Importance of integrin LFA-1 deactivation for the generation of immune responses (original) (raw)
Abstract
The dynamic regulation of ligand binding is considered crucial for integrin function. However, the importance of activity regulation for integrin function in vivo is largely unknown. Here, we have applied gene targeting to delete the GFFKR sequence of the lymphocyte function-associated antigen–1 (LFA-1) αL subunit cytoplasmic domain in mouse germline. Lymphocytes from Lfa-1 d/d mutant mice showed constitutive activation of LFA-1–mediated cell adhesion and impaired de-adhesion from intercellular adhesion molecule-1 that resulted in defective cell migration. In contrast, signaling through LFA-1 was not affected in Lfa-1 d/d cells. T cell activation by superantigen-loaded and allogeneic APCs, cytotoxic T cell activity, T-dependent humoral immune responses, and neutrophil recruitment during aseptic peritonitis were impaired in Lfa-1 d/d mice. Thus, deactivation of LFA-1 and disassembly of LFA-1–mediated cell contacts seem to be vital for the generation of normal immune responses.
Many integrins do not exhibit constitutive ligand binding capacity, but are expressed in an inactive state (1, 2). Integrin ligand binding is activated rapidly and transiently by cytoplasmic signals initiated by the stimulation of diverse cell surface receptors (inside-out signaling). The integrin, lymphocyte function-associated antigen-1 (LFA-1; αLβ2), is expressed on all leukocytes and is considered to play an important role in host defense (3). LFA-1 shares the β2 subunit with Mac-1 (αMβ2), p150/95 (αXβ2), and αDβ2. Binding of LFA-1 to its natural ligands, intercellular adhesion molecule (ICAM)-1, ICAM-2, or ICAM-3, mediates interactions between T cells and blood vessel endothelium, target cells, and APCs. LFA-1 stabilizes intercellular adhesion between T cells and APCs, which seems to be particularly relevant for facilitating T cell activation at low antigen concentration (4). In addition, binding of LFA-1 to its cognate ligands directly activates the Jnk and Erk-1/2 pathways, and thereby decreases the threshold of T cell activation (5, 6). LFA-1–deficient mice exhibit reduced lymph node cellularity and splenomegaly which is consistent with a major role of LFA-1 in normal lymphocyte recirculation (7–10). T cells from LFA-1−/− mice also displayed an impaired proliferation in response to allogeneic cells and mitogen. However, they mounted normal cytotoxic T cell responses during viral infections, but failed to reject immunogenic tumors (7).
The ligand-binding capacity of leukocyte integrins can be activated by signaling pathways emanating from T and B cell antigen receptors, chemokine receptors, and Toll-like receptors within a time-scale of seconds to minutes (1, 11–13). Integrin activation through these receptors causes large-scale conformational changes in integrin extracellular domains that are associated with increased ligand-binding affinity and leads to receptor clustering at cell contact areas. The signaling pathways that control LFA-1 activation are not understood completely, but involve protein kinase C; phosphatidylinositol 3-kinase; cytohesin-1; small GTPases; and the adaptor protein, SLAP-130/Fyb (14–20).
The cytoplasmic domains of α and β subunits are crucial for the regulation of integrin ligand-binding activity (1). Thus, changes in the cytoplasmic tails of integrins affect the structure and function of their extracellular domains. Mutational analyses have shown that interactions between membrane-proximal sequences of integrin cytoplasmic domains restrain integrins in an inactive conformation. For example, deletions of the entire α subunit cytoplasmic domain or of the highly conserved membrane-proximal GFFKR sequence disrupt interactions of α and β subunit tails, and produce constitutively active integrin in vitro. Consistent with this model, the talin head domain activates integrins by binding to the cytoplasmic domain of β subunits and—as a result of the high affinity of this interaction—ablates interactions between α and β cytoplasmic domains. Recent studies that measured fluorescence resonance energy transfer confirmed these changes of cytoplasmic domain interactions during integrin activation in living cells in vitro (21).
Although numerous studies have investigated the consequences of integrin deficiencies in mouse models, the in vivo relevance of restraining LFA-1 in an inactive state on resting leukocytes and rapidly deactivating LFA-1 after cell stimulation is unknown. In the present study, we mutated the αL subunit cytoplasmic tail in mouse germline leading to the expression of constitutively active LFA-1. We show that this mutation severely impairs immune responses in vitro and in vivo, and thereby demonstrate that complete deactivation is essential for the normal function of LFA-1.
RESULTS
Generation of a mutant mouse strain (Lfa-1 d/d) expressing constitutively active integrin LFA-1
Although the lack of LFA-1–mediated cell adhesion has been demonstrated to impair immune responses (7), the functional consequences of defective LFA-1 deactivation are unknown. To address this question, a mutant mouse strain (Lfa-1 d/d) that exhibits a constitutive germline deletion of the conserved amino acid motif GFFKR in the membrane-proximal segment of the LFA-1 αL chain cytoplasmic domain was generated by gene targeting (Fig. 1). This mutation was chosen because deletion of the GFFKR sequence is known to cause separation of the αL and β2 cytoplasmic tails, and to result in the constitutive activation of integrins, including LFA-1 (21–23). Correct introduction of the GFFKR deletion was confirmed by cDNA cloning and nucleotide sequence analysis of αL transmembrane and cytoplasmic domains from Lfa-1 d/d mice (unpublished data). Mutant Lfa-1 d/d mice were born at normal Mendelian ratios, were fertile, and did not exhibit any gross anatomic or behavioral abnormalities (unpublished data).
Figure 1.

Generation of Lfa-1 d/d mutant mice. (a) Organization of the murine Lfa-1 genomic locus (top), targeting vector (second from top), targeted Lfa-1 d allele (second from bottom), and Lfa-1 d allele following intercross with the cre deleter strain (bottom). (b) Southern blot analysis of genomic DNA derived from a wild-type mouse (lane 1) and a heterozygous Lfa-1 d-neo/+ mouse (lane 2) before deletion of the neocassette using the 3′ flanking probe. (c) PCR analysis of genotypes from wild-type (+/+), heterozygous Lfa-1 d/+ (d/+), and homozygous Lfa-1 d/d (d/d) mice after deletion of the neocassette. To detect the mutant allele, a genomic fragment containing the newly introduced HpaI site in exon 31 was amplified by PCR and digested with HpaI.
To control whether the Lfa-1 d/d mutation may affect integrin expression, flow cytometry analyses were performed. The results in Fig. 2 a show that cell surface expression of mutant LFA-1 was reduced as compared with the wild-type integrin. However, analysis of permeabilized cells revealed that the levels of total cellular LFA-1 protein were not altered by the Lfa-1 d/d mutation (Fig. 2 a). Moreover, splenocyte mRNA levels for mutant and wild-type LFA-1 did not differ significantly (unpublished data), and indicated that expression of the mutant Lfa-1 d gene was not impaired. Instead, these findings are consistent with previous observations showing that deletion of the membrane-proximal GFFKR sequence in the LFA-1 αL subunit affects surface expression by reducing heterodimer formation with the β2 subunit (23). Additional flow cytometry analyses of B and T cells and neutrophils demonstrated that the integrins, αXβ2 and Mac-1, which share the β2 subunit with LFA-1, were expressed at similar levels on Lfa-1 d/d and wild-type cells (Fig. 2 b). Moreover, Lfa-1 d/d and wild-type cells also did not differ in their expression of unrelated α4β1 and α2β1 integrins (Fig. 2 b); this indicates that the Lfa-1 d/d mutation does not alter the global expression of leukocyte integrins.
Figure 2.


