Functional Roles of Nonconserved Structural Segments in CFTR's NH2-terminal Nucleotide Binding Domain (original) (raw)
Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR), encoded by the gene mutated in cystic fibrosis patients, belongs to the family of ATP-binding cassette (ABC) proteins, but, unlike other members, functions as a chloride channel. CFTR is activated by protein kinase A (PKA)-mediated phosphorylation of multiple sites in its regulatory domain, and gated by binding and hydrolysis of ATP at its two nucleotide binding domains (NBD1, NBD2). The recent crystal structure of NBD1 from mouse CFTR (Lewis, H.A., S.G. Buchanan, S.K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W.B. Guggino, W.A. Hendrickson, et al. 2004. EMBO J. 23:282–293) identified two regions absent from structures of all other NBDs determined so far, a “regulatory insertion” (residues 404–435) and a “regulatory extension” (residues 639–670), both positioned to impede formation of the putative NBD1–NBD2 dimer anticipated to occur during channel gating; as both segments appeared highly mobile and both contained consensus PKA sites (serine 422, and serines 660 and 670, respectively), it was suggested that their phosphorylation-linked conformational changes might underlie CFTR channel regulation. To test that suggestion, we coexpressed in Xenopus oocytes CFTR residues 1–414 with residues 433–1480, or residues 1–633 with 668–1480, to yield split CFTR channels (called 414+433 and 633+668) that lack most of the insertion, or extension, respectively. In excised patches, regulation of the resulting CFTR channels by PKA and by ATP was largely normal. Both 414+433 channels and 633+668 channels, as well as 633(S422A)+668 channels (lacking both the extension and the sole PKA consensus site in the insertion), were all shut during exposure to MgATP before addition of PKA, but activated like wild type (WT) upon phosphorylation; this indicates that inhibitory regulation of nonphosphorylated WT channels depends upon neither segment. Detailed kinetic analysis of 414+433 channels revealed intact ATP dependence of single-channel gating kinetics, but slightly shortened open bursts and faster closing from the locked-open state (elicited by ATP plus pyrophosphate or ATP plus AMPPNP). In contrast, 633+668 channel function was indistinguishable from WT at both macroscopic and microscopic levels. We conclude that neither nonconserved segment is an essential element of PKA- or nucleotide-dependent regulation.
Keywords: ABC transporters, crystal structure, chloride ion-channel gating, phosphorylation, domain boundaries
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) is expressed in the apical membrane of epithelial cells where its dysfunction, due to mutations, causes cystic fibrosis (Riordan et al., 1989). Topologically (cartoon in Fig. 1, left), CFTR comprises two roughly homologous halves, each consisting of a transmembrane domain (TMD1 or TMD2) that likely includes six membrane-spanning helices and is followed (in the primary sequence) by a cytosolic nucleotide binding domain (NBD1 or NBD2); these two halves are linked by a regulatory domain (R domain). This domain organization identifies CFTR as a member of the large family of ATP-binding cassette (ABC) proteins (Riordan et al., 1989), transporters that occur in the cell membranes of all organisms from bacteria to humans. However, unique among ABC proteins, CFTR is a Cl− channel, and it requires phosphorylation of multiple R domain serines by cAMP-dependent protein kinase (PKA) before it can open (Tabcharani et al., 1991). But even once phosphorylated, a CFTR channel is able to open and close normally only in the presence of hydrolyzable nucleotide (Anderson et al., 1991), and gating of the channel is mediated by nucleotide interactions with CFTR's two NBDs (e.g., Anderson and Welsh, 1992; Vergani et al., 2003).
Figure 1.

Crystal structure of NBD1 of murine CFTR. (Left) Domain topology of CFTR comprising two transmembrane domains (TMD1 and 2), two cytoplasmic nucleotide binding domains (NBDs, marked 1 and 2), and unique cytosolic R domain. (Right) Ribbon diagrams (created using Swiss-PdbViewer v3.7b2) of crystal structure of mouse CFTR NBD1 with bound MgATP (Lewis et al., 2004; accession code 1R0X), in two orientations: top, view from the side of the ATP-binding pocket (top view); bottom, view from an angle roughly perpendicular to the top and along the plane of the F1-like parallel β-sheet (back view). Color coding: ABC-specific NH2-terminal antiparallel β-sheet, cyan; F1-like ATP-binding core, green; ABC-specific α-helical domain, gold; nonconserved insertion and extension, red; ATP, magenta; side chains of serines 660 and 670, blue. Dotted red line within the nonconserved insertion represents disordered residues 412–428, not resolved in the crystal structure, and is simply intended to indicate the continuity of the peptide chain. β-Strands and α-helices discussed in the text are marked by arrows.
The overall topological design of ABC proteins appears conserved, regardless of whether their domains are all translated from a single gene, as in the case of CFTR, or are translated individually and then associate, as in the case of many prokaryotic ABC transporters. Yet, despite this conserved architecture, primary sequence homology between ABC proteins is largely restricted to their NBDs. The structural consequences of that homology have been revealed by the several crystal structures of NBDs solved within the past few years, mostly from prokaryotic ABC transporters (Armstrong et al., 1998; Hung et al., 1998; Diederichs et al., 2000; Hopfner et al., 2000; Karpowich et al., 2001; Yuan et al., 2001; Locher et al., 2002; Smith et al., 2002; Chen et al., 2003; Schmitt et al., 2003; Verdon et al., 2003a), but also that from human TAP1 (Gaudet and Wiley, 2001). Consistent with their sequence similarity, these NBD structures all share a common fold with three distinct regions: a central F1-ATPase–like ATP-binding core (colored green in the mouse CFTR NBD1 structure in Fig. 1) built from a six-membered parallel β-sheet attached to a conserved α-helix, connected on one side to an ABC-specific antiparallel β-region (cyan, Fig. 1), which also contributes to the binding site for ATP (magenta, Fig. 1), and connected on the opposite side to an ABC-specific α-helical domain (gold, Fig. 1).
The much anticipated first glimpse of the structure of a CFTR domain was afforded by the recently determined crystal structure of NBD1 from mouse CFTR (Lewis et al., 2004). Surprisingly, although this CFTR NBD1 structure broadly conformed to the common architecture established by the previous structures, it differed from all other NBDs in two regions (Lewis et al., 2004). It contained an ∼30–amino acid insertion near its NH2 terminus (residues 404–435, between strands 1 and 2 of the antiparallel β-domain; Fig. 1, red), and it also included a similar-length extension (residues 639–670, α-helices 9 and 9b; Fig. 1, also red) at its COOH-terminal end. Because both these segments harbor serine residues in consensus sequences for phosphorylation by PKA, and both exhibited conformational flexibility, they were called “regulatory insertion” and “regulatory extension,” and it was suggested that their movement upon phosphorylation might provide a mechanism for PKA-mediated regulation of CFTR channel gating (Lewis et al., 2004).
There is now a growing consensus that in all functioning ABC proteins, pairs of NBDs act in concert. Recent evidence of coupled interactions between NBDs, in the form of NBD dimers, has come from functional measurements (Moody et al., 2002; Janas et al., 2003; Urbatsch et al., 2003; Vergani et al., 2003; Verdon et al., 2003b) as well as from structural studies (Hopfner et al., 2000; Fetsch and Davidson, 2002; Locher et al., 2002; Loo et al., 2002; Smith et al., 2002; Chen et al., 2003). Dimeric crystal structures have confirmed that NBDs dimerize in a head-to-tail orientation, with an ATP molecule sandwiched at each of two composite catalytic sites in the dimer interface (Hopfner et al., 2000; Locher et al., 2002; Smith et al., 2002; Chen et al., 2003). Each bound ATP molecule forms contacts with conserved residues of the Walker A and B sequences (Walker et al., 1982) found in the F1-like domain of one NBD, and across the interface with conserved residues of the “ABC signature” sequence (LSGGQ) in the α-helical domain of the partner NBD.
