment suggest that turnover rates for both CD4+ and CD8+ lymphocytes decline, promptly and substantially, as HIV-1 replication is suppressed.
When the mathematical model was used to analyze the results of these sequential studies during treatment, a good fit to the data was observed once again (Fig. 4 B). Estimates of p and d for CD4+ and CD8+ lymphocytes were then derived for each episode of labeling. Fig. 4 C shows that substantial decreases in both p and d for both T cell populations were seen by the second study, ∼5–11 wk into antiretroviral therapy. Further reductions in these turnover rates were noted for the third study of P1 and P2, ∼46–53 wk into treatment. The decreases in p reached statistical significance by the paired Student's t test for both CD4+ and CD8+ populations (P values of 0.03 and 0.006, respectively), but due to the sample size only approached significance by the more appropriate Wilcoxon signed rank test (0.0625, the smallest two-tailed P value obtainable with 5 data points). The decreases in d were not statistically significant. Nevertheless, these quantitative findings again confirm the qualitative impression that the heightened T lymphocyte turnover in HIV-1 infection is normalized, albeit incompletely, by effective antiretroviral therapy for about a year.
Proliferation Rates of CD4+ and CD8+ T Cells Correlate with Ki67 and TUNEL Expression
We next compared the values of p and d derived for all subjects from the three labeling studies to the expression of two lymphocyte markers: Ki67, a nuclear antigen expressed exclusively in cells in late G1, S, G2, and M phases of the cell cycle (20), and TUNEL, an indicator of apoptosis (21). As shown in Fig. 5 A, there was a direct correlation between the value of p and the fraction of cells positive for Ki67 or TUNEL. These correlations, seen in both CD4+ and CD8+ lymphocytes, were strong (correlation coefficients between 0.79 and 0.91) and statistically significant (P values <0.002). The value of d, however, was only weakly correlated with these markers (P value <0.05), and in CD8+ T cells the correlation between d and TUNEL was not significant (data not shown). Nevertheless, these results suggest that Ki67 and TUNEL expression in blood cells could serve as simple and reasonably reliable surrogate markers for lymphocyte proliferation in vivo.
Figure 5.

Correlation among parameters. (A) Correlation of the fraction of CD4+ and CD8+ lymphocytes expressing Ki67 antigen or TUNEL with p, the lymphocyte proliferation rate. Significant correlations were observed between values of p and fractions of Ki67+ CD4+ T cells (r = 0.91, P value <0.00001) or CD8+ T cells (r = 0.90, P value <0.00001), as well as between values of p and fractions of TUNEL+ CD4+ T cells (r = 0.79, P value = 0.00006) or CD8+ T cells (r = 0.87, P value <0.00001). Note that these regression lines are parallel in CD8+ lymphocytes (P value = 0.86), but not in CD4+ lymphocytes (P value = 0.0017). (B) Significant correlation between baseline CD4+ T cell count or plasma viral load and values of p, fraction of Ki67+ cells, or fraction of TUNEL+ cells in both CD4+ and CD8+ cell populations (P values <0.05). Results of patients with undetectable plasma viral load are plotted as of 50 copies/ml (detection limit).
Kinetic Parameters Correlate Positively with Plasma Viral Loads, and Inversely with CD4 Cell Counts
We also compared levels of Ki67 and TUNEL expression, as well as values of p and d, to the CD4+ T cell count and plasma viral load of each subject at the initiation of each labeling study, which we term baseline measurements (Fig. 5 B). An inverse correlation between the baseline CD4+ lymphocyte count and the fraction of CD4+ or CD8+ T cells positive for Ki67 or TUNEL, was evident, as was reported (5, 22). An inverse relationship was also noted for p, but not for d (data not shown). On the other hand, a direct correlation was observed between the baseline plasma viral load and each of the following parameters: p, Ki67 expression, and TUNEL positivity. A direct correlation was also found between d and baseline viral load, but only in the CD4+ T cell population (r = 0.61, P value = 0.016, data not shown). That such a correlation was not found for CD8+ cells suggests that the rate of death of proliferating CD8+ T cells was the same in uninfected and infected subjects with different viral loads. Moreover, because the linear regression lines for CD8+ lymphocytes are nearly parallel in Fig. 5 B, it appears that the parameters, p, Ki67, and TUNEL, are coupled not only to the amount of HIV-1 replication in vivo, but also to each other. Note that such a relationship also holds true for p and Ki67 expression in CD4+ lymphocytes. However, the linear regression line for TUNEL positivity takes on a significantly different slope (P value < 0.02), indicating that the coupling of cellular proliferation to apoptosis is distinguishable between CD4+ and CD8+ T cells. This difference may be due to the preferential infection and elimination of CD4+ lymphocytes by nonapoptotic mechanisms.