Integrin expression in Lfa-1 d/d mice. (a) Cell surface levels of LFA-1 and total cellular LFA-1 protein were measured by flow cytometry analyses of Lfa-1 d/d (red lines) and wild-type (blue lines) splenocytes. (b) Cell surface integrin expression was determined on Lfa-1 d/d (red lines) and wild-type (blue lines) splenic T cells (CD3+ cells), B cells (B220+ cells), and bone marrow neutrophils (Gr-1+ cells). Staining with isotype-matched controls is indicated as dotted lines (n = 3 independent experiments).
The effect of the Lfa-1 d/d mutation on cell adhesion was investigated using a recombinant ICAM-1(D1-2)-Fc protein. The results in Fig. 3 a demonstrate that adhesion of unstimulated Lfa-1 d/d thymocytes to ICAM-1(D1-2)-Fc was significantly elevated as compared with cells that expressed wild-type LFA-1. Exposure to the integrin activator, Mn2+, or incubation with phorbol ester further increased binding of Lfa-1 d/d and wild-type thymocytes to ICAM-1(D1-2)-Fc. However, even under these conditions, Lfa-1 d/d cells adhered more strongly to ICAM-1(D1-2)-Fc than did wild-type cells. Notably, ICAM-1(D1-2)-Fc adhesion of unstimulated Lfa-1 d/d cells was similar to that of Mn2+- or PMA-stimulated wild-type cells (Fig. 3 a). Maximal adhesion of wild-type thymocytes required stimulation with Mn2+ and PMA, whereas for Lfa-1 d/d cells, incubation with a single stimulus was sufficient. The absolute levels of maximal adhesion to ICAM-1(D1-2)-Fc did not differ between Lfa-1 d/d and wild-type cells (Fig. 3 a). To control whether the GFFKR deletion in the mouse αL gene specifically affects the integrin, LFA-1, thymocyte adhesion to vascular cell adhesion molecule (VCAM)-1-Fc, which interacts with α4β1 and α4β7 integrins, was examined. As depicted in Fig. 3 b, binding of Lfa-1 d/d and wild-type thymocytes to VCAM-1-Fc was comparable for all conditions tested. These results demonstrate that the Lfa-1 d/d mutation results in a constitutive, but partial, activation of LFA-1. Moreover, the maximal LFA-1–mediated adhesive capacity of cells was not altered by this mutation, and suggests that cell surface expression of mutant LFA-1 is sufficient for its functionality. Consistent with this notion, previous work showed that only a small subset of the integrin, Mac-1 (αMβ2), is sufficient to mediate the full adhesive capacity of neutrophils (24).
Figure 3.

Lfa-1 d/d mutation enhances cell adhesion but impairs endothelial transmigration. Thymocytes were incubated with plates coated with purified ICAM-1(D1-2)-Fc (a) or VCAM-1-Fc (b) and the fraction of adherent cells was determined (n = 4–8 independent experiments). (c) Lymph node cells stimulated with PMA and ionomycin and rested in IL-2 were incubated with CD3 mAbs for the indicated periods (10 and 40 min) and adhesion to ICAM-1(D1-2)-Fc was determined. Results are presented as percentage increase of ICAM-1(D1-2)-Fc adhesion of CD3-stimulated as compared with unstimulated cells. (d) Splenocytes (SPL) or lymph node (LN) cells were plated on bEnd5 endothelioma cells that were unstimulated (medium) or treated with TNF for 16 h. The number of cells adherent per unit area were determined (n = 5 independent experiments). (e) Transmigration of unstimulated (medium) or TNF-treated bEnd5 endothelioma cells by splenocytes or lymph node cells (right panel) was measured (n = 6 independent experiments). *P < 0.05; #P < 0.01 (Student's t test).
In contrast to stimulation with PMA, activation of integrin ligand-binding by the T cell antigen receptor and chemokines is mediated by Rap1, together with other signaling molecules (14, 25, 26). The critical cytoplasmic region of αL that is required for Rap1 responsiveness is adjacent to the GFFKR sequence (27); this raises the possibility that deletion of the GFFKR sequence in Lfa-1 d/d mice would alter activation of LFA-1 through the T cell receptor. Therefore, we investigated LFA-1 activation in response to T cell receptor signaling. Lymph node cells from wild-type and Lfa-1 d/d mice that were stimulated with PMA and ionomycin, and maintained in IL-2, were stimulated with CD3 antibodies and adhesion to ICAM-1(D1-2)-Fc was measured. The results in Fig. 3 c show that CD3 treatment induced adhesion of wild-type and Lfa-1 d/d cells to a similar extent; the adhesion of Lfa-1 d/d cells was accelerated slightly 10 min after addition of CD3 antibodies. Therefore, these data indicate that the αL-GFFKR deletion did not impair activation of LFA-1 in response to T cell receptor ligation.
LFA-1 is crucial for the firm adhesion of leukocytes to endothelial cells and the subsequent process of transendothelial migration. Therefore, we investigated the effects of the Lfa-1 d/d mutation on leukocyte endothelial interactions. Fig. 3 d shows that binding of resting splenocytes and lymph node cells to unstimulated bEnd5 endothelioma cells was not influenced by the Lfa-1 d/d mutation. However, when bEnd5 cells were stimulated with TNF to up-regulate ICAM-1, binding of Lfa-1 d/d splenocytes and lymph node cells was increased significantly as compared with wild-type cells. In marked contrast to cell adhesion, Lfa-1 d/d lymphocytes that were derived from spleen or lymph nodes were impaired significantly in their capacity to transmigrate bEnd5 endothelial cell monolayers (Fig. 3 e).
Impaired de-adhesion and migration of lymphocytes expressing mutant LFA-1
To investigate further why the αL-GFFKR deletion conferred enhanced ability of T cells to adhere to ICAM-1, but retarded their ability to migrate across stimulated endothelium, we turned to videomicroscopy to investigate directly the effects of the LFA-1 mutation on T cell migration on immobilized ICAM-1-Fc. T cells used in these experiments were stimulated with ConA and rested in IL-2 before examining their migratory capacity. The results show that the average migratory speed of Lfa-1 d/d T cells was reduced severely to ∼20% of the speed of wild-type T cells, whereas ICAM-1-Fc adhesion of Lfa-1 d/d T cells was increased slightly as compared with wild-type cells (Fig. 4, a and b). Tracking of individual T cells revealed that the distance migrated by each individual T cell was reduced greatly by the αL-GFFKR mutation (Fig. 4 c and d). Time-lapse videomicroscopy analysis of migrating T cells demonstrated that Lfa-1 d/d T cells were unable to translocate because they were attached firmly at their trailing edges (Videos 1 and 2, available at http://www.jem.org/cgi/content/full/jem.20041850/DC1). Therefore, these results indicate that the αL-GFFKR mutation results in impaired de-adhesion at trailing edges of migrating cells, and, consequently, inefficient cell translocation. Thus, complete deactivation of the integrin, LFA-1, may be required to mediate sequential adhesion and de-adhesion events during cell migration.
Figure 4.
