Recruitment of latent pools of high-avidity CD8+ T cells to the antitumor immune response (original) (raw)
Abstract
A major barrier to successful antitumor vaccination is tolerance of high-avidity T cells specific to tumor antigens. In keeping with this notion, HER-2/neu (neu)-targeted vaccines, which raise strong CD8+ T cell responses to a dominant peptide (RNEU420-429) in WT FVB/N mice and protect them from a neu-expressing tumor challenge, fail to do so in MMTV-neu (_neu_-N) transgenic mice. However, treatment of _neu_-N mice with vaccine and cyclophosphamide-containing chemotherapy resulted in tumor protection in a proportion of mice. This effect was specifically abrogated by the transfer of _neu_-N–derived CD4+CD25+ T cells. RNEU420-429-specific CD8+ T cells were identified only in _neu_-N mice given vaccine and cyclophosphamide chemotherapy which rejected tumor challenge. Tetramer-binding studies demonstrated that cyclophosphamide pretreatment allowed the activation of high-avidity RNEU420-429-specific CD8+ T cells comparable to those generated from vaccinated FVB/N mice. Cyclophosphamide seemed to inhibit regulatory T (T reg) cells by selectively depleting the cycling population of CD4+CD25+ T cells in _neu_-N mice. These findings demonstrate that _neu_-N mice possess latent pools of high-avidity neu-specific CD8+ T cells that can be recruited to produce an effective antitumor response if T reg cells are blocked or removed by using approaches such as administration of cyclophosphamide before vaccination.
Central and peripheral mechanisms of T cell tolerance must exist to prevent autoimmunity (1–3). However, these same mechanisms are likely contributors to ineffective T cell responses often observed in cancer patients (4). CD8+ T cells specific for tumor antigens have been detected in patients. In particular, HER-2/neu (neu)-specific T cells have been isolated from patients with breast and ovarian cancer (5). In addition, p53-specific T cells have been isolated from patients with colorectal carcinomas, human papilloma virus–specific T cells have been isolated from patients with cervical cancer, and T cells specific for more than 10 different antigens have been isolated from patients with melanoma (6). Although these T cell responses are often observed in patients who have been treated with an antigen-specific vaccine, most are weak and ineffective in controlling tumor growth. In some instances, this ineffectiveness may result from an ineffective vaccine approach. However, because the majority of tumor-associated antigens are either overexpressed or reactivated developmental self-antigens, tolerance to specific tumor antigens is a likely mechanism for blunted T cell responsiveness in many cases (7, 8).
Intrathymic expression of self-antigens often leads to central deletion of T cells that express high-avidity TCR specific for these antigens. However, T cells that express lower-avidity TCR for these antigens can escape into the periphery (9). In addition, both high-avidity and low-avidity T cells specific for peripherally expressed self-antigens will leave the thymus rather than undergo deletion (10). Therefore, peripheral (extrathymic), nondeletional mechanisms exist that render T cells specific for self-tissues ignorant or functionally impaired (11–13). Understanding the mechanisms of peripheral tolerance in the context of tumor antigens is critical for the development of interventions that can reverse the tolerant state and allow these T cells to respond more effectively to tumors. In particular, it is critical to determine whether there are latent pools of tumor-specific T cells in tumor-tolerant individuals that are capable of being coaxed into a functionally active state.
Several murine models are available for dissecting mechanisms of peripheral tolerance (14). Extending this approach to a relevant tumor antigen, we recently described the existence of immune tolerance in the HER-2/neu transgenic (_neu_-N) mouse model of breast cancer. These mice overexpress the rat protooncogene neu and develop spontaneous mammary tumors between 4 and 8 mo of age (15). Similar to observations in patients with breast and ovarian cancers (5), _neu_-N mice exhibit down-regulated immune responses after neu-targeted vaccination. Specifically, _neu_-N mice develop poor antibody and T cell responses to neu regardless of the neu-targeted vaccination approach used when compared with parental FVB/N mice receiving the same vaccine (16). The presence of the transgene does not result in complete central deletion of neu-specific T cells in _neu_-N mice, because anti-neu activity is detected. However, the possibility remains that subpopulations of high-avidity neu-specific T cells are deleted centrally, whereas T cells with lower avidity can leave the thymus but are subject to peripheral mechanisms of tolerance. Alternatively, tolerization of all neu-specific T cells may take place completely in the periphery. Various models of antigen-specific tolerance support all of these hypotheses.
We previously reported that treatment of _neu_-N mice with immune modulating doses of cyclophosphamide-containing chemotherapy before vaccination resulted in tumor protection in 10–30% of mice (17). Here we show that the vaccine-enhancing effect of cyclophosphamide is mediated through selectively inhibiting the cycling population of CD4+CD25+ regulatory T (T reg) cells in _neu_-N mice. Furthermore, high-avidity RNEU420-429-specific CD8+ T cells are not deleted in the periphery, as previously thought. Instead, these high-avidity T cells can be recruited and activated to provide a potent antitumor immune response if T reg cells are inhibited.
Results
Adoptive transfer of CD4+CD25+ regulatory T cells inhibits the antitumor immune response induced by cyclophosphamide given with vaccine
We previously demonstrated that 10–30% of tolerized _neu_-N mice can be cured of neu-expressing tumors when treated with immune-modulating doses of cyclophosphamide chemotherapy in sequence with neu-targeted vaccination. In contrast, all _neu_-N mice given vaccine alone develop tumors (17). T reg cells probably play a role in suppressing antigen-specific T cell responses (3). It has been suggested for more than 20 yr, and addressed more recently, that immune-modulating doses of cyclophosphamide may function by inhibiting suppressor T cell activity (18–21). We therefore evaluated T reg cells as a mechanism by which vaccine-induced immunity is suppressed in _neu_-N mice. _Neu_-N mice were given vaccine, alone or with cyclophosphamide, and then tumor challenged 2 wk later. Pretreatment with cyclophosphamide significantly enhanced the vaccine effect (P = 0.001) compared with mice given vaccine alone (Fig. 1 A). To determine if cyclophosphamide pretreatment eliminates a suppressive T cell population, _neu_-N mice that were given cyclophosphamide and vaccine received adoptively transferred CD8+ T cells (either 106 or 5 × 106 total per mouse; Fig. 1 B), or CD4+ T cells (either 106 or 107 total per mouse; Fig. 1 C) 1 d before tumor challenge. Initial experiments used T cells isolated from the spleens of tumor-bearing donor _neu_-N mice vaccinated 14 d before adoptive transfer. Because no enrichment of the transferred cells based on antigen specificity could be performed, we reasoned that any suppressive population would be activated in the vaccinated tumor-bearing donors. Neither dose of CD8+ T cells abrogated the antitumor response in the cyclophosphamide- and vaccine-treated mice (Fig. 1 B). However, CD4+ T cells from donor _neu_-N mice significantly suppressed the antitumor responses induced by cyclophosphamide and vaccine (P = 0.005) when 107 cells were transferred (Fig. 1 C). In subsequent experiments, T cells isolated from naive _neu_-N mice were used for the adoptive transfer, yielding the same results (unpublished data). These data suggest that there is a subset of CD4+ T cells with T reg cell function that inhibit the activity of tumor-rejecting, neu-specific T cells. In a second set of studies, mice received CD4+CD25− T cells (107 total per mouse) or CD4+CD25+ T cells (106 total per mouse). The CD4+CD25− T cell population failed to inhibit cyclophosphamide-modulated antitumor immunity (P = 0.611), whereas the CD4+CD25+ T cell population completely abrogated the antitumor immune response even at a 10-fold-lower dose (P = 0.001; Fig. 1 D). The suppressive activity of the CD4+CD25+ T cell subset was confirmed by in vitro proliferation assay in coculture with CD4+CD25− T cells (22) and detection of foxp3 expression by RT-PCR (reference 23 and unpublished data).
Figure 1.

