Gene Dosage–limiting Role of Aire in Thymic Expression, Clonal Deletion, and Organ-specific Autoimmunity (original) (raw)
Abstract
Inactivation of the autoimmune regulator (Aire) gene causes a rare recessive disorder, autoimmune polyendocrine syndrome 1 (APS1), but it is not known if Aire-dependent tolerance mechanisms are susceptible to the quantitative genetic changes thought to underlie more common autoimmune diseases. In mice with a targeted mutation, complete loss of Aire abolished expression of an insulin promoter transgene in thymic epithelium, but had no effect in pancreatic islets or the testes. Loss of one copy of Aire diminished thymic expression of the endogenous insulin gene and the transgene, resulting in a 300% increase in islet-reactive CD4 T cells escaping thymic deletion in T cell receptor transgenic mice, and dramatically increased progression to diabetes. Thymic deletion induced by antigen under control of the thyroglobulin promoter was abolished in Aire homozygotes and less efficient in heterozygotes, providing an explanation for thyroid autoimmunity in APS1. In contrast, Aire deficiency had no effect on thymic deletion to antigen controlled by a systemic H-2K promoter. The sensitivity of Aire-dependent thymic deletion to small reductions in function makes this pathway a prime candidate for more subtle autoimmune quantitative trait loci, and suggests that methods to increase Aire activity would be a potent strategy to lower the incidence of organ-specific autoimmunity.
Keywords: diabetes mellitus type I, autoimmune diseases, clonal deletion, immune tolerance, thymus
Introduction
Organ-specific tolerance was originally thought to depend primarily on a passive process of immunological “ignorance” due to sequestration of the antigen from naive T cells (1), or upon peripheral tolerance mechanisms such as peripheral deletion (2–4), anergy (4), or suppression by regulatory T cells (5, 6). However, a growing body of evidence indicates that the thymus establishes organ-specific tolerance via promiscuous expression of organ-specific antigens and deletion of forbidden clones with high avidity TCRs against these antigens (7–15). There is also increasing evidence that a failure in promiscuous thymic expression may result in autoimmunity. Thymic expression of myelin proteolipid protein induces clonal deletion of proteolipid protein–reactive T cells (16), but the thymus of SJL mice (like the DBA/2 and B6 strains) expresses a splice variant from this gene lacking a dominant epitope that coincides with a high frequency of encephalitogenic T cells in the periphery (17). In human type 1 diabetes, a frequent susceptibility allele of IDDM2 corresponds to a polymorphism in the insulin gene promoter that lowers thymic expression of pro-insulin mRNA only approximately two- to threefold (18, 19), raising the question of whether or not thymic deletion is sensitive to such subtle changes in thymic antigen levels.
A major advance establishing the significance of thymic tolerance to organ-specific gene products has come through the study of autoimmune polyendocrine syndrome 1 (APS1), a rare monogenic human disorder caused by homozygous mutations in autoimmune regulator (AIRE; references 20 and 21). Clinical manifestations of APS1 include a constellation of organ-specific autoimmune diseases (22). Autoimmune thyroid disease occurs as part of the spectrum in ∼13% of APS1 patients, and type 1 diabetes is present at a similar rate, potentially dependent on the MHC. The Aire protein localizes in nuclear speckles (23, 24), interacts with CBP (transcriptional coactivator CREB-binding protein; reference 25), and is able to activate transcription in luciferase assays (25, 26). Aire is expressed most highly in scattered thymic medullary epithelial cells (23, 27, 28). Aire knockout mice develop a range of organ-specific autoimmune manifestations comparable to human APS1 (27, 29). Anderson et al. (29) showed that autoimmunity is conferred by transplantation of an _Aire_-deficient thymus, and thymic medullary epithelial cells isolated from Aire knockout mice had lost promiscuous expression of a large number of organ-specific mRNAs including proinsulin mRNA. Liston et al. (30) showed that the thymus in Aire knockout mice had lost the ability to delete high avidity CD4+ T cells recognizing a transgenic antigen controlled by the insulin promoter.
So far, no information exists regarding Aire's role in thymic expression of thyroid-specific antigens, despite the fact that autoimmune thyroid disease is the most common autoimmune disease and subclinical thyroiditis occurs in up to 10% of women. Moreover, it is unknown if _Aire_-dependent thymic deletion is a well-buffered process that is only disrupted in this rare monogenic autoimmune disease, or alternatively, if it is a process prone to failure through the actions of more subtle, quantitative reductions in the pathway as are more likely to occur in common polygenic autoimmune diseases. Here, we show that Aire is essential for thymic expression and thymic deletion to antigen under islet- and thyroid-specific promoters, but not under a systemic promoter, and that _Aire_-induced elimination of forbidden clones of organ-specific cells is exquisitely sensitive to small functional reductions.
Materials and Methods
Mice.
3A9 TCR transgenic (31), ILK-3 Ins–hen egg lysozyme (HEL) transgenic (32), TLK3 Tg:mHEL transgenic (32, 33), and KLK4 H-2k:mHEL transgenic mice (34) produced in C57BL/6J mice were backcrossed more than seven generations to B10.Br/SgSnJ (JAX). (B6 × 129/Sv)F2 Aire +/0 mice (27) were backcrossed to the three strains of HEL:3A9 B10.Br mice for two generations. Backcross offspring were typed for TCR, HEL, H-2 (32), and Aire (27) by PCR, and H-2k Aire +/0 mice of appropriate HEL:TCR genotype were intercrossed to produce Aire 0/0 H-2k HEL:TCR double transgenic mice. Additional crosses (e.g., HEL Aire +/0 × TCR Aire 0/0) were used to increase the numbers of rare genotypes for experimental groups. Additional crosses were not considered for analysis of breeding performance. Experimental mice were age/sex matched, housed under identical conditions, and confirmed twice for genotype by PCR. All animal experiments were approved by the Animal Ethics and Experimentation Committee of the Australian National University.
Immunofluorescence Analysis.
Rat anti-AIRE mAbs were produced in the Monoclonal Antibody Facility at Walter and Eliza Hall Institute of Medical Research by standard polyethylene glycol fusion. Clone 5H12 (IgG2c) was generated to a 21–amino acid peptide corresponding to the 20 COOH-terminal amino acids of Aire with a C residue at the NH2 terminus for KLH conjugation. Thymi frozen in OCT compound were sectioned (8 μm), stained with rat anti-Aire mAb and rabbit anti-cytokeratin (DakoCytomation), followed by Alexa 488 anti–rat IgG and Alexa 568–conjugated anti–rabbit IgG, and mounted with fluorescent mounting medium (DakoCytomation). Images were acquired on a confocal microscope (MRC 1024; Bio-Rad Laboratories) with a three-line Kr/Ar laser (excitation lines 488, 568, and 647 nm).
HEL Expression on Pancreatic β Cells and in Serum.
Pancreatic islets were extracted from mice using a protocol modified from Bowen et al. (35), by infusing the common bile duct with Liberase R1 (Boehringer) and hand-picking islets. From purified islets, a single cell suspension was prepared by incubation with 0.25% trypsin (Boehringer) in HBSS at 37°C for 10 min, and stained with HyHEL9-tricolor. Serum HEL measurement was performed by capture on Ig-HEL-Tg spleen cells (36), staining with HyHEL9-tricolor (37), and flow cytometry as described in Zhang et al. (38).
Thymic Stroma Preparation.
Thymic stroma was enriched from the thymus of 6–12-wk-old transgenic mice as described previously (39). After enzymatic enrichment, CD45− thymic stromal cells were purified using CD45 microbeads (Miltenyi Biotec) and the AutoMACS system (Miltenyi Biotec), as per the manufacturer's recommendations. Purity was >98% as assessed by staining with FITC-conjugated anti-CD45.2 (clone 104; BD Biosciences).
HEL mRNA Measurement.
RNA was purified using the Trizol reagent (Invitrogen) and reverse transcribed into cDNA using oligo-dT and Superscript III (Invitrogen), as per the manufacturer's recommendations. Quantitative real-time PCR was performed using the Applied Biosystems Sybr Green master-mix system, primers for HEL (caacacccaggctacaaacc and gtttccatcgctgacgatct), insulin I (aaaggctctttacctggtgtgtgg and actgatccacaatgccacgcttct), or β actin (cgtgaaaagatgacccagatca and tggtacgaccagaggcatacag), and an ABI SDS7700 real-time PCR analyzer (Applied Biosystems). Cycle conditions were as follows: HEL: 95°C for 15 min followed by 40 cycles at 94°C for 15 s, 65°C for 30 s, and 72°C for 30 s; β actin/insulin: 95°C 15 min followed by 40 cycles at 94°C for 15 s, 60°C for 30 s, and 72°C for 30 s. Units of relative expression were calculated using the exponential of the difference in the cycle number at which the HEL and β actin reactions crossed the threshold value, relative to the nontransgenic sample.
