Reciprocal Changes in Tumor Antigenicity and Antigen-specific T Cell Function during Tumor Progression (original) (raw)
Abstract
Two seemingly incompatible models exist to explain the progression of cancers in immunocompetent hosts. The cancer immunosurveillance hypothesis posits that recognition of transformed cells by the immune system results in the generation of an effector response that may impede tumor growth. Clinically detectable cancer results from the emergence of tumor variants that escape this selective pressure. Alternatively, induction of immune tolerance to tumor antigens may enable cancer progression. We established a model where changes in the function of tumor-specific T cells and in tumor antigen expression could be followed during cancer progression. Early recognition of antigen led to activation, expansion, and effector function in tumor-specific CD4+ T cells resulting in the outgrowth of tumors expressing substantially reduced levels of antigen. Antigen loss was not complete, however, and levels remained above the threshold required for tumor-specific T cell recognition in vivo. In the face of persisting antigen, T cell tolerance ensued, leading to an impaired ability to mediate further antigen loss. Together, these studies establish that the processes of immunosurveillance and tumor editing coexist with a process in which the functional tumor-specific T cell repertoire is also “edited,” reconciling two hypotheses historically central to our attempts to understand host antitumor immunity.
Keywords: immunosurveillance, immunoediting, immune tolerance, T lymphocyte, tumor escape
Introduction
The role played by host immunity on the development and progression of cancer has been a subject of speculation and experimentation for over 50 yr (1–3). In animal models, deficiencies in both innate and adaptive immunity have been associated with an increased incidence and accelerated kinetics of tumor development induced by carcinogens, transgenic expression of oncogenes, or even of cancers that arise spontaneously (4, 5). These results support the hypothesis that the normal immune system plays a physiologic role in surveying for events associated with malignant transformation of host tissues. In immunocompetent mice, immune-mediated antitumor effector responses have been demonstrated in the early phases of tumor growth. In mice with established tumor, such responses are even capable of rejecting a small challenge of the same cancer type injected at a distant anatomical site—so called “concomitant immunity” (6). The identification of tumor escape variants that have lost expression of dominant antigens, have altered antigen processing machinery, or have lost components required for sensitivity to immune-mediated killing is compatible with successful evasion of immunologic selective pressure, a process termed “immunoediting” by Schreiber and colleagues (5). Consistent with this picture of tumor progression requiring adaptation and selection by host immunity, tumors arising in immunodeficient animals (i.e., “unedited” tumors) are frequently rejected when transplanted into syngeneic, immunocompetent recipients but not when implanted into secondary immunodeficient hosts (4).
Whereas expanded tumor-specific T cell populations can often be detected in the blood or tumors of cancer patients, consistent with host immunosurveillance, not infrequently, cancer cells isolated from progressing tumors can still be recognized by these T cells after their in vitro activation and expansion. These findings suggest that selection of less antigenic or immunogenic tumor subclones cannot be the sole mechanism involved in tumor evasion of host immunity. In fact, even though clonal expansion of tumor-specific T cells is sometimes demonstrated in vivo, such cells have often been shown to have impaired functional responses to antigen, especially when studied after minimal manipulation (7). Mouse models have also demonstrated that although early recognition of tumor antigen leads to clonal expansion of tumor-specific T cells, such cells have impaired responses to antigen in vitro, and their ability to be primed in vivo is substantially diminished compared with mice with established tumor not expressing the relevant antigen, or to mice without tumor (8–10). These observations have led to the argument that tumor progression is accompanied by the development of tumor antigen–specific T cell tolerance, perhaps through mechanisms akin to those that regulate responses to normal self-antigens (11). Together, these results support the hypothesis that the development of tumor-specific T cell tolerance is a key event in the course of tumor progression. From the perspective of a “failed” host response to tumor, it would appear that the ultimate outcome of an antigenic encounter by tumor-specific T cells is the induction of specific unresponsiveness—a conclusion seemingly at odds with the tenor of the immune surveillance hypothesis.
To more fully examine the immunological events that accompany tumor progression, we established a model that employs a defined antigen and a defined population of antigen-specific T cells. By fixing these two parameters, serial quantitative measurements of tumor antigenicity and tumor-specific T cell function could be made in a single system during tumor progression. This analysis demonstrated that the initial recognition of an immunogenic tumor by tumor-specific CD4+ T cells led to their activation, expansion, and acquisition of effector function capable of selecting for the outgrowth of tumors with substantially reduced levels of antigen expression. However, antigen loss was not complete and levels presented to the immune system by edited tumor remained above the threshold required for recognition by tumor-specific T cells in vivo. In the face of this persistent exposure to antigen, T cell effector function waned, leading to T cell tolerance, and these tumor antigen–experienced T cells were no longer capable of exerting selective pressure sufficient for mediating tumor antigen loss. Together, these studies establish that cancer progression involves reciprocal changes in the antigenic profile of the evolving tumor and in the functional capacity of the tumor antigen–specific T cells. These changes result in a homeostasis in which a less immunogenic tumor emerges in the face of an immune system less capable of responding to it.
Materials and Methods
Mice.
Male BALB/c (Thy1.2+/+) 6–8 wk old mice were purchased from the National Cancer Institutes. TCR transgenic mice on a BALB/c background expressing an αβ TCR specific for amino acids 110–120 from influenza haemaglutinin (HA) presented by I-Ed (6.5 transgenic mice) were a gift from H. von Boehmer (Harvard Medical School, Dana Farber Cancer Institute, Boston, MA) (12). TCR transgenic mice used in experiments were heterozygous for the transgene and were also Thy1.1+/1.2+ unless otherwise specified. Transgenic mice expressing an αβ TCR specific for amino acids 518–527 from HA presented by Kd (CL4 mice) were a gift from L. Sherman (The Scripps Research Institute, La Jolla, CA) (13). CL4 mice congenic for Thy1.1+/+ were used. BALB/c Rag 2−/− mice were purchased from The Jackson Laboratory. Experiments using mice were conducted in accordance with protocols approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine.
Tumor Cells.
Renal cell carcinoma cells (Renca) were obtained from the American Type Culture Collection. Cells were cultured in vitro in RPMI 1640 media, supplemented with 10% FCS, 50 U/ml penicillin/streptomycin, 2 mM l-glutamine, and 50 mM β-mercaptoethanol (complete media), and were grown at 37°C, 5% CO2. RencaHA was generated as described previously (14). 1 × 106 tumor cells in a total volume of 0.2 ml HBSS were injected into each mouse i.v.
Adoptive Transfer.