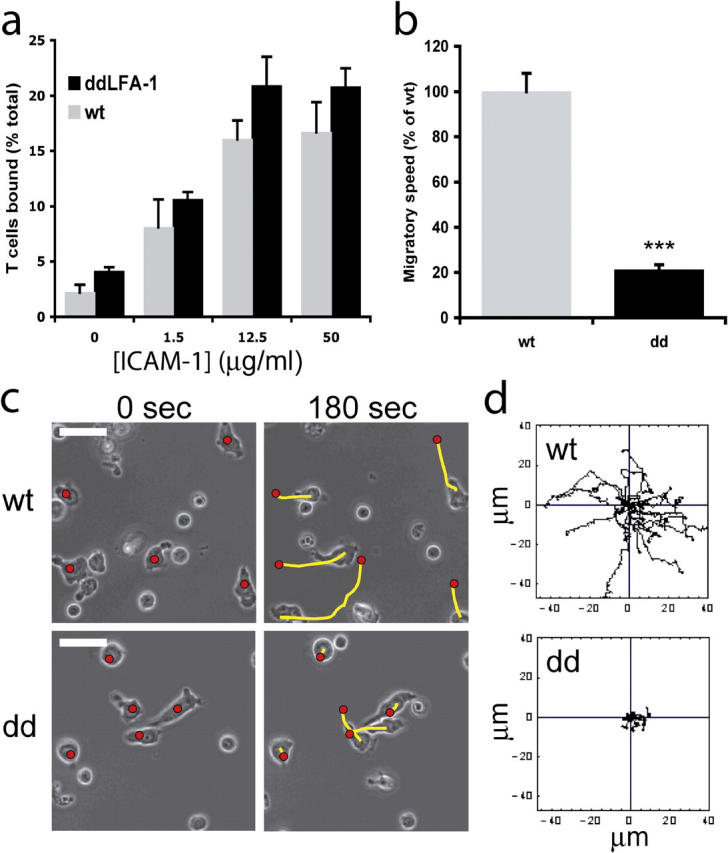
T cell adhesion and migration on immobilized ICAM-1-Fc. (a) Adhesion of spleen T cells onto immobilized ICAM-1 in the presence of 6 mM Mg2+ was determined after 30 min (mean + SD). (b) T cells migrating on 50 μg/ml immobilized ICAM-1-Fc with calculation of speed over 10 min. The speed of Lfa-1 d/d T cells is expressed as percentage of wt T cell speed + SEM (***P < 0.01). (c) Phase images of migrating T cells. The red circle indicates the position of the T cell at 0 s; the yellow line details the T cell trajectory over 180 s. White bars, 10 μm. (d) Migration of T cells was tracked over 600 s; each line represents one T cell (wt, n =20; Lfa-1 d/d, n = 21). Experiments are representative of n =2 (a) and n = 6 (b–d).
Influence of the Lfa-1 d/d mutation on lymphoid organogenesis and lymphocyte development
LFA-1 deficiency was found to alter the cellularity of peripheral lymphoid organs (7, 9). Therefore, we investigated the influence of constitutive LFA-1 activation on lymphoid organogenesis. Analysis of different lymphoid organs revealed that total cell numbers in peripheral lymph nodes and Peyer's patches were reduced substantially in Lfa-1 d/d mutant mice when compared with wild-type littermates (Table I). In contrast, spleen cell numbers were significantly greater in Lfa-1 d/d mice than in wild-type mice. Bone marrow and thymus cellularity as well as peripheral blood leukocyte numbers were not altered by the Lfa-1 d/d mutation. This phenotype of Lfa-1 d/d mice is similar that of Lfa-1 −/− mice, and suggests that the presence and the full conformational flexibility of LFA-1 are required for the normal development of secondary lymphoid organs.
Table I.
Lymphoid organ development in Lfa-1 d/d mice
| Cell numbers (×106) | |||
|---|---|---|---|
| Organ | Wild type | Lfa-1 d/d | p-value |
| Thymusa | 70.4 ± 4.8 | 61.8 ± 4.3 | NS |
| Bone marrowb | 35.0 ± 7.3 | 29.0 ± 4.1 | NS |
| Spleena | 71.9 ± 7.0 | 95.1 ± 8.4 | 0.040 |
| Lymph nodesa | 1.31 ± 0.10 | 0.54 ± 0.09 | <0.0001 |
| Peyer's patchesa | 0.39 ± 0.04 | 0.11 ± 0.02 | <0.0001 |
| Bloodc | 2.2 ± 0.5 | 2.7 ± 0.4 | NS |
Based on these results, we next examined the effects of the Lfa-1 d/d mutation on B and T cell development. The results showed that ratios of major B and T lymphocyte subsets in bone marrow (B220loIgM−, B220loIgM+, and B220hiIgMhi cells), thymus (CD4 and CD8 double negative, double positive, and single positive cells), spleen (IgMhiIgDlo and IgMloIgDhi cells), and lymph nodes (CD4+CD8− and CD8+CD4− cells) were not affected in Lfa-1 d/d mice (unpublished data). To investigate T cell development further, the T cell receptor Vβ repertoire was determined in thymocytes. The results demonstrated that the Vβ repertoire did not differ significantly between Lfa-1 d/d mutant and wild-type mice (unpublished data). Thus, constitutive activation of LFA-1 alters the cellularity of secondary lymphoid organs, but does not seem to have a major influence on B and T cell development.
Lfa-1 d/d mutation impairs activation of T cells through APCs
LFA-1 on T cells mediates contacts with APCs, and thereby, decreases the amount of antigen that is required for T cell activation (4). To investigate the influence of the Lfa-1 d/d gain-of-function mutation on T cell responses directly, we stimulated splenocyte cultures by incubation with the bacterial superantigen, Staphylococcal enterotoxin B (SEB), which activates Vβ8+ T cells of CD4 and CD8 subsets. The dose–response curve depicted in Fig. 5 a demonstrates that proliferation of Lfa-1 d/d T cells was attenuated significantly as compared with wild-type cells at all concentrations of SEB tested. Moreover, in allogeneic mixed lymphocyte reactions with irradiated BALB/c splenic stimulator cells, the proliferation of Lfa-1 d/d T cells was reduced significantly as compared with wild-type T cells (Fig. 5 b). To analyze further the effects of the Lfa-1 d/d mutation on T cell activation, the induction of CD25 and CD69 expression on SEB-stimulated T cells was studied. The results in Fig. 5 c demonstrate significantly reduced expression of CD25 and CD69 on Lfa-1 d/d as compared with wild-type T cells. Together, these results indicate that T cell activation by superantigen-pulsed or allogeneic APCs is impaired markedly by the Lfa-1 d/d mutation.
Figure 5.

Lfa-1 d/d mutation impairs T cell proliferation stimulated by APCs. Splenocytes from wild-type or Lfa-1 d/d mice were stimulated with the indicated concentrations of SEB (a) or irradiated BALB/c splenocytes or autologous cells (b). 3[H]-thymidine incorporation was measured 72 h later (n = 6 independent experiments). (c) Splenocytes were incubated with SEB (10 μg/ml) for 16 h. The percentage of CD4 T cells from wild-type and Lfa-1 d/d mice expressing CD25 and CD69 was determined by flow cytometry (n = 4–5 independent experiments). (d) Enriched splenic T cells were stimulated with plate-bound CD3 antibody, and 3[H]-thymidine incorporation was measured after 72 h (n = 4 independent experiments). *P < 0.05; #P < 0.01 (Student's t test).
To distinguish whether reduced activation of Lfa-1 d/d T cells may result from altered interactions with APCs or from T cell intrinsic defects, purified splenic T cells were stimulated with a range of concentrations of plate-bound CD3 antibody. The results in Fig. 5 d clearly show that Lfa-1 d/d T cells proliferate as efficiently as do wild-type T cells in response to plate-bound CD3, which suggests that the Lfa-1 d/d mutation does not cause a general defect of T cells to respond to antigen receptor–mediated stimulation.
In additional experiments, potential mechanisms underlying impaired activation of Lfa-1 d/d T cells were investigated. Expression of activated LFA-1 may improve and extend stable contact formation, which may lead to hyperactivation of T cells and accelerated apoptosis. To address this question directly, T cells were stimulated with SEB for various periods and the fraction of apoptotic Vβ8+ T cells was determined by annexin V staining. The results depicted in Fig. 6 a show that under the experimental conditions that were used, activation-induced cell death did not occur in wild-type or Lfa-1 d/d T cells. Instead, when compared with unstimulated Vβ8+ T cells, stimulation with SEB reduced the fraction of apoptotic wild-type and Lfa-1 d/d T cells to a similar extent (Fig. 6 a). To investigate further the influence of the Lfa-1 d/d mutation on T cell apoptosis after antigen receptor engagement, SEB was injected into hind footpads of mice. The results in Fig. 6 b show that deletion of Vβ8+ T cells caused by in vivo SEB administration was similar in wild-type and Lfa-1 d/d mice. Thus, the αL-GFFKR deletion of LFA-1 does not seem to lead to increased T cell apoptosis after antigen receptor stimulation.