Flow Cytometry.
6–12-wk-old mice were analyzed as described previously (40) using the following antibodies: 1G12 anti-clonotype (41) culture supernatant followed by rat anti–mouse IgG1 allophycocyanin; anti–CD8α-PerCP; anti–CD4-FITC or PE, anti–CD3-PE, and anti–CD69-PE (all from BD Biosciences); and anti–CD5-FITC and anti–CD25-PE (both from Caltag). Pancreatic islet cells were stained using HyHEL9-TC (purified and conjugated in our laboratory), with β islet cells distinguished by forward and side scatter.
Diabetes Incidence Study.
Urine glucose was tested using Testape/Glucostix at biweekly intervals or when the cage was wet. Mice with two successive positive Testapes were called diabetic, killed, and diabetes was confirmed by blood glucose measurement using a standard glucometer. Nondiabetic mice were culled at 24 wk. From each mouse, the pancreas was fixed in 10% formalin, paraffin embedded, and stained with hematoxylin and eosin.
Statistical Analysis.
A Student's t test was used for all statistical analyses referred to in the text, except for the diabetes incidence study, which was analyzed by a log rank test and fitting to the cox proportional hazards model. Results were considered significant if P < 0.05.
Results
AIRE Expression in the Thymus.
The effects of a targeted mutation in Aire (27) on thymic Aire protein was measured by immunofluorescent staining with an Aire mAb in thymic sections from Aire +/+, Aire +/0, and Aire 0/0 mice. Aire was observed in relatively high abundance in the thymic medulla of Aire +/+ (Fig. 1 A) and Aire +/0 (Fig. 1 B) mice, but not in Aire 0/0 mice (Fig. 1 C). The number of Aire-expressing cells was not changed by the loss of a single copy of Aire, with 85 ± 15.5 Aire-expressing cells per 0.196 mm2 in the medulla of B10.Br (Aire +/+) mice, and 89 ± 16.7 Aire-expressing cells per 0.196 mm2 in the medulla of Aire +/0 mice (average and standard deviation of 16 areas from two different thymi for each group). However, loss of a single copy of Aire did appear to reduce the amount of Aire per cell, as Aire-expressing cells in the Aire +/+ medulla exhibited a more solid pattern of staining (Fig. 1 D), whereas Aire-expressing cells in the Aire +/0 medulla had a more grainy pattern, which is likely to reflect less protein per cell (Fig. 1 E). The difference in appearance of Aire-expressing cells was distinct enough to allow discrimination between Aire +/+ and Aire +/0 sections by blind analysis.
Figure 1.
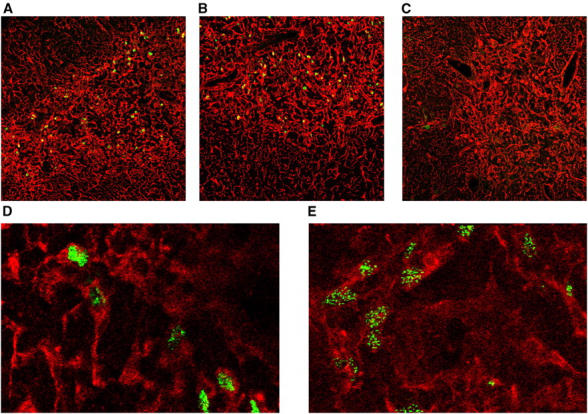
Aire expression in the thymus. Immunofluorescent staining of thymic sections showing broad spectrum cytokeratin staining (red) and AIRE protein levels (green). (A) B10.Br (Aire +/+) thymus at a magnification of 20. (B) Aire +/0 thymus at a magnification of 20. (C) Aire 0/0 thymus at a magnification of 20. (D) B10.Br (Aire +/+) thymus at a magnification of 60. (E) Aire +/0 thymus at a magnification of 60.
Effect of Aire Mutation on Insulin Promoter–driven HEL Expression.
Next, we investigated Aire's role in expression of the well-characterized, organ-specific promoter from the insulin gene in pancreatic islet β cells and in two sites of promiscuous expression, the thymus and testes. For this purpose, Aire knockout mice were crossed with a transgenic mouse strain carrying one copy of a gene comprising the rat insulin promoter linked to the sequence for membrane-bound HEL (insHEL; reference 32). The transgene was expressed at high levels in β cells from the islets of Langerhans, as determined by flow cytometry using the HEL-recognizing antibody, Hy9 (Fig. 2). Mutation of one or both copies of Aire, in Aire +/0 or Aire 0/0 transgenic mice, had no effect on expression in β cells (Fig. 2). Circulating HEL antigen in the serum, which we assume is released from β cells, was also not different in insHEL transgenic Aire +/+, Aire +/0, and Aire 0/0 mice (Fig. 2). Thus, Aire is not required for organ-specific expression of the insulin promoter.
Figure 2.
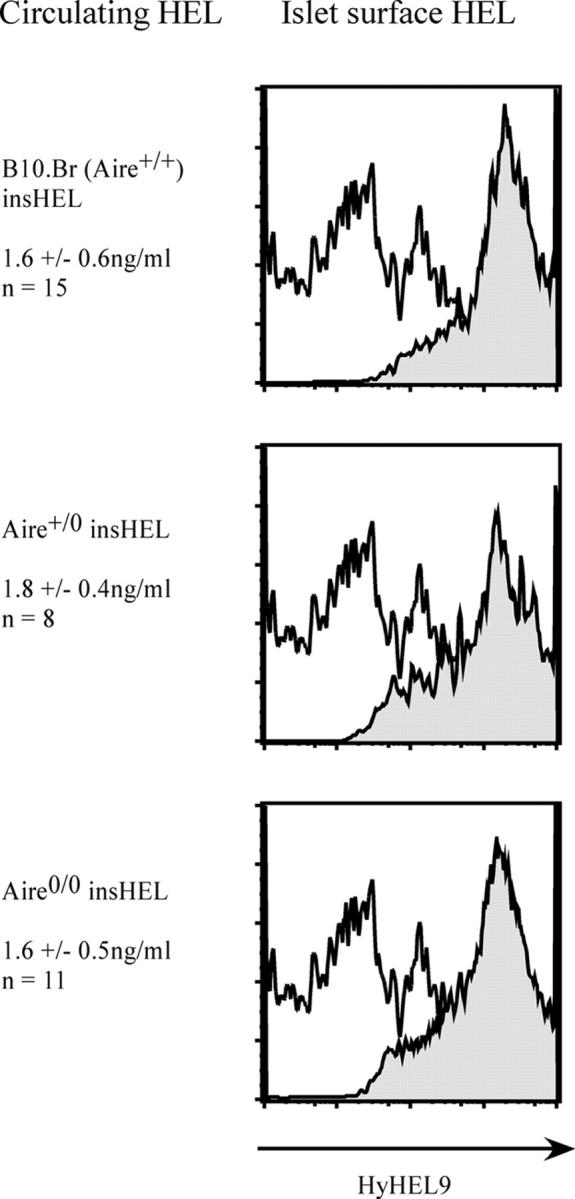
Absence of any effect of Aire mutation on insHEL expression in the β islets and serum. HEL protein concentration in the serum of 6–12-wk-old insHEL transgenic mice of the indicated Aire genotypes, and surface expression of HEL protein on pancreatic islet cells. HEL surface staining of insHEL transgenic islet is shown in gray, and background control staining of nontransgenic B10.Br islets is shown in white (representative histogram, n = 2).
In contrast, Aire was essential and gene dosage dependent for activation of the insulin promoter in thymic epithelial cells. As HEL protein levels were too low for reliable detection by immunofluorescence or flow cytometry in thymic epithelium (unpublished data), HEL mRNA was quantitated by real-time PCR. HEL mRNA was below the limit of detection in whole thymus preparations; however, it was present in purified CD45− thymic stroma from B10.Br Aire +/+ insHEL transgenic mice (Fig. 3 A). Thymic HEL mRNA was absent in Aire 0/0 insHEL transgenic mice and, surprisingly, was reduced to an intermediate level in Aire +/0 heterozygotes (Fig. 3 A). Aire dose dependence was not unique to the insulin promoter fragment in the transgene, but applied equally to thymic epithelial expression of mRNA from the endogenous insulin gene (Fig. 3 B). It should be noted that these Aire +/0 and Aire 0/0 mice show all the normal thymic epithelial cell subsets as determined by FACS analysis with stromal markers, indicating that loss of insulin promoter activity was not due to any effect on gross thymic epithelium cell differentiation (unpublished data).
