CD1d-dependent Activation of NKT Cells Aggravates Atherosclerosis (original) (raw)
Abstract
Adaptive and innate immunity have been implicated in the pathogenesis of atherosclerosis. Given their abundance in the lesion, lipids might be targets of the atherosclerosis-associated immune response. Natural killer T (NKT) cells can recognize lipid antigens presented by CD1 molecules. We have explored the role of CD1d-restricted NKT cells in atherosclerosis by using apolipoprotein E–deficient (apoE−/−) mice, a hypercholesterolemic mouse model that develops atherosclerosis. ApoE−/− mice crossed with CD1d−/− (CD1d−/−apoE−/−) mice exhibited a 25% decrease in lesion size compared with apoE−/− mice. Administration of α-galactosylceramide, a synthetic glycolipid that activates NKT cells via CD1d, induced a 50% increase in lesion size in apoE−/− mice, whereas it did not affect lesion size in apoE−/−CD1d−/− mice. Treatment was accompanied by an early burst of cytokines (IFNγ, MCP-1, TNFα, IL-2, IL-4, IL-5, and IL-6) followed by sustained increases in IFNγ and IL-4 transcripts in the spleen and aorta. Early activation of both T and B cells was followed by recruitment of T and NKT cells to the aorta and activation of inflammatory genes. These results show that activation of CD1d-restricted NKT cells exacerbates atherosclerosis.
Keywords: α-galactosylceramide, cytokines, inflammation, apolipoprotein E, mice
Introduction
Atherosclerosis is a slowly progressive disease that develops at sites of lipid accumulation in large- and medium-sized arteries, which can ultimately lead to infarction of the heart and brain. Studies in man and experimental models have shown an involvement of innate and adaptive immune mechanisms in the disease process (1, 2). Indeed, atherosclerotic lesions are infiltrated by activated T cells and professional APCs. It has been proposed that the cell-mediated immune response in atherosclerosis is directed against peptide antigens (2). However, given their abundance in the atherosclerotic plaque, lipid antigens may also be targets of disease-associated immune responses. The apolipoprotein E–deficient (apoE−/−) mouse suffers from hypercholesterolemia and develops atherosclerosis spontaneously (3). The distribution of the atherosclerotic lesions and their composition is similar to those observed in humans, and lesion formation proceeds from fatty streak to advanced atheroma with infiltrating inflammatory cells (2, 4).
Lipid antigens can be presented to T cells as part of a complex with the CD1 molecule, which is displayed on certain APCs (5). CD1d-restricted NKT cells constitute a unique subpopulation of T cells with surface expression of proteins commonly expressed by NK cells, such as the NK1.1 receptor (CD161) (6). NKT cells in mice express TCRs with a semiinvariant Vα14-Jα281 α-chain associated with either Vβ2, β7, or β8 β-chains. Upon stimulation, these cells have the capacity to rapidly secrete large amounts of cytokines, including IL-4 and IFNγ and to cause bystander activation of NK cells, B cells, DCs (7), and CD4+ and CD8+ T cells (8). The natural ligand(s) for CD1d-restricted NKT cells remains to be characterized, but the synthetic glycolipid α-galactosylceramide (αGalCer) (9) has been shown to specifically activate these cells, influence the Th effector response (6), and modulate T cell–dependent autoimmune diseases (10–12). Atherosclerosis bears several features of such diseases and is affected by Th modulation (2, 13). An oligoclonal expansion of Vα14+ (also called Vα34s) cells has been observed in the lesions of apoE−/− mice (14), and both CD1+ and NKT cells have been detected in human lesions (15, 16), underlining their possible involvement in the disease. These findings, and the capacity of NKT cells to produce large amount of cytokines, suggest that inflammation in the atherosclerotic plaque, and thus atherogenesis, might be modulated by CD1d-restricted NKT cells.
In this report, we studied the effect of CD1d-dependent stimulation of NKT cells on atherosclerosis by using αGalCer treatment in apoE−/− and apoE−/−CD1d−/− mice. The present results show that CD1d deficiency reduces atherosclerosis, whereas αGalCer treatment significantly increases disease in apoE−/− mice. Together, these data imply that CD1d-restricted NKT cell activation promotes development of atherosclerosis and suggests that lipid antigen presentation may be an important contributor to the pathogenesis of the disease.
Materials and Methods
Animals.