We have proposed that formation of such an ATP-bound head-to-tail NBD1–NBD2 dimer might be required for a CFTR channel to open (Vergani et al., 2003). With such a gating scheme in mind, Lewis et al. (2004) attempted to model the hypothetical head-to-tail NBD1–NBD2 heterodimer by docking a homology model of CFTR NBD2 onto their NBD1 structure, but were thwarted by steric hindrance arising from both NBD1 insertion and extension. Although the insertion was incompletely resolved in the NBD1 structure, both the insertion and the extension exhibited large B-factors, indicating disorder and hence flexibility. These findings led to the suggestion that these two nonconserved NBD1 segments might keep a nonphosphorylated CFTR channel closed by preventing formation of the NBD1–NBD2 heterodimer, but that, once phosphorylated, the segments might move aside, allowing that dimerization and hence facilitating channel gating (Lewis et al., 2004).
In the present work, we have tested the postulated regulatory roles of NBD1's insertion and extension by examining in detail the gating kinetics of CFTR channels engineered to lack the bulk of one or other of those segments. To delete those elements while avoiding the distortion of the remaining structure that could attend covalent linkage of normally nonadjacent residues, we exploited the ability of coexpressed hemi-channels to associate and form mature CFTR channels with essentially normal function (Ostedgaard et al., 1997; Chan et al., 2000; Csanády et al., 2000); that ability derives naturally from the modular construction of ABC proteins described above. We found that in patches excised from Xenopus oocytes, split CFTR channels lacking amino acids 415–432 or 634–667, most of the insertion or extension of NBD1, respectively, displayed phosphorylation- and ATP-dependent regulation of gating that differed little from that of intact wild-type (WT) CFTR channels. Our results suggest that neither the insertion nor the extension is a key element in phosphorylation-dependent regulation, contrary to the expectation from interpretation of the first high-resolution structure of an essential part of CFTR.
MATERIALS AND METHODS
Molecular Biology
pGEMHE-WT, pGEMHE-1-633, pGEMHE-Flag3-633, pGEMHE-Flag3-414, pGEMHE-433-1480, and pGEMHE-668-1480 were constructed as previously described (Chan et al., 2000). pGEMHE-1-414 was constructed by cutting pGEMHE-WT and pGEMHE-Flag3-414 with BsrGI (which cuts at nucleotide position 1238 of pGEMHE-WT, at amino acid 357 in CFTR) and BsrFI (which cuts at nucleotide 6411, downstream of the CFTR coding region), and ligating the 1.97-kB BsrGI/BsrFI fragment of pGEMHE-Flag3-414 to the 2.56-kB BsrGI/BsrFI fragment of pGEMHE-WT. pGEMHE-Flag3-633(S422A) was constructed by introducing the S422A point mutation into pGEMHE-Flag3-633, using Stratagene's QuickChange Mutagenesis Kit, and primers S422A-FW (5′-TAACAATAGAAAAACTGCTAATGGTGATGACAGCCTCT) and S422A-RW (5′-AGAGGCTGTCATCACCATTAGCAGTTTTTCTATTGTTA). All constructs were confirmed by automated sequencing.
Isolation and Injection of Xenopus Oocytes
Isolation and injection of Xenopus laevis oocytes was as previously described (Chan et al., 2000). Oocytes were isolated, treated with collagenase in a Ca2+-free oocyte Ringer's solution, and stored at 18°C in Ringer's supplemented with 1.8 mM CaCl2. Depending on the required expression level, 0.1–7 ng of WT cRNA, or 0.1–7.5 ng of cRNA encoding appropriate CFTR segments (premixed for coexpression), was injected in a fixed 50-nl total volume. Oocytes were further incubated at 18°C, and used for recordings 2–3 d after injection.
Excised Patch Recording
Patch pipettes were pulled from borosilicate glass, fire-polished, and had tip resistances of ∼2 MΩ and 4–7 MΩ for recording macroscopic and single-channel currents, respectively. Pipette solution contained (in mM) 136 NMDG-Cl, 2 MgCl2, 5 HEPES, pH 7.4 with NMDG. Bath solution contained (in mM) 134 NMDG-Cl, 2 MgCl2, 5 HEPES, 0.5 EGTA, pH 7.1 with NMDG. After manual removal of the vitelline layer, oocytes expressing CFTR constructs were put in a 35-mm Petri dish on the stage of an inverted microscope (Nikon Eclipse, TS 100). Membrane seals in the 100-GΩ range were formed by gentle suction, and patches were excised and transferred to a flow chamber in which the continuously flowing bath solution could be exchanged using electronic valves. The time constant for solution exchange, measured from the rate of decay of Ca2+-activated Cl− current upon rapid removal of 2 mM CaCl2, was 110 ± 10 ms (n = 10). MgATP (Sigma-Aldrich) was added from a 400 mM aqueous stock solution (pH 7.1 with NMDG). Li4-AMPPNP (Sigma-Aldrich) and Na4-pyrophosphate (PPi) were added from 400 mM and 200 mM aqueous stock solutions, respectively, each supplemented with equimolar MgCl2. CFTR channels were activated by 300 nM catalytic subunit of PKA, purified from bovine heart (Kaczmarek et al., 1980). Recordings were made at ∼25°C. Inward membrane currents were recorded at a pipette holding potential of +80 mV (Vm = −80 mV), amplified with an Axopatch 200B amplifier (Axon Instruments, Inc.), digitized at 1 kHz (Digidata 1322A; Axon Instruments, Inc.), and recorded to disk with pCLAMP 8 software (Axon Instruments, Inc.) using on-line Gaussian filtering at 50 Hz.
Kinetic Analysis
Current records in which single-channel transitions could be clearly resolved were baseline subtracted to remove slow drifts, idealized using half-amplitude threshold crossing combined with imposition of a fixed dead time of 6.5 ms (Csanády, 2000), and the resulting events lists subjected to kinetic analysis. Open probability (Po) was calculated from events lists as Σk(n k t k/(NT), where n k and t k denote the number of open channels and the duration of the _k_th event, respectively, N is the number of active channels in the patch, and T = Σk t k is the total duration of the record.
CFTR channel gating shows typical bursting behavior, with brief “flickery” closures interrupting bursts of openings flanked by long (“interburst”) closures. Because phosphorylation and [MgATP] regulate the lengths of bursts and interbursts (Gunderson and Kopito, 1994; Hwang et al., 1994; Winter et al., 1994; Csanády et al., 2000; Vergani et al., 2003), we extracted mean single-channel burst (τb) and interburst (τib) durations as previously described (Csanády et al., 2000). ATP-dependent gating transitions were pooled into a simple closed-open scheme and flickery closures modeled as pore-blockage events, resulting in a simple three-state Closed-Open-Blocked scheme. Rate constants (r CO, r OC, r OB, and r BO) were extracted by a simultaneous maximum likelihood fit to the dwell-time histograms of all conductance levels, using an algorithm that includes a correction for the filter dead time (Csanády, 2000). Mean burst and interburst durations were then calculated as τib = 1/r CO and τb = (1/r OC)(1+r OB/r BO).
Statistical tests (Csanády et al., 2000) were applied to evaluate whether the number of active channels in the patch (N) was likely equal to the maximum number of simultaneously open channels seen in the record (N'). Extracted τb, as well as τib and Po, values were included into our statistics if N > N' could be excluded with >90% confidence (N was between 1 and 6 for these patches). If transitions were well resolved (N' ≤ 10), but N > N' could not be excluded with confidence, we still used the absolute value of τb (which is little sensitive to N), but only relative values of Po and τib (both normalized to the analogous parameters obtained from control segments of record in the same patch).
Closing rate and opening rate were defined as 1/τb, and 1/τib, respectively. To determine [ATP] dependence of these rates, patches were superfused with test concentrations of MgATP in between bracketing exposures to 2 mM MgATP. Po, closing, and opening rates in test [ATP] were determined and normalized to the average of the same parameters extracted from the bracketing segments in 2 mM ATP.