Figure 6.

Lfa-1d/d mutation does not alter T cell apoptosis and LFA-1–mediated signaling. (a) Splenocytes from wild-type or Lfa-1 d/d mice were stimulated with SEB (10 μg/ml) and the percentage of Vβ8+ T cells that stained positive for annexin V was determined by flow cytometry (n = 4 independent experiments). (b) The fraction of Vβ8+ CD4 and CD8 T cells was determined in popliteal lymph nodes of mice left untreated (Ctrl) and 24 h after s.c. injection of 10 μg SEB into each hind footpad. n = 4 mice per group; #P < 0.01, *P < 0.05 (ctrl vs. SEB). (c) Lymph node cells from Lfa-1 d/d (d/d) and wild-type mice (+/+) were unstimulated or incubated with 10 μg/ml purified soluble ICAM-1(D1-2)-Fc for 30 min in the presence of 1 mM Mn2+. Phosphorylation and total protein levels of Jnk and Erk-1/2 were determined by Western blot analyses of cell lysates. (d) Nuclear extracts were prepared from thymocytes of Lfa-1 d/d (d/d) and wild-type mice (+/+), and protein levels of p27Kip1 and laminA/C were determined by Western blot analysis.
Lfa-1 d/d mutation does not influence signaling through LFA-1
Integrins may function as signaling receptors, and integrin ligation may trigger signaling processes that influence cellular activation and proliferation (1). Therefore, we addressed the question of whether partial activation of LFA-1 through the Lfa-1 d/d mutation may result in constitutive signaling activity or enhanced ligand-induced signaling. LFA-1 ligation was shown previously to induce activation of JNK and Erk1/2 MAP kinases (5). As shown in Fig. 6 c, constitutive levels of phosphorylated JNK or Erk1/2 did not differ between Lfa-1 d/d and wild-type cells. Exposure of cells to recombinant ICAM-1(D1-2)-Fc induced robust phosphorylation of JNK, which was not influenced by the expression of activated LFA-1d. In addition, Lfa-1 d/d and wild-type cells exhibited low, but comparable, levels of Erk1/2 phosphorylation following incubation with ICAM-1(D1-2)-Fc. Enforced expression of constitutively active Rap1, which functions as a potent integrin activator, impaired T cell activation by elevating cellular levels of the cyclin-dependent kinase inhibitor, p27Kip1 (28). In addition, LFA-1 may influence cellular p27Kip1 levels by its capacity to signal through JAB1 (5, 6), which is involved in the control of p27Kip1 degradation (29). Therefore, we compared p27Kip1 protein levels in nuclear extracts of Lfa-1 d/d and wild-type thymocytes. However, the results in Fig. 6 d demonstrate that the Lfa-1 d/d mutation did not alter p27Kip1 levels. Thus, constitutive or ligand-induced signaling through LFA-1 does not seem to be influenced by the Lfa-1 d/d mutation.
Lfa-1 d/d mutation reduces lytic activity but not generation of cytotoxic T lymphocytes
To investigate the effects of the Lfa-1 d/d mutation on T cell functions in more detail, mice were immunized with ovalbumin and CpG-DNA as adjuvant and cytotoxic T cell activity was determined in vitro. These conditions of immunization result in a strong activation of APCs and render the generation of antigen-specific cytotoxic cells independent of CD4 T cells (30, 31). The results in Fig. 7 a show that the lytic activity of Lfa-1 d/d cells against EL4 target cells pulsed with the ovalbumin-derived peptide, SIINFEKL, was reduced greatly when compared with wild-type controls. These results may be due to an impaired generation of cytotoxic T cells in Lfa-1 d/d mutant mice or a reduced capacity of these cells to kill target cells. To distinguish between these possibilities, antigen-specific CD8 T cells were quantitated with SIINFEKL-loaded H-2Kb tetramers after immunization of mice with ovalbumin and CpG-DNA. As depicted in Fig. 7 b, the proportion of tetramer-binding CD62lo CD8 T cells was greater in Lfa-1 d/d mice than in wild-type mice (0.81 ± 0.13% for Lfa-1 d/d mice versus 0.34 ± 0.08% for wild-type mice; n = 6; P < 0.01), whereas comparable tetramer staining was observed in the CD62hi subset of CD8 T cells (0.36 ± 0.08% for Lfa-1 d/d mice versus 0.53 ± 0.11% for wild-type mice; n = 6; P = 0.394). These results suggest that the Lfa-1 d/d mutation impairs the lytic activity, but not the generation, of CD8 T cells.
Figure 7.

Distinct effects of the Lfa-1d/d mutation on the lytic activity and generation of cytotoxic T cells. (a) Lfa-1d/d and wild-type mice were immunized with OVA using CpG-DNA as adjuvant. Draining lymph node cells were harvested 4 d later and cultured with rIL-2 for an additional 4 d. CTL activity was assayed using untreated (dotted lines) or SIINFEKL-pulsed (solid lines) EL4 cells (n = 3–4 independent experiments). (b) Lfa-1d/d (d/d) and wild-type mice (wt) were immunized with OVA and CpG-DNA. Draining lymph nodes were harvested 9 d later and CD8 T cells were analyzed for SIINFEKL/H-2Kb tetramer and CD62L antibody staining (n = 6 independent mice per group). *P < 0.05; #P < 0.01 (Student's t test).
Impaired humoral immune response in Lfa-1 d/d mice
The Lfa-1 d/d mutation was found to perturb cell contact–dependent responses of immune cells in vitro. To investigate further the effects of the Lfa-1 d/d mutation on immune responses in vivo, the development of T cell–dependent humoral immunity was studied. Mice were immunized with TNP-CGG adsorbed to alum as an adjuvant; TNP-specific antibody production was measured 21 d later. The results in Fig. 8 demonstrate that the production of TNP-specific IgM as well as IgG1 and IgG2a antibodies were impaired in Lfa-1 d/d mice; this suggests that T helper 1- and T helper 2-type responses were affected. Therefore, these results indicate that T cell–dependent humoral immune responses are attenuated substantially in Lfa-1 d/d mutant mice.
Figure 8.

Impaired T cell–dependent antibody responses in Lfa-1 d/d mice. T cell-dependent production of TNP-specific antibody isotypes was measured in serum of wild-type (diamonds) and Lfa-1 d/d mice (circles) after immunization of mice with TNP-CGG adsorbed to alum (n = 7–9 independent mice per group). *P < 0.05; #P < 0.01 (Student's t test).
Delayed recruitment of Lfa-1 d/d neutrophils in inflammation
LFA-1 is crucial for the recruitment of leukocytes from the blood circulation to inflamed tissues. In vitro experiments have shown that the Lfa-1 d/d mutation enhances adhesion to endothelial cells, but impairs transendothelial migration and cell motility on ICAM-1-Fc (Figs. 3 and 4). To elucidate the relevance of the Lfa-1 d/d mutation for inflammatory responses in vivo, aseptic peritonitis was induced by thioglycollate injection, and the recruitment of neutrophils (Gr-1hiMac-1hi cells) was monitored. In wild-type mice, i.p. injection of thioglycollate caused a marked and rapid influx of neutrophils (Fig. 9). Peritoneal neutrophil numbers were highest after 6 h and declined thereafter. However, in Lfa-1 d/d mutant mice, neutrophil accumulation was delayed substantially. Thus, neutrophil numbers were reduced by >60% in Lfa-1 d/d mice as compared with wild-type mice when analyzed 6 h after thioglycollate injection (Fig. 9). At the 12-h time point, comparable numbers of Lfa-1 d/d and wild-type neutrophils were observed in inflamed peritoneal cavities. Therefore, these results show that the Lfa-1 d/d mutation substantially impairs inflammatory cell recruitment.