Double deficient apoE−/−CD1d−/− mice were generated by crossing CD1d−/− mice (17), backcrossed at least six times to the C57BL/6J background, with apoE−/− mice (Taconic M&B). The offspring was intercrossed to produce mice with homozygous deletion in both apoE and CD1d genes (apoE−/− CD1d−/−). For experiments, 5-wk-old female apoE−/− and apoE−/−CD1d−/− mice were injected twice a week with PBS (controls) or αGalCer (Kirin Brewery Company). The first injection was performed i.v. and the following i.p. For the long-term protocol (20 injections), 12 mice per group were used. For shorter protocols (one, three, or five injections), groups of five to six mice received αGalCer and groups of three to five mice received PBS. Animals were kept on chow diet, and all experiments were approved by the institutional ethical committee for animal welfare.
Sample Preparation and Lesion Analysis.
Animals were anesthetized with carbon dioxide and killed by exsanguination through cardiac puncture. After vascular perfusion with sterile RNase-free PBS, the heart, spleen, and liver were removed. The descending aorta was immediately and carefully dissected free from surrounding tissue under a dissection microscope and snap frozen for mRNA analysis. Lipoprotein profiles were determined in serum by fast protein liquid chromatography (18). The heart and proximal aorta were embedded in OCT compound (Tissue-tek) and frozen. 10-μm cryosections of the aortic root were analyzed for lesion size. Lesion size was defined as the cross section surface area of Oil Red O staining within the aortic intima plus media. Mean lesion size for each mouse was calculated from measurement of cryosections taken from every 100 μm of the first 600 μm in the ascending aorta, starting from the aortic cusps. Sections were also used for immunohistochemistry to identify I-Ab (KH74; BD Biosciences), VCAM-1 (429; BD Biosciences), and αSM-actin (A5691; Sigma-Aldrich). The expression of VCAM-1 was quantitated by dividing the stained surface area by the total surface area of the vessel. The expression of I-Ab was quantitated by dividing the number of I-Ab+ cells by the total number of hematoxylin-stained cells per lesion. All histological analyses were done in a blinded fashion.
Flow Cytometry.
Spleen and hepatic mononuclear cells were prepared as described previously (13, 19). NKT cells were detected either by immunofluorescent double staining with PE–anti-NK1.1 (PK136) and FITC–anti-TCRβ (H57–597) antibodies or double staining with αGalCer-loaded CD1d-dimer (PElabeled) (Becton Dickinson) and FITC–anti-TCRβ. Activated T cells were detected by staining spleen mononuclear cells with PE–anti-CD69 (H1–2F3) and FITC–anti-TCRβ antibodies, whereas activated B cells were detected by PE–anti-CD19 (1D3) and FITC–anti-B7.2 (GL1) antibodies. All antibodies were from BD Biosciences, and the analyses performed on a FACSCalibur® (Becton Dickinson).
Cytometric Bead Array.
IFNγ, MCP-1, TNFα, IL-2, IL-4, IL-5, IL-6, IL-10, and IL-12 concentrations in sera were analyzed with the Mouse Th1/Th2 Cytokine (no. 551287) and the Mouse Inflammation (no. 552364) Cytometric Beads Array kits from BD Biosciences according to the manufacturer's instructions. Analyses were run on a FACSCalibur®.
RNA Preparation and RT-PCR.
RNA was prepared from spleen and aorta after homogenizing tissue in FastRNA® Pro Green (Q-Biogene) with equal volumes of RLT lysis buffer (QIAGEN) and phenol-CHISAM (Sigma-Aldrich). Purification of total RNA was performed using the RNeasy system (QIAGEN) including a DNase step. RNA concentration and quality were assessed on a Bioanalyzer capillary electrophoresis system (Agilent). Real-time PCR was performed in a TaqMan 7700 (Applied Biosystems) according to the manufacturer's instructions. cDNA was synthesized from total RNA using Superscript II (Invitrogen).
Statistics.
Data are presented as mean ± SEM. Differences between the means were evaluated by using the Mann-Whitney test and were considered significant when P < 0.05.
Online Supplemental Material.
The supplemental Materials and Methods (available at http://www.jem.org/cgi/content/full/jem.20030997/DC1) provides details on the real-time RT-PCR analyses.