Analysis of Macroscopic Currents
Fitting of decaying macroscopic currents with single or double exponential functions was done using pCLAMP 8 software (Axon Instruments, Inc.). Fractional currents in the presence of various [MgATP] were obtained by dividing the mean of the steady current in test [ATP] by the average of the mean currents in 2 mM MgATP immediately before and after the test.
Fitting of Dose–Response Curves
Plots of fractional macroscopic current (Fig. 3 E), Po (Fig. 5 D), and opening rate (Fig. 5 F) as a function of [MgATP] were fitted with the Michaelis-Menten equation x = x max([ATP]/([ATP]+K m)), where x and x max are the measured parameter and its (fitted) maximum value, and K m is the (fitted) Michaelis-Menten constant.
Figure 3.
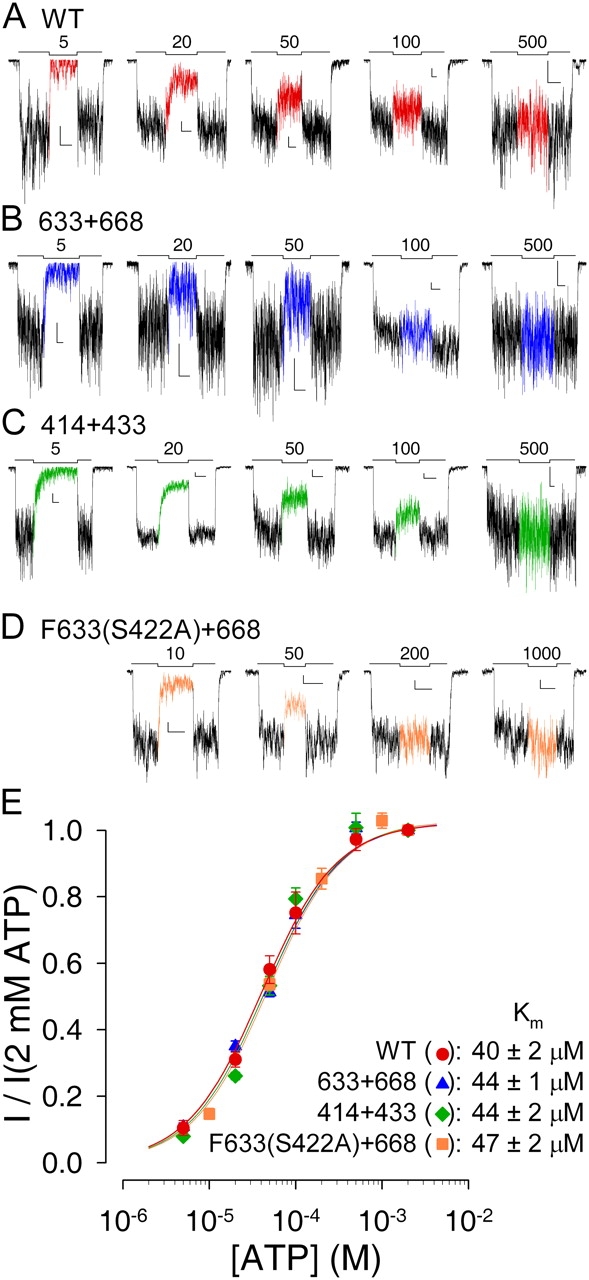
ATP dependence of macroscopic current is intact for severed CFTR constructs. Currents from macropatches containing (A) WT, (B) 633+668, (C) 414+433, and (D) Flag-tagged 633(S422A)+668 channels superfused with test concentrations of MgATP ranging from 5 μM to 1 mM, bracketed by exposures to 2 mM MgATP (bars; numbers indicate test [ATP] in μM). L-shaped scale bars in each panel indicate 2 pA and 10 s. (E) Mean currents in test [MgATP], normalized to the average of the currents in the 2 mM MgATP bracketing segments, plotted against test [ATP]. Lines show fits to the Michaelis-Menten equation, giving Km values printed.
Figure 5.
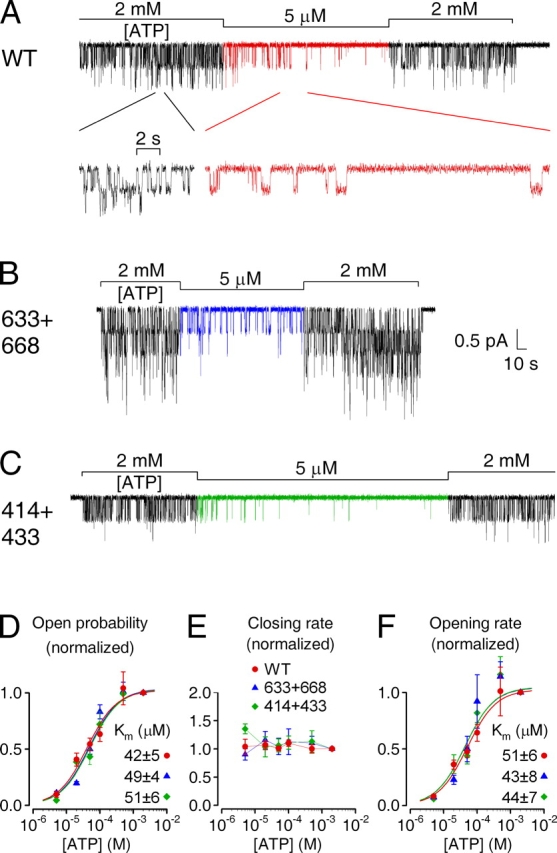
ATP dependence of single-channel gating kinetics is intact for severed CFTR constructs. Currents from patches with few active (A) WT, (B) 633+668, and (C) 414+433 channels exposed to 5 μM MgATP, bracketed by superfusion with 2 mM MgATP (bars). Note prolonged closed (interburst) periods in 5 μM ATP, evident in the inset with expanded time scale in A. (D–F) Mean burst (τb) and interburst (τib) durations were extracted (materials and methods) from segments of record in various [ATP], and closing and opening rates defined as 1/τb and 1/τib. Po (D), closing (E) and opening rates (F) in test [ATP], normalized to the average of the same parameters in the bracketing control segments, are plotted against [ATP]. Curves show fits of Po (D) and opening rate (F) to the Michaelis-Menten equation, giving Km values printed.
Single-channel Conductances
Unitary current sizes were extracted by fitting all-points histograms of segments of record, obtained at fixed membrane voltages between −120 and +80 mV, with sums of Gaussians. Plots of unitary current against membrane voltage were fitted by linear regression to determine slope conductances.
Statistics
Reported parameters represent mean ± SEM. Statistical significance was assessed using Student's t test. Differences are reported as significant for P < 0.05.
RESULTS
Unaltered Phosphorylation Dependence of Macroscopic Current in Severed CFTR Channels Lacking NBD1 Insertion or Extension
WT CFTR channel activity is strictly regulated by PKA (for reviews see Gadsby and Nairn, 1999; Sheppard and Welsh, 1999). Gating by binding/hydrolysis of MgATP at CFTR's two cytosolic NBDs requires prior phosphorylation of the R domain (cartoon in Fig. 1, left) by PKA. When inside-out patches containing large numbers of WT human epithelial CFTR are excised from resting Xenopus oocytes and superfused with MgATP (2 mM) alone, only tiny channel currents are elicited compared with those that are activated by subsequent exposure of the same channels to 300 nM PKA catalytic subunit in the continued presence of ATP (Fig. 2 A; see also Csanády et al., 2000). This indicates that the low level of activation of CFTR channels by endogenous kinases in resting oocytes (Chan et al., 2000) is largely reversed after patch excision due to strong membrane-bound phosphatase activity. Under identical conditions, a 1-min exposure to MgATP alone of severed channels lacking most of the extension of NBD1, formed from coexpressed segments 1–633 plus 668–1480 (called 633+668), elicited similarly small currents, although subsequent exposure to PKA resulted in macroscopic current activation (Fig. 2 B). Identical results were obtained for severed channels lacking much of the NBD1 insertion, formed from segments 1–414 plus 433–1480 (called 414+433; Fig. 2 C). In contrast, even when we injected large amounts (5–10 ng) of cRNA for any of the individual CFTR segments, 1–633, 1–414, 433–1480, or 668–1480 alone, we never saw unitary channel openings upon exposure to 2 mM MgATP with or without 300 nM PKA: 0 channels observed in 5 patches for each segment (all patches nevertheless displayed robust Ca2+-activated Cl− currents), compared with averages of 400–600 channels per patch for coexpressed complementary NH2- plus COOH-terminal segments, in the same batches of oocytes, from ≥2-fold smaller amounts of cRNA. This is consistent with our previous studies in which these half constructs failed to produce detectable membrane currents in intact oocytes (Chan et al., 2000).