For transfer of transgenic CD4+ T cells, single cell suspensions were generated from both LNs and spleens of TCR transgenic donors. CD4+ T cells were enriched by depleting CD8+ T cells and B cells as described (15). Briefly, cells were incubated with biotinylated antibody against CD8+ (53–6.7) and CD45R/B220 (RA3-6B2). The biotin antibody–coated cells were then removed by streptavidin-conjugated magnetic beads (Dynal). The percentage of lymphocytes positive for CD4 and the clonotypic TCR (mAb 6.5) was determined by flow cytometry. Cells were washed, and 2.5 × 106 CD4+ anti-HA TCR+ T cells were injected i.v. into each recipient. Thy1.1+/+CD8+ T cells from CL4 transgenic mice were purified similarly except for the replacement of anti-CD8+ with anti-CD4+ antibody. For experiments involving a cotransfer of both HA-specific CD4+ and CL4CD8+ T cells, HA-specific CD4+ T cells were Thy1.2+/+. The purity of total CD4+ or CD8+ T cells after enrichment was usually 92 and 85%, respectively. For 5,6-carboxy-fluorescein succinimidyl ester (CFSE) (Molecular Probes) labeling, purified CD4+ T cells were incubated with 1 μM CFSE at 37°C for 10 min and then washed with ice cold HBSS before transfer.
To assess the tumor-editing capability of the tumor-experienced CD4+ T cells, 2.5 × 106 clonotypic HA-specific CD4+ (Thy1.1+) T cells were transferred into tumor-free mice or mice with 10-d established RencaHA. 3 wk after transfer, mice were killed and LNs (including draining and nondraining nodes) and spleens were recovered. Total CD4+ T cells were purified using CD4+ T cell isolation kit (Miltenyi). These purified T cells were transferred again into mice challenged with RencaHA 10 d earlier. Each mouse received 2.6 × 106 Thy1.1+ CD4+ T cells contained within the total purified CD4+ T cell population. 3 wk after T cell transfer, tumor explants were prepared, and HA levels on tumor explants were measured by RT-PCR as described below.
Flow Cytometric Analysis.
All antibodies were purchased from BD Biosciences unless otherwise specified. HA-specific CD4+ TCR transgenic T cells were stained with biotinylated rat anti–clonotypic TCR antibody 6.5 (prepared from a hybridoma) followed by PE-conjugated streptavidin. Single cell populations from the LNs and spleens were stained with the indicated mAbs for cell surface markers. For analysis, 30,000 gated events were collected on a FACSCAN (Becton Dickinson) and analyzed using CellQuest software (Becton Dickinson).
Bromodeoxyuridine Experiments.
After T cell transfer, recipient mice were fed with bromodeoxyuridine (BrdU)-containing water (0.8 mg/ml) at specified intervals for seven consecutive days. BrdU water was changed daily. For analysis, cells from the hilar LNs were isolated and stained for BrdU using the BD Biosciences BrdU flow kit following the manufacturer instructions. For better resolution, anti-Thy1.1 antibody was used instead of the anti-TCR clonotypic antibody to gate on the transferred CD4+ T cells. Since not all Thy1.1+CD4+ cells were transgenic cells, in parallel to BrdU staining, each sample was stained with anti–Thy1.1-FITC, anti–CD4-Cyc, and anti-TCR clonotypic antibodies followed by streptavidin-PE to obtain the percentage of HA-specific cells in the Thy1.1+CD4+ population. BrdU staining on cells from tumor-free mice was taken as background and was subtracted from the percentage of the BrdU+ cells in the Thy1.1+CD4+ population in tumor-bearing mice. The net value was then normalized to the percentage of HA-specific CD4+ cells to get the percentage of BrdU+ cells in the HA-specific CD4+ population.
Immunohistochemistry.
Lung tissues were harvested from mice. Tissue was fixed for 3 d at 4°C and then embedded in ImmunoHistoWax (A Phase sprl). Serial sections were stained using biotin-labeled anti-HA mAb (H18). The Vectastain ABC kit (Vector Laboratories) and NovaRed (Vector Laboratories) were used for development. Sections were counterstained with hematoxylin QS (Vector Laboratories) and analyzed using a Nikon Eclipse E400. Final image processing was performed using Adobe Photoshop.
Real-Time RT-PCR Analysis of Explants.
Mice were killed 31 d after tumor injection. Lungs were dissected and rinsed with HBSS. Single cell suspensions were made by mechanical dissociation and passage through nylon mesh. Nonadherent cells were removed after overnight culture, and adherent tumor cells were grown without drug selection for 1–2 wk to allow the tumor cells to expand and any contaminating, nonadherent cells to be removed. Cultures were washed twice with HBSS, and adherent cells were then collected after trypsinization. For experiments in which explanted tumors were passaged into secondary recipients, the growth kinetics were noted to be slightly faster than that of cultured RencaHAhi cells, reflecting the selection and/or adaptation of the explants for in vivo growth. However, all groups had significant tumor burdens by 17 d after tumor challenge when T cell recognition was assessed.
For experiments where HA message was measured, RNA was extracted from 1 × 106 cells with Trizol (Invitrogen). Reverse transcription was performed with the SuperScript First-Strand Synthesis System (Invitrogen). cDNA amounts were analyzed by real-time quantitative PCR with the Taqman System (Applied Biosystems). Each sample was assayed in triplicate for HA together with the internal reference, HPRT, using the Taqman Universal PCR Master Mix and the ABI Prism 7700 Sequence Detection System (Applied Biosystems). The relative HA mRNA frequencies were determined by normalization to HPRT. Fold differences between samples were calculated as follows: one cycle difference between samples is equivalent to a twofold difference in transcript. cDNA from RencaWT cells was included in each set of experiments as a negative control. The primer sequences for HA were 5′-CGCCGGATGGCTCTTG-3′ (forward) and 5′-ACAATGTAGGACCATGATCTCACTG-3′ (reverse). The HA-specific probe sequence was 5′-6FAM-AAACCCAGAATGCGACCCACTGCTTTAMRA-3′
In Vivo Priming with Vaccinia-HA.
A recombinant vaccinia virus encoding HA from the 1934 PR8 strain of influenza (vacHA) was a gift from F. Guarnieri (Johns Hopkins University, Baltimore, MD). VacHA was amplified as described previously (9). On the days indicated, mice were primed by i.p. inoculation with 1 × 107 plaque-forming units of recombinant vacHA suspended in HBSS.
IFN-γ ELISPOT.
5 × 105 cells from hilar LNs were plated into wells with or without the addition of 10 μg/ml of synthetic HA110–120 peptide (SFERFEIFPKE), incubated at 37°C overnight, and assayed as described previously (15).
Results
Tumor-specific CD4+ T Cells Acquire Effector Function during Initial Encounter with Antigen.