Results and Discussion
Two approaches were used in order to determine the role of the CD1d-NKT cells in atherosclerosis: generation of double deficient apoE−/−CD1d−/− mice and specific NKT cell stimulation by αGalCer treatment. Total cholesterol and triglyceride levels did not differ between apoE−/− CD1d−/− double deficient and apoE−/− single deficient mice, treated or not with αGalCer, nor did plasma lipoprotein profiles determined by fast protein liquid chromatography (not depicted).
ApoE−/−CD1d−/− mice exhibited significantly smaller atherosclerotic lesions in the aortic root than apoE−/− single knockout mice (74,638 ± 5.9 μm2 versus 100,548 ± 5.6 μm2; P = 0.0087) (Fig. 1, a–c). This was apparent throughout the aortic root (Fig. 1 b). When CD1d-restricted NKT cells were activated by administration of αGalCer over a 10-wk period, lesion size increased by 50% in apoE−/− mice compared with PBS-treated control mice (151,276 ± 9.377 μm2 versus 100,548 ± 5.572 μm2; P < 0.0001). In apoE−/− CD1d−/− mice, αGalCer did not affect lesion size (Fig. 1, a and c), demonstrating that the effect of αGalCer on atherosclerosis depends on CD1d. Together, these findings indicate that CD1d-dependent activation of NKT cells aggravates atherosclerosis and that lack of CD1d, the restriction element for presentation of lipid antigens to NKT cells, leads to reduced lesions in a mouse model of human atherosclerosis.
Figure 1.


Effects of CD1d deficiency and αGalCer treatment on atherosclerosis. 5-wk-old female apoE−/− and apoE−/−CD1d−/− mice were injected twice a week for 10 wk with αGalCer or PBS and killed 48 h after the last injection (n = 12 for each group). (a) Mean lesion size in Oil Red O–stained aortic root sections. Mean ± SEM (***P < 0.001 versus apoE−/−-PBS and all apoE−/−CD1d−/−; §§P < 0.01 versus apoE−/−-PBS mice). (b) Lesion size at every 100 μm for the first 600 μm of the aortic root in apoE−/− and apoE−/−CD1d−/− mice. (c) Representative Oil Red O–stained cryosections of aortic roots (magnification ×50).
To assess inflammatory activation, lesions in the aortic root were stained for the adhesion molecule VCAM-1 and the MHC class II protein I-Ab. VCAM-1 was expressed in the lesion and in the media underneath the lesion (Fig. 2, a–c). αSM-actin staining of adjacent aortic root sections confirmed that VCAM-1 was mainly expressed by smooth muscle cells (not depicted). I-Ab was expressed by inflammatory cells in the lesions (Fig. 2, d and e). ApoE−/− CD1d−/− mice expressed significantly less VCAM-1 than apoE−/− mice (P = 0.027) (Fig. 2 a). The proportion of I-Ab–expressing cells did not differ between apoE−/− and apoE−/−CD1d−/− mice (Fig. 2 d); however, the decrease in lesion area in the CD1d−/− mice resulted in a reduced number of I-Ab cells per section when compared with apoE−/− mice. αGalCer treatment increased VCAM-1 and I-Ab expression in apoE−/− mice (P = 0.036) but not in apoE−/− CD1d−/− mice (Fig. 2, a–e), implying that αGalCer induction of these genes was dependent on CD1d-restricted NKT cells.
Figure 2.

Effects of CD1d deficiency and αGalCer treatment on the expression of VCAM-1 and I-Ab in atherosclerotic lesions. Experimental groups were the same as in Fig. 1. (a) VCAM-1 quantitation (VCAM-1+ area/vessel area); (b and c) representative sections of aortic root stained for VCAM-1 by avidin-biotin-immunoperoxidase (brown) (×50 and ×400). (d) I-Ab quantitation (I-Ab+ cells/total hematoxylin+ cells) and (e) representative sections of aortic roots stained for I-Ab by avidin-biotin-immunoperoxidase (brown) (×400). Arrows point at I-Ab+ cells. Mean ± SEM (*P < 0.05 versus apoE−/− treated with PBS and versus all apoE−/−CD1d−/−; **P < 0.01 versus all apoE−/−CD1d−/− mice; §P < 0.05 versus apoE−/−-PBS).