Figure 2.

Obligatory dependence on phosphorylation by PKA of macroscopic currents of severed CFTR constructs. Macropatches containing tens or hundreds of (A) WT, (B) 633+668, (C) 414+433, or (D) F633(S422A)+668 (with NH2-terminal Flag tag), channels were superfused with 2 mM MgATP and, after ∼1 min, transiently with 300 nM PKA catalytic subunit (bars); the 20-s time bar applies to all four panels A–D, which show recordings obtained at −80 mV. (E) Mean currents in MgATP alone, before (pre, black bars) or after (post, gray bars) exposure to PKA, normalized to the mean of the steady current in the presence of PKA (PKA, striped bars). Neither pre-PKA (P > 0.32) nor post-PKA (P > 0.11) currents were significantly different from those of WT for any of the three severed constructs.
To address the possibility of redundancy between the insertion and extension in mediating the response of channel gating to phosphorylation, we tested a construct in which the extension was omitted and in which the sole phosphorylatable serine in the insertion, serine 422, was mutated to alanine. Coexpression of segment Flag-3-633(S422A) (tagged at its NH2 terminus with a Flag epitope) with segment 668–1480 gave rise to severed CFTR channels (called F633 (S422A)+668) whose activity was as strictly phosphorylation dependent (Fig. 2 D) as WT. Relative to the current elicited by addition of PKA (Fig. 2 E, PKA, striped bars), that during prior exposure to ATP alone (pre, black bars) was negligibly small, 0.014 ± 0.005 (n = 21) for 633+668, 0.007 ± 0.002 (n = 21) for 414+433, and 0.014 ± 0.003 (n = 12) for F633 (S422A)+668, the same as found for WT (0.012 ± 0.005, n = 13).
Once CFTR channels have been phosphorylated, they require only MgATP for gating (Anderson et al., 1991), and channel open probability (Po) increases with the extent of phosphorylation of the ∼10 consensus serines in the R domain known to be substrates of PKA (unpublished data; see also Chang et al., 1993). In membrane patches from oocytes, a subset of these phosphoserines appears to be dephosphorylated rapidly by membrane-bound phosphatases, as witnessed by an immediate, ∼50%, decline in WT CFTR channel current upon PKA removal (Fig. 2 A; see also Csanády et al., 2000). The residual activity is then relatively stable, and declines over the course of several minutes, presumably due to slower dephosphorylation of the remaining phosphoserines. All three severed constructs lacking the NBD1 insertion or extension replicated this characteristic behavior (Fig. 2, B–D). Residual current in ATP alone, a few seconds (∼15–35 s) after PKA removal (Fig. 2 E, post, gray bars), was 0.46 ± 0.03 (n = 7) for 633+668, 0.56 ± 0.03 (n = 11) for 414+433, and 0.55 ± 0.02 (n = 13) for F633(S422A)+668, of that in the presence of PKA, just as it was for WT (0.50 ± 0.02, n = 9).
These results suggest that the strict regulation of channel activity by phosphorylation and dephosphorylation remains intact for CFTR channels that lack most of the insertion (residues 415–432) or the extension (residues 634–667) of NBD1, and even for channels lacking both the extension and serine 422 of the insertion. Because 414+433 channels (Fig. 2 C) seemed to activate more slowly than the others (Figs. 2, A, B, and D), for each construct we measured the time taken (t1/2) to reach half the maximum current amplitude upon activation by PKA. Activation of 414+433 channels (t1/2 = 33 ± 3 s, n = 18) was slightly but significantly (P = 0.007) slower than for WT (t1/2 = 22 ± 2 s, n = 24), which was comparable to the others; t1/2 was 16 ± 1 s (n = 25) for 633+668, and 21 ± 3 s (n = 7) for F633(S422A)+668. This slightly slower activation of 414+433 channels might reflect, at least partly, simply severing in that region as it was not significantly (P = 0.17) slower than that of 432+433 channels (t1/2 = 25 ± 1 s, n = 7).
Unaltered [ATP] Dependence of Macroscopic Current in Severed CFTR Channels Lacking NBD1 Insertion or Extension
CFTR channel current is a saturable function of [ATP] (Anderson and Welsh, 1992; Venglarik et al., 1994). This dependence on [ATP] is a sensitive indicator of NBD integrity and it is altered by several catalytic site mutations (Anderson and Welsh, 1992; Vergani et al., 2003). We therefore compared the apparent affinity for ATP of WT CFTR (Fig. 3 A) with that of the three severed constructs lacking the NBD1 insertion or extension (Fig. 3, B–D) by exposing prephosphorylated macropatches to test concentrations of ATP ranging from 5 μM to 1 mM, bracketed by exposures to saturating (2 mM) ATP. Fractional currents at the test [ATP], normalized to the average of the bracketing currents and plotted against test [ATP], were well fit by the Michaelis-Menten equation (Fig. 3 E), yielding Km values of 40–50 μM for 633+668, 414+433, and F633(S422A)+668, the same as that found for WT channels. Thus, the overall [ATP] dependence of channel gating was intact for these severed constructs lacking the NBD1 insertion or extension.
Shorter Bursts Cause Slightly Smaller Maximal Open Probability of Severed CFTR Channels Lacking the NBD1 Insertion
To look more closely at channel function, we examined single-channel gating kinetics in patches containing few enough (1–10) channels to allow resolution of individual gating transitions. Using protocols like those in Fig. 2, we extracted (see materials and methods) Po (Fig. 4 D), and mean burst (Fig. 4 E) and interburst durations (Fig. 4 F), in the presence of 300 nM PKA and shortly after its removal (i.e., ∼15–150 s post PKA), all at 2 mM [MgATP], for WT (Fig. 4 A), 633+668 (Fig. 4 B), and 414+433 (Fig. 4 C) CFTR constructs.
Figure 4.