Figure 9.

Delayed neutrophil recruitment during peritonitis in Lfa-1 d/d mice. Lfa-1 d/d and wild-type mice were injected i.p. with thioglycollate and infiltrating neutrophils were identified by their Gr-1hiMac-1hi-staining profile in flow cytometry analysis n = 4–6 independent mice per group. *P < 0.05 (Student's t test).
DISCUSSION
It is well-established that many integrins are preformed as inactive cell surface receptors that are responsive to diverse cellular signals, and result in rapid activation of ligand-binding capacity followed by deactivation of integrins to a resting state (1, 32). Although numerous studies have investigated the molecular mechanisms of integrin activation in vitro, the relevance of dynamic activity regulation for integrin function in vivo remains largely unknown. In the present study, we have addressed this question by the generation and analysis of a mouse strain exhibiting a constitutive germline deletion of the conserved GFFKR sequence in the LFA-1 αL subunit. The consequences of this mutation for the LFA-1–mediated adhesion of normal mouse cells were consistent with in vitro studies using cell transfectants (23). Cells from Lfa-1 d/d mutant mice exhibited an increased constitutive adhesion to purified LFA-1 ligand and endothelial cells and were more responsive to integrin-activating stimuli. However, the maximal adhesive capacity of lymphocytes to ICAM-1 was not influenced by the Lfa-1 d/d mutation. Importantly, videomicroscopy analyses demonstrated a defective detachment of the trailing edge of Lfa-1 d/d cells migrating on an ICAM-1-Fc–coated surface, and thereby indicates that the Lfa-1 d/d mutation impairs de-adhesion of cells from LFA-1 ligands. Consistent with these results, it was shown previously that stabilizing the active form of LFA-1 with mAb24 causes impaired de-adhesion of T cells from ICAM-1 (33). In contrast to the effects on cell adhesion, constitutive or ligand-induced signaling of LFA-1 was not altered in Lfa-1 d/d cells. Therefore, these results demonstrate that the Lfa-1 d/d mutation resulted in a constitutive, but partial, activation of LFA-1–mediated cell adhesion that was associated with defective disassembly of LFA-1–mediated cell adhesions.
Lack of LFA-1–mediated cellular interactions in αL-deficient mice was shown to cause multiple immune defects (7). Thus, Lfa-1 –/– mice showed splenomegaly and reduced lymph node sizes, impaired proliferative response of T cells to allogeneic stimulators, attenuated thioglycollate-induced peritonitis, and reduced NK cell cytotoxicity and delayed-type hypersensitivity responses. Therefore, it was conceivable that enhancing interactions of immune cells through constitutive activation of LFA-1 may cause hyperinflammatory responses. However, analysis of Lfa-1 d/d mice revealed opposite results, and demonstrated that the Lfa-1 d/d mutation also impaired immune responses. Similar to the phenotype of Lfa-1 –/– mice, we found Lfa-1 d/d mice to exhibit enlarged spleens and hypoplastic lymph nodes, to show impaired mixed lymphocyte responses, and to have a reduced recruitment of neutrophils to the inflamed peritoneal cavity. In addition, T cell activation by superantigen-loaded APCs, cytotoxic T cell activity, and T cell-dependent humoral immune responses were impaired markedly by the Lfa-1 d/d mutation. Therefore, these results suggest that the capacity to deactivate LFA-1 fully may be as important for the generation of normal immune responses as the presence of LFA-1 and the ability to activate its functions.
Cell contacts mediated by LFA-1 are transient; perturbing the kinetics or duration of these cellular interactions may impair recruitment or responses of immune cells (11). The orderly regulation of cell adhesion and de-adhesion is particularly relevant for cell migration (3, 34). Formation of adhesion sites at the leading edge of the cell is required to provide traction sites, whereas cell adhesions have to be disassembled at the rear of the cell to allow it to detach and move forward. Thus, the lack of integrin activation and the inability to deactivate integrins are expected to impair cell migration. In vitro studies using activating antibodies (33) or transfectants expressing LFA-1 mutants (35) have confirmed this hypothesis for lymphoid cells. The results of the present report extend this concept to normal leukocytes and in vivo conditions. We show that lymphocytes from Lfa-1 d/d mice are impaired severely in their capacity to migrate on ICAM-1-Fc and to translocate through endothelial monolayers in vitro, whereas in vivo, the recruitment of Lfa-1 d/d neutrophils in a model of aseptic peritonitis was delayed strongly. In addition, the reduced migratory capacity of lymphocytes may contribute to alterations of secondary lymphoid organ cellularity in Lfa-1 d/d mice. It is conceivable that the hypoplasia of lymph nodes and Peyer's patches in Lfa-1 d/d mice results from the partial inability of lymphocytes to cross high endothelial venules, and that cells that are unable to enter these organs accumulate in spleen.
Immune responses involving cell contact–dependent activation and effector function of T cells were impaired markedly by the Lfa-1 d/d mutation. Thus, T cell proliferation in response to superantigen-loaded and allogeneic APCs, as well as cytotoxic T cell activity, were reduced. These findings are consistent with previous work that showed that stabilizing the active form of LFA-1 by mAb24 inhibited the proliferative response of T cells to antigen and the cytolytic activity of lymphokine-activated killer cells (33). Because the Lfa-1 d/d mutation and mAb24 treatment disable complete deactivation of LFA-1, impaired resolution of LFA-1–mediated cell adhesions may be a major mechanism that underlies these defective responses. For example, cytotoxic T cells exert their full cytolytic activity by successive interactions with multiple target cells. Therefore, defective de-adhesion of cytotoxic T cells from target cells would reduce the number of target cells encountered and killed. Furthermore, the process of T cell activation by APCs involves successive phases of transient and long-term cellular contacts, followed by the detachment of T cells before they undergo cell division and regain a migratory behavior (36–38). Therefore, defective de-adhesion of Lfa-1 d/d T cells may reduce cell proliferation and the subsequent formation of new cell contacts by antigen-activated T cells.
In contrast to the capacity of cytotoxic T cells to lyse target cells, generation of antigen-specific CD8 T cells was not impaired in Lfa-1 d/d mice using an immunization protocol that renders cytotoxic T cell induction independent of CD4 T cell help (30, 31). A possible explanation for this result may be provided by studies with αL-deficient mice which showed that LFA-1 is not required for the generation of cytotoxic T cells (7). Alternatively, or in addition, CD8 T cells may not be sensitive to delayed de-adhesion from APCs, or activation of APCs by a strong Toll-like receptor agonist, such as CpG-DNA, may dampen the influence of LFA-1 through induction of additional cell adhesion receptors and costimulatory molecules. Moreover, T cells that are activated in local lymph nodes are known to resume their migratory behavior and continue to recirculate after a short stationary period. Therefore, it also is possible that the defect of Lfa-1 d/d T cells in cell migration impairs the exit of activated Lfa-1 d/d T cells and, as opposed to wild-type T cells, results in their prolonged accumulation in lymph nodes.
T helper cell–dependent humoral immune responses were impaired substantially in Lfa-1 d/d mice. Generation of T-dependent antibody responses involves movement of antigen-specific B cells to the edges of B cell follicles where they present antigen to specific CD4 T cells that have migrated to this site after activation by, and detachment from, dendritic cells (39). Activated B cells expand and remain in the follicles to become germinal center cells, or differentiate into plasma cells that migrate to the lymph node medulla and other sites (39, 40). Defective resolution of cell adhesions also may contribute, at least in part, to the reduced T-dependent antibody production in _Lfa_-1d/d mice. This mechanism may attenuate activation and proliferation of antigen-specific T cells, but also may retain these cells in the T cell area, and thereby, prevent interaction with specific B cells. Efficient movement of T and B cells between different anatomic compartments of lymphoid organs may be impaired further by the reduced migratory capacity of Lfa-1 d/d cells.