NKT cell activation is likely to enhance activation of macrophages, endothelial cells, and other cells capable of secreting proinflammatory cytokines. Such a cascade may be responsible for the increased expression of VCAM-1 and I-Ab observed in αGalCer-treated mice. VCAM-1 expression by vascular smooth muscle cells is a characteristic feature of atherosclerosis, where it reflects inflammatory activation of lesion cells; however, its role in the recruitment and activation of inflammatory cells remains unclear. Endothelial VCAM-1 expression contributes to atherosclerosis by promoting recruitment of mononuclear cells to forming lesions (20).
An early burst of inflammatory cytokines was detected in sera after an injection of αGalCer; both typical Th1 (IFNγ, TNFα, IL-2) and Th2 (IL-4, IL-5) cytokines were increased as well as IL-6 and MCP-1 (Fig. 3 a). However, neither IL-10, which has antiinflammatory and atheroprotective properties (18, 25), nor IL-12 was detected in any of the groups (not depicted). The increase was remarkable, e.g., 5,000-fold for IFNγ and 250-fold for MCP-1. Bystander activation of T and B cells was registered by an increased number of cells double positive for CD69/TCRβ and B7.2/CD19, respectively (Fig. 3 b). This early burst of cytokines might explain the increased expression of VCAM-1, which can be induced by proinflammatory cytokines, and I-Ab, which is induced by IFNγ. The increased levels of circulating MCP-1 might be significant for the exacerbated lesion development in αGalCer-treated mice, since this chemokine has important proatherogenic effects (21, 22). High levels of IL-6 could also have effects on atherosclerosis, since early lesions in apoE−/− mice are exacerbated by recombinant IL-6 (23). Elevated circulating IL-6 concentrations is correlated with increased risk of coronary and peripheral atherosclerosis in man (24). After repeated injections, serum levels of cytokines fell below the detection limit, and no signs of increased T or B cell activation could be detected anymore. Real-time RT-PCR analysis (see Supplemental Materials and Methods, available at http://www.jem.org/cgi/content/full/jem.20030997/DC1) of spleen mRNA confirmed the increase in IFNγ (85-fold) and IL-4 (25-fold) mRNA in mice injected once with αGalCer (Fig. 3 c) and demonstrated that increased IFNγ and IL-4 expression in the spleen was still detectable after repeated injections (Fig. 3 c).
Figure 3.

Systemic effects of αGalCer in apoE−/− mice. 5-wk-old apoE−/− mice were injected one, three, or five times, twice a week, with αGalCer or PBS. The first injection was i.v. (n = 6 for αGalCer and n = 5 for PBS) and the following ones i.p (n = 5 for αGalCer and n = 3 for PBS). Mice were killed 12 h after the last injection. (a) Serum levels of cytokines after one injection of αGalCer or PBS. In control (PBS) mice, levels of IFNγ, TNFα, and IL-6 were not detectable (nd). (b) Activated spleen T (CD69+TCRαβ+) and B (CD19+B7.2+) cells determined by flow cytometry. (c) IFNγ and IL-4 mRNA in spleens measured by quantitative real-time RT-PCR and normalized to β-actin mRNA after one, three, and five injections. The values for IFNγ are represented in a logarithmic scale. Mean ± SEM (*P < 0.05; **P < 0.01 versus PBS-injected mice).
Direct evidence for a proatherogenic action of IFNγ has been provided by several studies in gene-targeted mice (2, 26), and analysis of human lesions has revealed expression of IFNγ by activated immune cells (2). Therefore, it is likely that the early burst of IFNγ upon αGalCer treatment contributed to the proatherogenic effect observed in our study. The increased expression of IFNγ-inducible I-Ab protein in aortic lesions and the demonstration of elevated aortic IFNγ mRNA (see below) in αGalCer-treated mice support this notion. The effects of the sustained production of IL-4 are more difficult to interpret since the role of IL-4 in atherosclerosis is still under debate (24). However, our data show a correlation between increased IL-4 secretion and increased lesion size. These findings are in line with those in a recent study of atherosclerosis in double-deficient apoE−/−IL-4−/− mice (27).
αGalCer injections led to dramatic systemic changes in the NKT cell population. When comparing apoE−/− mice injected once or repeatedly with αGalCer, a transient decrease followed by recovery was observed for TCR+ NK1.1+ cells in the spleen (Fig. 4 a) and liver (not depicted). This could either be due to activation-induced apoptosis or down-regulation of the NK1.1 receptor. To address this issue, NKT cells were also detected using CD1d-dimer staining in mice injected once i.v. with αGalCer and killed 12 or 72 h later. In the spleen, activation through αGalCer injection led to an initial reduction of NKT cells, but after 72 h the spleen had five times more NKT cells than spleens from PBS-injected controls (Fig. 4 b). In the liver, the NKT cell population decreased 12 h after the injection but returned to normal levels after 72 h (Fig. 4 b).