Discussion
The dynamics of lymphocyte turnover in HIV-1 infection remained unresolved despite numerous studies over the past 6 y. We addressed this controversy by conducting a clinical study to directly measure rates of cellular proliferation and death by adapting a recently developed technique using the stable isotope deuterium incorporated into glucose to label the DNA of dividing cells (9, 10, 17). Important modifications in our approach included the extension of D-glucose infusion to 7 d to increase the chance of capturing rare events, as well as the frequent sampling (7–12 data points per study) of blood to carefully define the profile of label uptake and loss. In addition, a mathematical model was developed to interpret the results quantitatively. We now report findings that conclusively demonstrate heightened lymphocyte turnover in HIV-1 infection. As shown in Table I, compared with normal controls, the mean death rate of CD4+ T cells was elevated by ∼3.0-fold, which was somewhat balanced by a 6.3-fold increase in the mean proliferation rate and a 3.6-fold increase in the mean rate of cell infusion from a source (see Discussion below). In CD8+ T cells, the mean death rate was minimally higher (∼1.2-fold) in infected patients, which was accompanied by a 7.7-fold rise in mean proliferation rate and a small decrease in the mean source rate. Even though we have small sample sizes, these results are in good agreement with data from direct labeling studies in infected humans (10, 17) or SIV-infected monkeys (7, 8), or from indirect assessments of cellular proliferation (5, 6, 23–25).
The model that the primary effect of HIV-1/SIV infection is to increase T cell destruction and that the major consequence of antiretroviral therapy is to decrease that destruction was recently called into question (10–12, 17). Lymphocyte turnover was reported to fall only after 12–18 mo of effective treatment (17), but not after 12 wk (10, 17). These results thus raised the specter that the major defect in lymphocyte dynamics during HIV-1 infection is a block in the regenerative capacity. In this study, we have reexamined this important issue by carrying out longitudinal labeling studies in patients before and during successful treatment with drug regimens that are not known to have any direct effects on lymphocytes. It is clear from results shown in Fig. 4 that lymphocyte turnover rates decreased substantially by 5–11 wk into therapy. There was no evidence in any case to indicate that these rates increased as HIV-1 replication came under control. With continuation of antiretroviral agents for ∼1 y, lymphocyte turnover rates dropped further to values only slightly higher than those of normal subjects.
While the small sample size did not allow us to statistically test if the proliferation rates are identical in controls and in infected individuals after long-term therapy, the regression line of p versus viral load in Fig. 5 B suggests that with continued potent therapy the proliferation rates will normalize. This finding is in reasonable agreement with D-glucose-labeling data from two patients treated for 18 mo (17), as well as with those from surrogate measurements (24, 25). However, our results cannot be reconciled with those that showed unchanged (17) or increased (10) lymphocyte turnover with short-term antiretroviral therapy. A possible explanation may be that the previous conclusion was based on a cross-sectional comparison of treated and untreated patient groups that are not well matched clinically. Also, if we only look at single time points after infusion, as was done in previous studies (10, 17), there are several cases where the fraction of labeled DNA is higher in our treated patients than in the nontreated patients. But such a cursory examination is inadequate in accurately capturing the labeling profile of a cell population of interest. Thus, given the strength of our new longitudinal data, there should be little doubt that a model of high lymphocyte turnover and normalization upon treatment correctly depicts the situation in HIV-1 infection.