Adoptive transfer of CD4**+CD25+** T reg cells abrogates the immune modulatory effect of cyclophosphamide. 10 _neu_-N mice/group were vaccinated with either 3T3neuGM alone on day 0 or with cyclophosphamide on day −1 and 3T3neuGM on day 0. Adoptively transferred T cells were given on day 13, as described in Materials and methods. On day 14, all mice were challenged with NT2 tumor cells. Mice were monitored for tumor outgrowth two times/wk. (A) cyclophosphamide given before vaccine results in significant protection (P = 0.001) from NT tumor challenge. Adoptively transferred, purified CD8+ splenocytes (B) were not capable of suppressing the effect of cyclophosphamide, whereas CD4+ splenocytes (C) did significantly inhibit the immune modulatory effects of cyclophosphamide in a dose-dependent fashion (P = 0.005 for 107 CD4+). (D) The experiment was repeated using purified CD4+CD25− splenocytes and purified CD4+CD25+ splenocytes. A significant suppressive effect of the T cells localized to the CD4+CD25+ splenocytes (P = 0.001), and not to the CD4+CD25− splenocytes (P = 0.611). Each study was repeated at least twice with similar results. Cy, cyclophosphamide.
Vaccine combined with cyclophosphamide chemotherapy uncovers T cells specific for the immunodominant epitope RNEU420-429, directly correlating RNEU420-429 T cell activity with in vivo tumor rejection
In nontolerized FVB/N mice, neu-targeted vaccine will completely cure mice from a neu-expressing tumor challenge. In vivo rejection of tumor is associated with a neu-specific CD8+ T cell repertoire that is directed at the immunodominant T cell epitope RNEU420-429 (24). We have shown that the RNEU420-429-specific CD8+ T cells are capable of lysing neu-expressing tumors in vitro and eradicating neu-expressing tumors in FVB/N mice in vivo. However, activated RNEU420-429-specific T cells are rarely detected in vaccinated _neu_-N mice (24). Because cyclophosphamide chemotherapy combined with vaccine cures 10–30% of _neu_-N mice, we hypothesized that the curative effect was caused by the cyclophosphamide chemotherapy regimen activating RNEU420-429-specific T cells. Tumor-challenged _neu_-N mice were given vaccine with or without cyclophosphamide chemotherapy and followed for development of neu-expressing mammary tumors. As previously reported, cyclophosphamide chemotherapy resulted in a significant enhancement of the vaccine effect (P = 0.002), and 25% of mice receiving vaccine and cyclophosphamide chemotherapy remained tumor-free (Fig. 2 A; reference 17). These mice were killed on day 50, and intracellular cytokine staining (ICS) was performed to assess recognition of RNEU420-429 in both tumor-bearing and tumor-free mice. As shown in Fig. 2 B, the two tumor-free _neu_-N mice (mouse E and mouse F) showed the highest percentage of RNEU420-429-specific CD8+ T cells. The tumor-bearing mice (mice A–D) showed no activation above the background observed in the _neu_-N mice given vaccine alone. Thus, cyclophosphamide chemotherapy combined with vaccine seems to activate RNEU420-429-specific T cells in the _neu_-N mice, enabling them to reject the neu-expressing tumor challenge. Similar activation of RNEU420-429-specific T cells was observed in tumor-free _neu_-N mice given cyclophosphamide alone with vaccine (unpublished data).
Figure 2.

RNEU420-429-specific T cells can be identified in polyclonal T cell populations from _neu_-N mice given vaccine and cyclophosphamide chemotherapy that rejected neu-expressing tumors. (A) Eight _neu_-N mice/group were tumor challenged on day −3, followed on day 0 by vaccination with or without cyclophosphamide chemotherapy. Mice were monitored for tumor outgrowth two times/wk. All mice that received mock vaccine with or without cyclophosphamide chemotherapy developed tumor by day 35 (not depicted). (B) At the end of the experiment in (A), splenic T cells were isolated, and reactivity to RNEU420-429 was determined by ICS. Plotted is the percentage of CD8+ T cells that secreted IFN-γ in response to RNEU420-429 minus the percentage responding to NP118-126. This experiment was repeated at least three times with similar results. Cy, cyclophosphamide.
Because the presence of RNEU420-429-specific T cells could result simply from a boosting effect of having successfully rejected the neu-expressing tumor, we determined if RNEU420-429-specific T cells could be detected in _neu_-N mice given cyclophosphamide chemotherapy in the absence of a tumor challenge. RNEU420-429-specific CD8+ T cell responses were detected in 20–30% of these mice (unpublished data), correlating well with our previous data measuring the antitumor effect of vaccine and cyclophosphamide chemotherapy in tumor-bearing _neu_-N mice (17). Thus, T cells specific for the immunodominant epitope, RNEU420-429, can be detected after treatment with cyclophosphamide chemotherapy and vaccine in tumor naive _neu_-N mice.
Comparison of RNEU420-429-specific CD8+ T cell lines from vaccinated FVB/N and _neu_-N mice reveal differences in T cell avidity that may explain the enhanced therapeutic effect of cyclophosphamide chemotherapy and vaccine in the _neu_-N mice
CD8+ T cell lines were generated after neu-targeted vaccination in FVB/N and _neu_-N mice. All lines were generated by repeated in vitro stimulation with 3T3neuB7-1 target cells that contain the entire neu cDNA. Initial analysis of T cell lines derived from the vaccinated FVB/N and _neu_-N mice, as well as _neu_-N mice given vaccine and cyclophosphamide chemotherapy, revealed that all are specific for the RNEU420-429 peptide (Fig. 3 A). In contrast, these lines did not recognize the irrelevant H-2Dq-binding peptide NP118-126 (25). To characterize functional differences between the FVB/N-derived and the _neu_-N–derived T cell lines, a more rigorous analysis directly comparing the three T cell lines was performed. Each T cell line was evaluated for the ability to lyse NT2 mammary tumor cells, which express naturally processed neu peptides. The RNEU420-429-specific T cell line derived from vaccinated _neu_-N mice was less effective at lysing tumor cells than the lines derived from vaccinated FVB/N mice and from _neu_-N mice given vaccine and cyclophosphamide chemotherapy (Fig. 3 B). The T cell lines were all >99% CD8+ and expressed comparable surface levels of TCR, CD8α, and CD3 (Fig. 3 C). Because these lines are comparable in the degree of peptide specificity and TCR, CD8α, and CD3 levels, we hypothesized that the difference in tumor lysis could be caused by a difference in avidity of TCR for the H-2Dq/RNEU420-429 MHC/peptide complex.
Figure 3.

FVB/N and _neu_-N–derived CD8**+** T cell lines are specific for RNEU420-429 but differentially lyse neu-expressing mammary tumors despite expressing similar levels of cell surface markers. (A) T cell lines were derived from vaccinated FVB/N mice (FVB/N line), _neu_-N were mice given vaccine alone (_neu_-N line), or _neu_-N mice were given vaccine and cyclophosphamide chemotherapy that had rejected an NT tumor challenge (_neu_-N plus chemotherapy line). IFN-γ ICS was performed after T cells were stimulated overnight with equal numbers of T2Dq cells pulsed with either the irrelevant peptide NP118-126 (shaded histogram) or with RNEU420-429 (black line). (B) CTL assay using the three T cell lines and neu-expressing NT2 tumor targets. These experiments were repeated more than six times with similar results. (C) Cells were stained with antibodies to CD3, CD8α, TCR Vβ2, TCR Vβ4, and TCR Vβ6 as described in Materials and methods. Black line, FVB/N line; dashed line, _neu_-N line; gray line, _neu_-N plus chemotherapy line.