Figure 4.
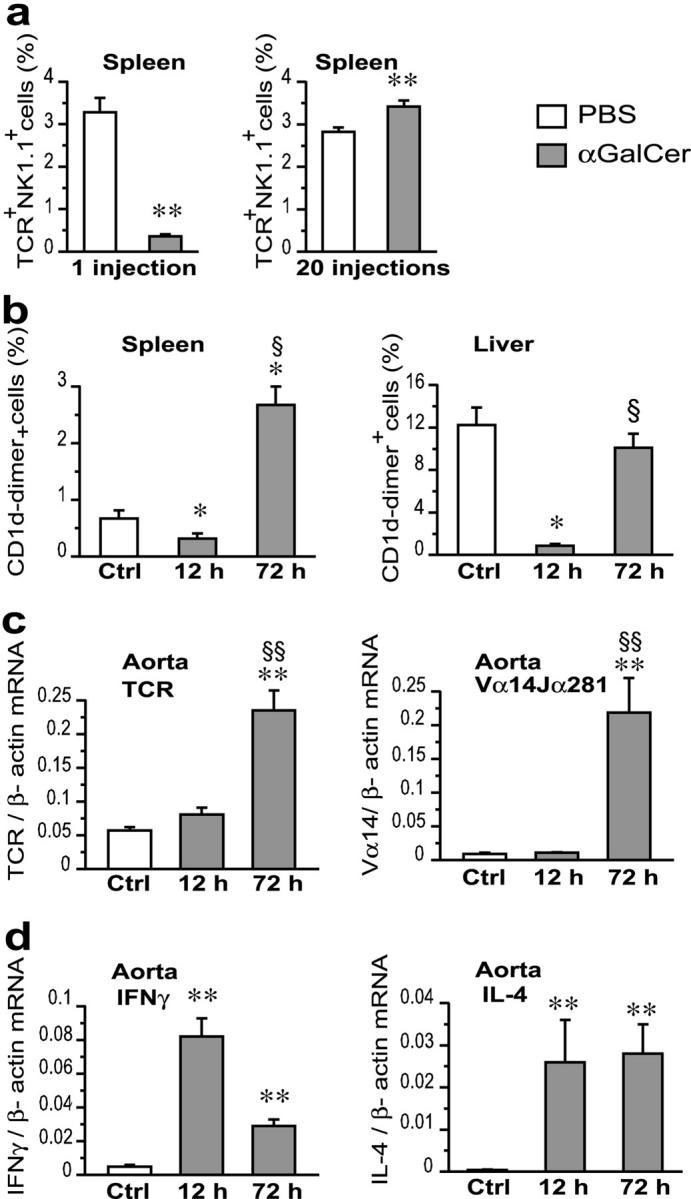
Effect of αGalCer injection on cytokine and TCR transcripts in spleen and aorta of apoE−/− mice. (a) Proportion of spleen cells double positive for TCRβ and NK1.1 in mice injected once and killed at 12 h and in mice injected 20 times, starting at 5 wk of age, and killed 48 h after the last injection. FACS® data expressed as the percentage of forward/side scatter-gated lymphocytes. (b) CD1d-dimer+ NKT cells in spleen and liver of 5-wk-old mice injected once i.v. with PBS (Ctrl) (n = 6) or αGalCer and killed 12 h (n = 5) or 72 h (n = 6) later. FACS® data expressed as the percentage of forward /side scatter-gated lymphocytes. (c and d) Levels of total TCRβ and Vα14Jα281- specific TCR mRNA (c) and of IFNγ and IL-4 mRNA (d) in the aorta of mice treated with PBS or αGalCer as described for panel b. Real-time RT-PCR data are expressed as relative mRNA units normalized to β-actin mRNA. Mean ± SEM (*P < 0.05; **P < 0.01 versus PBS-injected mice; §P < 0.05; §§P < 0.01 versus αGalCer-injected mice, killed at 12 h).