Figure 3.
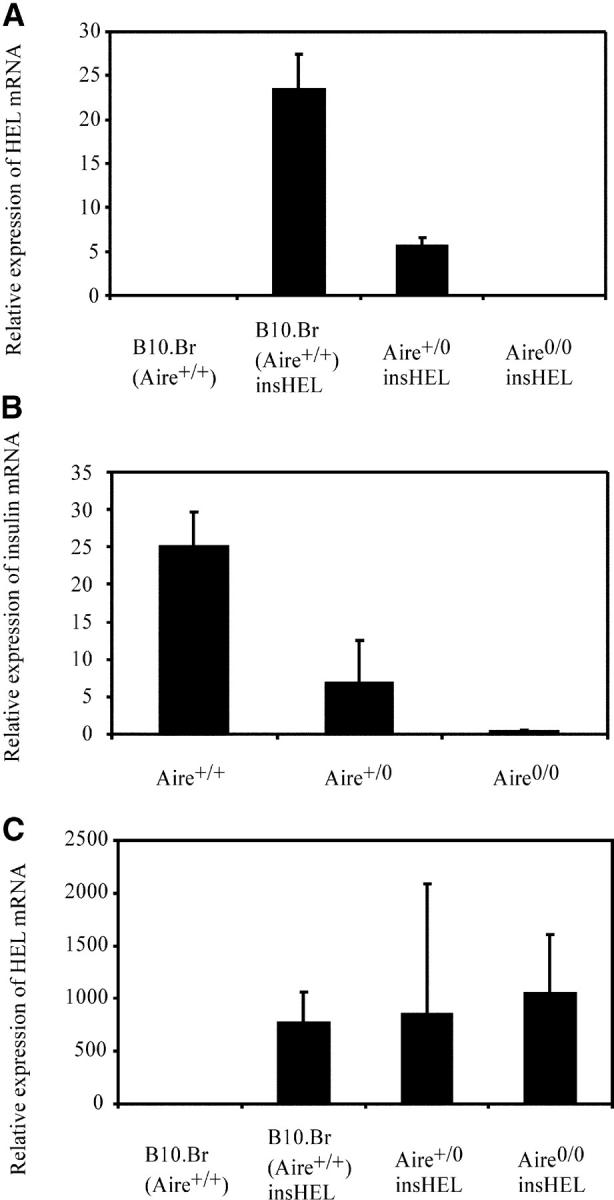
Activation of the insulin promoter is dependent on Aire in the thymic stroma and independent of Aire in the testes. (A) Thymic stroma was purified from pools of 10 thymi of nontransgenic or insHEL transgenic mice of the indicated Aire genotypes. HEL mRNA, normalized to β actin mRNA, was quantified by mRNA extraction, reverse transcription, and real-time PCR. The mean and standard deviation of three separate reactions is shown. (B) Independent samples of thymic stroma was purified from pools of 10 thymi of Aire+/+, Aire+/0, and Aire0/0 mice (all nontransgenic for HEL). Endogenous insulin mRNA, normalized to β actin mRNA, was quantified by mRNA extraction, reverse transcription, and real-time PCR. The mean and standard deviation of three separate reactions is shown. (C) Testes were removed from insHEL transgenic mice of the indicated Aire genotypes, and real-time PCR was used to measure HEL mRNA displayed in arbitrary units normalized to the level of β actin mRNA. No HEL mRNA was detected in nontransgenic mice (n = 1); however, HEL mRNA was ectopically expressed in B10.Br (Aire +/+; n = 4), Aire +/0 (n = 5), and Aire 0/0 (n = 6) insHEL transgenic mice. The mean and standard error of biological replicates (each the average of three technical replicates) is shown.
Promiscuous gene expression is also observed in the testes, and the fact that Aire is weakly expressed in the testes (24) and that male Aire 0/0 mice have reduced fertility (27) raise the possibility that Aire might also be responsible for ectopic expression in the testes. To test this hypothesis, mRNA was purified from insHEL transgenic Aire +/+, Aire +/0, and Aire 0/0 testes, and HEL mRNA levels were measured by real-time PCR. The insulin promoter HEL transgene was expressed as mRNA in the testes, but Aire was not required for testicular ectopic expression (Fig. 3 C), indicating that a separate mechanism accounts for ectopic expression in the testes.
Impact of Aire Dose-dependent Thymic Activation of the Insulin Promoter on Thymic Deletion.
Next, we asked whether or not the intermediate decrease in thymic HEL mRNA in heterozygous Aire +/0 insHEL transgenic mice had any significant impact on the efficiency of thymic deletion. A large cohort of Aire mutant and control mice were bred to be doubly transgenic for the insHEL transgene and the 3A9 TCR transgene, which causes most developing T cells to carry a well-characterized TCR recognizing HEL46-61/I-Ak. HEL-reactive T cells were tracked through thymic development using a clonotypic antibody to the 3A9 TCR, 1G12, and a variety of developmental markers (Figs. 4–6). The results of this analysis showed a profound defect in thymic deletion in _Aire_-null homozygotes and an intermediate defect in Aire heterozygotes, thus mirroring the mRNA data. By staining for the most mature subset of CD4+ CD8− 1G12hi CD69− thymocytes (Fig. 4 B), the number of cells in this subset was dramatically reduced in B10.Br double transgenic thymus to 3% of those in TCR-only controls with no insHEL transgene (Fig. 5 B). Mutation of one or both Aire copies interfered with deletion of mature cells, their frequencies rising 370% in Aire +/0 (P < 0.005 vs. B10.Br double transgenic mice) and 1,900% in Aire 0/0 mice (P < 10−11 vs. B10.Br double transgenic mice) compared with Aire +/+ controls.
Figure 4.
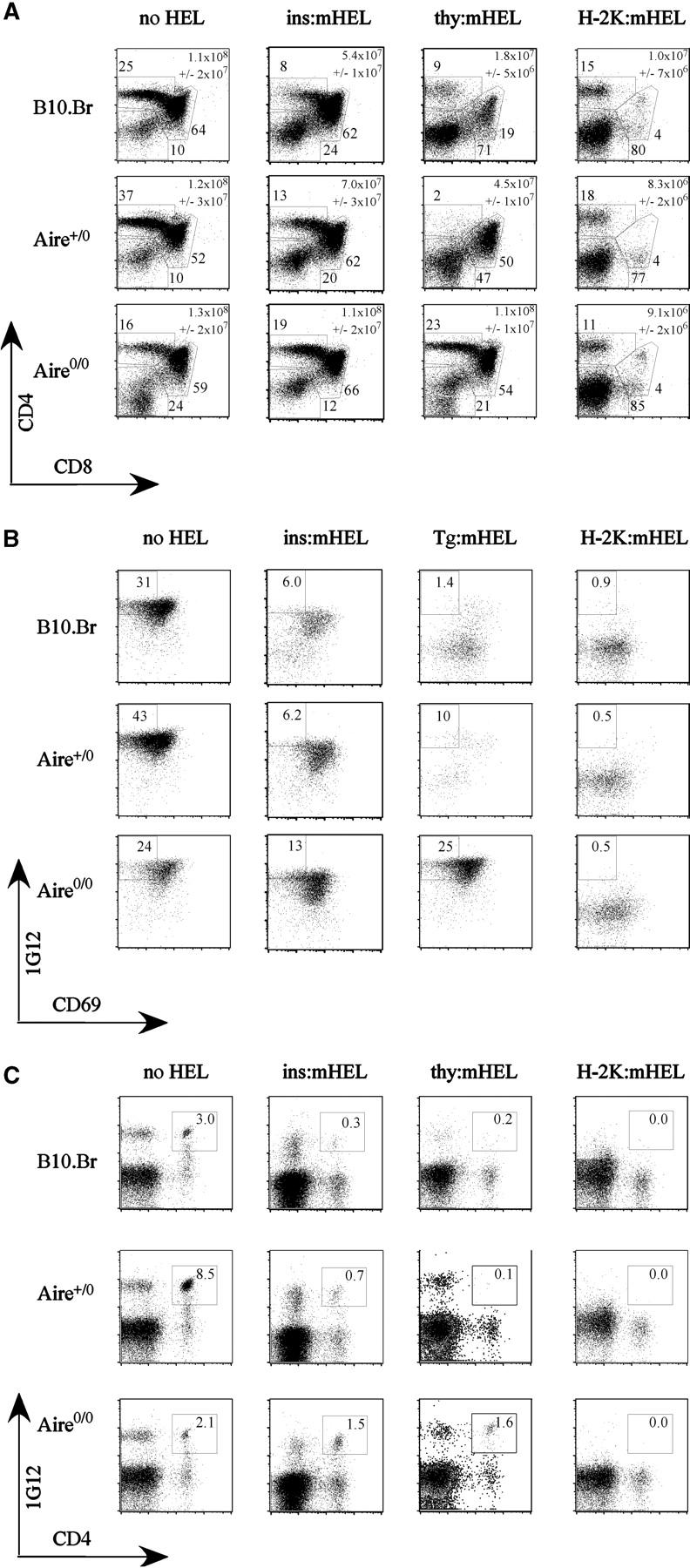
Effect of Aire deficiency on thymic deletion to HEL antigen controlled by the insulin, thyroglobulin, and H-2k promoters. Representative flow cytometric profiles from 3A9 TCR transgenic mice carrying the indicated HEL transgenes, and of the indicated Aire genotypes. Numbers indicate the percentage of plotted cells falling within the gates shown. (A) CD4 versus CD8 staining of thymocytes, showing gates used for double negative, DP, and CD4 SP cells. Mean absolute numbers of thymocytes with standard deviation is indicated on each representative profile. (B) Staining for CD69 and clonotypic 1G12 antibody to the 3A9 TCR, after gating on CD4+ CD8− SP thymocytes. Gates show the most mature subset of 1G12+ CD69− cells. (C) Staining for CD4 and clonotypic 1G12 on splenic lymphocytes.