Some may continue to argue that the rapid turnover of lymphocytes in this disease is accompanied by a diminished fractional regenerative capacity as well. But one must keep in mind that such a scenario would lead to a constant loss of lymphocytes, and could not possibly account for the prolonged quasisteady-state in T cell counts seen in most cases of HIV-1 infection. To maintain a stable lymphocyte count from day to day, a rapid cell death rate must be balanced by an equally rapid replacement rate. In fact, such steady-state considerations have long argued against regenerative failure as the principal cause of CD4+ lymphocyte depletion seen in AIDS, because it necessarily implies a slower death rate for the cell population. That CD4+ T cells would have a longer than normal survival time in the presence of rampant HIV-1 replication is difficult to fathom.
Several of our findings warrant additional comments. First, when the mean values of p in Table I were transformed into doubling times (t2 = ln2/p), averages of 173 and 231 d were obtained in normal persons for CD4+ and CD8+ T cells, respectively. These numbers are in line with prior estimates in normal monkeys (7, 8) and determinations of intermitotic intervals based on the persistence of chromosomal defects in human T lymphocytes (26, 27). Mean values of t2 in infected patients were, as expected, considerably shorter for both CD4+ (28 d) and CD8+ lymphocytes (31 d). When the mean values of d were similarly transformed, an average t1/2 of 16 d was obtained for both T cell populations in normal individuals. In infected patients, a mean t1/2 of 6 d was noted for CD4+ lymphocytes, whereas a mean t1/2 of 14 d was observed for CD8+ lymphocytes. As these survival times are strikingly short, it is important to point out that they refer only to lymphocytes that are amenable to study by the D-glucose labeling technique. But it is clear from our model (Fig. 1) that there could exist a source population, such as resting cells, that could input cells into the compartment under study (7, 18, 19). Since the relative proportion of these two separate cellular compartments is not known, it would be inappropriate to apply these short t1/2's to the entire CD4+ or CD8+ T cell population to derive absolute rates of lymphocyte death per day. In fact, it should be emphasized that only fractional turnover rates are presented and discussed in this study. Any comparison with absolute rates of lymphocyte proliferation and death must be done cautiously. A further source of heterogeneity in the CD4+ and CD8+ T cell populations that is not taken into consideration is their subdivision into naive and memory populations. Memory populations turnover more rapidly than naive populations (17, 26, 27) and thus one might speculate that the kinetic parameters measured in this short-term labeling study are more indicative of memory than naive cells. However, because the fractions of naive and memory cells were not measured and since such fractions vary among individuals and change under treatment, we have refrained from making any such interpretations of our data.
Second, HIV-1 infection is associated with a ∼6-8–fold elevation in the mean values of p in both CD4+ and CD8+ lymphocytes (Table I), consistent with changes seen in SIV infection of macaques (7, 8). A greater increase in the mean value of d, however, was found for CD4+ T cells (approximately threefold) than in CD8+ T cells (∼1.2-fold). These findings show that while there are similarities in the HIV-1–induced generalized activation of both lymphocyte populations, as discussed previously (7, 8, 28), there are also discernible differences in the kinetics of cell death in this study, with the death rate being significantly faster in CD4+ lymphocytes than in CD8+ lymphocytes (P value <0.02). This conclusion is supported by the results presented in Fig. 5 B. In the CD8+ population, both cellular proliferation (p and Ki67 expression) and apoptosis (TUNEL positivity ex vivo) are directly proportional to the degree of virus replication in vivo. On the other hand, in the CD4+ population, the relationship of viral load to apoptosis appears to be different, although the relationship to cellular proliferation remains the same as that in CD8+ T cells. Thus, the combination of a higher death rate (Table I) and a proportionally lower frequency of apoptosis in CD4+ versus CD8+ T cells (Fig. 5 B) suggests that many CD4+ lymphocytes may be dying by a pathway other than generalized T cell activation. We speculate that there is a sizeable component of nonapoptotic virus-mediated cell killing.