To address this issue, we stained the three T cell lines with decreasing concentrations of the H-2Dq-RNEU420-429 tetramer. This study revealed that the T cells derived from _neu_-N mice given vaccine alone stained with a much lower intensity than the FVB/N-derived T cells. T cells derived from _neu_-N mice given vaccine and cyclophosphamide chemotherapy showed a staining profile similar to the FVB/N-derived line (Fig. 4 A). Even a 10-fold dilution of the tetramer resulted in loss of staining of the _neu_-N T cell line, whereas the other two lines showed staining even at a 100-fold dilution of the tetramer. Further quantitation revealed that at least 16 μM of tetramer complex was required to stain the T cell line derived from vaccinated _neu_-N mice. In contrast, only 0.16 μM of tetramer was required to demonstrate similar staining of the FVB/N-derived T cell line and of the T cell line derived from _neu_-N mice given vaccine and cyclophosphamide chemotherapy. Additionally, binding kinetics studies were performed by binding tetramer to T cell lines and competing it off with the H-2Dq-specific antibody 30–5-7S (26). As shown in Table I, the dissociation rate constant (koff) for the _neu_-N line was roughly sixfold higher than that of the FVB/N line and the T cell line derived from _neu_-N mice given vaccine and cyclophosphamide chemotherapy. Other reports have indicated that the expression level of CD8β influences functional avidity (27). However, in our studies, no difference in CD8β staining was observed between the T cell lines from the _neu_-N mice given vaccine alone and the _neu_-N mice given vaccine and cyclophosphamide chemotherapy (Fig. 4 B). These data strongly support our hypothesis that the _neu-_N–derived T cell line has a lower avidity for the MHC class I/RNEU420-429 epitope complex than the FVB/N-derived T cell line or the T cell line derived from vaccine and cyclophosphamide chemotherapy treated _neu_-N mice. Importantly, low avidity correlated with reduced lysis of the neu-expressing mammary tumor (Fig. 3 B).
Figure 4.

RNEU420-429–specific CD8**+** T cell lines derived from FVB/N and _neu_-N mice demonstrate quantitatively different avidities for the H-2Dq/RNEU420-429 MHC/peptide complex but no difference in CD8β staining. (A) The three T cell lines were stained with decreasing amounts of H-2Dq/ RNEU420-429 tetramer. Shown is the tetramer staining of gated CD8+ T cells. Black solid line, 1:5 tetramer dilution; dashed line, 1:50 tetramer dilution; gray line, 1:500 tetramer dilution; shaded, no tetramer. (B) T cell lines from _neu_-N mice given vaccine alone (dashed line) and _neu_-N mice given cyclophosphamide chemotherapy (gray line) were stained for CD8β on day 6 after stimulation.
Table I.
koff for RNEU420-429-specific T cell linesa
| T cell line | koff | SD |
|---|---|---|
| (min−1) | ||
| FVB/N | 41.5 × 10−3 | ±10.1 × 10−3 |
| _neu_-N | 272.7 × 10−3 | ±164.1 × 10−3 |
| _neu_-N + chemotherapy | 17.5 × 10−3 | ±3.9 × 10−3 |
High-avidity RNEU420-429-specific T cells are detected in a polyclonal population of splenic T cells isolated from _neu_-N mice given vaccine and cyclophosphamide chemotherapy
The data analyzing T cell lines suggest that treatment with immune-modulatory doses of cyclophosphamide chemotherapy before vaccination results in the recruitment of functional high-avidity neu-specific T cells in up to 30% of _neu_-N mice. The studies analyzing splenic T cells from _neu_-N mice demonstrate that RNEU420-429-specific T cells can be isolated from a polyclonal T cell response after vaccine combined with cyclophosphamide chemotherapy (Fig. 2 B). To demonstrate that these RNEU420-429-specific T cells are high avidity, 42 additional tumor-challenged _neu_-N mice were vaccinated in sequence with cyclophosphamide chemotherapy and monitored for tumor development. As expected, 20% of the mice remained tumor-free (unpublished data). On day 56, splenocytes were isolated from tumor-bearing and tumor-free mice, and reactivity to RNEU420-429 was assessed by ICS after one in vitro stimulation (unpublished data). TCR avidity was determined by dilutional tetramer staining of splenocytes from vaccinated FVB/N mice, tumor-free _neu_-N mice given vaccine and cyclophosphamide chemotherapy, and tumor-bearing _neu_-N mice given vaccine alone (included as a negative control to show background tetramer staining because no reactivity to RNEU420-429 was observed by ICS). RNEU420-429-specific T cells from the vaccinated FVB/N mice and cyclophosphamide chemotherapy–treated _neu_-N mice exhibited equivalent tetramer staining, comparable to the tetramer staining observed in the high-avidity T cell lines (Fig. 5). These staining studies were performed by first gating on CD8+ T cells that are CD62Llo, demonstrating that the tetramer-positive RNEU420-429-specific T cells are activated. We therefore conclude that the CD8+ T cell responses to neu in parental FVB/N mice, but not in tolerized _neu_-N mice, are dominated by high-avidity T cells specific for RNEU420-429. In addition, immune-modulating doses of cyclophosphamide chemotherapy given in sequence with neu-targeted vaccination can uncover activated, high-avidity T cells specific for the immunodominant neu epitope in _neu_-N tolerized mice.
Figure 5.

High-avidity, RNEU420-429-specific T cells can be identified in polyclonal T cell populations from _neu_-N mice given vaccine and cyclophosphamide chemotherapy that rejected neu-expressing tumors. CD8+ enriched splenocytes were stained with decreasing amounts of H-2Dq-RNEU420-429 tetramer. Three representative samples are shown. Plots on the left are gated on CD8+, CD62Llo lymphocytes and show the 1:50 dilution of tetramer. Corresponding plots on the right show histograms gating on the CD8+, tetramer+ cells stained with decreasing amounts of tetramer.
Cyclophosphamide exerts its effect predominantly on cycling CD4+CD25+ T cells
To understand the mechanism of action of cyclophosphamide on CD4+CD25+ T cells better, the total number of CD4+CD25+ T cells was monitored in cyclophosphamide-treated naive _neu_-N mice. A significant decrease (P ≤ 0.0001) in the total number of CD4+CD25+ T cells was observed in the LN 2 d after cyclophosphamide treatment (Fig. 6 A). This decrease was detected in the LNs at the time when T cell induction would be occurring in vaccinated mice. The percentage of CD4+ T cells that are CD25+ was also monitored for 2 wk in cyclophosphamide-treated _neu_-N mice. A significant drop (P = 0.0007) in the percent of CD4+CD25+ was also seen within 2 d after cyclophosphamide treatment, which then recovered over the next 2 wk (Fig. 6 B).
Figure 6.