Figure 6.

Effect of Aire deficiency on proportions of autoreactive CD4**+**cells expressing CD25+. Mean percentage of thymic CD4+ CD8− 1G12hi cells that are CD25+ with standard deviation. B10.Br (Aire+/+) mice are shown in black, Aire+/0 mice are shown in gray, and Aire0/0 mice are shown in white.
Figure 5.
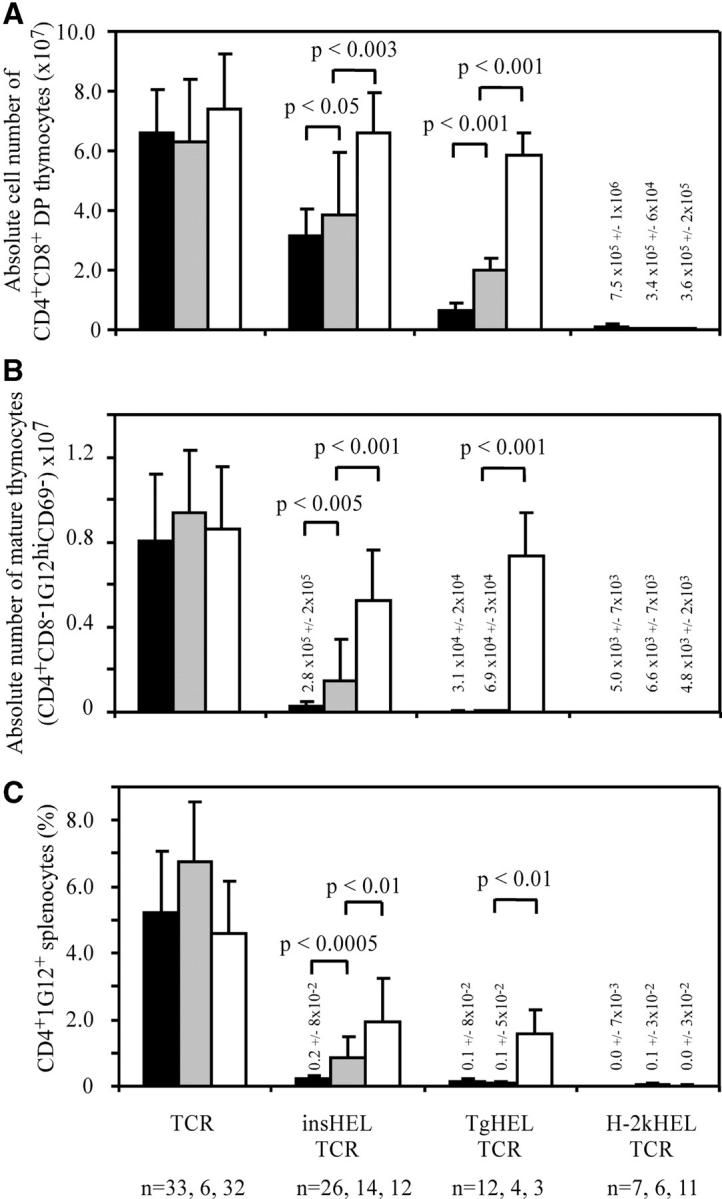
Quantitation of the effects of Aire deficiency on thymic deletion. Thymic and splenic subsets were measured by flow cytometry in TCR transgenic mice with the indicated HEL transgenes as shown in Fig. 4. Columns show mean number of cells. B10.Br (Aire+/+) mice are shown in black, Aire+/0 mice are shown in gray, and Aire0/0 mice are shown in white. Error bars indicate standard deviation. Groups with low values have the mean absolute cell number indicated above the column. (A) Mean number of CD4+ CD8+ DP thymocytes. (B) Mean number of mature HEL-reactive CD4 thymocytes (CD4+ CD8− 1G12hi CD69−). (C) Frequency of HEL-reactive 1G12+ CD4+ T cells in the peripheral repertoire of the spleen.
The defect in thymic HEL expression in double transgenic Aire +/0 and Aire 0/0 mice also prevented the increase in proportion of CD4+ CD8− 1G12hi thymocytes expressing CD25 that normally occurs in HEL-expressing animals (Fig. 6). In B10.Br TCR transgenic mice, the presence of the insHEL transgene increased the percentage of CD4+ CD8− 1G12+ cells that are CD25+ from 3.6 to 25.2%. In Aire +/0 mice, the increase in CD25+ cells was significantly lower at 9.6% and in Aire 0/0 mice, the percentage of CD25+ cells actually dropped to 2.3%. When the absolute numbers of CD4+ CD8− 1G12+ CD25+ cells are considered, however, B10.Br TCR transgenic mice had slightly higher numbers than B10.Br double transgenic mice (not depicted). Thus, small numbers of CD4+ CD25+ 3A9 T cells are produced at basal levels in TCR transgenic mice, but these appear refractory to deletion when antigen is produced in the thymus through the action of Aire, resulting in their relative enrichment among mature thymocytes in Aire +/+ double transgenic animals.
Consequence of Heterozygous or Homozygous Aire Mutation on Circulating Islet-reactive T Cells and Diabetes.
The defective negative selection in the thymus of Aire +/0 and Aire 0/0 insHEL double transgenic mice is accompanied by higher numbers of HEL-reactive CD4+ T cells in the spleen compared with B10.Br (Aire +/+) double transgenic mice (Figs. 4 C and 5 C). Compared with double transgenic control mice, the frequency of 1G12+ CD4+ cells was increased by 300% in Aire +/0 mice (P < 0.0005) and 700% in Aire 0/0 mice (P < 10−6). The small number of HEL-reactive CD4+ T cells that escape thymic deletion in control insHEL double transgenic mice initiate subclinical insulitis. Comparable insulitis was also observed in Aire +/0 and Aire 0/0 double transgenic mice (unpublished data). In the presence of both functioning copies of Aire, this insulitis rarely progresses to autoimmune diabetes (24% for B10.Br insHEL double transgenic mice and 23% for Aire +/+ sibling insHEL double transgenic mice). However, loss of a single copy of Aire increased the diabetes incidence in insHEL:TCR mice to 55%. Diabetes incidence rose to 67% in mice missing both copies of Aire (Fig. 7).
Figure 7.

Aire +/0 and Aire 0/0 double transgenic mice show an enhanced susceptibility to diabetes. Double transgenic mice of B10.Br (n = 40), Aire +/+ (n = 23), Aire +/0 (n = 22), and Aire 0/0 (n = 18) genotypes were tracked for diabetes incidence from weaning to 24 wk of age. There was a significant difference between the groups with the log rank test (P < 0.0005). By fitting to the Cox proportional hazards model, the incidence of diabetes in Aire +/0 and Aire 0/0 double transgenic mice was significantly greater than that in B10.Br (Aire +/+) and Aire +/+ double transgenic mice (P < 0.01 and P < 0.001). There was no significant difference between diabetes incidences in Aire +/0 and Aire 0/0 double transgenic mice.