RencaHA is a stable transfectant of a renal cell carcinoma that expresses influenza HA as a model tumor antigen. i.v. injection of RencaHA results in the formation of multiple pulmonary tumor nodules that are lethal after ∼5 wk. Previous studies demonstrated that although this tumor fails to metastasize from the lungs to regional lymphatics, HA antigen is recognized by HA-specific T cells in the hilar LNs in a process that requires antigen processing and presentation by BM-derived APCs (16). To determine the impact of antigen recognition on the function of tumor-specific T cells, CD4+ T cells were purified from TCR transgenic mice specific for an MHC class II epitope of HA, labeled with CFSE, and transferred either into mice that had been challenged with RencaHA 10 d earlier or into nontumor bearing mice (Fig. 1). 7 d after T cell transfer, clear evidence of antigen recognition was observed in the hilar LNs of tumor-bearing mice, as indicated by the increased frequency (1.27 vs. 0.02%) and CFSE dilution (79.5 vs. 3%) of HA-specific T cells compared with nontumor bearing mice. Given the regional distribution of the tumor, these changes were much more modest in the spleens. 2 wk later (31 d after tumor challenge), gross examination of the lungs revealed extensive studding with macroscopic tumor nodules bilaterally. The frequency of HA-specific CD4+ T cells in the hilar nodes was still expanded relative to that observed in nontumor bearing mice, although some degree of contraction had occurred during this interval in mice with progressing RencaHA (1.27% on day 7 vs. 0.87% on day 21). For comparison, nontumor bearing mice that had received the same number of CFSE-labeled HA-specific CD4+ T cells were challenged i.p. with a recombinant vaccinia virus encoding HA (vacHA) and analyzed 6 d later. The magnitude of HA-specific CD4+ T cell expansion in the hilar nodes was comparable to that seen in mice with RencaHA lung metastases (1.32 vs. 1.27%, respectively), although the accumulation of HA-specific T cells that underwent extensive division was greater in response to virus than in response to tumor. Not surprisingly, in the context of systemic infection with vacHA the total HA-specific T cell expansion was also greater than in response to localized tumor, as indicated by their increased frequency in the spleens (and other LNs [unpublished data]) of virally infected mice.
Figure 1.

Recognition of antigen by tumor-specific CD4+ T cells. CFSE-labeled HA-specific CD4+ T cells were transferred into mice inoculated with RencaHA 10 d earlier. On days 7 and 21 after T cell transfer, expansion of the transgenic CD4+ T cells was revealed by staining with anti-TCR clonotypic antibody and anti-Thy1.1. (A) FACS profiles of the transgenic CD4+ T cells in hilar draining LNs. CFSE histograms of the gated cells are also shown. Percentage of the gated population is displayed in each dot plot. For CFSE profiles, percentage of the divided cells in the gated population is indicated in each histogram. For comparison, CFSE-labeled HA-specific CD4+ T cells were transferred into tumor-free mice. Some were given vacHA the next day and analyzed 6 d later. (B) Percentage of the transgenic CD4+ T cells in spleen. Spleen cells from the same mice as in A were stained and analyzed by flow cytometry. The results are representative of three experiments with three mice per group.
During the early phases of antigen recognition, the expanded HA-specific T cells in the hilar nodes of RencaHA-bearing mice differentiated into cells capable of making interferon gamma (IFN-γ) (Fig. 2 A), albeit to a lesser extent than T cells from vacHA-primed, nontumor bearing mice. Interestingly, 2 wk after this initial endogenous activation of tumor-specific T cells the frequency of HA-specific IFN-γ–producing T cells fell toward baseline in association with tumor progression.
Figure 2.

Early recognition of HA results in effector function of tumor-specific T cells. (A) T cell transfer was conducted as described in Fig. 1. IFN-γ ELISPOT was performed with cells from hilar LNs in each well ± HA peptide. The number of IFN-γ–positive spots was obtained by subtracting the number of spots in the no peptide group from the number of spots in the peptide group. The results are mean ± SD of pooled data from two separate experiments. (B) Activated CD4+ T cells provide help for CTL priming. Purified CD8+ T cells from CL4 transgenic mice on Thy1.1 background were transferred either alone or together with HA-specific CD4+ T cells (Thy1.2+) to tumor-free or tumor-bearing mice (10 d tumor). 7 d after T cell transfer, cells from draining LNs were stained for the transferred CD8+ T cells with antibody to Thy1.1 and CD8. For CD4+ T cells, cells were stained with anti-TCR clonotypic antibody and anti-CD4. Percentage of the gated population is given in each dot plot. The results are representative of two experiments with three mice per group.
Early Recognition of Antigen by Tumor-specific CD4+ T Cells Augments the Expansion of Tumor-specific CD8+ T Cells.
Theses changes observed during the initial phases of antigen recognition by tumor-specific CD4+ T cells (entry into the cell cycle, clonal expansion, and the production of IFN-γ) are all features associated with CD4+ T cell effector function. To further evaluate if these changes truly reflect a developing effector response, we examined another important parameter of CD4+ T cell function, i.e., the ability to provide help for the activation of MHC class I–restricted CD8+ T cells (Fig. 2 B). As seen above with CD4+ T cells, the transfer of MHC class I–restricted, HA-specific CD8+ T cells from TCR transgenic mice (CL4 mice) into mice with established RencaHA resulted in their expansion during the first week after transfer (no tumor 0.03% vs. RencaHA 0.16%). However, the magnitude of the CD8+ T cell “burst” in response to recognition of tumor antigen was increased threefold (0.16–0.48%) when these cells were cotransferred with HA-specific CD4+ T cells. Together, therefore, the initial recognition of tumor antigen by tumor-specific CD4+ T cells is accompanied by their clonal expansion (0.13–1.6% in this experiment), Th1 cell differentiation, and the delivery of help for the activation of tumor-specific CD8+ T cells, all consistent with an effector response.
Early Activation of Tumor-specific CD4+ T Cells Alters the Antigenic Profile of Progressing Tumor.
We therefore wished to examine whether this initial effector response led to measurable antitumor immunity. Previous studies demonstrated that the transfer of HA-specific CD4+ transgenic T cells into mice 10 d after an i.v. challenge with RencaHA led to a slight prolongation of survival (7–10 d) over mice not receiving T cell transfer (16). The effect of T cell transfer on the level of HA expression by the progressing tumor was examined by immunohistochemistry (Fig. 3). Whereas HA was uniformly expressed by the cells comprising the tumor nodules arising in RAG knockout mice challenged with RencaHA, the level of HA expression was considerably reduced and was more heterogeneous in the tumors found in immunocompetent BALB/c recipients, demonstrating that the endogenous adaptive immune response was capable of some degree of immunoediting of this antigenic tumor. The transfer of HA-specific CD4+ T cells into BALB/c mice with established RencaHA further reduced the number and size of the tumor nodules compared with no T cell transfer. Strikingly, HA expression was undetectable in such nodules as measured by this technique. Indeed, the level of HA expression on tumor explants from mice that received transgenic T cells was indistinguishable from background staining by flow cytometric analysis as well (unpublished data). Therefore, the initial activation resulted in the selection of tumor escape variants that lost expression of the target antigen.