Third, the labeling profiles in Figs. 3 and 4 B showed that labeled lymphocytes appeared in blood after only a short lag time. In fact, a detailed analysis using our mathematical model revealed that the average lag was ∼0.5 d (data not shown). Moreover, every determination made on samples taken at the time point immediately after the end of D-glucose administration demonstrated a drop in label incorporation, including timepoints taken only 2–3 d after the infusion period (P5 and P7 in Fig. 3). Taken together, these results argue for rapid transport and equilibration of T lymphocytes between blood and tissue, where much of the cellular proliferation takes place. Models that assume a long delay (10, 17) would yield misleading conclusions. Furthermore, due to this rapid equilibration, the parameter d should reflect true loss of labeled T cells and not simply transport out of the blood as was the case for monocytes.
Lastly, we note that the labeling profiles in Figs. 3 and 4 B cannot be fitted by analytical models (10, 17) that assume p = d and s = 0 in Fig. 1 (data not shown). Infusion of lymphocytes from a source is in fact required. As shown in Table I, a significant increase in the source rate for CD4+ T cells is seen with HIV-1 infection. The thymus is, of course, one such possible source. Several studies have assessed the number of recent thymic emigrants in the blood of HIV-1–infected persons by measuring the concentration of T cell receptor excision circles (TREC) (29–31) as a surrogate. Abnormally low TREC numbers were found in all infected individuals in one study (29), but only in a subset of patients in another study (30). Nevertheless, a low TREC content does not necessarily imply a decreased thymic output, since TREC could be diluted by enhanced lymphocyte proliferation or lost by cell death (30, 31). Thus, it remains possible that the thymus is serving as one source of lymphocytes in our model. However, given that we find that the source rates for CD8 cells are slightly decreased by HIV-1 infection, while CD4+ T cell source rates are increased 3.6-fold (Table I), we favor the hypothesis that a population of resting or slowly dividing T cells in secondary lymphoid tissues is acting as an additional source (7, 19, manuscript in preparation). With infrequent cell divisions, such lymphocytes are essentially invisible to our short-term labeling study. Upon activation, these lymphocytes would undergo clonal expansion and enter the pool of cells acquiring label during the experiment, thereby appearing like a source. With this interpretation, the parameter p that we have estimated could then be interpreted as the average proliferation rate of the resting and activated populations and hence well approximated by the fraction of activated cells times the proliferation rate of an activated cell. Since we only follow the loss of labeled DNA, the estimated death rate, d, would then be interpreted as the death rate of cells that have acquired label, i.e., activated cells. This would also explain why in the CD8+ T cell population there is no difference between death rates of controls and infected individuals, since in both cases we are estimating d for activated cells, and why the cellular half-lives that we have computed from the estimates of d are so short.
In summary, our new findings document the increased turnover of T cells in HIV-1 infection, and reject the notion of a global lymphocyte regenerative defect in this disease.
Acknowledgments
We thank S. Monard, S. Furlan, K. Abe, M. Davidian, and S. Siler for technical assistance, and Glaxo Wellcome, Dupont Pharmaceutical, and Agouron Pharmaceuticals for providing antiretroviral drugs. Portions of this work were performed under the auspices of the U.S. Department of Energy.
This study was supported by The Rockefeller University Hospital General Clinical Research Center, Columbia-Rockefeller Center for AIDS Research (AI42848), the Irene Diamond Fund, Bristol Myers Squibb Foundation, and National Institutes of Health grant AI40387. H. Mohri was a recipient of the CR-CFAR Developmental Award in 1999. L. Weiberger was supported by a predoctoral fellowship from the Howard Hughes Medical Institute.
The preliminary results of this study were presented at the 7th Conference on Retroviruses and Opportunistic Infections from January 30–February 2, 2000, San Francisco, CA (abstract #652) and at the 8th Conference on Retroviruses and Opportunistic Infections from February 4–8, 2001, Chicago, IL (abstract #269).
L. Weinberger's current address is Biophysics Graduate Group, University of California, Berkeley, CA 94720.
Footnotes
*
Abbreviations used in this paper: dA, deoxyadenosine; D-glucose, deuterated glucose; TUNEL, TdT-mediated dUTP nick-end labeling; SIV, simian immunodeficiency virus; TREC, T cell receptor excision circles.