The fact that some Aire mutant insHEL:TCR mice were diabetic at weaning (Fig. 7), together with a lower than expected frequency of mice of this genotype (Table I), raised the possibility that some Aire +/0 and Aire 0/0 double transgenic mice were developing diabetes and dying before weaning. Double transgenic mice were generated by crossing an Aire +/0 insHEL transgenic mouse to an Aire +/0 TCR transgenic mouse. Of the double transgenic offspring, a 25:50:25% composition of Aire +/+:Aire +/0:Aire 0/0 mice was expected. However, genotyping after weaning (Table I) indicated that there was an overrepresentation of Aire +/+ double transgenic mice, with numbers of double transgenic Aire +/0 mice being reduced by 20% and numbers of double transgenic Aire 0/0 mice being reduced by 40% relative to expected. To observe if this distortion of the expected Mendelian ratio was due to increased mortality between birth and weaning, a series of litters were monitored closely for numbers born and subsequent ill health, and any pups found dead were genotyped by PCR. The expected composition of nontransgenic:insHEL transgenic:TCR transgenic:double transgenic pups is 25:25:25:25%. The preweaning fatality rate was 8% and deaths were acute without any signs of preceding illness. Despite cannibalism, tissue from half of these acute deaths could be recovered for genotyping, biased toward pups of older ages, as pups dying soon after birth were often eaten before recovery of a tissue sample. Although the expected frequency of double transgenic pups is 25%, 91% of typable deaths occurred in double transgenic mice, with 94% of these being either Aire +/0 or Aire 0/0 (Table I). Thus, Aire mutant double transgenic pups have an increased neonatal mortality compared with sibling controls.
Table I.
Breeding Results for insHEL Aire+/0 × TCR Aire+/0 Intercross (Gender Not Constant)
| No. mice borna | 177 | |||
|---|---|---|---|---|
| No. mice weaneda | 163 (8% fatality rate) | |||
| No. dead pups unrecovereda | 7 | |||
| Genotype of recovered dead pupsa | 4× Aire0/0 double transgenic | |||
| 3× Aire+/0 double transgenic | ||||
| Total no. dead pups recoveredb | 35 | |||
| Aire+/+ | Aire+/0 | Aire0/0 | ||
| Genotype of recovered pups | Nontransgenic | 0 | 2 | 0 |
| insHEL | 0 | 0 | 1 | |
| TCR | 0 | 0 | 0 | |
| Double transgenic | 2 | 21 | 9 | |
| Aire+/+ | Aire+/0 | Aire0/0 | ||
| Total no. weaned mice genotyped as double transgenicb | 36 | 11 | 18 | 7 |
| Observed | 31% | 50% | 19% | |
| Expected | 25% | 50% | 25% |
The pancreas was able to be obtained from two dead pups and analyzed histologically, revealing very few islets and those remaining being severely inflamed with mononuclear cells. Based on the expected frequency of Aire mutant double transgenic animals (25%), the selective reduction in frequency of this class when genotyped at weaning (to 19%), and the 8% preweaning mortality concentrated in the same class of animals, it appears that ∼40% of Aire mutant double transgenic animals die between birth and weaning. The fact that the losses are confined to mice with high frequencies of islet-reactive T cells makes it likely that mortality is due to acute insulin deficiency, although the rapid cannibalism makes it difficult to determine the cause of death.
Essential Role for Aire in Thymic Tolerance to Thyroid-specific Antigen.
Because the action of Aire in tolerance to thyroid-specific or systemic autoantigens is not known, we next investigated how complete or partial Aire deficiency affected thymic deletion in transgenic mice expressing HEL controlled by the thyroglobulin (TgHEL) or H-2Kb (H-2kHEL) promoters. 3A9 TCR transgenic Aire 0/0 mice were bred to the TgHEL and H-2kHEL strains, and progeny were backcrossed to produce Aire 0/0 and Aire +/0 double transgenic mice. The TgHEL transgenic strain produces membrane-bound HEL protein at high levels in the thyroid epithelium, undetectable levels of sHEL present in the circulation (32, 33), and protein expression is readily detected by immunofluorescence in scattered thymic epithelial cells, primarily in the medulla (unpublished data). Interestingly, detectable levels of endogenous thyroglobulin mRNA have been detected in cortical epithelial cells, albeit at lower levels than medullary epithelial cells (42). It is unknown whether cortical transcripts of endogenous thyroglobulin are productive and tolerogenic. The H-2kHEL strain expresses membrane-bound HEL in all tissues, with high expression on radioresistant cells and thymocytes (34).
In the thymus of Aire +/+ TgHEL:TCR mice, double positive (DP) and single positive (SP) cells were deleted to a greater degree than in corresponding insHEL double transgenic mice (Figs. 4, A and B, and 5, A and B), consistent with higher levels of HEL protein in thymic medullary epithelial cells of TgHEL mice. Thymic deletion in TgHEL double transgenic mice was completely abolished by homozygous loss of Aire, so that the number of CD4+ CD8+ DP cells and CD4+ CD8− 1G12hi CD69− mature thymocytes became indistinguishable from TCR transgenic controls with no HEL transgene (Fig. 5, A and B). Peripheral CD4+ T cells bearing the HEL-reactive TCR were also greatly increased in Aire 0/0 TgHEL double transgenic mice (Fig. 5 C), although not attaining the levels of TCR-only controls presumably because of peripheral tolerance mechanisms that are Aire independent. In Aire +/0 heterozygotes, thymic deletion of DP cells was also significantly less efficient, with DP cell numbers intermediate between Aire +/+ and Aire 0/0 TgHEL double transgenic counterparts (Fig. 5 A). Although the loss of one copy of Aire decreased the efficiency of deletion to TgHEL, the process nevertheless reached completion during the progression from DP to mature CD4+ CD8− 1G12hi CD69− cells (Fig. 5 B) because these were reduced to 0.9% of TCR control numbers in Aire +/0 animals (compared with 0.4% of TCR controls in Aire +/+ TgHEL double transgenic mice).
In contrast to the striking failure of thymic deletion to antigen controlled by islet- or thyroid-specific promoters, Aire deficiency had no effect on thymic deletion toward the systemically expressed H-2kHEL (Figs. 4 and 5). It is unlikely that this reflects saturation of thymic deletion in these double transgenic mice, as a marked increase in DP cells in H-2KHEL double transgenic mice occurs when thymic deletion is impaired by non-MHC genes from the nonobese diabetic strain yields (unpublished data).
Discussion
The data above define the critical role for Aire in thymic mRNA expression, T cell deletion, and autoimmunity to organ-specific gene products. The findings clarify the pathogenesis of APS1, providing direct evidence at mRNA, cellular, and whole system levels for the simple interpretation that APS1 results from failure of a specialized mechanism for thymic expression and thymic deletion to gene products that would otherwise only be encountered extrathymically. At each of these levels, the data show that _Aire_-induced deletion of organ-specific clones is not a highly buffered process that can only be disrupted by rare homozygous mutations, but is in fact remarkably sensitive to small reductions in function in _Aire_-heterozygous carriers. The fact that _Aire_-induced thymic deletion is prone to failure as a result of small quantitative variations makes this mechanism a prime candidate for the actions of quantitative susceptibility genes in common organ-specific autoimmune diseases.
The data here provide direct evidence that Aire's primary function is to induce ectopic expression of organ-specific gene products in thymic medullary epithelium, but not in the peripheral organ or other ectopic sites. The data establish that the thymic activity of Aire crosses the species barrier between rat and mouse and that sequences within the 600-bp insulin promoter fragment are sufficient for this activity. The β cell–specific transcription factor Pdx-1 binds to elements within this insulin promoter fragment and plays a key role in promoting expression in pancreatic islet β cells. Ectopic expression of Pdx-1 in Isl-1–overexpressing cells is sufficient to activate insulin gene expression in other epithelial cell types (43). The _Aire_-induced activity of the promoter in the thymus suggests the following two testable hypotheses: (a) Aire substitutes for Pdx-1 in the thymus, or (b) Aire acts by inducing Pdx-1 in thymic epithelial cells.