Figure 3.
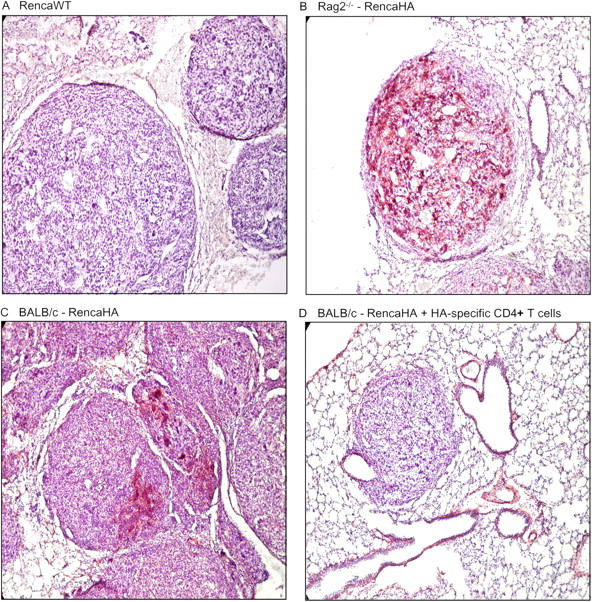
Immunoediting of RencaHA by the endogenous repertoire and by transferred, HA-specific CD4+ T cells. Mice were injected with 106 tumor cells i.v. Mice in group D received HA-specific CD4+ T cells 10 d posttumor. All mice were killed at 25 d posttumor inoculation. Lung tissue was harvested, and sections were stained with anti-HA antibody and developed with NovaRed (red staining). Sections were counterstained with hematoxylin. (A, C, and D) BALB/c mice; (B) BALB/c Rag 2−/− mice.
From the perspective of assessing the outcome of antigen recognition by HA-specific CD4+ T cells, complete eradication of cells expressing the target antigen could be viewed as a successful effector response, in spite of the ultimate progression of tumor. Therefore, it was important to determine if the tumors that progressed in mice receiving transgenic T cells had any residual HA expression as detected by more sensitive techniques. RencaHA was excised from the lungs of BALB/c mice that either had or had not previously received HA-specific CD4+ T cells. RNA was isolated from the tumor cells, and the relative amount of messenger RNA encoding HA was determined by quantitative real-time RT-PCR (qRT-PCR) (Fig. 4). Compared with cultured RencaHA cells, tumor cells obtained from the lungs of immunocompetent mice had about an eightfold reduction in HA message. Importantly, tumors isolated from mice that had received HA-specific CD4+ T cells still had detectable HA mRNA, in spite of the loss of detectable HA protein as measured by antibody staining. T cell transfer resulted in at least a 180-fold reduction of HA message below what was achieved with the endogenous BALB/c T cell repertoire alone (range 180–1,448 fold).
Figure 4.

HA mRNA frequencies on tumor explants as revealed by qRT-PCR. Tumor explants from mice with and without HA-specific T cell transfer were generated as described. Each explant or RencaHA cell line was assayed in triplicate using probe/primer set specific for HA. Samples were simultaneously assayed for HPRT which was used as an internal reference. The cDNA concentration of each sample was adjusted so that HPRT amplification was equivalent in all samples. Data shown are representative of two separate reactions with similar results.
Edited Tumor Is Still Detectable by Tumor Antigen–specific T Cells.
Although PCR was able to detect HA message from tumor that had been edited by HA-specific CD4+ T cells, the most relevant parameter pertaining to the function of the responding T cells is whether edited tumor expresses protein antigen at levels sufficient for continued recognition by the antigen-specific T cells. To determine this, we evaluated the T cell recognition of RencaHA cells that had never been passaged in vivo (RencaHAhi) versus explanted tumor cells that had been edited by the endogenous BALB/c immune system (RencaHAendog), versus explanted tumor cells edited in BALB/c mice adoptively transferred with HA-specific CD4+ transgenic T cells (RencaHATCR-Tg) (Fig. 5). In the absence of antigen, transgenic T cells fail to divide, as seen in mice with Renca wild-type tumor. Not surprisingly, extensive division of HA-specific T cells is seen in mice with both RencaHAhi and RencaHAendog. However, although the transfer of HA-specific CD4+ T cells results in substantial reduction in the level of HA expressed by the antigen loss variants (Figs. 3 and 4), the results of this experiment clearly demonstrate that the amount of HA produced by edited tumor (RencaHATCR-Tg) is still sufficient to be detected by T cells having the identical specificity and affinity for antigen as the cells responsible for the initial editing.
Figure 5.

Edited tumor cells are recognized by naïve HA-specific CD4+ T cells. Mice were challenged with RencaHA. 10 d later, half of the mice received HA-specific CD4+ T cells. 21 d after T cell transfer, the tumors were harvested from the lungs and briefly expanded in vitro in the absence of drug selection. A second cohort of mice was then injected with either Renca WT cells, RencaHA cells that had never been passaged in vivo (RencaHAhi), RencaHA cells explanted from BALB/c mice that did not receive T cell transfer (RencaHAendog), or RencaHA cells explanted from mice that had received HA-specific CD4+ T cells (RencaHATCR-Tg). 10 d after tumor challenge, all mice were injected with naïve CFSE-labeled HA-specific CD4+ T cells. 7 d later cells were isolated from hilar LNs. Lymphocytes were stained with anti-Thy1.1 antibody and anti-TCR clonotypic antibody and analyzed by flow cytometry. Results are gated on Thy1.1+ cells.
Kinetics of Tumor-specific T Cell Proliferation during Tumor Progression.
Given the changes in HA expression by RencaHA during tumor progression, we wished to examine the kinetics of HA-specific T cell recognition in vivo beyond the initial events captured by CSFE dilution profiling. Mice with or without established RencaHA were injected with HA-specific CD4+ T cells and then placed on drinking water containing BrdU during the intervals outlined in Fig. 6. Consistent with the earlier results using CFSE, T cell recognition of HA during the first week led to significant incorporation of BrdU by dividing HA-specific CD4+ T cells (69.7%). Interestingly, a sizable fraction of this tumor-specific T cell population that responded to antigen during the first week persisted throughout the course of tumor progression, as indicated by the continued detection of BrdU-positive cells in the hilar LNs 2 wk after removing BrdU from the drinking water (32.5%). In contrast to the robust proliferative response observed during the initial encounter with antigen, there was a sharp decline in the proliferation of HA-specific CD4+ T cells during the third week after T cell transfer, as measured by BrdU labeling during this interval (12.4%).
Figure 6.