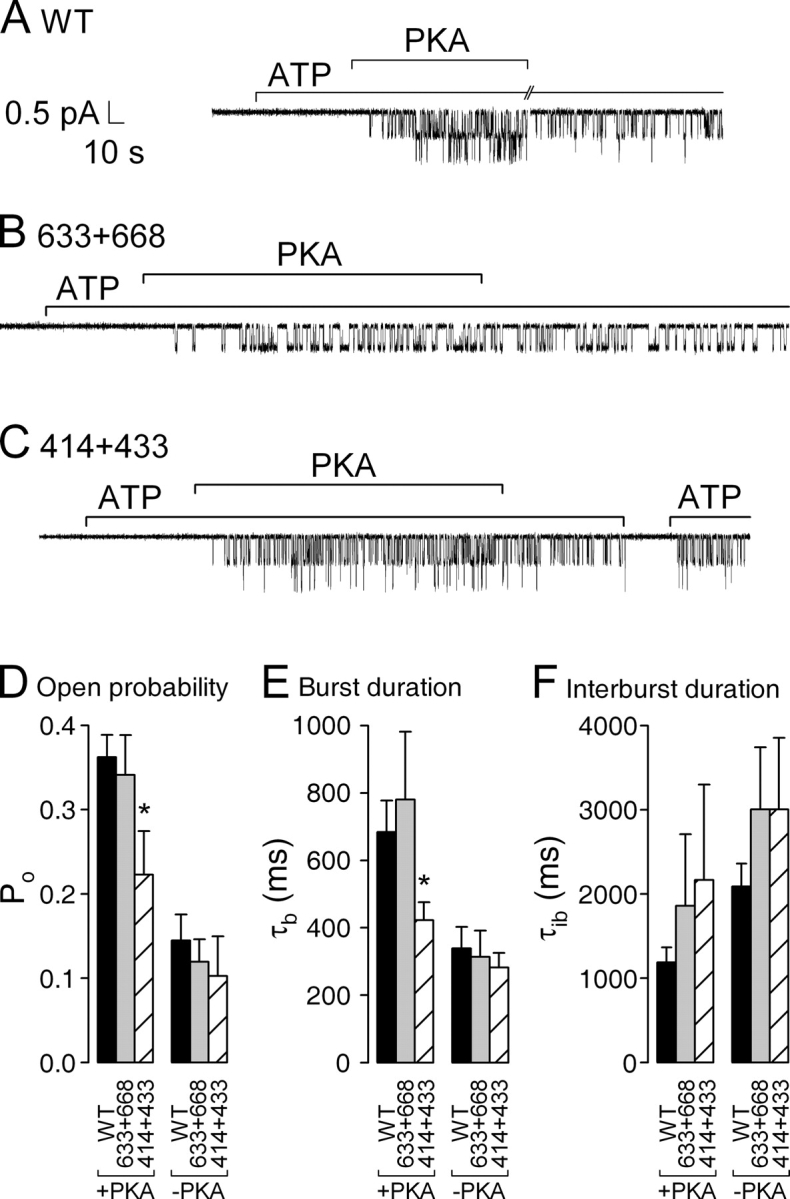
Single-channel kinetics in the presence of PKA and following its removal. Patches containing (A) two WT, (B) a single 633+668, or (C) two 414+433 channels were superfused with 2 mM MgATP; after ∼1 min 300 nM PKA catalytic subunit was transiently added (bars). Note absence of openings during exposure to ATP alone, before addition of PKA. (D) Open probabilities, (E) mean burst (τb), and (F) interburst durations (τib), in the presence of PKA+ATP (left) or in just ATP after PKA removal (right), extracted from multichannel patches (materials and methods). Po and τb values in PKA are given in the text; τib in PKA was 1190 ± 177 ms (n = 11) for WT, 1861 ± 850 ms (n = 5) for 633+668, and 2165 ± 1136 ms (n = 6) for 414+433. After PKA removal, Po was 0.14 ± 0.03 (n = 7), 0.12 ± 0.03 (n = 4), and 0.10 ± 0.05 (n = 5); τb was 339 ± 64 ms (n = 8), 314 ± 77 ms (n = 6), and 282 ± 44 ms (n = 10); and τib was 2090 ± 268 ms (n = 7), 3004 ± 736 ms (n = 4), and 3005 ± 847 ms (n = 5) for WT, 633+668, and 414+433, respectively. Asterisks in D and E indicate significantly smaller Po (P = 0.017) and significantly shorter bursts (P = 0.03), respectively, for 414+433 compared with WT.
Maximal Po in the presence of PKA (Fig. 4 D, left) was similar for WT (0.36 ± 0.03, n = 11, black bar) and 633+668 channels (0.34 ± 0.05, n = 5, gray bar), but it was slightly, though significantly, smaller for 414+433 channels (0.22 ± 0.05, n = 6, striped bar) due to their shorter burst durations in the presence of PKA (423 ± 53 ms, n = 10; Fig. 4 E, left, striped bar) compared with those of WT (684 ± 93 ms, n = 12, black bar) or 633+668 (780 ± 201 ms, n = 7, gray bar) channels.
Upon PKA removal, Po declined ≥2-fold for all three constructs (Fig. 4 D, right; see also Fig. 2) due to both a shortening of burst durations (Fig. 4 E, right) as well as a lengthening of interburst durations (Fig. 4 F, right; Csanády et al., 2000).
Unaltered [ATP] Dependence of Single-channel Gating Kinetics in Severed CFTR Channels Lacking NBD1 Insertion or Extension
We also examined in detail the ATP dependence of gating of these constructs in patches containing few channels, using the protocol of Fig. 3. For each experimental record we extracted mean burst (τb) and interburst (τib) durations and then plotted Po, closing (1/τb), and opening rate (1/τib) in test [ATP], normalized to that in 2 mM ATP, against test [ATP] (Fig. 5, D–F).
The [ATP] dependence of Po (Fig. 5 D) replicated that of macroscopic current (Fig. 3 E), Michaelis fits again yielding Km values of 40–50 μM for WT, 633+668, and 414+433 channels. For WT CFTR channels, [ATP] is known to affect Po predominantly through opening rate (Gunderson and Kopito, 1994; Venglarik et al., 1994; Winter et al., 1994; Csanády et al., 2000; Vergani et al., 2003; see inset with expanded time scale in Fig. 5 A). The same is true for both 633+668 and 414+433 channels which, like WT, had rates of closing from bursts that were largely insensitive to [ATP] over the whole range tested (5 μM to 2 mM; Fig. 5 E), whereas [ATP] stimulated opening rate of all three channel types with Km values of 40–50 μM (Fig. 5 F).
Deletion of the Insertion, but not the Extension, of NBD1 Speeds Closing from Locked-open Bursts
Pyrophosphate (PPi) and the poorly hydrolyzable ATP analogue AMPPNP, when added together with ATP, can markedly prolong open burst durations of WT CFTR channels (Hwang et al., 1994; Gunderson and Kopito, 1994). The lifetime of this “locked-open” state is on the order of tens of seconds, and is conveniently measured as the decay time constant of macroscopic current observed upon sudden removal of all nucleotides (Csanády et al., 2000). Because mutations of certain catalytic sites (Vergani et al., 2003), or severing of the CFTR backbone at particular locations, e.g., at the COOH terminus of the R domain (Csanády et al., 2000), can destabilize such locked-open states, we tested PPi and AMPPNP on our severed constructs lacking NBD1 insertion or extension. PPi (2 mM), added with ATP (0.1 mM), robustly stimulated macroscopic currents for 633+668 (Fig. 6 B), just as for WT (Fig. 6 A); although fractional enhancement of current by PPi was variable, it was perhaps slightly larger for 633+668 (2.32 ± 0.28, n = 11) than for WT (1.84 ± 0.13, n = 14), but this difference was not significant (P = 0.11). In both cases, the current relaxation upon removal of nucleotides was slow and could be reasonably fit by a single exponential (fit lines in Fig. 6, A and B), with a time constant (τ) averaging 32 ± 4 s (n = 14) for WT and 27 ± 5 s (n = 12) for 633+668 (Fig. 6 D). In contrast, PPi only marginally enhanced current amplitude in 414+433 channels (Fig. 6 C), yielding a fractional increase (1.31 ± 0.14, n = 11) significantly (P = 0.01) smaller than found for WT. Upon PPi removal from 414+433 channels, about half of the current decayed rapidly, with a time constant similar to that seen after removal of just ATP (Fig. 6 C, first fit line) or of ATP+PKA (Fig. 6 C, third fit line), largely reflecting the speed of solution exchange, while the other half declined >10-fold more slowly than solution exchange (Fig. 6 C, second fit line; see inset with expanded time scale). The time constant (τ2) and fractional amplitude (a2) of this slower component, from double-exponential fits, averaged τ2 = 7.8 ± 1.0 s and a2 = 0.65 ± 0.05 (n = 12), respectively. Even this slower component of the decay of PPi-stimulated 414+433 current was therefore substantially faster than the decay of PPi-stimulated WT current. This shorter burst duration of 414+433 channels locked open by PPi (Fig. 6 D) is fully compatible with the smaller current increase seen on adding PPi (Fig. 6 C).