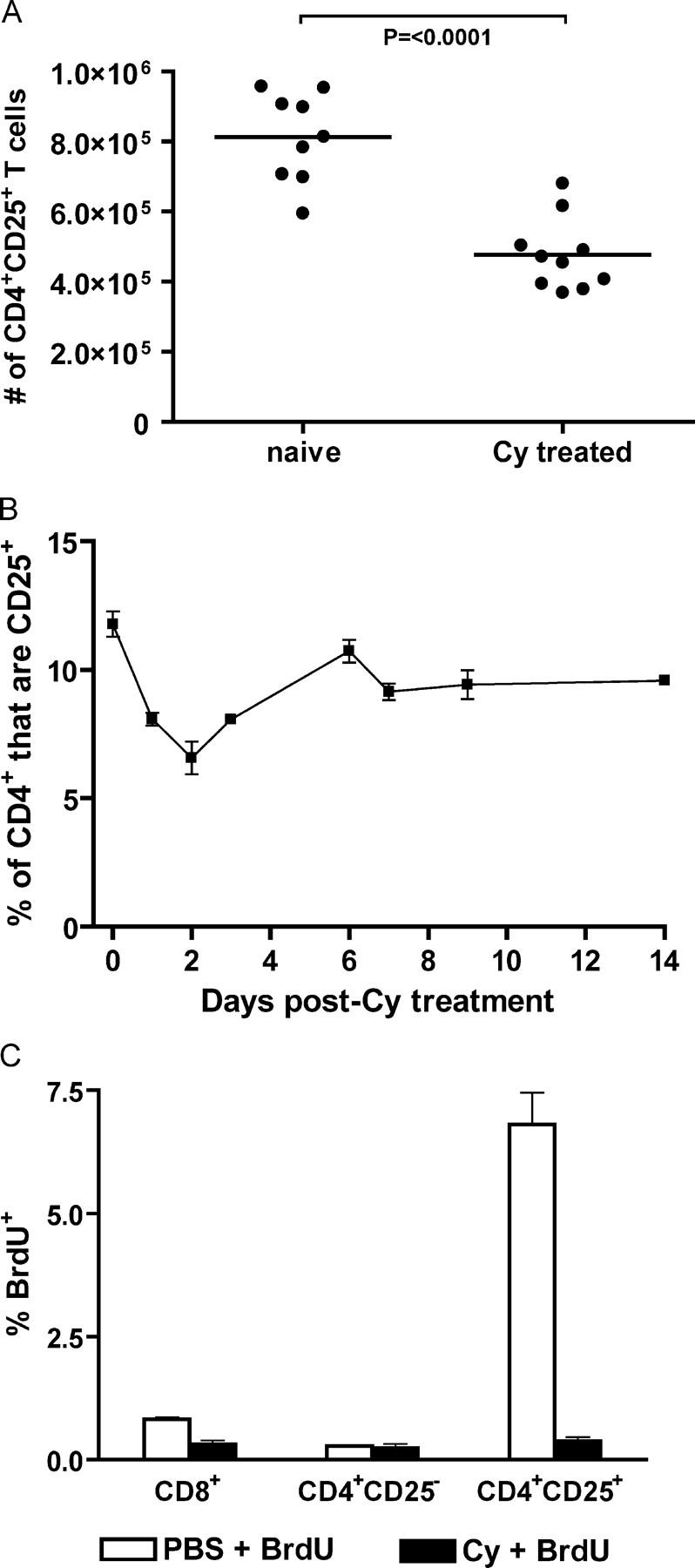
Cyclophosphamide depletes cycling CD4**+CD25+** T cells. (A) Axillary LNs from PBS- or cyclophosphamide-treated _neu_-N mice (10/group) were isolated 48 h after cyclophosphamide administration. Total cell number in each LN was determined, and the number of CD4**+CD25+** T cells was calculated based on the percent of CD4**+CD25+** T cells determined by flow cytometry. (B) LN cells from _neu_-N mice (4/group) given cyclophosphamide were isolated and analyzed for the number of CD4**+CD25+** T cells on the indicated days. The experiment was repeated three times. (C) _Neu_-N mice were given cyclophosphamide or PBS on day 0. On day 1, the mice were given a 2-mg dose of BrdU. On day 2, LNs were harvested, and the lymphocytes were stained for CD4, CD8, CD25, and BrdU. This experiment was repeated three times with similar results. Cy, cyclophosphamide.
Because cyclophosphamide has a relatively modest effect on total CD4+CD25+ T cell numbers, and the population of CD4+CD25+ T cells in whole LNs is polyclonal, it is possible that cyclophosphamide affects a specific subset of CD4+ CD25+ T cells. Others have shown that CD4+CD25+ T cells proliferate in the steady state in response to self-antigen (28). Thus, cyclophosphamide may act on the CD4+CD25+ T reg cell population that is proliferating to self-antigen in our model. To test this possibility, _neu_-N mice were given cyclophosphamide or PBS on day 0 and then pulsed with a 2-mg dose of bromodeoxyuridine (BrdU) on day 1. On day 2, LNs were harvested, and lymphocytes were stained for CD4, CD8, CD25, and BrdU. In the groups injected with PBS and BrdU, <1% of CD8+ T cells and <0.3% of CD4+CD25− T cells incorporated BrdU. In contrast, 6.8% of CD4+CD25+ T cells incorporated BrdU. When treated with cyclophosphamide, the percent of CD4+CD25+ T cells that incorporated BrdU was not above background (Fig. 6 C). Thus, cyclophosphamide seems to eliminate actively cycling CD4+CD25+ T cells. These data provide strong evidence that cyclophosphamide administration depletes cycling T reg cells, allowing the generation and activation of high-avidity RNEU420-429-specific T cells.
Direct depletion of CD4+CD25+ T cells with anti-CD25 antibody allows the detection of high-avidity RNEU420-429-specific T cells
Based on these studies, cyclophosphamide seems to act on T reg cells that suppress high-avidity RNEU420-429-specific T cell activity in _neu_-N mice. However, it is also possible that cyclophosphamide enhances high-avidity RNEU420-429-specific CD8+ T cells through a T reg cell–independent mechanism. To address this issue, T reg cells were targeted directly with the anti-CD25–depleting antibody, PC61, given 4 d before vaccination (29). We first confirmed that a single 1.0-mg dose of PC61 decreased the total number of CD4+CD25+ T cells from 12% of total CD4+ T cells to less than 3% in _neu_-N mice (Fig. 7 A). Importantly, direct depletion of CD4+CD25+ T cells with PC61 resulted in the ability to isolate RNEU420-429-specific T cells from 10–20% of mice (Fig. 7 B). This finding correlated with enhanced vaccine-induced prevention of tumor progression that was similar to observations with cyclophosphamide-modulated vaccine therapy (unpublished data). Furthermore, RNEU420-429-specific T cells isolated from mice depleted of CD4+CD25+ T cells with PC61 before vaccination were high avidity, because they could be detected with multiple dilutions of the H-2Dq-RNEU420-429 tetramer (Fig. 7 C).
Figure 7.
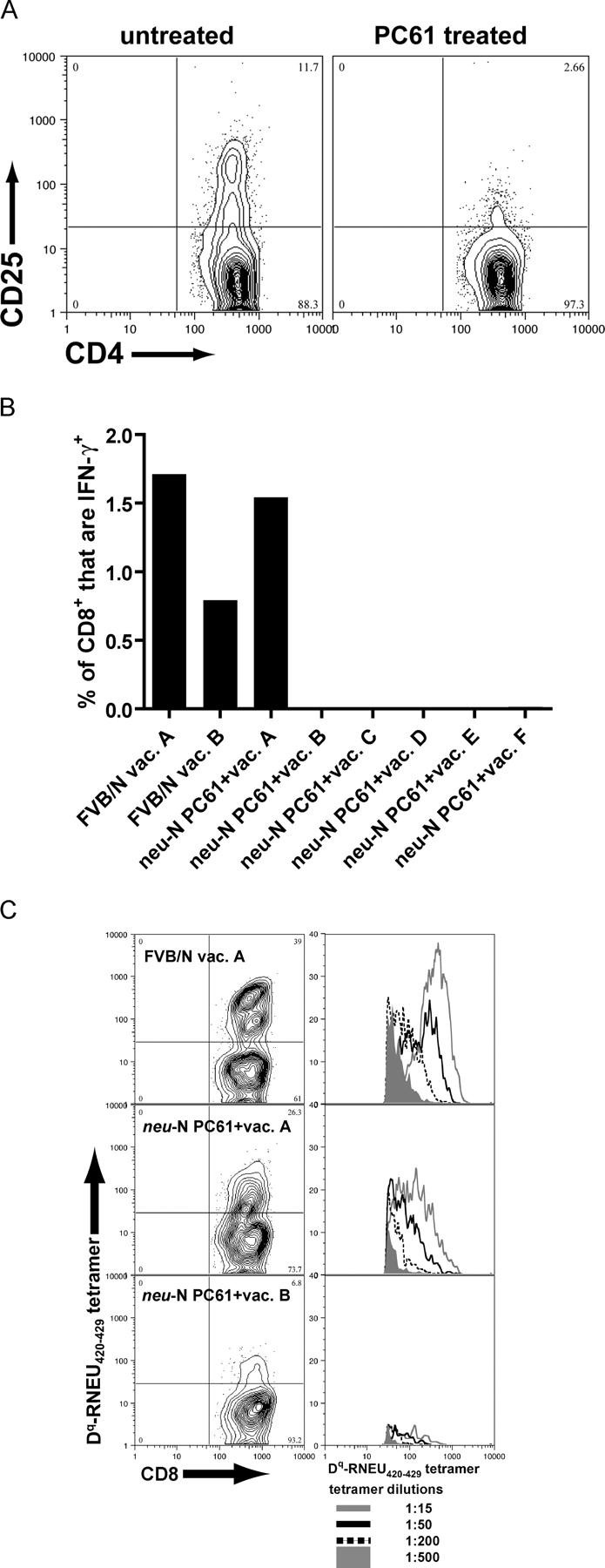
High-avidity RNEU420-429-specific CD8**+** T cells are detected after depletion of CD4**+CD25+** T reg cells with the CD25**+** T cell-depleting antibody, PC61. (A) Depletion was obtained using a single 1-mg dose of PC61 given 4 d before analysis. Splenocytes were isolated and stained with the noncompeting anti-CD25 antibody 7D4. (B) Mice were given 1 mg of PC61 4 d before vaccination. 2 wk after vaccination, splenocytes were isolated, and ICS was performed. Shown are the percent of CD8+ T cells that were RNEU420-429-specific. Similar results have been seen in more than six independent experiments. (C) Splenocytes from (B) were stimulated for 1 wk with RNEU420-429 and then stained with decreasing amounts of H-2Dq/RNEU420-429 tetramer. Three representative samples are shown. Plots on the left are gated on CD8+, CD62Llo lymphocytes and show the 1:50 dilution of tetramer. Corresponding plots on the right show histograms gating on the CD8+, tetramer+ cells stained with decreasing amounts of tetramer.