To elucidate further the mechanisms underlying impaired immune responses of Lfa-1 d/d cells and mice, we investigated whether the Lfa-1 d/d mutation may cause increased apoptosis as a result of hyperactivation of T cells, or alter LFA-1–mediated signaling. However, T cell apoptosis following in vitro SEB stimulation or in vivo administration of SEB did not differ between wild-type and Lfa-1 d/d T cells. In addition, neither the constitutive nor the ICAM-1–induced activation of Jnk and Erk-1/2 MAP kinases was augmented in Lfa-1 d/d cells. Finally, the cyclin-dependent kinase inhibitor, p27Kip1, that was shown to be up-regulated by overexpression of the integrin activator, Rap1 (28), and may be targeted by LFA-1 signaling through JAB1 (29), was expressed at normal levels in Lfa-1 d/d cells. Activation of purified T cells by plate-bound CD3 antibody also was normal, and thereby confirmed that Lfa-1 d/d T cells do not exhibit an intrinsic defect to be activated through the antigen receptor.
In summary, we have generated a mutant mouse strain that shows expression of constitutively active integrin LFA-1, and thereby disables complete deactivation of LFA-1. The results demonstrate that deactivation of LFA-1 and resolution of LFA-1–mediated cell adhesion are vital for the generation of normal immune responses in vitro and in vivo. Therefore, the present study supports the concept that dynamic regulation of ligand-binding capacity is a key element of integrin function.
MATERIALS AND METHODS
Antibodies and reagents.
Antibodies against the surface markers CD3ɛ (145-2C11), CD4 (GK1.5), CD8 (53–6.7), CD11a (2D7), CD11b (M1/70), CD11c (HL3), CD18 (C61/17), CD25 (3C7), CD29 (HMβ1-1), CD49b (Ha1/29), CD49d (R1-2), CD62 (MEL-14), CD69 (H1.2F3), B220 (RA3-6B2), Gr1 (RB6-8C5), T cell receptor Vβ8 (F23.1), and annexin V were from BD Biosciences. Antibodies against p27Kip1, laminA/C, (p)ERK, and (p)JNK were from Cell Signaling. Thioglycollate was from Difco Labs and SEB was from Toxin Technology. TNP-CGG and TNP-BSA were from Biosearch Technologies, and alum was obtained from Serva. ConA, PMA, and ionomycin were purchased from Sigma-Aldrich.
Gene targeting and mice.
A murine genomic ES-129 BAC library (Genome Systems, Inc.) was screened by hybridization with a 225-bp murine Lfa-1 cDNA fragment encompassing exon 31 as a probe. BAC fragments were cloned into pBluescript (Stratagene) and fully sequenced. The targeting vector was constructed in pBluescript such that the specific mutation and an additional HpaI-cut were introduced into the short arm. The long arm was flanked by a neomycin resistance cassette and a HSV–thymidine kinase cassette. E14.1 embryonic stem cells were electroporated with the ClaI-linearized targeting vector, and the transfected cells were subjected to G418 and gancyclovir selection. Homologous recombinated clones were detected by genomic PCR and confirmed by Southern blot hybridization after digestion of embryonic stem cell DNA with HpaI. Single integration was verified by probing the Southern blot with the neomycin resistance cassette. Germline transmission of the targeted allele was confirmed by Southern blot analysis and the neogene was removed from germline by crossing mutant mice with the cre deleter strain (41).
The mice used in these experiments were homozygous for the mutation and had lost the neocassette. Wild-type littermates were used as controls. Genotyping for the lfa-1 d/d mutation was performed by PCR (forward: 5′-AGCCCTGGCTATCCTAGACTC-3′; reverse: 5′-GGTCCTGAGATGGCAGTTTCTCCG-3′) followed by HpaI digestion of the PCR product. Mice were kept according to national guidelines for animal care in a specific pathogen-free animal facility. Animal studies were approved by the Regierung von Oberbayern.
Adhesion assays.
ICAM-1(D1-2)-Fc and VCAM-1-Fc fusion proteins containing the human Fcγ1 fragment were purified from supernatants of transfected 293T using protein A–sepharose columns. For cell adhesion assay, 96-well plates were coated with 1.5 μg/well purified ICAM-1(D1-2)-Fc, VCAM-1-Fc (0.8 μg/well), or Fcγ1 fragments (1.0 μg/well) overnight at 4°C. The plates were washed with HBSS and blocked with 1% BSA for 1 h at 37°C. Cells were labeled for 30 min at 37°C with 6 μg/ml H33342 dye (Calbiochem) in HBSS and washed twice with HBSS. The cells (2 × 106 cells/ml) were resuspended in HBSS and preincubated with 1 mM Mn2+, 20 ng/ml PMA, or a combination of both for 10 min at 37°C. Subsequently, cells were allowed to adhere for 30 min at 37°C, and nonadherent cells were removed by inverse centrifugation for 10 min at 50 g. Adhesion assays were quantified by fluorimetry using a Biolumin 960 (Molecular Dynamics). For all experiments, nonspecific adhesion to Fcγ1 fragments was subtracted from adhesion to ICAM-1(D1-2)-Fc or VCAM-1-Fc to calculate the fraction of cells adhering specifically to integrin ligands.
To stimulate T cell adhesion through the antigen receptor pathway, lymph node T cells were stimulated with PMA (10 ng/ml) and ionomycin (1 μg/ml) for 2 d. After washing to remove PMA and ionomycin, T cells were maintained in IL-2 (20 ng/ml) for 2 d. T cell adhesion was stimulated by addition of CD3 antibodies (2 μg/ml) for the indicated time periods.
Adhesion assays with bEnd.5 endothelial cells were performed using 16-well glass chamber slides (Nunc). Chamber slides were coated with 2.5 μg/well fibronectin (Roche Diagnostics) and cultured with endothelial cells (2 × 105/well) for 2 d. Depending on the experiment, endothelial cells were incubated with 5 nM TNF per well for the last 16 h of culture. After removal of the cytokine, endothelial cells were coincubated with 5 × 105 lymphocytes in 100 μl adhesion medium (DMEM containing 5% FCS and 25 mM Hepes) for 30 min on a rocking platform. Assays were washed twice in PBS and fixed in 2.5% glutaraldehyde in PBS at 4°C for 2 h. Bound cells per predefined field were determined by counting five fields per well. Assays were performed in triplicates for each value.
Videomicroscopy.
Concanavalin A (2.5 μg/ml, 16 h; Sigma-Aldrich)–activated splenic T cells were isolated on a Nycomed gradient and cultured for 24 h in 20 ng/ml rIL-2/10% FCS/RPMI 1640 before use. For videomicroscopy, 35-mm glass bottom microwell dishes (MatTek Corp) were coated overnight with 50 μg/ml murine ICAM-1-Fc, then blocked with 2% BSA in PBS. T cells (4 × 105/ml in HBSS buffer plus 6 mM MgCl2) were added and allowed to settle for 10 min at 37°C; nonattached cells were removed by gentle washing. Images were taken using a Nikon Diaphot 300 microscope, 40× lens, and AQM2001 Kinetic Acquisition Manager software (Kinetic Imaging Ltd.). Cells were tracked at 5-s intervals using Motion Analysis software (Kinetic Imaging Ltd.) and the data were analyzed using a Mathematica notebook (Wolfram Research Inc.) developed by Daniel Zicha (Cancer Research UK, London. England, UK). Migration speeds were expressed as mean velocity over 10 min ± SEM. Statistical analysis was performed using the ANOVA test.
Endothelial transmigration assay.