Figure 6.

Relaxation of macroscopic current upon removal of PPi+ATP or AMPPNP+ATP is faster for severed 414+433 channels. Macropatches expressing hundreds of (A) WT, (B) 633+668, or (C) 414+433 channels were superfused with either 2 mM or 0.1 mM (together with PPi) MgATP, 300 nM PKA catalytic subunit, and 2 mM Mg-PPi, as indicated by bars; the 40-s time bar applies to all three panels A–C. In A and B, smooth lines through data are single-exponential fits, τ = 56 s for WT (A), τ = 30 s for 633+668 (B). In C, all three instances of current decay were fitted (smooth green lines); first and third relaxations were fit with single exponentials yielding τ = 422 ms and τ = 353 ms, respectively; the relaxation following PPi removal was fit with two exponentials yielding time constants and fractional amplitudes of τ1 = 400 ms, τ2 = 6.5 s, a1 = 0.56, a2 = 0.44. (D) Average τ for WT and 633+668 from single-exponential fits, and τ2 for 414+433 from double-exponential fits, to decaying currents after removal of PPi+ATP. (E) Average τ2 for WT, 633+668, and 414+433 from double-exponential fits to decaying currents after removal of AMPPNP+ATP. Asterisks indicate significantly shorter slow time constants for 414+433 compared with WT, in D (P = 10−5) and in E (P = 0.049).
Similar results were obtained when AMPPNP (1 mM) was used with ATP (0.1 mM) to lock channels in the open burst state. For WT and 633+668, the slow component accounted for a fractional amplitude of 0.83 ± 0.10 and 0.89 ± 0.05, respectively, of the total decaying current, and slow time constants (Fig. 6 E), from double-exponential fits, were 23 ± 8 s (n = 5) for WT and 33 ± 7 s (n = 9) for 633+668; for 414+433, the slow component had a fractional amplitude of 0.33 ± 0.03 (n = 7) and a time constant of 8.0 ± 1.4 s (Fig. 6 E).
Unaltered Unitary Conductance of Severed CFTR Channels Lacking NBD1 Insertion or Extension
As for WT, the single-channel current–voltage relationships in symmetrical 140 mM [Cl−] were linear for both 633+668 and 414+433 channels (Fig. 7), and single-channel conductances were 6.4 ± 0.3 pS (n = 5) for 633+668 and 6.3 ± 0.2 pS (n = 5) for 414+433, not significantly different (P > 0.48) from WT (6.2 ± 0.2 pS, n = 6). Thus, our data provide no indication that any major perturbation of the pore architecture is caused by omission of the NBD1 insertion or extension.
Figure 7.

Single-channel conductance is unaltered in severed-NBD1 constructs. Representative examples of single-channel current–voltage (I–V) relationships in symmetrical 140 mM Cl− for WT, 633+668, and 414+433 CFTR channels. Slopes of straight lines fitted by linear regression yield single-channel conductances; mean values are given in the text.
DISCUSSION
Does NBD1 Insertion or Extension Mediate Phosphorylation-dependent Regulation of Gating?
We have investigated the proposed roles of the insertion and extension of CFTR's NBD1 (not found so far in other NBD structures) in phosphorylation-dependent regulation and ATP-dependent gating of CFTR channels, by examining split channel constructs from which the bulk of those regions was omitted. Because our severed half molecules produced no measurable membrane currents when RNA encoding any one of them was injected alone, we can be certain that all currents analyzed here flowed through channels built from two coexpressed, complementary, polypeptide chains and that therefore contained all CFTR residues (1–1480) other than the regions we deleted from the two short nonconserved segments.
WT channel gating is under strict regulation by phosphorylation; Po of WT channels in excised patches exposed to ATP before addition of PKA is only ∼1% of that of phosphorylated channels (Fig. 2, A and E). This PKA-dependent regulation of channel gating, believed mediated largely through the R domain, reflects predominantly an inhibitory influence of the dephosphorylated R domain, because a contiguous construct with a large part (residues 708–835) of the R domain deleted (Rich et al., 1991) and a split CFTR construct lacking the entire R domain (residues 634–836; cut-ΔR; Csanády et al., 2000) both gave rise to channels that were constitutively activated, i.e., they opened and closed in the presence of MgATP without prior exposure to PKA. Although the activity of cut-ΔR channels was slightly stimulated by exposure to PKA, Po of nonphosphorylated cut-ΔR channels exposed to MgATP alone was already at least half that of phosphorylated WT channels (Csanády et al., 2000).
The insertion and extension of CFTR's NBD1, both of which contain serines in consensus sequences for phosphorylation by PKA, have been suggested to play a role in keeping nonphosphorylated channels shut, possibly by interfering with the formation of the NBD1–NBD2 dimer (Lewis et al., 2004) proposed to represent the channel open state (Vergani et al., 2003). We considered the four following a priori possibilities for the postulated inhibitory roles of these CFTR-specific segments. Prevention of opening (i.e., of NBD dimer formation) of nonphosphorylated channels could be mediated by (1) the insertion alone, or (2) the extension alone, or (3) both the insertion and the extension working in concert. Alternatively, there might be redundancy, such that (4) either the insertion or the extension is sufficient to keep nonphosphorylated channels shut. Our results clearly rule out channel inhibition via the insertion alone (possibility 1) since 414+433 channels (lacking the bulk of the insertion) require PKA for opening (Fig. 2, C and E), or via the extension alone (possibility 2) since nonphosphorylated 633+668 channels (lacking the extension) are also shut before exposure to PKA (Fig. 2, B and E), and hence also via both insertion and extension working together (possibility 3). Lack of success to date in coassembling CFTR channels by coexpression of three separate polypeptide chains has precluded a direct test, by double deletion, of possibility 4 that the presence of either of these two structural segments is sufficient to inhibit NBD dimer formation in nonphosphorylated channels. We could, however, simultaneously delete the extension and render the insertion unresponsive to phosphorylation by mutating its only phosphorylatable residue, serine 422, to alanine. The resulting construct, F633(S422A)+668, yielded channels that, like WT, remained closed until they were phosphorylated, and then activated normally upon phosphorylation (Fig. 2, D and E). We therefore conclude that neither the insertion nor the extension of NBD1 is a key element essential for PKA-dependent regulation. What, then, are the functions of these two CFTR-specific NBD1 segments?
Functional Influence of NBD1 Insertion
When we examined the gating of 414+433 channels, which lack most of the NBD1 insertion, we found that [ATP] dependence of macroscopic current (Fig. 3, C and E) and [ATP] dependence of single-channel gating kinetics (Fig. 5, C–F) all remained unaltered. Although ATP binding at both NBD sites seems necessary, presumably to allow formation of the ATP-bound NBD1–NBD2 dimer, before a CFTR channel can open (Vergani et al., 2003), the NBD1 composite site (formed by NBD1 Walker A and B sequences together with the NBD2 ABC signature sequence) of WT channels binds ATP extremely tightly, with a high apparent affinity (Aleksandrov et al., 2001, 2002; Basso et al., 2003). The lower apparent affinity (∼50 μM) for ATP stimulation of CFTR channel opening rate (and Po) is therefore proposed to reflect rate-limiting binding of ATP at the NBD2 composite catalytic site (formed by the NBD2 Walker sequences plus the NBD1 signature sequence), unless ATP binding at the NBD1 site is drastically impaired by mutation (Vergani et al., 2003). Our results indicate that deletion of the CFTR-specific NBD1 insertion, anchored in close proximity to the NBD1 composite site, nevertheless does not impair ATP binding there sufficiently for it to limit channel opening, as the [ATP] dependence of 414+433 channel opening matches that of WT, and hence still reflects the (unaltered) affinity of the NBD2 composite site for ATP.