References
- 1.Wei, X., S.K. Ghosh, M.E. Taylor, V.A. Johnson, E.A. Emini, P. Deutsch, J.D. Lifson, S. Bonhoeffer, M.A. Nowak, B.H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 373:117–122. [DOI] [PubMed] [Google Scholar]
- 2.Ho, D.D., A.U. Neumann, A.S. Perelson, W. Chen, J.M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 373:123–126. [DOI] [PubMed] [Google Scholar]
- 3.Sprent, J., and D. Tough. 1995. HIV results in the frame (CD4+ cell turnover). Nature. 375:198. [DOI] [PubMed] [Google Scholar]
- 4.Wolthers, K.C., G. Bea, A. Wisman, S.A. Otto, A.M. de Roda Husman, N. Schaft, F. de Wolf, J. Goudsmit, R.A. Coutinho, A.G. van der Zee, et al. 1996. T cell telomere length in HIV-1 infection: no evidence for increased CD4+ T cell turnover. Science. 274:1543–1547. [DOI] [PubMed] [Google Scholar]
- 5.Sachsenberg, N., A.S. Perelson, S. Yerly, G.A. Schockmel, D. Leduc, B. Hirschel, and L. Perrin. 1998. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 187:1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenner-Racz, K., H.J. Stellbrink, J. van Lunzen, C. Schneider, J.P. Jacobs, B. Raschdorff, G. Grosschupff, R.M. Steinman, and P. Racz. 1998. The unenlarged lymph nodes of HIV-1–infected, asymptomatic patients with high CD4 T cell counts are sites for virus replication and CD4 T cell proliferation. The impact of highly active antiretroviral therapy. J. Exp. Med. 187:949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohri, H., S. Bonhoeffer, S. Monard, A.S. Perelson, and D.D. Ho. 1998. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science. 279:1223–1227. [DOI] [PubMed] [Google Scholar]
- 8.Rosenzweig, M., M.A. DeMaria, D.M. Harper, S. Friedrich, R.K. Jain, and R.P. Johnson. 1998. Increased rates of CD4+ and CD8+ T lymphocyte turnover in simian immunodeficiency virus-infected macaques. Proc. Natl. Acad. Sci. USA. 95:6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macallan, D.C., C.A. Fullerton, R.A. Neese, K. Haddock, S.S. Park, and M.K. Hellerstein. 1998. Measurement of cell proliferation by labeling of DNA with stable isotope-labeled glucose: studies in vitro, in animals, and in humans. Proc. Natl. Acad. Sci. USA. 95:708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellerstein, M., M.B. Hanley, D. Cesar, S. Siler, C. Papageorgopoulos, E. Wieder, D. Schmidt, R. Hoh, R. Neese, D. Macallan, S. Deeks, and J.M. McCune. 1999. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 5:83–89. [DOI] [PubMed] [Google Scholar]
- 11.Pantaleo, G. 1999. Unraveling the strands of HIV's web. Nat. Med. 5:27–28. [DOI] [PubMed] [Google Scholar]
- 12.Rowland-Jones, S. 1999. HIV infection: where have all the T cells gone? Lancet. 354:5–7. [DOI] [PubMed] [Google Scholar]
- 13.Collins, M.L., B. Irvine, D. Tyner, E. Fine, C. Zayati, C. Chang, T. Horn, D. Ahle, J. Detmer, L.P. Shen, et al. 1997. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucl. Acids Res. 25:2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perelson, A.S., A.U. Neumann, M. Markowitz, J.M. Leonard, and D.D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 271:1582–1586. [DOI] [PubMed] [Google Scholar]
- 15.Efron, B., and R. Tibshirani. 1986. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat. Sci. 1:54–77. [Google Scholar]
- 16.Whitelaw, D.M. 1972. Observations on human monocyte kinetics after pulse labeling. Cell Tiss. Kinet. 5:311–317. [DOI] [PubMed] [Google Scholar]
- 17.McCune, J.M., M.B. Hanley, D. Cesar, R. Halvorsen, R. Hoh, D. Schmidt, E. Wieder, S. Deeks, S. Siler, R. Neese, and M. Hellerstein. 2000. Factors influencing T-cell turnover in HIV-1-seropositive patients. J. Clin. Invest. 105:R1–R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.18. Perelson, A.S., S. Bonhoeffer, H. Mohri, and D.D. Ho. 1999. Science. 284:555b–555d.