Endothelial transmigration assays were performed as described previously (42) using 6.5-mm transwells (Corning) with 5 μm pore size. Inserts were coated with 50 μg/ml laminin (Roche Diagnostics). bEnd5 endothelioma cells (5 × 104) were seeded on the top of each porous filter and cultured for 2 d. Depending on the experiment, the cells were stimulated with 5 nM TNF per well for the last 16 h of culture. On day 3, 106 lymphocytes were added on the top of each filter and the assays were run for 4 h at 37°C and 7% CO2. Migrated lymphocytes were collected for cell counting using CASY (Schärfe System). Confluency of the endothelial monolayer was confirmed by Giemsa staining of inserts. Assays were performed as triplicates for each value.
Lymphocyte proliferation and allogeneic mixed lymphocyte reaction.
Splenocytes were incubated at 2 × 105 cells per well in 96-well round-bottom plates with soluble SEB in 200 μl media. Alternatively, B cell–depleted splenocytes were incubated in wells that were coated with anti-CD3 antibody. To measure cell proliferation, 1 μCi [3H]-thymidine (Amersham Biosciences) was added after 48 h of culture and incubated for 16 h.
For induction of mixed lymphocyte reactions, allogeneic splenocytes (H-2d) were used as stimulator cells and cocultured with responder cells at concentrations of 4 × 106 cells/ml. The stimulator population was irradiated with 30 Gy before coculture. Proliferation was measured after 88 h by addition of 1 μCi/well 3[H]-thymidine (Amersham Biosciences) during the last 16 h of culture.
Cytotoxic lymphocyte assay.
Mice were immunized s.c. into the hind footpad with ovalbumin (150 μg/footpad; OVA grade IV, Sigma-Aldrich) using CpG-DNA 1668 (5 nmol/footpad) as an adjuvant. Draining lymph nodes were removed 4 d later and cells were cultured for an additional 4 d in medium supplemented with 10 U/ml rIL-2, which allows expansion of in vivo primed CTL precursors. 51Cr-release assays were performed with EL4 target cells pulsed with 0.1 μM of the OVA-derived peptide, SIINFEKL. Serial dilutions of effector CTL were added to target cells and 51Cr-release was measured after 4 h of coculture. Specific lysis was calculated according to the formula: %specific lysis = [cpm (sample) − cpm (spontaneous release)/cpm (maximal release) − cpm (spontaneous release)] × 100. For control, unpulsed EL4 cells were used as targets. Specific lysis of unpulsed EL4 cells was <2% in all experiments.
Ex vivo tetramer staining of CD8 T cells.
To investigate generation of cytotoxic T cells, mice were immunized with OVA and CpG-DNA 1668 as described above. 9 d later, draining lymph nodes were analyzed for presence of SIINFEKL tetramer binding, and thus, peptide-specific CD8-positive, CD62L low T cells as described (43). In brief, cells were stained with anti-CD8 APC (clone CD8α; Caltag), anti-CD62L FITC (clone MEL-14), and MHC SIINFEKL tetramer PE (H-2Kb/SIINFEKL [257–264/murine β2-microglobulin streptavidin-PE]) for 1 h at 4°C. Additionally, an Fc block (CD16/CD32, 2.4G2; BD Biosciences) was used to avoid unspecific antibody binding. For life/death cell discrimination, cells were incubated with ethidium monoazide bromid (Molecular Probes).
Thioglycollate-induced peritonitis.
Mice were injected i.p. with 0.8 ml thioglycollate (Difco Labs; 40.5 mg/ml). After 0 h, 6 h, and 12 h, peritoneal lavage cells were collected and neutrophils were identified by flow cytometry analysis with Gr-1 and Mac-1 antibodies.
Humoral immune responses.
Mice were immunized i.p. with 5 μg TNP-CGG (NP19-CGG) adsorbed to alum. Sera were collected before and 21 d after immunization. Total NP-specific immunoglobulin isotypes (IgM, IgG1, and IgG2a) were detected using an ELISA with NP17-BSA–conjugated plates (10 μg/ml in carbonate-buffer, pH 9.5). Sera were diluted 1:500. Murine NP-specific antibodies were detected with alkaline phosphatase–conjugated isotype-specific detection antibodies that were obtained from BD Biosciences (anti-IgG1, anti-IgG2a) or Jackson ImmunoResearch Laboratories (anti-IgM).
Online supplemental materials.
Activated wild-type (Video 1) or LFA-1d/d (Video 2) splenic T cells were plated on immobilized ICAM-1-Fc (50 μg/ml) in the presence of 6 mM Mg2+. A phase image was taken every 10 s. Thus, 1 s of film equals 1 min real time. Images were taken with a 40× lens and AQM2001 Kinetic Acquisition Manager software (Kinetic Imaging Ltd.). Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041850/DC1.
Acknowledgments
This work was supported by the SFB576, as well as grants HO 1015/6-1 and 6-2 (to B. Holzmann) and PF 259/2-6 (to K. Pfeffer) of the Deutsche Forschungsgemeinschaft, and a grant from the Kommission für Klinische Forschung, Klinikum rechts der Isar (to B. Holzmann) and the Cancer Research UK (to A. Smith and N. Hogg).
The authors have no conflicting financial interests.
Abbreviations used: ICAM, intercellular adhesion molecule; LFA-1, lymphocyte function- associated antigen-1; SEB, Staphylococcal enterotoxin B; VCAM, vascular cell adhesion molecule.
References
- 1.Hynes, R.O. 2002. Integrins: bidirectional allosteric signaling machines. Cell. 110:673–687. [DOI] [PubMed] [Google Scholar]
- 2.Bouvard, D., C. Brakebusch, E. Gustafsson, A. Aszódi, T. Bengtsson, A. Berna, and R. Fässler. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89:211–223. [DOI] [PubMed] [Google Scholar]
- 3.Hogg, N., M. Laschinger, K. Giles, and A. McDowall. 2003. T-cell integrins: more than just sticking points. J. Cell Sci. 116:4695–4705. [DOI] [PubMed] [Google Scholar]
- 4.Bachmann, M.F., K. McKall-Faienza, R. Schmits, D. Bouchard, J. Beach, D.E. Speiser, T.W. Mak, and P.S. Ohashi. 1997. Distinct roles for LFA-1 and CD28 during activation of naive T cells: adhesion versus costimulation. Immunity. 7:549–557. [DOI] [PubMed] [Google Scholar]
- 5.Perez, O.D., D. Mitchell, G.C. Jager, S. South, C. Murriel, J. McBride, L.A. Herzenberg, S. Kinoshita, and G.P. Nolan. 2003. Leukocyte functional antigen 1 lowers T cell activation thresholds and signaling through cytohesin-1 and Jun-activating binding protein 1. Nat. Immunol. 4:1083–1092. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi, E., S. Denti, A. Granata, G. Bossi, J. Geginat, A. Villa, L. Rogge, and R. Pardi. 2000. Integrin LFA-1 interacts with the transcriptional co-activator JAB1 to modulate AP-1 activity. Nature. 404:617–621. [DOI] [PubMed] [Google Scholar]
- 7.Schmits, R., T.M. Kündig, D.M. Baker, G. Shumaker, J.J.L. Simard, G. Duncan, A. Wakeham, A. Shahinian, A. van der Heiden, M.F. Bachmann, et al. 1996. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J. Exp. Med. 183:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrew, D.P., J.P. Spellberg, H. Takimoto, R. Schmits, T.W. Mak, and M.M. Zukowski. 1998. Transendothelial migration and trafficking of leukocytes in LFA-1-deficient mice. Eur. J. Immunol. 28:1959–1969. [DOI] [PubMed] [Google Scholar]