The data establish a critical role for Aire in thymic deletion to thyroid-specific gene products. Transgenic mice carrying HEL antigen controlled by the thyroglobulin promoter display higher levels of antigen in thymic medullary epithelium, and thymic deletion is concomitantly much greater than in corresponding insHEL animals. Despite this greater activity in Aire +/+ animals, thymic deletion was more completely abolished by homozygous loss of Aire (Figs. 4 and 5). This finding explains the high incidence of thyroiditis and thyroglobulin-specific autoantibodies in APS1 patients (44) and Aire-deficient mice (29). In contrast, thymic deletion to HEL controlled by the H-2K promoter was entirely unaffected by loss of Aire. There are two possible interpretations of this result: (a) Any role for Aire in epithelial differentiation or antigen presentation might be masked in these animals by the high levels of expression or expression on nonepithelial cells, or (b) the function of Aire is limited solely to inducing organ-specific mRNAs in thymic medullary epithelium. Although we cannot exclude the first possibility, the latter is favored by the data here and by evidence that thymic deletion to the H-2K–controlled antigen is not saturated based on its sensitivity to genetic defects caused by non-MHC genes from the nonobese diabetic mouse (unpublished data).
Based on the strictly recessive inheritance pattern of APS1, it might have been concluded that _Aire_-induced thymic deletion is an efficient, highly buffered mechanism that is only disrupted in rare individuals homozygous for severe loss of function mutations. The findings here show that this tolerance mechanism is in fact prone to failure as a result of small quantitative genetic effects. Heterozygosity for the targeted Aire mutation caused a marked decrease in thymic expression of the insulin promoter construct and the endogenous insulin promoter, indicating a semi-dominant effect at this level of gene action. The targeted allele truncates the encoded Aire protein at amino acid 218 at the end of exon 5 with a neomycin cassette eliminating exon 6 (27), thus mirroring the most frequent human R257X nonsense mutation. The targeted allele nevertheless appears to be a null mutation because immunohistochemical staining of homozygous mutant tissues failed to detect immunoreactivity with a polyclonal antibody raised to a segment encoded by exon 4 (27). Nevertheless, it cannot be ruled out that the residual fragment might have a subtle dominant negative effect because the NH2-terminal portion is stable in vitro (unpublished data) and contains a nuclear localization signal and an HRS/ASS domain responsible for homodimerization. The immunofluorescence analysis of heterozygotes shown in Fig. 1 establishes that the reduced insulin promoter activity is not due to a reduction in numbers of Aire-expressing cells, but may reflect a reduction in the amount of Aire protein per cell.
The Aire gene dosage–dependent loss of insHEL mRNA in the thymus was mirrored by a semi-dominant failure of thymic deletion in insHEL:3A9 TCR double transgenic animals, resulting in a 300% increase in HEL-specific CD4 T cells escaping the thymus in heterozygotes and a 700% increase in homozygotes (Fig. 5 C). The fact that thymic deletion efficiency parallels insHEL mRNA expression in thymic epithelial cells argues that the primary role of Aire is to induce thymic expression of organ-specific mRNAs, rather than an indirect role in antigen presentation by these cells. The demonstration of gene dose–dependent effects on thymic expression of the endogenous insulin gene, and on thymic deletion induced by the thyroglobulin promoter, excludes the possibility that the transgene is uniquely sensitive to gene dose because of insertion site or elements missing from the promoter fragment, and indicates that dosage sensitivity is likely to be a general characteristic of Aire-induced thymic expression.
The peripheral consequence of this semi-dominant defect in thymic deletion was a clear increase in susceptibility to autoimmune islet destruction in insHEL:3A9 TCR double transgenic mice. The occurrence of diabetes in Aire heterozygous double transgenic mice (55%) appears to be intermediate between that of the Aire 0/0 mice (65%) and of wild-type controls (25%). However, although the incidence in both the heterozygous and knockout strains are significantly greater than that of wild-type siblings, demonstrating the exquisite sensitivity of thymic tolerance to small changes in Aire expression, the difference between the Aire +/0 and Aire 0/0 strains is not significant. One interpretation of this result is that susceptibility to diabetes in this model follows a bipolar distribution rather than a linear progression, and that the “autoimmune threshold” has already been crossed by the loss of one copy of Aire. Preweaning mortality is elevated in Aire +/0 and Aire 0/0 double transgenic animals (Table I), and although the basis for mortality could not be determined, the fact that it is concentrated exclusively in the double transgenic pups, where there are very large numbers of islet-reactive CD4+ T cells made in the thymus, suggests an autoimmune basis for these neonatal deaths. Although this is the simplest interpretation of the data, we nevertheless cannot exclude more complicated explanations such as increased frequency of cyotoxic T cells or failure of regulatory T cells. It is striking that the Aire mutation acts semi-dominantly in terms of thymic expression, thymic deletion, and autoimmunity in the sensitized background of insHEL:3A9 TCR double transgenic mice. It is likely that the direction of the immune system against the pancreas results in borderline autoimmunity, where even a small failure in thymic deletion is able to induce the cascade to autoimmunity.
Aire heterozygosity had contrasting effects on thymic deletion induced by insulin- or thyroglobulin-regulated HEL, indicating that quantitative changes in Aire activity may only allow increasing numbers of T cells to escape deletion within a defined “risk zone” of TCR avidity and thymic peptide/MHC density. Thymic expression in Aire+/0 insHEL animals falls below levels needed to prevent escape of 3A9 T cells to the periphery. In comparison, more HEL is expressed in the thymus of TgHEL animals, and although thymic deletion of DP cells is markedly less efficient in TgHEL Aire+/0 heterozygotes than in Aire+/+ counterparts (Fig. 5 A), it nevertheless remains more efficient than in the DP compartment of Aire+/+ insHEL animals. At the TCRhi CD69− CD4 SP stage, thymic deletion in TgHEL Aire heterozygotes has progressed to levels comparable to wild-type controls. This contrast suggests that deletion to organ-specific antigens depends on probabilistic contacts with rare cells displaying sufficient autoantigen to induce apoptosis. When antigen/MHC densities fall, the frequency of apoptosis-inducing contacts falls proportionally as reflected in the intermediate reduction in DP cells in TgHEL Aire+/0 thymi. However, it is only when antigen/MHC densities lie within a limiting risk zone, as exists in insHEL:3A9 TCR animals, that the frequency of above threshold contacts falls so low that significant numbers of high avidity T cells successfully complete their maturation and escape the thymus. The comparatively long process of T cell positive selection and maturation in the thymic medulla may exist primarily to maximize the chance of rare apoptosis-inducing contacts and thus minimize the effects of quantitative variation in _Aire_-induced expression of organ-specific antigens.
Based on the data of Anderson et al. (29) and for the thyroglobulin promoter here, a large number of organ-specific gene products are likely to depend on Aire for thymic expression. For each of these there will be a range of potential epitopes, depending on the MHC haplotype, and a diversity of TCR avidities produced in the thymus against each of these peptide/MHC epitopes. Hence, it seems likely that appreciable numbers of forbidden clones in any individual may fall into the risk zone for the quantitative defects in thymic deletion observed in the insHELx3A9TCR combination. Consistent with this view, the strongest inherited susceptibility locus in human type 1 diabetes other than the MHC maps to a common variant of the insulin gene promoter that decreases thymic expression by two- to threefold (18, 19). Similarly, mice with fewer copies of the proinsulin gene and lower thymic proinsulin mRNA display heightened spontaneous T cell reactivity to proinsulin in vitro (18, 19, 45). Although differences in insulin promoter efficiency could act directly on β cells by forcing them to compensate, leading to β cell stress and autoimmunization, the data here provide a firm basis for the view that subtle reductions in thymic insulin mRNA may indeed increase the frequency of insulin-reactive T cells escaping thymic deletion and predispose to diabetes. Along similar lines, mice missing one copy of the myelin protein P0 gene have half the normal P0 mRNA in the thymus and exhibit heightened in vitro T cell recall responses that are determined by the thymus P0 genotype in grafting experiments (46). Finally, there is an intermediate effect of myelin basic protein gene heterozygosity on thymic deletion at specific ages in transgenic mice expressing a high avidity myelin basic protein–reactive TCR (47). The findings here draw these different observations together by showing that the specialized process of _Aire_-induced thymic deletion to organ-specific gene products is exquisitely sensitive to quantitative perturbations.