The kinetics of tumor-specific CD4+ T cell proliferation during tumor progression. Mice that received HA-specific CD4+ (Thy1.1+/−) T cells were provided with BrdU-containing water at the specified interval. On the day of analysis, cells from the hilar LNs were isolated and stained for surface markers CD4 and Thy1.1, followed by intercellular BrdU staining. The data are representative of two separate experiments with similar results.
Loss of the Capacity for Tumor Editing by Antigen Experienced, Tumor-specific T Cells.
On one level, these results indicate that the kinetics of T cell proliferation during tumor progression reflect the changing antigen density of the tumor cells to which they are responding. However, this interpretation likely oversimplifies the complexity of assessing regional antigen density of a logarithmically expanding tumor burden and the parameters that govern how much antigen is captured and concentrated by APCs. In fact, in experiments where the HA-specific T cell transfer was delayed until 3 wk after RencaHA was established (a time when the endogenous host response has selected for growth of lower HA expressing tumor), extensive expansion and CFSE dilution of HA-specific T cells was observed (unpublished data). Therefore, an alternative interpretation of the declining proliferation rate of HA-specific T cells measured during the latter stages of tumor progression is that there is an intrinsic change in the functional responsiveness of the tumor-specific CD4+ T cells over time. Consistent with this hypothesis, previous results in this system demonstrated that HA-specific CD4+ T cells in mice with established RencaHA tumor have a markedly impaired response to vacHA immunization compared with what is observed in vaccinated, nontumor bearing mice (16).
In reconciling these intrinsic changes in tumor-specific T cell function with the demonstrated capacity of such cells to edit tumor antigenicity, we wished to examine whether antigen-experienced T cells from tumor-bearing mice were altered in their capacity to exert immunologic selective pressure on progressing tumor (Fig. 7). HA-specific CD4+ T cells were transferred into mice with or without established RencaHA. 3 wk after T cell transfer, total CD4+ T cells were purified from the spleens and LNs and then transferred into secondary RencaHA-bearing recipients. 3 wk later, tumor was harvested from the lungs and the relative level of HA mRNA was determined by qRT-PCR. As seen previously, the endogenous host response of BALB/c mice resulted in an approximate 8–16-fold reduction in HA message of explanted RencaHA compared with cultured RencaHA. Transfer of CD4+ T cells from nontumor bearing mice that had previously received HA-specific CD4+ T cells resulted in a >500-fold further reduction of HA message by the selected tumor (Fig. 7, dark green curve versus dark blue). In contrast, CD4+ T cells from RencaHA-bearing mice were significantly less effective in editing RencaHA in the secondary recipients (16–64-fold reduction in HA message compared with no T cell transfer; Fig. 7, dark green curve versus light green). This result demonstrates that changes in tumor-specific T cell function occur over time in the face of persisting antigen, impairing the capacity of host immunity to exert selective pressure on tumor growth. Ultimately, equilibrium is achieved in which the antigenic profile of the resulting tumor and the functional capacity of the tumor antigen–specific T cell repertoire are reciprocally shaped allowing for tumor progression.
Figure 7.

Antigen-experienced, tumor-specific T cells have a decreased ability to edit tumor. HA-specific CD4+ T cells were transferred into tumor-free or tumor-bearing mice. 21 d later, spleens and LNs were harvested from the mice. CD4+ T cells were enriched and retransferred into mice with established RencaHA tumor. 3 wk after T cell transfer, tumor explants were harvested and processed. Shown are real-time RT-PCR results for HA mRNA frequencies on tumor explants. Each sample was assayed in triplicate, with HPRT included as internal reference. Each group consisted of two to three mice.
Discussion
The cancer immunosurveillance hypothesis, which had been largely discarded for many years, has recently undergone an extensive reexamination with the current tools of molecular immunology (17). Studies in mice with selective defects in innate and/or adaptive immunity have yielded strong evidence that host immunity plays a significant role in altering the incidence and kinetics of cancer development and in shaping the immunogenicity of tumors that arise. In humans, histopathologic and molecular profiling of tumor specimens have revealed significant correlations between the presence of immune cell infiltrates and disease outcome, supporting the view that the host response can have a meaningful impact on the clinical behavior of malignancy (18). Similarly, the pathology literature provides ample documentation of metastases frequently displaying altered expression of immunologically relevant targets compared with early stage primary cancers as evidence for tumor adaptation to immunologic selective pressure (19).
The reciprocal question of how tumor progression influences the function of tumor antigen–specific T cells has also been examined in several experimental model systems and in patients with cancer. These studies have demonstrated diverse outcomes ranging from T cell ignorance (20, 21) to induction of effector function (22, 23) to the deletion or development of anergy of tumor-specific T cells (8, 9, 24–26). Several parameters are likely to influence which of these outcomes predominate. Anatomic localization of tumor has been shown to alter not only the likelihood of tumor antigen recognition but also the pathways of presentation (directly by tumor cells versus indirectly by APCs) to tumor-specific T cells (27, 28). For nonhematopoietic cancers, disruption of normal tissue architecture, hypoxia-induced cell death, tumor transit across basement membranes, and systemic dissemination increase the probability of detection. These very processes are also thought to provide signals to innate immunity which detects a “stress signature” by cancer cells and supporting stroma. In fact, one of the earliest adaptations by tumor to this response may be the elaboration of signals to suppress the stress signature or the response to it (29), such as those mediated by STAT-3 (30, 31), TGF-β (32), and vascular endothelial growth factor (33) in a pattern analogous to what is seen with wound healing. Consistent with this notion, cancer cells that display blunted stress responses, such as reduced induction of heat shock proteins, have been shown to be relatively less immunogenic (34, 35).
Ultimately, eliciting T cell immunity in settings such as those studied here requires tumor antigen capture and processing by BM-derived APCs (16, 36). Again, the likelihood of T cell recognition and the outcome of that recognition are influenced by several parameters, including features intrinsic to the antigen such as the presence of candidate T cell epitopes capable of being liberated by the cells' antigen processing machinery and fitting the host's MHC binding motifs (37), tumor antigen density (38), the rate of antigen liberation by dying tumor cells, the presence of inflammatory signals to influence the function of antigen-loaded APCs (39), and the frequency (40) and avidity of functionally competent tumor-specific T cells.
In this paper, we examined both sides of this process in a single model system in which two of the key variables listed above were defined (i.e., tumor antigen– and antigen-specific T cells), enabling a quantitative assessment of changes in each during tumor progression. Operationally, RencaHA is an immunogenic tumor. We have reported previously that it is spontaneously rejected when given as a s.c. tumor challenge in BALB/c mice but not in mice tolerant of HA by virtue of its transgenic expression as a self-antigen (14). The level of HA expression on RencaHA tumor cells is heterogeneous (in spite of limiting dilution cloning), and i.v. injection into immunocompetent mice results in the outgrowth of lung nodules that express less HA than the parental clone or than the nodules that develop in RAG−/− mice (Figs. 3 and 4). This result provides a quantitative demonstration of the endogenous response altering the antigenic profile of tumor escape variants (Fig. 4), as has been reported qualitatively in other systems. By transferring a fixed number of HA-specific CD4+ transgenic T cells having a given affinity for HA peptide–MHC class II complexes, we were able to follow the fate of this tumor-specific CD4+ T cell population through distinct phases of the response to tumor antigen and superimpose these events on the changing antigenic profile of the tumor.