The local NKT cell response in the aorta was detected by TCR mRNA analysis (see Supplemental Materials and Methods, available at http://www.jem.org/cgi/content/full/jem.20030997/DC1). Both total TCR mRNA and NKT cell–specific Vα14-Jα281 transcripts were already present in the aorta of PBS-injected apoE−/− control mice (Fig. 4 c). This is in line with our previous finding of Vα14 (also called Vα34s) when analyzing TCR transcripts in advanced lesions of apoE−/− mice (14). After one i.v. injection of αGalCer, an increase in TCR mRNA in the aorta could be detected as early as 12 h after injection and continued to increase at 72 h. NKT cell–specific Vα14-Jα281 mRNA increased dramatically 72 h after treatment also. IFNγ and IL-4 mRNA increased in the aorta of αGalCer-injected mice (Fig. 4 d). This rise peaked at 12 h after injection, and transcripts remained elevated after 72 h. The early increases in IFNγ and IL-4 mRNA in the aorta is likely due to the NKT cells already present in the lesion, which may in turn activate other cytokine-producing cells such as classical T cells and macrophages. Our data also suggest that systemic NKT cell activation induced by αGalCer injection leads to increased recruitment and, probably, activation of NKT cells in lesions. Such a scenario is also supported by the increased I-Ab and VCAM-1 expression in lesions of mice with αGalCer-activated NKT cells.
The finding of a proatherogenic role for NKT cells suggests that antigen-specific activation of these cells could contribute to disease development. The natural antigens for CD1d-restricted NKT cells are still unknown, despite numerous studies, but are thought to include lipid antigens. Atherosclerosis is strongly linked to lipid metabolic disturbances, and therefore our results suggest that lipid antigens may be involved in the disease process (2, 28). Further studies will be needed to identify such disease-related lipid antigens, clarify the precise role of NKT cells in the different phases of disease development, and determine the role of the CD1d-NKT cells in human atherosclerosis. In conclusion, controlling the activation state of NKT cells may represent a new therapeutic approach for atherosclerosis.
Acknowledgments
We are grateful to Kirin Brewery Company for providing us with αGalCer, and to Dr. Y.J. Geng for helpful discussion.
This work was supported by the Swedish Research Council (projects 14106, 14121, and 6816), the Swedish Heart-Lung Foundation, the ARCOL Foundation, Torsten and Ragnar Söderberg Foundation, and Arbetsmarknadens Försäkrings AB National Institutes of Health grant HL69509. G. Paulsson Berne and M. Rudling are investigators of the Swedish Research Council. E. Tupin is the recipient of a research stipend from the Swedish Heart-Lung Foundation.
The online version of this article contains supplemental material.
References
- 1.Binder, C.J., M.K. Chang, P.X. Shaw, Y.I. Miller, K. Hartvigsen, A. Dewan, and J.L. Witztum. 2002. Innate and acquired immunity in atherogenesis. Nat. Med. 8:1218–1226. [DOI] [PubMed] [Google Scholar]
- 2.Hansson, G.K. 2001. Immune mechanisms in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 21:1876–1890. [DOI] [PubMed] [Google Scholar]
- 3.Piedrahita, J.A., S.H. Zhang, J.R. Hagaman, P.M. Oliver, and N. Maeda. 1992. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 89:4471–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breslow, J.L. 1996. Mouse models of atherosclerosis. Science. 272:685–688. [DOI] [PubMed] [Google Scholar]
- 5.Joyce, S., and L. Van Kaer. 2003. CD1-restricted antigen presentation: an oily matter. Curr. Opin. Immunol. 15:95–104. [DOI] [PubMed] [Google Scholar]
- 6.Kronenberg, M., and L. Gapin. 2002. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2:557–568. [DOI] [PubMed] [Google Scholar]
- 7.Fujii, S., K. Shimizu, C. Smith, L. Bonifaz, and R.M. Steinman. 2003. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J. Exp. Med. 198:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godfrey, D.I., K.J. Hammond, L.D. Poulton, M.J. Smyth, and A.G. Baxter. 2000. NKT cells: facts, functions and fallacies. Immunol. Today. 21:573–583. [DOI] [PubMed] [Google Scholar]