CD4+CD25+ T cells from _neu_-N mice suppress vaccine-induced RNEU420-429-specific CD8+ T cells in vaccinated, nontolerized FVB/N mice
Additional studies were performed to confirm further that CD4+CD25+ T reg cells directly suppress RNEU420-429-specific CD8+ T cell responses. Specifically, groups of parental nontolerized FVB/N mice were vaccinated either alone or 1 d after receiving 5 × 105 adoptively transferred CD4+ CD25+ T cells. Because the vaccine is extremely immunogenic in the nontolerant FVB/N mice, the CD4+CD25+ T cells were isolated from tumor-bearing, vaccinated _neu_-N mice with the goal of transferring an activated T reg cell population. After 2 wk, mice receiving vaccine alone demonstrated RNEU420-429-specific CD8+ T cells ranging from 1–3%. However, mice receiving adoptively transferred CD4+ CD25+ T cells demonstrated a significant (P = 0.016) decrease in RNEU420-429-specific CD8+ T cell responses ranging from 0.25–1% (Fig. 8). These data provide additional evidence that CD4+CD25+ T reg cells have a direct suppressing effect on RNEU420-429-specific CD8+ T cell responses. Despite the decrease in the percent of RNEU420-429-specific T cells, the T cells that were activated seemed to be functional, because no suppression of the antitumor response was observed when the FVB/N mice that received the T reg cell transfer were given a neu-expressing tumor challenge (unpublished data).
Figure 8.

Adoptive transfer of CD4**+CD25+** T cells from vaccinated, tumor-bearing _neu_-N mice suppresses the activation of RNEU420-429-specific CD8**+** T cells in FVB/N mice. CD4+CD25+ T cells from vaccinated, tumor-bearing _neu_-N mice were isolated by cell sorting. 5 × 105 CD4+CD25+ T reg cells were transferred into naive FVB/N mice that were vaccinated the following day. On day 14 after vaccination, splenic CD8+ T cells were isolated, and ICS was performed. Shown are the percent of total CD8+ T cells that produced IFN-γ in response to RNEU420-429. This study was repeated twice with similar results. •, individual mice; solid line, average.
Discussion
We previously identified RNEU420-429 as the immunodominant epitope encoded by the rat neu gene and recognized by the majority of T cell lines and clones derived from vaccinated FVB/N mice. We now report three new findings that give insight into the mechanisms of CD8+ T cell tolerance in _neu_-N transgenic mice. First, treatment of _neu_-N mice with immunomodulating doses of chemotherapy in sequence with neu-targeted vaccination uncovers high-avidity RNEU420-429-specific CD8+ T cell activity that is associated with more effective eradication of neu-expressing tumors in vivo. Second, one mechanism by which cyclophosphamide chemotherapy enhances vaccine-induced RNEU420-429-specific T cells is through deletion of cycling CD4+CD25+ T reg cells. Third, unlike FVB/N mice, neu-targeted vaccine given alone to _neu_-N mice induces lower-avidity RNEU420-429-specific T cell responses.
To our knowledge, this is the first report demonstrating the unmasking of high-avidity CD8+ T cell responses against a naturally expressed tissue-specific tumor antigen in a murine model of tolerance. Others have reported the induction of low-avidity antigen-specific T cells upon vaccination of transgenic mice for other model tumor antigens as compared with nontransgenic mice (8, 30, 31). In some cases, T cell avidity can be improved by repeated in vitro stimulation of T cells (11) or repeated antigen exposure in vivo (32). However, the initial generation of high-avidity T cells after in vivo vaccination regimens has not been previously demonstrated.
In several models of tumor tolerance, low-avidity T cells induced to recognize self-antigens were sufficient to suppress growth of tumors expressing the antigen (8). However, most other studies have shown that high-avidity T cells are superior antigen-recognition and lytic agents than their low-avidity counterparts (33–36). In one study, low-avidity CTL generated against the melanoma antigen gp100 were capable of lysing only peptide-pulsed cells, whereas high-avidity CTL efficiently lysed endogenously expressed levels of peptide presented by gp100-expressing tumor cells (37). Similarly, high-avidity CTL isolated from B16–GM-CSF–vaccinated mice showed superior in vitro and in vivo antitumor activity when compared with lower-avidity CTL (38). In our mammary tumor model, the development of high-avidity T cells is associated with protection against tumor outgrowth and an apparent abatement of the tolerance exhibited in _neu_-N mice. We have shown that high-avidity neu-specific T cells can be recovered in 10–30% of _neu_-N mice treated with immune-modulating doses of chemotherapy. In addition to the T cell line shown in this paper, we have isolated other high-avidity RNEU420-429-specific T cell lines from _neu-_N mice that have eradicated established transplanted mammary tumors after vaccination and cyclophosphamide chemotherapy. It is not possible, however, to isolate high-avidity RNEU420-429-specific T cells from tumor-bearing _neu_-N mice given vaccine alone. These data provide additional evidence that optimal antitumor immunization will depend in part on the ability to induce high-avidity T cells specific for immunodominant epitopes contained in tumor antigens.
Our data also illustrate the importance of using the optimal vaccine strategy when targeting tumor antigens. We show that unless vaccine is combined with deletion or inhibition of T reg cells, the most potent CD8+ T cells will not be recruited to the antitumor immune response. Similar findings have been reported recently by Antony et al. (39) showing in an adoptive transfer model that optimal vaccine against melanoma antigens could be achieved only when T reg cells were removed. Other recent reports suggest that the vaccine itself can overcome the influence of T reg cells if signaling through Toll-like receptors is involved (e.g., a vaccinia-based vaccine) (40). However, our experience in the _neu_-N mice suggests otherwise. Our early reports studying the neu-specific immune responses in the _neu_-N mice showed no difference between our GM-CSF–secreting vaccine and a neu-expressing vaccinia vaccine (16). Furthermore, the high potency of our GM-CSF vaccine approach is supported by our earlier report that this vaccine given as a single agent results in the regression of 40-mm2 tumors in the FVB/N nontolerized mice (17).
The studies presented here focused on the mechanism of cyclophosphamide in potentiating the immune response in vaccinated _neu_-N mice. We are currently investigating the mechanism of doxorubicin (Dox) in the combined chemotherapy regimen. However, our previous data do not support a role for Dox in eliminating T reg cell activity, because Dox inhibits vaccine-induced immune responses when given at the time of T cell priming (17). Dox enhances the effect of vaccine only when given at the time of T cell expansion, probably by enhancing the T cell's cytolytic activity (41). Thus, the ability to uncover high-avidity RNEU420-429-specific CD8+ T cells in the _neu_-N mice is not dependent on Dox, because similar results were seen in mice given cyclophosphamide and Dox, cyclophosphamide alone, or PC61 in combination with vaccine.
In this study avidity is defined in part as the degree to which a T cell binds MHC/peptide tetramer. Although numerous studies have shown a correlation between tetramer binding and T cell function (36, 42, 43), others have not (34, 43, 44). Here, the T cell lines that showed high-intensity tetramer staining were able to lyse neu-expressing tumor cells to a greater degree than the T cell line that displayed poor tetramer binding. Many investigations have also measured avidity of T cells by their ability to lyse peptide-pulsed cells (7, 8, 30, 37, 45). We confirmed these differences in T cell avidity for the H-2Dq/RNEU420-429 MHC/peptide complex by determining TCR koff rates for each T cell line. The T cell line that showed the lowest amount of surface tetramer binding had the fastest koff rate of the three lines analyzed. To the best of our knowledge, this is the first time the TCR koff rate has been quantitated for tolerized versus nontolerized T cells in a murine tumor model.