The early events after HA-specific CD4+ T cell transfer clearly demonstrate tumor antigen recognition in the regional (hilar) LNs draining the lungs, which is the major site of tumor growth. Previous studies in this model established that RencaHA in the lungs fails to metastasize to the hilar LNs (as measured by HA-specific PCR) and that host APCs are required for CD4+ T cell recognition to occur (16). Therefore, this initial recognition suggests that some degree of tumor antigen liberation and capture by APCs occurs in the early stages of tumor growth. During this phase, tumor-specific CD4+ T cell recognition results in cell division (CFSE dilution), clonal expansion in the regional LNs, differentiation into cells capable of producing IFN-γ, and the provision of help for CD8+ T cell activation (Figs. 1 and 2). Whereas these events are typically associated with a developing effector response, some of these features are also observed during the development of CD4+ T cell anergy in response to tumor (9), self antigen (41), or even to experimental injection with peptide antigen (42, 43). Ultimately, the gold standard for ascribing the functional significance of the observed changes in tumor-specific CD4+ T cells is their impact on tumor growth. RencaHA-bearing mice that received HA-specific CD4+ T cells had diminished tumor burdens compared with those without T cell transfer (as measured by lung weight and the number of tumor nodules [unpublished data]). More significantly, the expression of the target antigen by progressing tumor was dramatically reduced in recipients of HA-specific CD4+ T cells (Figs. 3 and 4; a two to three log reduction in HA message below the levels expressed by tumors arising in immunocompetent mice without T cell transfer).
The mechanism(s) responsible for the CD4+ T cell–dependent effector response seen in this model are not yet fully defined. RencaHA is constitutively MHC class II negative, but its expression is induced by IFN-γ, so direct recognition by CD4+ T cells in the effector phase has not been formally ruled out. Studies in parent → F1 chimeras have shown a critical role for APCs in the initiation phase of T cell recognition (16). During this phase, early activation of HA-specific CD4+ T cells can provide help for CD8+ T cell priming (Fig. 2 B). Although CD8+ T cell–dependent mechanisms may contribute to tumor rejection, the transfer of purified HA-specific CD4+ T cells into RAG−/− mice with established RencaHA lung masses results in the near complete eradication of the tumor (unpublished data), demonstrating that CD8+ T cell independent mechanisms may also be operative as has been previously reported in related systems (44–46).
In spite of the significant antitumor effector response initially generated against this immunogenic tumor, tumors that escape this response continue to express low levels of the model antigen (Fig. 4). More importantly, the amount of antigen produced by edited tumor was sufficient to be recognized in vivo by CD4+ HA-specific T cells expressing the same clonotypic TCR as the cells responsible for the initial editing (Fig. 5). Therefore, in spite of the changing antigen load it is likely that antigen-experienced tumor-specific T cells continue to be presented antigen throughout the course of tumor progression, never achieving sterile immunity.
In several in vivo systems, the persistence of antigen has been shown to correlate with the induction of antigen-specific T cell tolerance—either through anergy or deletion (47, 48). In the current model, several lines of evidence suggested that in the face of continued exposure to tumor antigen, the initial effector response elicited by CD4+ HA-specific T cells decayed. First, although the early expansion of this population was accompanied by a fraction of these cells acquiring the capacity to produce IFN-γ, the detection of IFN-producing cells was transient (Fig. 2 A), even though a sizable population of expanded HA-specific CD4+ T cells remained (Fig. 1). Second, the proliferation of tumor-specific CD4+ T cells fell dramatically between the first and third weeks after T cell transfer (Fig. 6). Whereas this may be partly due to falling antigen load, this parameter is difficult to quantify given the opposing influences of decreasing expression of HA on a per cell basis but increasing overall tumor burden with time. Third, previous studies in this system have demonstrated that tumor antigen–experienced HA-specific T cells had diminished responses to systemic challenge with vacHA(16), as opposed to what might be expected for true memory cells that survive the contraction phase of an effector response. Finally, CD4+ T cells from RencaHA bearing mice were significantly less potent than those from nontumor bearing mice at exerting selective pressure for HA loss by progressing tumor in secondary recipients (Fig. 7). Together, recognition of tumor antigen led to expansion and contraction of an acute response but without the development of memory. This decay in endogenous effector function and diminished responsiveness to exogenous priming correlated with a loss of editing capacity.
There are now several studies examining the pathways by which a population of effector and/or memory T cells may become tolerant (41, 49–51). In considering the mechanisms underlying this transition, it is important to emphasize that the operational definition of tolerance in such settings applies to the functional capacity of a population of T cells of a given specificity. Much like the difficult problem of tracking the precursor–product relationship between effector cells and memory T cells (52), it is unclear whether tumor-specific effector cells in this model are directly rendered unresponsive, or are deleted, leaving behind a population of anergized cells that never fully differentiated into effector cells when initially activated. Alternatively, a third possibility is that some fraction of the effector cells do survive the contraction to become memory cells (as in antiviral responses) but that their function is masked by an emerging population of tumor-specific suppressor cells. One of the unexpected findings in the BrdU experiment (Fig. 6) was that tumor-specific T cells that had responded to antigen in the first week after transfer persisted at a relatively high frequency (32.5%) in the hilar nodes for the duration of the experiment. In a related (though less immunogenic) tumor model, we now have evidence that such long-lived tumor antigen–experienced CD4+ T cells are potent suppressors of both naïve and Th1 effector cells specific for tumor antigen (unpublished data). Accordingly, the population of CD4+ T cells specific for a given tumor antigen may be functionally heterogeneous, containing a mixture of naïve, effector–memory, and suppressor T cell subpopulations. In such a model, the overall functional responsiveness may reflect the relative abundance of each.
Over 20 yr ago, Robert North and colleagues performed an extensive analysis of the host response to immunogenic tumors and demonstrated the early induction of an effector T cell response capable of rejecting a contra lateral tumor challenge in mice in which the initial tumor grows progressively (53, 54). Further evidence for this concomitant immunity was demonstrated by the passive transfer of Ly-1-2+ (i.e., CD8+) T cells isolated from mice with early stage tumor into irradiated tumor-bearing recipients, which led to tumor rejection. However the capacity to transfer immunity decayed when taken from donors bearing later stage tumor, and this decay was shown to be mediated by the progressive accumulation of a population of Ly-1+2− (i.e., CD4+) suppressor T cells. Like the immunosurveillance hypothesis before it, the significance of concomitant immunity and tumor-specific suppressor T cells was largely disputed in the absence of more defined mechanisms responsible for these observations. In the face of renewed interest in active T cell regulation and with the availability of new tools with which to approach the problem, the identification of such mechanisms may soon be forthcoming.