- 9.Kawano, T., J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, et al. 1997. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 278:1626–1629. [DOI] [PubMed] [Google Scholar]
- 10.Sharif, S., G.A. Arreaza, P. Zucker, Q.S. Mi, J. Sondhi, O.V. Naidenko, M. Kronenberg, Y. Koezuka, T.L. Delovitch, J.M. Gombert, et al. 2001. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune type 1 diabetes. Nat. Med. 7:1057–1062. [DOI] [PubMed] [Google Scholar]
- 11.Hong, S., M.T. Wilson, I. Serizawa, L. Wu, N. Singh, O.V. Naidenko, T. Miura, T. Haba, D.C. Scherer, J. Wei, et al. 2001. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat. Med. 7:1052–1056. [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto, K., S. Miyake, and T. Yamamura. 2001. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 413:531–534. [DOI] [PubMed] [Google Scholar]
- 13.Laurat, E., B. Poirier, E. Tupin, G. Caligiuri, G.K. Hansson, J. Bariety, and A. Nicoletti. 2001. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 104:197–202. [DOI] [PubMed] [Google Scholar]
- 14.Paulsson, G., X. Zhou, E. Törnquist, and G.K. Hansson. 2000. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 20:10–17. [DOI] [PubMed] [Google Scholar]
- 15.Melian, A., Y.J. Geng, G.K. Sukhova, P. Libby, and S.A. Porcelli. 1999. CD1 expression in human atherosclerosis. A potential mechanism for T cell activation by foam cells. Am. J. Pathol. 155:775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan, W.L., N. Pejnovic, T.V. Liew, C.A. Lee, R. Groves, and H. Hamilton. 2003. NKT cell subsets in infection and inflammation. Immunol. Lett. 85:159–163. [DOI] [PubMed] [Google Scholar]
- 17.Mendiratta, S.K., W.D. Martin, S. Hong, A. Boesteanu, S. Joyce, and L. Van Kaer. 1997. CD1d1 mutant mice are deficient in natural T cells that promptly produce IL-4. Immunity. 6:469–477. [DOI] [PubMed] [Google Scholar]
- 18.Caligiuri, G., M. Rudling, V. Ollivier, M.P. Jacob, J.B. Michel, G.K. Hansson, and A. Nicoletti. 2003. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol. Med. 9:10–17. [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe, H., K. Ohtsuka, M. Kimura, Y. Ikarashi, K. Ohmori, A. Kusumi, T. Ohteki, S. Seki, and T. Abo. 1992. Details of an isolation method for hepatic lymphocytes in mice. J. Immunol. Methods. 146:145–154. [DOI] [PubMed] [Google Scholar]
- 20.Cybulsky, M.I., and M.A. Gimbrone, Jr. 1991. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 251:788–791. [DOI] [PubMed] [Google Scholar]
- 21.Gu, L., Y. Okada, S.K. Clinton, C. Gerard, G.K. Sukhova, P. Libby, and B.J. Rollins. 1998. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 2:275–281. [DOI] [PubMed] [Google Scholar]
- 22.Boring, L., J. Gosling, M. Cleary, and I.F. Charo. 1998. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 394:894–897. [DOI] [PubMed] [Google Scholar]
- 23.Huber, S.A., P. Sakkinen, D. Conze, N. Hardin, and R. Tracy. 1999. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 19:2364–2367. [DOI] [PubMed] [Google Scholar]
- 24.Von Der Thusen, J.H., J. Kuiper, T.J. Van Berkel, and E.A. Biessen. 2003. Interleukins in atherosclerosis: molecular pathways and therapeutic potential. Pharmacol. Rev. 55:133–166. [DOI] [PubMed] [Google Scholar]
- 25.Mallat, Z., S. Besnard, M. Duriez, V. Deleuze, F. Emmanuel, M.F. Bureau, F. Soubrier, B. Esposito, H. Duez, C. Fievet, et al. 1999. Protective role of interleukin-10 in atherosclerosis. Circ. Res. 85:e17–e24. [DOI] [PubMed] [Google Scholar]
- 26.Gupta, S., A.M. Pablo, X. Jiang, N. Wang, A.R. Tall, and C. Schindler. 1997. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Invest. 99:2752–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davenport, P., and P.G. Tipping. 2003. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 163:1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binder, C.J., S. Hörkkö, A. Dewan, M.K. Chang, E.P. Kieu, C.S. Goodyear, P.X. Shaw, W. Palinski, J.L. Witztum, and G.J. Silverman. 2003. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 9:736–743. [DOI] [PubMed] [Google Scholar]