The sensitivity of the _Aire_-induced deletion process to quantitative effects establishes this pathway as a prime target for quantitative trait genes, providing a firm biological basis to guide the design of future gene association studies in more common organ-specific autoimmune diseases. The original studies analyzing the penetrance of APS1 found no evidence for polyendocrine disease in heterozygotes, as an analysis of 58 patients showed a sibling penetrance of 24–25%, consistent with a fully recessive inheritance (48), although there are reports of patients with mutations in both copies of AIRE and autoimmunity, but without the full APS1 clinical disease (49). It is therefore unlikely that mutation in a single copy of AIRE can be sufficient to lead to the unique clinical manifestations in APS1 syndrome. However, no direct studies have been done on the incidence of single autoimmune diseases in human heterozygotes for AIRE mutations, although there are anecdotal reports of autoimmunity that do not meet the criteria for APS1 in AIRE heterozygotes (50). The only systematic evaluation of carriers of AIRE mutations analyzed eight parents of APS1 patients and found that five had subclinical immune disorders (51).
Moderate elevation in the frequency of forbidden clones escaping the thymus, as observed here in Aire heterozygotes, may alone be insufficient to precipitate the MHC-independent polyendocrine autoimmune disease typical of APS1, but may nevertheless be sufficient to precipitate single autoimmune diseases in conjunction with other susceptibility factors. Additive interactions between genetic loci are difficult to detect by genome-wide scans without a specific hypothesis because of the problems of correction for multiple testing and complicating effects of population stratification. Based on the growing understanding of the pathway for thymic deletion to organ-specific antigens, the data here provide a firm basis for testing specific interactions between candidate genes, including Aire, polymorphisms in organ-specific genes such as insulin (18, 19), MHC haplotypes that may present specific autoantigens less efficiently, variants in genes that modulate TCR signaling or TCR apoptosis induction such as CD28, CTLA4, or Bim, and other yet to be defined components of the _Aire_-induced thymic deletion pathway. By the same token, it is reasonable to predict that a relatively modest increase in AIRE activity, if this could be induced by some means, would markedly lower the incidence of type 1 diabetes and other organ-specific autoimmune diseases even in individuals with wild-type AIRE. For both of these reasons, it will be important for future work to define the other components in the pathway for _AIRE_-induced thymic deletion.
Acknowledgments
The authors would like to thank E. Unanue and D. Peterson for their gifts of staining reagents; K. Sullivan, R. Gambell, and the staff of Medical Genome Centre for curating the mouse colony; A. Prins for histology; and K. Pulsford, D. Howard, and S. Ewing for genotyping.
A. Liston is a recipient of an Australian Postgraduate Award. S. Lesage is a recipient of a Canadian Institutes of Heath Research scholarship. We acknowledge the funding for the Center of Excellence of Disease Genetics of the Academy of Finland and the Sigrid Juselius Foundation. This work was supported by grants from the Juvenile Diabetes Research Foundation and the National Health and Medical Research Council.
The authors have no conflicting financial interests.
Abbreviations used in this paper: Aire, autoimmune regulator; APS1, autoimmune polyendocrine syndrome 1; DP, double positive; HEL, hen egg lysozyme; SP, single positive.
References
- 1.Ohashi, P.S., S. Oehen, K. Buerki, H. Pircher, C.T. Ohashi, B. Odermatt, B. Malissen, R.M. Zinkernagel, and H. Hengartner. 1991. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 65:305–317. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambolez, F., K. Jooss, F. Vasseur, and A. Sarukhan. 2002. Tolerance induction to self antigens by peripheral dendritic cells. Eur. J. Immunol. 32:2588–2597. [DOI] [PubMed] [Google Scholar]
- 4.Greenwald, R.J., V.A. Boussiotis, R.B. Lorsbach, A.K. Abbas, and A.H. Sharpe. 2001. CTLA-4 regulates induction of anergy in vivo. Immunity. 14:145–155. [DOI] [PubMed] [Google Scholar]
- 5.Shevach, E. 2002. CD4+CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 6.Le Douarin, N., C. Corbel, A. Bandeira, V. Thomas-Vaslin, Y. Modigliani, A. Coutinho, and J. Salaun. 1996. Evidence for a thymus-dependent form of tolerance that is not based on elimination or anergy of reactive T cells. Immunol. Rev. 149:35–53. [DOI] [PubMed] [Google Scholar]
- 7.Fritz, R.B., and M.L. Zhao. 1996. Thymic expression of myelin basic protein (MBP). Activation of MBP-specific T cells by thymic cells in the absence of exogenous MBP. J. Immunol. 157:5249–5253. [PubMed] [Google Scholar]
- 8.Kojima, K., M. Reindl, H. Lassmann, H. Wekerle, and C. Linington. 1997. The thymus and self-tolerance: co-existence of encephalitogenic S100 beta-specific T cells and their nominal autoantigen in the normal adult rat thymus. Int. Immunol. 9:897–904. [DOI] [PubMed] [Google Scholar]
- 9.Kirchner, T., S. Tzartos, F. Hoppe, B. Schalke, H. Wekerle, and H.K. Muller-Hermelink. 1988. Pathogenesis of myasthenia gravis. Acetylcholine receptor-related antigenic determinants in tumor-free thymuses and thymic epithelial tumors. Am. J. Pathol. 130:268–280. [PMC free article] [PubMed] [Google Scholar]
- 10.Egwuagu, C.E., P. Charukamnoetkanok, and I. Gery. 1997. Thymic expression of autoantigens correlates with resistance to autoimmune disease. J. Immunol. 159:3109–3112. [PubMed] [Google Scholar]
- 11.Martens, H., B. Goxe, and V. Geenen. 1996. The thymic repertoire of neuroendocrine self-antigens: physiological implications in T-cell life and death. Immunol. Today. 17:312–317. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan, D. 1998. Peripheral-antigen-expressing cells in thymic medulla: factors in self-tolerance and autoimmunity. Curr. Opin. Immunol. 10:656–662. [DOI] [PubMed] [Google Scholar]
- 13.Heath, V.L., N.C. Moore, S.M. Parnell, and D.W. Mason. 1998. Intrathymic expression of genes involved in organ specific autoimmune disease. J. Autoimmun. 11:309–318. [DOI] [PubMed] [Google Scholar]
- 14.Werdelin, O., U. Cordes, and T. Jensen. 1998. Aberrant expression of tissue-specific proteins in the thymus: a hypothesis for the development of central tolerance. Scand. J. Immunol. 47:95–100. [DOI] [PubMed] [Google Scholar]
- 15.Sospedra, M., X. Ferrer-Francesch, O. Dominguez, M. Juan, M. Foz-Sala, and R. Pujol-Borrell. 1998. Transcription of a broad range of self-antigens in human thymus suggests a role for central mechanisms in tolerance toward peripheral antigens. J. Immunol. 161:5918–5929. [PubMed] [Google Scholar]
- 16.Klein, L., M. Klugmann, K.A. Nave, V.K. Tuohy, and B. Kyewski. 2000. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6:56–61. [DOI] [PubMed] [Google Scholar]
- 17.Anderson, A.C., L.B. Nicholson, K.L. Legge, V. Turchin, H. Zaghouani, and V.K. Kuchroo. 2000. High frequency of autoreactive myelin proteolipid protein-specific T cells in the periphery of naive mice: mechanisms of selection of the self-reactive repertoire. J. Exp. Med. 191:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vafiadis, P., S.T. Bennett, J.A. Todd, J. Nadeau, R. Grabs, C.G. Goodyer, S. Wickramasinghe, E. Colle, and C. Polychronakos. 1997. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 15:289–292. [DOI] [PubMed] [Google Scholar]
- 19.Pugliese, A., M. Zeller, A. Fernandez Jr., L.J. Zalcberg, R.J. Bartlett, C. Ricordi, M. Pietropaolo, G.S. Eisenbarth, S.T. Bennett, and D.D. Patel. 1997. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 15:293–297. [DOI] [PubMed] [Google Scholar]