Acknowledgments
We thank E. Fuchs, D. Pardoll, I. Borrello, and E. Sotomayor for critical review of the article, and H. von Boehmer and L. Sherman for originally providing the TCR transgenic mice.
This work was supported by grant nos. NIHP50CA96888-01 and NIH2P01CA15396.
The authors have no conflicting financial interests.
Abbreviations used in this paper: BrdU, bromodeoxyuridine; CFSE, 5,6-carboxy-fluorescein succinimidyl ester; HA, haemaglutinin; qRT-PCR, quantitative real-time RT-PCR.
References
- 1.Ehrlich, P. 1909. Ueber den jetzigen stand der Karzinomforschung. Ned. Tijdschr. Geneeskd. 5:273–290. [Google Scholar]
- 2.Thomas, L. 1959. Discussion for Reactions to homologous tissue antigens in relation to hypersensitivity. Cellular and Humoral Aspects of the Hypersensitive States. H.S. Lawrence, editor. Hoeber-Harper, New York. 529–532.
- 3.Burnet, F.M. 1970. The concept of immunological surveillance. Progr. Exp. Tumor Res. 13:1–27. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran, V., H. Ikeda, A.T. Bruce, J.M. White, P.E. Swanson, L.J. Old, and R.D. Schreiber. 2001. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 410:1107–1111. [DOI] [PubMed] [Google Scholar]
- 5.Dunn, G.P., A.T. Bruce, H. Ikeda, L.J. Old, and R.D. Schreiber. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3:991–998. [DOI] [PubMed] [Google Scholar]
- 6.North, R.J. 1984. The therapeutic significance of concomitant antitumor immunity. I. LY-1- 2+ T cells from mice with a progressive tumor can cause regression of an established tumor in gamma-irradiated recipients. Cancer Immunol. Immunother. 18:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee, P.P., C. Yee, P.A. Savage, L. Fong, D. Brockstedt, J.S. Weber, D. Johnson, S. Swetter, J. Thompson, P.D. Greenberg, et al. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5:677–685. [DOI] [PubMed] [Google Scholar]
- 8.Bogen, B. 1996. Peripheral T cell tolerance as a tumor escape mechanism: deletion of CD4+ T cells specific for a monoclonal immunoglobulin idiotype secreted by a plasmacytoma. Eur. J. Immunol. 26:2671–2679. [DOI] [PubMed] [Google Scholar]
- 9.Staveley-O'Carroll, K., E. Sotomayor, J. Montgomery, I. Borrello, L. Hwang, S. Fein, D. Pardoll, and H. Levitsky. 1998. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA. 95:1178–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sotomayor, E.M., I. Borrello, E. Tubb, F.M. Rattis, H. Bien, Z. Lu, S. Fein, S. Schoenberger, and H.I. Levitsky. 1999. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat. Med. 5:780–787. [DOI] [PubMed] [Google Scholar]
- 11.Sotomayor, E.M., I. Borrello, and H.I. Levitsky. 1996. Tolerance and cancer: a critical issue in tumor immunology. Crit. Rev. Oncog. 7:433–456. [DOI] [PubMed] [Google Scholar]
- 12.Kirberg, J., A. Baron, S. Jakob, A. Rolink, K. Karjalainen, and H. von Boehmer. 1994. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex–restricted receptor. J. Exp. Med. 180:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan, D.J., R. Liblau, B. Scott, S. Fleck, H.O. McDevitt, N. Sarvetnick, D. Lo, and L.A. Sherman. 1996. CD8(+) T cell-mediated spontaneous diabetes in neonatal mice. J. Immunol. 157:978–983. [PubMed] [Google Scholar]
- 14.Morgan, D.J., H.T. Kreuwel, S. Fleck, H.I. Levitsky, D.M. Pardoll, and L.A. Sherman. 1998. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 160:643–651. [PubMed] [Google Scholar]
- 15.Lu, Z., L. Yuan, X. Zhou, E. Sotomayor, H.I. Levitsky, and D.M. Pardoll. 2000. CD40-independent pathways of T cell help for priming of CD8(+) cytotoxic T lymphocytes. J. Exp. Med. 191:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuenca, A., F. Cheng, H. Wang, J. Brayer, P. Horna, L. Gu, H. Bien, I.M. Borrello, H.I. Levitsky, and E.M. Sotomayor. 2003. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res. 63:9007–9015. [PubMed] [Google Scholar]
- 17.Dunn, G.P., L.J. Old, and R.D. Schreiber. 2004. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22:329–360. [DOI] [PubMed] [Google Scholar]
- 18.Zhang, L., J.R. Conejo-Garcia, D. Katsaros, P.A. Gimotty, M. Massobrio, G. Regnani, A. Makrigiannakis, H. Gray, K. Schlienger, M.N. Liebman, et al. 2003. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348:203–213. [DOI] [PubMed] [Google Scholar]
- 19.Seliger, B., T. Cabrera, F. Garrido, and S. Ferrone. 2002. HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 12:3–13. [DOI] [PubMed] [Google Scholar]
- 20.Speiser, D.E., R. Miranda, A. Zakarian, M.F. Bachmann, K. McKall-Faienza, B. Odermatt, D. Hanahan, R.M. Zinkernagel, and P.S. Ohashi. 1997. Self antigens expressed by solid tumors do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J. Exp. Med. 186:645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochsenbein, A.F., P. Klenerman, U. Karrer, B. Ludewig, M. Pericin, H. Hengartner, and R.M. Zinkernagel. 1999. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc. Natl. Acad. Sci. USA. 96:2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen, L.T., A.R. Elford, K. Murakami, K.M. Garza, S.P. Schoenberger, B. Odermatt, D.E. Speiser, and P.S. Ohashi. 2002. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J. Exp. Med. 195:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marzo, A.L., B.F. Kinnear, R.A. Lake, J.J. Frelinger, E.J. Collins, B.W. Robinson, and B. Scott. 2000. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J. Immunol. 165:6047–6055. [DOI] [PubMed] [Google Scholar]
- 24.Shrikant, P., and M.F. Mescher. 1999. Control of syngeneic tumor growth by activation of CD8+ T cells: efficacy is limited by migration away from the site and induction of nonresponsiveness. J. Immunol. 162:2858–2866. [PubMed] [Google Scholar]
- 25.Colella, T.A., T.N. Bullock, L.B. Russell, D.W. Mullins, W.W. Overwijk, C.J. Luckey, R.A. Pierce, N.P. Restifo, and V.H. Engelhard. 2000. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med. 191:1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molldrem, J.J., P.P. Lee, S. Kant, E. Wieder, W. Jiang, S. Lu, C. Wang, and M.M. Davis. 2003. Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J. Clin. Invest. 111:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochsenbein, A.F., S. Sierro, B. Odermatt, M. Pericin, U. Karrer, J. Hermans, S. Hemmi, H. Hengartner, and R.M. Zinkernagel. 2001. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature. 411:1058–1064. [DOI] [PubMed] [Google Scholar]