It is still unexplained why adding cyclophosphamide chemotherapy to the vaccine regimen consistently overcomes tolerance in, at most, 30% of treated mice. Also, each mouse seems to respond in an all-or-none fashion. It is possible that improving the success of cyclophosphamide modulation depends on the long-term depletion or inhibition of T reg cell function. We have found that the T reg cell population in _neu_-N mice recovers within 2 wk of cyclophosphamide treatment. The kinetics is similar to T reg cell recovery after depletion with the CD25-targeted antibody, PC61 (29). It is more difficult to study recurrent treatment with cyclophosphamide, because lymphopenia can abrogate the vaccine-induced antigen-specific T cell response. CD25-targeted depleting antibodies are also difficult to give after an initial dose, because they also target vaccine-induced, activated T cells that up-regulate CD25. Furthermore, other systemic and local mechanisms within the tumor's microenvironment are probably active and require modulation to allow the long-term tumor trafficking and survival of high-avidity antigen-specific T cells. Studies are underway to elucidate additional mechanisms of peripheral tolerance in the _neu_-N mice.
Based on the data described in this report, we propose the following as a model to explain one mechanism of CD8+ T cell tolerance in _neu_-N mice. CTL from vaccinated _neu_-N mice that are specific for the immunodominant epitope are rare and are of lower avidity than T cells derived from parental FVB/N mice. These low-avidity T cells are also either weak lytic agents or anergized T cells. However, higher-avidity T cells (similar in avidity to those derived from nontolerant FVB/N mice) specific for the immunodominant epitope can escape thymic deletion and are actively suppressed in the periphery of these mice. Our data strongly support CD4+ CD25+ T reg cell involvement as one mechanism for the induction of peripheral tolerance. Central tolerance is unlikely to be a major mechanism, because high-avidity CD8+ T cells specific for RNEU420-429 can be recovered from vaccinated _neu_-N mice treated with T reg cell-depleting agents such as cyclophosphamide or PC61. It is possible that central tolerance does play a role because of uptake of neu antigen in the periphery by immature dendritic cells when mature mice begin to overexpress neu in the periphery. Without the proper maturation signals, the outcome is likely to be cross-tolerance of CTL rather than cross-priming (46). However, our data demonstrate that _neu_-N mice develop CD4+CD25+ T reg cells that actively suppress RNEU420-429-specific T cells (47). This is probably one mechanism of peripheral tolerance at work in our model, because treatment with cyclophosphamide or PC61, which have been reported to delete or inhibit T reg cells, reverses tolerance when combined with vaccine (18–21). This possibility is further supported by our data showing that cyclophosphamide selectively inhibits cycling CD4+CD25+ T reg cells in _neu_-N mice.
In summary, we have described one mechanism of CD8+ T cell tolerance to the protooncogene neu expressed in _neu_-N transgenic mice. Further investigation is needed to determine the additional mechanisms involved in the induction of neu-directed immune tolerance. However, our data strongly suggest that the most effective antitumor vaccine regimens must include a T reg cell-targeting agent to allow the most potent tumor-reactive T cells to be activated.
Materials and Methods
Mice.
All mice used were between 8 and 12 wk of age. FVB/N mice were purchased from the National Cancer Institute and Taconic Laboratories. _neu_-N mice (15), provided by William Muller, were bred to homozygosity as verified by Southern blot analysis (16) and bred and housed at Johns Hopkins University. All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine.
Cell lines and media.
The GM-CSF–secreting vaccine cell lines 3T3GM and 3T3neuGM (expressing neu) were generated and grown as previously described (24). The IT22, IT22neu, and T2Dq cell lines used for targets in T cell assays were also previously described (24). The NT2 and NT5 neu-expressing tumor lines were derived from spontaneously arising mammary tumors excised from _neu_-N mice. NT2 cells used in tumor-challenge experiments express stable neu and MHC class I. NT5 was retrovirally infected with the human B7-1 gene to generate NT5B7-1 cells as previously described (16, 17).
T cell lines were generated from FVB/N or _neu_-N mice given vaccine (3T3neuGM) or from _neu_-N mice that rejected an NT2 tumor challenge after being given vaccine and cyclophosphamide chemotherapy (see Immunization protocols). Splenocytes were initially stimulated every 5 d with irradiated, IFN-γ treated NT5B7-1 cells and then every 9 d by addition of irradiated 3T3neuB7-1 cells and FVB/N-derived splenocytes. T cells were maintained at 37°C and 5% CO2 in CTL media (RPMI 1640 supplemented with 10% FBS, 1% l-glutamine, 0.1 mM 2-mercaptoethanol (Sigma-Aldrich), and 0.5% penicillin/streptomycin supplemented with 10 cetus U/ml murine IL-2, a supernatant from B16 IL-2 line (17).
Peptides.
RNEU420-429 (PDSLRDLSVF) and NP118-126 (RPQASGVYM) peptides were synthesized at >95% purity and purchased from either the Johns Hopkins Biosynthesis and Sequence Facility or from Macromolecular Resources of Colorado State University. The NP118-126 peptide is from the lymphocytic choriomeningitis virus nucleoprotein (25) and is used as an irrelevant H2-Dq–binding peptide.
Antibodies and tetramer and flow cytometric analysis.
mAb hybridoma supernatants (all acquired from the American Type Culture Collection) were used for staining T cell lines: B20.6 (TCR Vβ2), KT4 (TCR Vβ4), and RR4-7 (TCR Vβ6). Directly conjugated antibodies anti-CD3 FITC, anti-CD4 CyChrome, anti-CD8α CyChrome, anti-CD8β FITC, anti-CD25 PE (PC61 and 7D4), anti-CD62L allophycocyanin, and anti-IFN-γ PE were purchased from BD Biosciences. The CD25-depleting antibody PC61 was purified from the supernatant of PC61.5.3 hybridoma (American Type Culture Collection) grown in Protein Free Hybridoma Media II (Invitrogen).
The H-2Dq/RNEU420-429 tetramer was constructed using previously described methods (48). Tetramer staining was performed by first staining only for CD8 (T cell lines) or CD8 and CD62L (ex vivo experiments). Cells were washed, and tetramer was added at varying dilutions and incubated for 30 min at 8–12°C. Cells were washed twice and immediately fixed with fresh 1% paraformaldehyde in PBS. Tetramer staining was analyzed by gating only on CD8+ cells (T cell lines) or CD8+, CD62Llo cells (ex vivo experiments).
ICS was performed using the mouse ICS kit from BD Biosciences for murine IFN-γ. Purified T cells were incubated for 6–12 h with an equal ratio of indicated targets (IT22neu, IT22, or T2Dq cells pulsed with either RNEU420-429 or NP118-126) in the presence of GolgiStop (BD Biosciences). Cells were then stained for CD8, fixed and permeabilized, and stained with anti-IFN-γ PE. Analysis was performed by gating on CD8+ cells and calculating the percent of total CD8+ cells that were also IFN-γ+. The percent of antigen-specific cells was calculated by subtracting the percent of IFN-γ+ cells in the irrelevant antigen sample from the percent of IFN-γ+ cells in the relevant antigen sample.
Flow cytometric data were collected using BD FACScan and BD FACSCalibur cytometers (BD Biosciences). Data were analyzed using CELLQuest (BD Biosciences) and FlowJo software (Tree Star, Inc.).
Immunization protocols.