However, the maximal open probability of 414+433 channels in the presence of PKA was ∼60% that of WT (Fig. 4 D), due to somewhat (∼1.6-fold) faster closing from bursts (Fig. 4 E). The impact of a structural perturbation on the free energy, relative to the open burst state, of the transition state for closure from a burst is given by ΔΔGT-O ‡=−kTln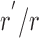 , where r' and r are the rates of closure from a burst (see materials and methods) for channels with or without the structural perturbation, respectively (k, Boltzmann's constant; T, temperature in °K). Expressed in these energetic terms, the effect of deleting the NBD1 insertion is a rather small lowering of the free-energy barrier for closure, as indicated by a ΔΔGT-O ‡ of only −0.5 ± 0.2 kT. The effects of the absence of the insertion on locked-open bursts in the presence of mixtures of ATP with PPi, or with AMPPNP, were only slightly larger. Deletion of residues 415–432 decreased the lifetime of locked-open bursts by 4.1-fold in the case of PPi (Fig. 6, C and D) and by 2.9-fold in the case of AMPPNP (Fig. 6 E), relative to WT. These reductions indicate a lowering of the free-energy barrier for closure from the locked-open state, ΔΔGT-Locked ‡, amounting to −1.4 ± 0.2 kT for the PPi-locked state, and −1.1 ± 0.4 kT for the AMPPNP-locked state.
, where r' and r are the rates of closure from a burst (see materials and methods) for channels with or without the structural perturbation, respectively (k, Boltzmann's constant; T, temperature in °K). Expressed in these energetic terms, the effect of deleting the NBD1 insertion is a rather small lowering of the free-energy barrier for closure, as indicated by a ΔΔGT-O ‡ of only −0.5 ± 0.2 kT. The effects of the absence of the insertion on locked-open bursts in the presence of mixtures of ATP with PPi, or with AMPPNP, were only slightly larger. Deletion of residues 415–432 decreased the lifetime of locked-open bursts by 4.1-fold in the case of PPi (Fig. 6, C and D) and by 2.9-fold in the case of AMPPNP (Fig. 6 E), relative to WT. These reductions indicate a lowering of the free-energy barrier for closure from the locked-open state, ΔΔGT-Locked ‡, amounting to −1.4 ± 0.2 kT for the PPi-locked state, and −1.1 ± 0.4 kT for the AMPPNP-locked state.
Most likely, both the normal open burst duration and the locked-open burst duration reflect stability of an NBD1–NBD2 dimer. During normal gating, evidence suggests that ATP is bound at both composite sites before hydrolysis of the ATP at the NBD2 site eventually leads to dissociation of the dimer, and hence to channel closure (Vergani et al., 2003). But, in locked-open channels, at least one of the sites is presumably occupied by the poorly hydrolyzable analogue, resulting in a more stable dimer whose slower dissociation is reflected in the slower channel closure. In this view, both the normal and the locked-open NBD1–NBD2 dimer appear somewhat destabilized in 414+433 channels. Alternatively, the structural perturbation might alter the strict coupling between the NBD dimer and the channel gate, allowing locked-open channels to close before dimer dissociation. Although we observed reductions in burst duration and locked-open lifetime, comparable to those shown here for 414+433 channels, for CFTR channels simply severed near the COOH terminus of the R domain (between residues 835 and 837; Csanády et al., 2000), the functional effects on 414+433 channels found here must be ascribed to the physical absence of residues 415–432 rather than to backbone discontinuity, as channels simply severed before residue 433 (from coexpression of segments 1–432 plus 433–1480) show normal burst durations (Chan et al., 2000) and normal locked-open lifetimes in PPi or AMPPNP. For instance, split 1–432 + 433–1480 CFTR channels gave a τ of 36 ± 7 s (n = 7) for unlocking after exposure to PPi.
Functional Influence of NBD1 Extension
Deletion of residues 634–667 of the extension (comprising mostly α-helices H9 and H9b; Fig. 1), which includes PKA substrate serine 660, neither prevented PKA-mediated regulation (Fig. 2, B and E; Fig. 4, B and D–F) nor modified the nucleotide dependence of gating (Fig. 3, B and E; Fig. 5, B and D–F) or the stability of locked-open bursts (Fig. 6, B, D, and E). In contrast, deletion of the 11 residues preceding amino acid 634 (residues 623–633, comprising β-strand S10 and most of α-helix H8; Fig. 1) resulted in complete loss of CFTR maturation and hence channel function (Chan et al., 2000). Considered together, these results imply that S10 and H8 are essential components of the core structure of NBD1, whereas helices H9 and H9b are not. More likely, then, the extension in fact belongs to the R domain (see below), which contains multiple sites for phosphorylation by PKA, most of which have yet to be assigned a specific regulatory role (for review see, e.g., Gadsby and Nairn, 1999). Indeed, the single point mutation S660A was found to have a negligible effect on the gating of phosphorylated CFTR channels in excised patches exposed to saturating [ATP] (Winter and Welsh, 1997). This echoes the lack of effect we find of deleting most of the “NBD1 extension,” including Ser 660, on the dependence of gating on phosphorylation at high [PKA] (Figs. 2, 4, and 6), or on the regulation of gating by nucleotides (Figs. 3 and 5). These results on gating of strongly phosphorylated CFTR channels in excised patches do not contradict the finding (Wilkinson et al., 1997) of a ≤2-fold reduction in sensitivity to activation by IBMX (hence presumably by PKA) of S660A or S670A CFTR channels in intact oocytes. Possibly, some corresponding modest impairment of gating of 633–668 channels might become evident if they were to be examined after partial activation by submaximal [PKA].
NBD1 Domain Boundaries Revisited
Given the new structural and functional data, it might be worth revisiting the definition of the NH2- and COOH-terminal boundaries of NBD1. Although we previously assigned the NH2-terminal boundary of NBD1 to near amino acid 433, because residues 415–432, but not residues 433–448, could be deleted without obvious loss of function (Chan et al., 2000), we acknowledged the conundrum these deletion results posed in light of the first published crystal structures of NBDs of the homologous ABC proteins RbsA (Armstrong et al., 1998) and HisP (Hung et al., 1998). The difficulty being that our NBD sequence alignments, hampered by low sequence homology near the NH2 terminus of NBD1, aligned omissible CFTR residues 415–432 with those of the first β-strand in HisP and RbsA, i.e., with the central strand of the ABC-specific antiparallel β-sheet (see Fig. 1, strand S1 in cyan β-sheet); we noted our reasonable concern that deletion of that central strand would be expected to disrupt the entire β-sheet. The recently solved structure of CFTR's NBD1 (Lewis et al., 2004) has resolved this apparent paradox, by demonstrating that the region we deleted comprises a surface segment inserted uniquely (among the many NBD structures solved to date) into NBD1 of CFTR (Fig. 1, red dotted line), whereas strand S1, encompassing CFTR residues 392–401 (Fig. 1, cyan), would have remained intact in our deletion construct. The new structure makes it clear that the NH2-terminal boundary of CFTR's NBD1 occurs around residue 390.
On the other hand, NBD1's COOH-terminal boundary is not so clear. Lewis et al. (2004) proposed that NBD1 extends to near amino acid 670 on the basis of optimal expression efficiency, in Escherichia coli, of mouse CFTR constructs extending to that region. Though it remains unclear why shorter constructs did not express in the bacteria, the robust expression and intact function of our 633+668 construct demonstrate clearly that NBD1 of human CFTR folds correctly in the vertebrate Xenopus cells when truncated at residue 633, after α-helix H8 (Fig. 1; see also Chan et al., 2000). Helix H9 and associated helix H9b both lie on the surface of NBD1 and appear flexible (Lewis et al., 2004). Moreover, there is little homology in the primary sequence of ABC proteins beyond helix H8 of CFTR (residue 635), and the close fit in three-dimensional space between the backbone of CFTR's NBD1 and those of solved NBD structures of other ABC proteins degrades in that region; e.g., before helix H8 for MJ0796 (Yuan et al., 2001), after helix H8 for BtuD (Locher et al., 2002), RbsA (Armstrong et al., 1998), MalK (Diederichs et al., 2000), and MJ1267 (Karpowich et al., 2001), or after helix H9 for HlyB (Schmitt et al., 2003), human TAP1 (Gaudet and Wiley, 2001), GlcV (Verdon et al., 2003a), and HisP (Hung et al., 1998). Thus, both structure conservation and our functional results argue that helix H9b (which includes phosphorylatable serines 660 and 670) is more likely part of the R domain, as previously proposed (e.g., Riordan et al., 1989; Gadsby and Nairn, 1999; Chan et al., 2000). Indeed, helix H9 might also be considered part of the R domain, given the favorable close packing (hydrophobic interactions) between H9 and H9b (Lewis et al., 2004). Alternatively, helix H9 could be viewed as a short linker between NBD1 and the R domain. These arguments place the COOH-terminal boundary of NBD1 at the end of helix H8, i.e., around CFTR residue 635, or, possibly, the end of helix H9, i.e., around residue 645.