- 20.Nagamine, K., P. Peterson, H.S. Scott, J. Kudoh, S. Minoshima, M. Heino, K.J. Krohn, M.D. Lalioti, P.E. Mullis, S.E. Antonarakis, et al. 1997. Positional cloning of the APECED gene. Nat. Genet. 17:393–398. [DOI] [PubMed] [Google Scholar]
- 21.The Finnish-German APECED Consortium. 1997. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 17:399–403. [DOI] [PubMed] [Google Scholar]
- 22.Betterle, C., N.A. Greggio, and M. Volpato. 1998. Clinical review 93: autoimmune polyglandular syndrome type 1. J. Clin. Endocrinol. Metab. 83:1049–1055. [DOI] [PubMed] [Google Scholar]
- 23.Heino, M., P. Peterson, J. Kudoh, K. Nagamine, A. Lagerstedt, V. Ovod, A. Ranki, I. Rantala, M. Nieminen, J. Tuukkanen, et al. 1999. Autoimmune regulator is expressed in the cells regulating immune tolerance in thymus medulla. Biochem. Biophys. Res. Commun. 257:821–825. [DOI] [PubMed] [Google Scholar]
- 24.Heino, M., P. Peterson, N. Sillanpaa, S. Guerin, L. Wu, G. Anderson, H.S. Scott, S.E. Antonarakis, J. Kudoh, N. Shimizu, et al. 2000. RNA and protein expression of the murine autoimmune regulator gene (Aire) in normal, RelB-deficient and in NOD mouse. Eur. J. Immunol. 30:1884–1893. [DOI] [PubMed] [Google Scholar]
- 25.Pitkanen, J., V. Doucas, T. Sternsdorf, T. Nakajima, S. Aratani, K. Jensen, H. Will, P. Vahamurto, J. Ollila, M. Vihinen, et al. 2000. The autoimmune regulator protein has transcriptional transactivating properties and interacts with the common coactivator CREB-binding protein. J. Biol. Chem. 275:16802–16809. [DOI] [PubMed] [Google Scholar]
- 26.Bjorses, P., M. Halonen, J.J. Palvimo, M. Kolmer, J. Aaltonen, P. Ellonen, J. Perheentupa, I. Ulmanen, and L. Peltonen. 2000. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am. J. Hum. Genet. 66:378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramsey, C., O. Winqvist, L. Puhakka, M. Halonen, A. Moro, O. Kampe, P. Eskelin, M. Pelto-Huikko, and L. Peltonen. 2002. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum. Mol. Genet. 11:397–409. [DOI] [PubMed] [Google Scholar]
- 28.Bjorses, P., M. Pelto-Huikko, J. Kaukonen, J. Aaltonen, L. Peltonen, and I. Ulmanen. 1999. Localization of the APECED protein in distinct nuclear structures. Hum. Mol. Genet. 8:259–266. [DOI] [PubMed] [Google Scholar]
- 29.Anderson, M.S., E.S. Venanzi, L. Klein, Z. Chen, S. Berzins, S.J. Turley, H. Von Boehmer, R. Bronson, A. Dierich, C. Benoist, and D. Mathis. 2002. Projection of an immunological self-shadow within the thymus by the Aire protein. Science. 298:1395–1401. [DOI] [PubMed] [Google Scholar]
- 30.Liston, A., S. Lesage, J. Wilson, L. Peltonen, and C.C. Goodnow. 2003. Aire regulates negative selection of organ-specific T cells. Nat. Immunol. 4:350–354. [DOI] [PubMed] [Google Scholar]
- 31.Ho, W.Y., M.P. Cooke, C.C. Goodnow, and M.M. Davis. 1994. Resting and anergic B cells are defective in CD28-dependent costimulation of naive CD4+ T cells. J. Exp. Med. 179:1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akkaraju, S., W.Y. Ho, D. Leong, K. Canaan, M.M. Davis, and C.C. Goodnow. 1997. A range of CD4 T cell tolerance: partial inactivation to organ-specific antigen allows nondestructive thyroiditis or insulitis. Immunity. 7:255–271. [DOI] [PubMed] [Google Scholar]
- 33.Akkaraju, S., K. Canaan, and C.C. Goodnow. 1997. Self-reactive B cells are not eliminated or inactivated by autoantigen expressed on thyroid epithelial cells. J. Exp. Med. 186:2005–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartley, S.B., J. Crosbie, R. Brink, A.B. Kantor, A. Basten, and C.C. Goodnow. 1991. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 353:765–769. [DOI] [PubMed] [Google Scholar]
- 35.Bowen, K.M., L. Andrus, and K.J. Lafferty. 1980. Successful allotransplantation of mouse pancreatic islets to nonimmunosuppressed recipients. Diabetes. 29:98–104. [DOI] [PubMed] [Google Scholar]
- 36.Goodnow, C.C., J. Crosbie, S. Adelstein, T.B. Lavoie, S.J. Smith-Gill, R.A. Brink, H. Pritchard-Briscoe, J.S. Wotherspoon, R.H. Loblay, K. Raphael, et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 334:676–682. [DOI] [PubMed] [Google Scholar]
- 37.Smith-Gill, S.J., A.C. Wilson, M. Potter, E.M. Prager, R.J. Feldmann, and C.R. Mainhart. 1982. Mapping the antigenic epitope for a monoclonal antibody against lysozyme. J. Immunol. 128:314–322. [PubMed] [Google Scholar]
- 38.Zhang, M., M.S. Vacchio, B.P. Vistica, S. Lesage, C.E. Egwuagu, C.R. Yu, M.P. Gelderman, M.C. Kennedy, E.F. Wawrousek, and I. Gery. 2003. T cell tolerance to a neo-self antigen expressed by thymic epithelial cells: the soluble form is more effective than the membrane-bound form. J. Immunol. 170:3954–3962. [DOI] [PubMed] [Google Scholar]
- 39.Gray, D.H., A.P. Chidgey, and R.L. Boyd. 2002. Analysis of thymic stromal cell populations using flow cytometry. J. Immunol. Methods. 260:15–28. [DOI] [PubMed] [Google Scholar]
- 40.Lesage, S., S.B. Hartley, S. Akkaraju, J. Wilson, M. Townsend, and C.C. Goodnow. 2002. Failure to censor forbidden clone of CD4 T cells in autoimmune diabetes. J. Exp. Med. 196:1175–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Parijs, L., D.A. Peterson, and A.K. Abbas. 1998. The Fas/Fas ligand pathway and Bcl-2 regulate T cell responses to model self and foreign antigens. Immunity. 8:265–274. [DOI] [PubMed] [Google Scholar]
- 42.Derbinski, J., A. Schulte, B. Kyewski, and L. Klein. 2001. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2:1032–1039. [DOI] [PubMed] [Google Scholar]
- 43.Kojima, H., T. Nakamura, Y. Fujita, A. Kishi, M. Fujimiya, S. Yamada, M. Kudo, Y. Nishio, H. Maegawa, M. Haneda, et al. 2002. Combined expression of pancreatic duodenal homeobox 1 and islet factor 1 induces immature enterocytes to produce insulin. Diabetes. 51:1398–1408. [DOI] [PubMed] [Google Scholar]
- 44.Perniola, R., A. Falorni, M.G. Clemente, F. Forini, E. Accogli, and G. Lobreglio. 2000. Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Eur. J. Endocrinol. 143:497–503. [DOI] [PubMed] [Google Scholar]
- 45.Chentoufi, A.A., and C. Polychronakos. 2002. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes. 51:1383–1390. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto, K., S. Miyake, M. Schachner, and T. Yamamura. 2003. Heterozygous null mutation of myelin P0 protein enhances susceptibility to autoimmune neuritis targeting P0 peptide. Eur. J. Immunol. 33:656–665. [DOI] [PubMed] [Google Scholar]
- 47.Huseby, E.S., B. Sather, P.G. Huseby, and J. Goverman. 2001. Age-dependent T cell tolerance and autoimmunity to myelin basic protein. Immunity. 14:471–481. [DOI] [PubMed] [Google Scholar]
- 48.Ahonen, P. 1985. Autoimmune polyendocrinopathy–candidosis–ectodermal dystrophy (APECED): autosomal recessive inheritance. Clin. Genet. 27:535–542. [DOI] [PubMed] [Google Scholar]
- 49.Boe, A.S., P.M. Knappskog, A.G. Myhre, J.I. Sorheim, E.S. Husebye, A. Soderbergh, F. Rorsman, M. Halonen, O. Ekwall, P. Bjorses, and O. Kampe. 2002. Mutational analysis of the autoimmune regulator (AIRE) gene in sporadic autoimmune Addison's disease can reveal patients with unidentified autoimmune polyendocrine syndrome type I. Eur. J. Endocrinol. 146:519–522. [DOI] [PubMed] [Google Scholar]
- 50.Buzi, F., R. Badolato, C. Mazza, S. Giliani, L.D. Notarangelo, G. Radetti, and A. Plebani. 2003. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome: time to review diagnostic criteria? J. Clin. Endocrinol. Metab. 88:3146–3148. [DOI] [PubMed] [Google Scholar]
- 51.Sediva, A., D. Cihakova, and J. Lebl. 2002. Immunological findings in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) and their family members: are heterozygotes subclinically affected? J. Pediatr. Endocrinol. Metab. 15:1491–1496. [DOI] [PubMed] [Google Scholar]