Mice were given vaccine (3T3neuGM), or mock vaccine (3T3GM cells) alone or in combination with cyclophosphamide, or cyclophosphamide plus Dox as described previously (17), or PC61 depletion. Mice given vaccine alone were injected s.c. with 3 × 106 total cells divided equally among one forelimb and two hind limbs. Mice given vaccine and cyclophosphamide received a single i.p. injection of cyclophosphamide, 100 mg/kg in 0.5 ml (Mead Johnson), on day −1 and vaccine (as described previously) on day 0. Vaccine given with cyclophosphamide and Dox included cyclophosphamide on day −1, vaccine on day 0, and Dox, 5 mg/kg given i.v. in 0.5 ml (Gensia Sicor Pharmaceuticals Inc.) on day 7 (this regimen is referred to as cyclophosphamide chemotherapy in the text). We and others previously found that Dox given at the time of T cell expansion can enhance the cytotoxic function of CD8+ T cells (17, 41). PC61-depletion studies were performed by giving one dose of PC61 (1 mg i.p.) on day −4 followed by vaccine on day 0. In tumor-challenge experiments, NT2 tumor cells (5 × 104 cells injected s.c. in the mammary fat pad) were injected either on day −3 or on day 14. Mice were monitored for tumor outgrowth twice per week.
Adoptive transfer experiments.
_neu_-N donor mice were either naive or given an NT2 tumor challenge on day −3 and 3T3neuGM vaccine on day 0. 2 wk after vaccination, donor CD4+ and CD8+ splenic T cells were isolated using MACS negative isolation kits (Miltenyi Biotec). Donor CD4+CD25+ and CD4+CD25− splenic subsets were isolated from purified CD4+ T cells by staining for CD25 and sorting the positive and negative fractions using a FACSVantage cell sorter (Becton Dickinson). All T cell subsets were analyzed by flow cytometry to confirm purity. T cells were washed with PBS and injected i.v. into recipient mice.
Bromodeoxyuridine incorporation assay.
_neu_-N mice were given cyclophosphamide or PBS i.p. on day 0. On day 1, mice were given a 2-mg dose of BrdU injected i.p. On day 2, axillary LN cells were isolated, washed, and stained using the FITC BrdU Flow Kit (BD Biosciences).
Chromium-release assays
Lysis assays were performed in triplicate in 96-well V-bottom plates as previously described (16). After 4-h incubation, supernatant was assayed for 51Cr release and the percentage of specific lysis was determined by the formula: (51Cr release sample − spontaneous 51Cr release)/(maximum 51Cr release − spontaneous 51Cr release) × 100.
Tetramer off-rate experiments.
T cells were washed and resuspended at 106 in 100-μl buffer. Cells were incubated with 8 × 10−3 M H-2Dq/RNEU420-429 tetramer for 1 h at 4°C and then 20-fold molar excess of the H-2Dq antibody 30–5-7S (American Type Culture Collection) was added to prevent tetramer from rebinding T cells. Mean fluorescence intensity of tetramer staining was determined by flow cytometry at various time points from 0 to 240 min. Resulting data were fit to a first-order exponential decay using the Origin program (Microcal Software, Inc.). Off-rates were calculated as the reciprocal of t1.
Statistical analysis.
Statistical significance of the tumor-free survival plots was determined using the log rank test. A Student's t test was applied to compare statistical significance between treatment groups. All analysis was performed using Prism 4 software (GraphPad Software).
Acknowledgments
We thank J. Flook for his technical assistance with cell sorting and Drs. D. Pardoll and J. P. Schneck for their critical review of the text. We would also like to recognize the National Tetramer Core Facility at Emory University for generation of the H-2Dq/RNEU420-429 MHC/peptide tetramer cDNA construct.
This work was supported by National Cooperative Drug Discovery Groups National Institutes of Health (NIH)/National Cancer Institute grant no. 2U19CA72108 and a Breast Cancer Research Foundation grant (to E.M. Jaffee); Specialized Program of Research Excellence in Breast Cancer grant no. 1P50CA88843-01 (to E.M. Jaffee and L.A. Emens); Department of Defense grant no. DAMD17-01-1-0281 and Maryland Cigarette Restitution Fund no. M020216 (to L.A. Emens); American Cancer Society Research Scholar grant no. RSG-01-080-01-LIB and Susan G. Komen Foundation grant no. BCTR00-00068 (to R.T. Reilly); NIH training grant no. CA09243 (to E.A. Manning); NIH/National Institute of Allergy and Infectious Diseases grant no. 5T32AI07247-21 and CRI grant 311-2007 (to A.M. Ercolini); and Department of Defense grant no. DAMD17-01-1-0282 (to A.M. Ercolini and B.H. Ladle). J.-P.H. Machiels is a Fulbright Scholar supported by a grant from Belgium Televie-FNRS (credit 7.4568.98) and Oeuvre Belge du Cancer. B.H. Ladle is a Howard Hughes Medical Institute Medical Student Research Training Fellow. E.M. Jaffee is the first recipient of the Dana and Albert “Cubby” Broccoli Professorship in Oncology.
This work describes the use of a GM-CSF–secreting tumor vaccine. Although none of the authors have financial interests in the work, the Johns Hopkins University receives milestone payments and has the potential to receive royalties in the future.
Abbreviations used: BrdU, bromodeoxyuridine; Dox, doxorubicin; ICS, intracellular cytokine stain; koff, dissociation rate constant; neu, HER-2/neu; _neu_-N, MMTV-HER-2/neu transgenic mice; T reg, regulatory T.
A.M. Ercolini and B.H. Ladle contributed equally to this work.
References
- 1.Sprent, J., and H. Kishimoto. 2002. The thymus and negative selection. Immunol. Rev. 185:126–135. [DOI] [PubMed] [Google Scholar]
- 2.Walker, L.S., and A.K. Abbas. 2002. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2:11–19. [DOI] [PubMed] [Google Scholar]
- 3.O'Garra, A., and P. Vieira. 2004. Regulatory T cells and mechanisms of immune system control. Nat. Med. 10:801–805. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll, D. 2003. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 21:807–839. [DOI] [PubMed] [Google Scholar]
- 5.Disis, M.L., and M.A. Cheever. 1997. HER-2/neu protein: a target for antigen-specific immunotherapy of human cancer. Adv. Cancer Res. 71:343–371. [DOI] [PubMed] [Google Scholar]
- 6.Novellino, L., C. Castelli, and G. Parmiani. 2005. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 54:187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez, J., A. Ko, and L.A. Sherman. 2001. CTLA-4 blockade enhances the CTL responses to the p53 self-tumor antigen. J. Immunol. 166:3908–3914. [DOI] [PubMed] [Google Scholar]
- 8.Morgan, D.J., H.T. Kreuwel, S. Fleck, H.I. Levitsky, D.M. Pardoll, and L.A. Sherman. 1998. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 160:643–651. [PubMed] [Google Scholar]
- 9.Liu, G.Y., P.J. Fairchild, R.M. Smith, J.R. Prowle, D. Kioussis, and D.C. Wraith. 1995. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 3:407–415. [DOI] [PubMed] [Google Scholar]
- 10.Sprent, J., and S.R. Webb. 1995. Intrathymic and extrathymic clonal deletion of T cells. Curr. Opin. Immunol. 7:196–205. [DOI] [PubMed] [Google Scholar]
- 11.Ohlen, C., M. Kalos, D.J. Hong, A.C. Shur, and P.D. Greenberg. 2001. Expression of a tolerizing tumor antigen in peripheral tissue does not preclude recovery of high-affinity CD8+ T cells or CTL immunotherapy of tumors expressing the antigen. J. Immunol. 166:2863–2870. [DOI] [PubMed] [Google Scholar]
- 12.Adler, A.J., D.W. Marsh, G.S. Yochum, J.L. Guzzo, A. Nigam, W.G. Nelson, and D.M. Pardoll. 1998. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow-derived antigen-presenting cells. J. Exp. Med. 187:1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Visser, K.E., T.A. Cordaro, H.W. Kessels, F.H. Tirion, T.N. Schumacher, and A.M. Kruisbeek. 2001. Low-avidity self-specific T cells display a pronounced expansion defect that can be overcome by altered peptide ligands. J. Immunol. 167:3818–3828. [DOI] [PubMed] [Google Scholar]