- 28.Spiotto, M.T., P. Yu, D.A. Rowley, M.I. Nishimura, S.C. Meredith, T.F. Gajewski, Y.X. Fu, and H. Schreiber. 2002. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 17:737–747. [DOI] [PubMed] [Google Scholar]
- 29.Groh, V., J. Wu, C. Yee, and T. Spies. 2002. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 419:734–738. [DOI] [PubMed] [Google Scholar]
- 30.Wang, T., G. Niu, M. Kortylewski, L. Burdelya, K. Shain, S. Zhang, R. Bhattacharya, D. Gabrilovich, R. Heller, D. Coppola, et al. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10:48–54. [DOI] [PubMed] [Google Scholar]
- 31.Cheng, F., H.W. Wang, A. Cuenca, M. Huang, T. Ghansah, J. Brayer, W.G. Kerr, K. Takeda, S. Akira, S.P. Schoenberger, et al. 2003. A critical role for Stat3 signaling in immune tolerance. Immunity. 19:425–436. [DOI] [PubMed] [Google Scholar]
- 32.Gorelik, L., and R.A. Flavell. 2001. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 7:1118–1122. [DOI] [PubMed] [Google Scholar]
- 33.Gabrilovich, D.I., H.L. Chen, K.R. Girgis, H.T. Cunningham, G.M. Meny, S. Nadaf, D. Kavanaugh, and D.P. Carbone. 1996. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 2:1096–1103 (published erratum appears in Nat. Med. 1996. 2:1267). [DOI] [PubMed] [Google Scholar]
- 34.Melcher, A., S. Todryk, N. Hardwick, M. Ford, M. Jacobson, and R.G. Vile. 1998. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat. Med. 4:581–587. [DOI] [PubMed] [Google Scholar]
- 35.Srivastava, P.K., and M. Heike. 1991. Tumor-specific immunogenicity of stress-induced proteins: convergence of two evolutionary pathways of antigen presentation? Semin. Immunol. 3:57–64. [PubMed] [Google Scholar]
- 36.Sotomayor, E.M., I. Borrello, F.M. Rattis, A.G. Cuenca, J. Abrams, K. Staveley-O'Carroll, and H.I. Levitsky. 2001. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 98:1070–1077. [DOI] [PubMed] [Google Scholar]
- 37.Yewdell, J.W., and J.R. Bennink. 1999. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 17:51–88. [DOI] [PubMed] [Google Scholar]
- 38.Spiotto, M.T., Y.X. Fu, and H. Schreiber. 2003. Tumor immunity meets autoimmunity: antigen levels and dendritic cell maturation. Curr. Opin. Immunol. 15:725–730. [DOI] [PubMed] [Google Scholar]
- 39.Medzhitov, R., and C.A. Janeway Jr. 1999. Innate immune induction of the adaptive immune response. Cold Spring Harb. Symp. Quant. Biol. 64:429–435. [DOI] [PubMed] [Google Scholar]
- 40.Lyman, M.A., S. Aung, J.A. Biggs, and L.A. Sherman. 2004. A spontaneously arising pancreatic tumor does not promote the differentiation of naive CD8(+) T lymphocytes into effector CTL. J. Immunol. 172:6558–6567. [DOI] [PubMed] [Google Scholar]
- 41.Huang, C.T., D.L. Huso, Z. Lu, T. Wang, G. Zhou, E.P. Kennedy, C.G. Drake, D.J. Morgan, L.A. Sherman, A.D. Higgins, et al. 2003. CD4+ T cells pass through an effector phase during the process of in vivo tolerance induction. J. Immunol. 170:3945–3953. [DOI] [PubMed] [Google Scholar]
- 42.Pape, K.A., R. Merica, A. Mondino, A. Khoruts, and M.K. Jenkins. 1998. Direct evidence that functionally impaired CD4+ T cells persist in vivo following induction of peripheral tolerance. J. Immunol. 160:4719–4729. [PubMed] [Google Scholar]
- 43.Perez, V.L., L. Van Parijs, A. Biuckians, X.X. Zheng, T.B. Strom, and A.K. Abbas. 1997. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 6:411–417. [DOI] [PubMed] [Google Scholar]
- 44.Greenberg, P.D., D.E. Kern, and M.A. Cheever. 1985. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2− T cells. Tumor eradication does not require participation of cytotoxic T cells. J. Exp. Med. 161:1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hung, K., R. Hayashi, A. Lafond-Walker, C. Lowenstein, D. Pardoll, and H. Levitsky. 1998. The central role of CD4(+) T cells in the antitumor immune response. J. Exp. Med. 188:2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mumberg, D., P.A. Monach, S. Wanderling, M. Philip, A.Y. Toledano, R.D. Schreiber, and H. Schreiber. 1999. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. USA. 96:8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsdell, F., and B.J. Fowlkes. 1992. Maintenance of in vivo tolerance by persistence of antigen. Science. 257:1130–1134. [DOI] [PubMed] [Google Scholar]
- 48.Redmond, W.L., J. Hernandez, and L.A. Sherman. 2003. Deletion of naive CD8 T cells requires persistent antigen and is not programmed by an initial signal from the tolerogenic APC. J. Immunol. 171:6349–6354. [DOI] [PubMed] [Google Scholar]
- 49.Kreuwel, H.T., S. Aung, C. Silao, and L.A. Sherman. 2002. Memory CD8(+) T cells undergo peripheral tolerance. Immunity. 17:73–81. [DOI] [PubMed] [Google Scholar]
- 50.Long, M., A.D. Higgins, M.A. Mihalyo, and A.J. Adler. 2003. Effector CD4 cell tolerization is mediated through functional inactivation and involves preferential impairment of TNF-alpha and IFN-gamma expression potentials. Cell. Immunol. 224:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Higgins, A.D., M.A. Mihalyo, and A.J. Adler. 2002. Effector CD4 cells are tolerized upon exposure to parenchymal self-antigen. J. Immunol. 169:3622–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacob, J., and D. Baltimore. 1999. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 399:593–597. [DOI] [PubMed] [Google Scholar]
- 53.North, R.J., and I. Bursuker. 1984. Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1-2+ effector T cells. J. Exp. Med. 159:1295–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bursuker, I., and R.J. North. 1985. Suppression of generation of concomitant antitumor immunity by passively transferred suppressor T cells from tumor-bearing donors. Cancer Immunol. Immunother. 19:215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]