- 9.Berlin-Rufenach, C., F. Otto, M. Mathies, J. Westermann, M.J. Owen, A. Hamann, and N. Hogg. 1999. Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J. Exp. Med. 189:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamann, A., D. Jablonski Westrich, A. Duijvestijn, E.C. Butcher, H. Baisch, R. Harder, and H.G. Thiele. 1988. Evidence for an accessory role of LFA-1 in lymphocyte-high endothelium interaction during homing. J. Immunol. 140:693–699. [PubMed] [Google Scholar]
- 11.Dustin, M.L., T.G. Bivona, and M.R. Philips. 2004. Membranes as messengers in T cell adhesion signaling. Nat. Immunol. 5:363–372. [DOI] [PubMed] [Google Scholar]
- 12.Hogg, N., R. Henderson, B. Leitinger, A. McDowall, J. Porter, and P. Stanley. 2002. Mechanisms contributing to the activity of integrins on leukocytes. Immunol. Rev. 186:164–171. [DOI] [PubMed] [Google Scholar]
- 13.Takagi, J., and T.A. Springer. 2002. Integrin activation and structural rearrangement. Immunol. Rev. 186:141–163. [DOI] [PubMed] [Google Scholar]
- 14.Katagiri, K., M. Hattori, N. Minato, S.-K. Irie, K. Takatsu, and T. Kinashi. 2000. Rap1 is potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol. Cell. Biol. 20:1956–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Constantin, G., M. Majeed, C. Giagulli, L. Piccio, J.Y. Kim, E.C. Butcher, and C. Laudanna. 2000. Chemokines trigger immediate β2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 13:759–769. [DOI] [PubMed] [Google Scholar]
- 16.Giagulli, C., E. Scarpini, L. Ottoboni, S. Narumiya, E.C. Butcher, G. Constantin, and C. Laudanna. 2004. RhoA and ζPKC control distinct modalities of LFA-1 activation by chemokines: critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity. 20:25–35. [DOI] [PubMed] [Google Scholar]
- 17.Weber, K.S.C., C. Weber, G. Ostermann, H. Dierks, W. Nagel, and W. Kolanus. 2001. Cytohesin-1 is a dynamic regulator of distinct LFA-1 functions in leukocyte arrest and transmigration triggered by chemokines. Curr. Biol. 11:1969–1974. [DOI] [PubMed] [Google Scholar]
- 18.Laudanna, C., J.J. Campbell, and E.C. Butcher. 1996. Role of Rho in chemoattractant-activated leukocyte adhesion through integrins. Science. 271:981–983. [DOI] [PubMed] [Google Scholar]
- 19.Griffiths, E.K., C. Krawczyk, Y.-Y. Kong, M. Raab, S.J. Hyduk, D. Bouchard, V.S. Chan, I. Kozieradzki, A.J. Oliveira-dos-Santos, A. Wakeham, et al. 2001. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science. 293:2260–2263. [DOI] [PubMed] [Google Scholar]
- 20.Peterson, E.J., M.L. Woods, S.A. Dmowski, G. Derimanov, M.S. Jordan, J.N. Wu, P.S. Myung, Q.-H. Liu, J.T. Pribila, B.D. Freedman, et al. 2001. Coupling of the TCR to integrin activation by SLAP-130/Fyb. Science. 293:2263–2265. [DOI] [PubMed] [Google Scholar]
- 21.Kim, M., C.V. Carman, and T.A. Springer. 2003. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 301:1720–1725. [DOI] [PubMed] [Google Scholar]
- 22.Lu, C., J. Takagi, and T.A. Springer. 2001. Association of the membrane proximal regions of the α and β subunit cytoplasmic domains contains an integrin in the inactive state. J. Biol. Chem. 276:14642–14648. [DOI] [PubMed] [Google Scholar]
- 23.Lu, C.-F., and T.A. Springer. 1997. The α subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J. Immunol. 159:268–278. [PubMed] [Google Scholar]
- 24.Diamond, M.S., and T.A. Springer. 1993. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120:545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katagiri, K., M. Shimonaka, and T. Kinashi. 2004. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-γ1. J. Biol. Chem. 279:11875–11881. [DOI] [PubMed] [Google Scholar]
- 26.Shimonaka, M., K. Katagiri, T. Nakayama, N. Fujita, T. Tsuruo, O. Yoshie, and T. Kinashi. 2003. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J. Cell Biol. 161:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tohyama, Y., K. Katagiri, R. Pardi, C. Lu, T.A. Springer, and T. Kinashi. 2003. The critical cytoplasmic regions of the αL/β2 integrin in Rap1-induced adhesion and migration. Mol. Biol. Cell. 14:2570–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katagiri, K., M. Hattori, N. Minato, and T. Kinashi. 2004. Rap1 functions as a key regulator of T-cell and antigen-presenting cell interactions and modulates T-cell responses. Mol. Cell. Biol. 22:1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomoda, K., Y. Kubota, Y. Arata, S. Mori, M. Maeda, T. Tanaka, M. Yoshida, N. Yoneda-Kato, and J. Kato. 2002. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J. Cell Biol. 277:2302–2310. [DOI] [PubMed] [Google Scholar]
- 30.Sparwasser, T., R.M. Vabulas, B. Villmow, G.B. Lipford, and H. Wagner. 2000. Bacterial CpG-DNA activates dendritic cells in vivo: T helper cell-independent cytotoxic T cell responses to soluble proteins. Eur. J. Immunol. 30:3591–3597. [DOI] [PubMed] [Google Scholar]
- 31.Cho, H.J., K. Takabayashi, P.M. Cheng, M.D. Nguyen, M. Corr, S. Tuck, and E. Raz. 2000. Immunostimulatory DNA-based vaccines induce cytotoxic lymphocyte activity by a T-helper cell-independent mechanism. Nat. Biotechnol. 18:509–514. [DOI] [PubMed] [Google Scholar]
- 32.Shimaoka, M., J. Takagi, and T.A. Springer. 2002. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31:485–516. [DOI] [PubMed] [Google Scholar]
- 33.Dransfield, I., C. Cabanas, J. Barrett, and N. Hogg. 1992. Interaction of leukocyte integrins with ligand is necessary but not sufficient for function. J. Cell Biol. 116:1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridley, A.J., M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T. Parsons, and A.R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- 35.Weber, C., C.-F. Lu, J.M. Casasnovas, and T.A. Springer. 1997. Role of αLβ2 integrin avidity in transendothelial chemotaxis of mononuclear cells. J. Immunol. 159:3968–3975. [PubMed] [Google Scholar]
- 36.Mempel, T.R., S.E. Henrickson, and U.H. von Andrian. 2004. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 427:154–159. [DOI] [PubMed] [Google Scholar]
- 37.Miller, M.J., S.H. Wei, I. Parker, and M.D. Cahalan. 2002. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 296:1869–1873. [DOI] [PubMed] [Google Scholar]
- 38.Stoll, S., J. Delon, T.M. Brotz, and R.N. Germain. 2002. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 296:1873–1876. [DOI] [PubMed] [Google Scholar]
- 39.Garside, P., E. Ingulli, R.R. Merica, J.G. Johnson, R.J. Noelle, and M.K. Jenkins. 1998. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 281:96–99. [DOI] [PubMed] [Google Scholar]
- 40.Slifka, M.K., R. Antia, J.K. Whitmore, and R. Ahmed. 1998. Humoral immunity due to long-lived plasma cells. Immunity. 8:363–372. [DOI] [PubMed] [Google Scholar]
- 41.Schwenk, F., U. Baron, and K. Rajewsky. 1995. A _cre_-transgenic mouse strain for the ubiquitous deletion of _loxP_-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23:5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiss, Y., G. Hoch, U. Deutsch, and B. Engelhardt. 1998. T cell interaction with ICAM-1-deficient endothelium in vitro: essential role for ICAM-1 and ICAM-2 in transendothelial migration of T cells. Eur. J. Immunol. 28:3086–3099. [DOI] [PubMed] [Google Scholar]
- 43.Busch, D.H., I.M. Pilip, S. Vijh, and E.G.P. Am. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 8:353–362. [DOI] [PubMed] [Google Scholar]