- 14.Klausner, R.D. 1999. Studying cancer in the mouse. Oncogene. 20;18:5249–5252. [DOI] [PubMed] [Google Scholar]
- 15.Guy, C.T., M.A. Webster, M. Schaller, T.J. Parsons, R.D. Cardiff, and W.J. Muller. 1992. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA. 89:10578–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reilly, R.T., M.B. Gottlieb, A.M. Ercolini, J.P. Machiels, C.E. Kane, F.I. Okoye, W.J. Muller, K.H. Dixon, and E.M. Jaffee. 2000. HER-2/neu is a tumor rejection target in tolerized HER-2/neu transgenic mice. Cancer Res. 60:3569–3576. [PubMed] [Google Scholar]
- 17.Machiels, J.P., R.T. Reilly, L.A. Emens, A.M. Ercolini, R.Y. Lei, D. Weintraub, F.I. Okoye, and E.M. Jaffee. 2001. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 61:3689–3697. [PubMed] [Google Scholar]
- 18.North, R.J. 1982. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 155:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoover, S.K., S.K. Barrett, T.M. Turk, T.C. Lee, and H.D. Bear. 1990. Cyclophosphamide and abrogation of tumor-induced suppressor T cell activity. Cancer Immunol. Immunother. 31:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghiringhelli, F., N. Larmonier, E. Schmitt, A. Parcellier, D. Cathelin, C. Garrido, B. Chauffert, E. Solary, B. Bonnotte, and F. Martin. 2004. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 34:336–344. [DOI] [PubMed] [Google Scholar]
- 21.Turk, M.J., J.A. Guevara-Patino, G.A. Rizzuto, M.E. Engelhorn, S. Sakaguchi, and A.N. Houghton. 2004. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J. Exp. Med. 20;200:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thornton, A.M., and E.M. Shevach. 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 24.Ercolini, A.M., J.P. Machiels, Y.C. Chen, J.E. Slansky, M. Giedlen, R.T. Reilly, and E.M. Jaffee. 2003. Identification and characterization of the immunodominant rat HER-2/neu MHC class I epitope presented by spontaneous mammary tumors from HER-2/neu-transgenic mice. J. Immunol. 170:4273–4280. [DOI] [PubMed] [Google Scholar]
- 25.Lee, D.R., R.J. Rubocki, W.R. Lie, and T.H. Hansen. 1988. The murine MHC class I genes, H-2Dq and H-2Lq, are strikingly homologous to each other, H-2Ld, and two genes reported to encode tumor-specific antigens. J. Exp. Med. 168:1719–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savage, P.A., J.J. Boniface, and M.M. Davis. 1999. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 10:485–492. [DOI] [PubMed] [Google Scholar]
- 27.Cawthon, A.G., H. Lu, and M. Alexander-Miller. 2001. Peptide requirement for CTL activation reflects the sensitivity to CD3 engagement: correlation with CD8αβ versus CD8αα expression. J. Immunol. 167:2577–2584. [DOI] [PubMed] [Google Scholar]
- 28.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 59:3128–3133. [PubMed] [Google Scholar]
- 30.Nugent, C.T., D.J. Morgan, J.A. Biggs, A. Ko, I.M. Pilip, E.G. Pamer, and L.A. Sherman. 2000. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J. Immunol. 164:191–200. [DOI] [PubMed] [Google Scholar]
- 31.Lustgarten, J., A.L. Dominguez, and C. Cuadros. 2004. The CD8+ T cell repertoire against Her-2/neu antigens in neu transgenic mice is of low avidity with antitumor activity. Eur. J. Immunol. 34:752–761. [DOI] [PubMed] [Google Scholar]
- 32.Busch, D.H., and E.G.P. Am. 1999. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 189:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander-Miller, M.A., G.R. Leggatt, and J.A. Berzofsky. 1996. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc. Natl. Acad. Sci. USA. 93:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dutoit, V., V. Rubio-Godoy, M.A. Doucey, P. Batard, D. Lienard, D. Rimoldi, D. Speiser, P. Guillaume, J.C. Cerottini, P. Romero, and D. Valmori. 2002. Functional avidity of tumor antigen-specific CTL recognition directly correlates with the stability of MHC/peptide multimer binding to TCR. J. Immunol. 168:1167–1171. [DOI] [PubMed] [Google Scholar]
- 35.Dutoit, V., V. Rubio-Godoy, P.Y. Dietrich, A.L. Quiqueres, V. Schnuriger, D. Rimoldi, D. Lienard, D. Speiser, P. Guillaume, P. Batard, et al. 2001. Heterogeneous T-cell response to MAGE-A10(254-262): high avidity-specific cytolytic T lymphocytes show superior antitumor activity. Cancer Res. 61:5850–5856. [PubMed] [Google Scholar]
- 36.Yee, C., P.A. Savage, P.P. Lee, M.M. Davis, and P.D. Greenberg. 1999. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J. Immunol. 162:2227–2234. [PubMed] [Google Scholar]
- 37.Yang, S., G.P. Linette, S. Longerich, and F.G. Haluska. 2002. Antimelanoma activity of CTL generated from peripheral blood mononuclear cells after stimulation with autologous dendritic cells pulsed with melanoma gp100 peptide G209-2M is correlated to TCR avidity. J. Immunol. 169:531–539. [DOI] [PubMed] [Google Scholar]
- 38.Zeh, H.J., III, D. Perry-Lalley, M.E. Dudley, S.A. Rosenberg, and J.C. Yang. 1999. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J. Immunol. 162:989–994. [PubMed] [Google Scholar]
- 39.Antony, P.A., C.A. Piccirillo, A. Akpinarli, S.E. Finkelstein, P.J. Speiss, D.R. Surman, D.C. Palmer, C.C. Chan, C.A. Klebanoff, W.W. Overwijk, et al. 2005. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 174:2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang, Y., C.T. Huang, X. Huang, and D.M. Pardoll. 2004. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 5:508–515. [DOI] [PubMed] [Google Scholar]
- 41.Nigam, A., R.F. Yacavone, M.L. Zahurak, C.M. Johns, D.M. Pardoll, S. Piantadosi, H.I. Levitsky, and W.G. Nelson. 1998. Immunomodulatory properties of antineoplastic drugs administered in conjunction with GM-CSF-secreting cancer cell vaccines. Int. J. Oncol. 12:161–170. [DOI] [PubMed] [Google Scholar]
- 42.Hernandez, J., P.P. Lee, M.M. Davis, and L.A. Sherman. 2000. The use of HLA A2.1/p53 peptide tetramers to visualize the impact of self tolerance on the TCR repertoire. J. Immunol. 164:596–602. [DOI] [PubMed] [Google Scholar]
- 43.Derby, M.A., J. Wang, D.H. Margulies, and J.A. Berzofsky. 2001. Two intermediate-avidity cytotoxic T lymphocyte clones with a disparity between functional avidity and MHC tetramer staining. Int. Immunol. 13:817–824. [DOI] [PubMed] [Google Scholar]
- 44.Rubio-Godoy, V., V. Dutoit, D. Rimoldi, D. Lienard, F. Lejeune, D. Speiser, P. Guillaume, J.C. Cerottini, P. Romero, and D. Valmori. 2001. Discrepancy between ELISPOT IFN-gamma secretion and binding of A2/peptide multimers to TCR reveals interclonal dissociation of CTL effector function from TCR-peptide/MHC complexes half-life. Proc. Natl. Acad. Sci. USA. 98:10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreuwel, H.T., J.A. Biggs, I.M. Pilip, E.G. Pamer, D. Lo, and L.A. Sherman. 2001. Defective CD8+ T cell peripheral tolerance in nonobese diabetic mice. J. Immunol. 167:1112–1117. [DOI] [PubMed] [Google Scholar]
- 46.Heath, W.R., C. Kurts, J.F. Miller, and F.R. Carbone. 1998. Cross-tolerance: a pathway for inducing tolerance to peripheral tissue antigens. J. Exp. Med. 187:1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakaguchi, S., N. Sakaguchi, J. Shimizu, S. Yamazaki, T. Sakihama, M. Itoh, Y. Kuniyasu, T. Nomura, M. Toda, and T. Takahashi. 2001. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 182:18–32. [DOI] [PubMed] [Google Scholar]
- 48.Altman, J.D., P.A. Moss, P.J. Goulder, D.H. Barouch, M.G. Heyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]