- 19.Bonhoeffer, S., H. Mohri, D. Ho, and A.S. Perelson. 2000. Quantification of cell turnover kinetics using 5-bromo-2′-deoxyuridine. J. Immunol. 164:5049–5054. [DOI] [PubMed] [Google Scholar]
- 20.Gerdes, J., H. Lemke, H. Baisch, H.H. Wacker, U. Schwab, and H. Stein. 1984. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 133:1710–1715. [PubMed] [Google Scholar]
- 21.Gavrieli, Y., Y. Sherman, and S.A. Ben-Sasson. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gougeon, M.L., H. Lecoeur, A. Dulioust, M.G. Enouf, M. Crouvoisier, C. Goujard, T. Debord, L. Montagnier. 1996. Programmed cell death in peripheral lymphocytes from HIV-infected persons: increased susceptibility to apoptosis of CD4 and CD8 T cells correlates with lymphocyte activation and with disease progression. J. Immunol. 156:3509–3520. [PubMed] [Google Scholar]
- 23.Fleury, S., G.P. Rizzardi, A. Chapuis, G. Tambussi, C. Knabenhans, E. Simeoni, J. Meuwly, J. Corpataux, A. Lazzarin, F. Miedema, and G. Pantaleo. 2000. Long-term kinetics of T cell production in HIV-infected subjects treated with highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA. 97:5393–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hazenberg, M.D., J.W. Stuart, S.A. Otto, J.C. Borleffs, C.A. Boucher, R.J. de Boer, F. Miedema, and D. Hamann. 2000. T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood. 95:249–255. [PubMed] [Google Scholar]
- 25.Lempicki, R.A., J.A. Kovacs, M.W. Baseler, J.W. Adelsberger, R.L. Dewar, V. Natarajan, M.C. Bosche, J.A. Metcalf, R.A. Stevens, L.A. Lambert, et al. 2000. Impact of HIV-1 infection and highly active antiretroviral therapy on the kinetics of CD4+ and CD8+ T cell turnover in HIV-infected patients. Proc. Natl. Acad. Sci. USA. 97:13778–13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michie, C.A., A. McLean, C. Alcock, and P.C. Beverley. 1992. Lifespan of human lymphocyte subsets defined by CD45 isoforms. Nature. 360:264–265. [DOI] [PubMed] [Google Scholar]
- 27.McLean, A.R., and C.A. Michie. 1995. In vivo estimates of division and death rates of human T lymphocytes. Proc. Natl. Acad. Sci. USA. 92:3707–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark, D.R., R.J. de Boer, K.C. Wolthers, and F. Miedema. 1999. T cell dynamics in HIV-1 infection. Adv. Immunol. 73:301–327. [DOI] [PubMed] [Google Scholar]
- 29.Douek, D.C., R.D. McFarland, P.H. Keiser, E.A. Gage, J.M. Massey, B.F. Haynes, M.A. Polis, A.T. Haase, M.B. Feinberg, J.L. Sullivan, et al. 1998. Changes in thymic function with age and during the treatment of HIV infection. Nature. 396:690–695. [DOI] [PubMed] [Google Scholar]
- 30.Zhang, L., S.R. Lewin, M. Markowitz, H.H. Lin, E. Skulsky, R. Karanicolas, Y. He, X. Jin, S. Tuttleton, M. Vesanen, H. Spiegel, et al. 1999. Measuring recent thymic emigrants in blood of normal and HIV-1-infected individuals before and after effective therapy. J. Exp. Med. 190:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hazenberg, M.D., S.A. Otto, J.W. Cohen Stuart, M.C. Verschuren, J.C. Borleffs, C.A. Boucher, R.A. Coutinho, J.M. Lange, T.F. Rinke de Wit, A. Tsegaye, et al. 2000. Increased cell division but not thymic dysfunction rapidly affects the T-cell receptor excision circle content of the naive T cell population in HIV-1 infection. Nat. Med. 6:1036–1042. [DOI] [PubMed] [Google Scholar]