Together, our results suggest that neither the insertion nor the extension of NBD1 is an essential component of the strict PKA-mediated disinhibition of CFTR channel gating. Perhaps, in the context of an intact CFTR channel, interactions with other structural elements cause both insertion and extension to adopt positions unlike those observed in the monomeric NBD1 crystals. However, our findings do not rule out other functional roles for the insertion and extension. It appears that the insertion can modulate the stability of the open-burst state of the channel, and so might help stabilize the proposed NBD1–NBD2 dimer, perhaps in a phosphorylation-dependent manner. The extension probably ought to be considered part of the R domain, and, through its two consensus PKA sites, it might contribute to the incremental regulation of CFTR channel open probability by phosphorylation.
Acknowledgments
We thank Dr. Paola Vergani for helpful discussions, David C. Kopsco, Nazim Fataliev, and Dr. Beáta Törőcsik for molecular biology support, and Dóra Takács for isolation and injection of oocytes.
This work was supported by National Institutes of Health (NIH) DK51767 and NIH Fogarty International Center award R03-TW05761.
William N. Green served as editor.
Abbreviations used in this paper: ABC, ATP-binding cassette; CFTR, cystic fibrosis transmembrane conductance regulator; NBD, nucleotide binding domain; WT, wild-type.
References
- Aleksandrov, L., A. Mengos, X. Chang, A. Aleksandrov, and J.R. Riordan. 2001. Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 276:12918–12923. [DOI] [PubMed] [Google Scholar]
- Aleksandrov, L., A.A. Aleksandrov, X.B. Chang, and J.R. Riordan. 2002. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277:15419–15425. [DOI] [PubMed] [Google Scholar]
- Anderson, M.P., and M.J. Welsh. 1992. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science. 257:1701–1704. [DOI] [PubMed] [Google Scholar]
- Anderson, M.P., H.A. Berger, D.P. Rich, R.J. Gregory, A.E. Smith, and M.J. Welsh. 1991. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 67:775–784. [DOI] [PubMed] [Google Scholar]
- Armstrong, S., L. Tabernero, H. Zhang, M. Hermodson, and C. Stauffacher. 1998. The 2.5 Å structure of the N-terminal ATP-binding cassette of the ribose ABC transporter. Biophys. J. 74:A338. [Google Scholar]
- Basso, C., P. Vergani, A.C. Nairn, and D.C. Gadsby. 2003. Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating. J. Gen. Physiol. 122:333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K.W., L. Csanády, D. Seto-Young, A.C. Nairn, and D.C. Gadsby. 2000. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol. 116:163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, X.B., J.A. Tabcharani, Y.X. Hou, T.J. Jensen, N. Kartner, N. Alon, J.W. Hanrahan, and J.R. Riordan. 1993. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem. 268:11304–11311. [PubMed] [Google Scholar]
- Chen, J., G. Lu, J. Lin, A.L. Davidson, and F.A. Quiocho. 2003. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 12:651–661. [DOI] [PubMed] [Google Scholar]
- Csanády, L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády, L., K.W. Chan, D. Seto-Young, D.C. Kopsco, A.C. Nairn, and D.C. Gadsby. 2000. Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs, K., J. Diez, G. Greller, C. Muller, J. Breed, C. Schnell, C. Vonrhein, W. Boos, and W. Welte. 2000. Crystal structure of MalK, the ATPase subunit of the trehalose/maltose ABC transporter of the archaeon Thermococcus litoralis. EMBO J. 19:5951–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetsch, E.E., and A.L. Davidson. 2002. Vanadate-catalyzed photocleavage of the signature motif of an ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA. 99:9685–9690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby, D.C., and A.C. Nairn. 1999. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79:S77–S107. [DOI] [PubMed] [Google Scholar]
- Gaudet, R., and D.C. Wiley. 2001. Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J. 20:4964–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson, K.L., and R.R. Kopito. 1994. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. J. Biol. Chem. 269:19349–19353. [PubMed] [Google Scholar]
- Hopfner, K.P., A. Karcher, D.S. Shin, L. Craig, L.M. Arthur, J.P. Carney, and J.A. Tainer. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 101:789–800. [DOI] [PubMed] [Google Scholar]
- Hung, L.W., I.X. Wang, K. Nikaido, P.Q. Liu, G.F. Ames, and S.H. Kim. 1998. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 396:703–707. [DOI] [PubMed] [Google Scholar]
- Hwang, T.C., G. Nagel, A.C. Nairn, and D.C. Gadsby. 1994. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 91:4698–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janas, E., M. Hofacker, M. Chen, S. Gompf, C. van der Does, and R. Tampe. 2003. The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J. Biol. Chem. 278:26862–26869. [DOI] [PubMed] [Google Scholar]
- Kaczmarek, L.K., K.R. Jennings, F. Strumwasser, A.C. Nairn, U. Walter, F.D. Wilson, and P. Greengard. 1980. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase enhances calcium action potentials of bag cell neurons in cell culture. Proc. Natl. Acad. Sci. USA. 77:7487–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowich, N., O. Martsinkevich, L. Millen, Y.R. Yuan, P.L. Dai, K. MacVey, P.J. Thomas, and J.F. Hunt. 2001. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure (Camb). 9:571–586. [DOI] [PubMed] [Google Scholar]
- Lewis, H.A., S.G. Buchanan, S.K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W.B. Guggino, W.A. Hendrickson, et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher, K.P., A.T. Lee, and D.C. Rees. 2002. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098. [DOI] [PubMed] [Google Scholar]
- Loo, T.W., M.C. Bartlett, and D.M. Clarke. 2002. The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing Walker A sequence. J. Biol. Chem. 277:41303–41306. [DOI] [PubMed] [Google Scholar]
- Moody, J.E., L. Millen, D. Binns, J.F. Hunt, and P.J. Thomas. 2002. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277:21111–21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard, L.S., D.P. Rich, L.G. DeBerg, and M.J. Welsh. 1997. Association of domains within the cystic fibrosis transmembrane conductance regulator. Biochemistry. 36:1287–1294. [DOI] [PubMed] [Google Scholar]
- Rich, D.P., R.J. Gregory, M.P. Anderson, P. Manavalan, A.E. Smith, and M.J. Welsh. 1991. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 253:205–207. [DOI] [PubMed] [Google Scholar]
- Riordan, J.R., J.M. Rommens, B. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, and J.L. Chou. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- Schmitt, L., H. Benabdelhak, M.A. Blight, I.B. Holland, and M.T. Stubbs. 2003. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J. Mol. Biol. 330:333–342. [DOI] [PubMed] [Google Scholar]
- Sheppard, D.N., and M.J. Welsh. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S45. [DOI] [PubMed] [Google Scholar]
- Smith, P.C., N. Karpowich, L. Millen, J.E. Moody, J. Rosen, P.J. Thomas, and J.F. Hunt. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani, J.A., X.B. Chang, J.R. Riordan, and J.W. Hanrahan. 1991. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 352:628–631. [DOI] [PubMed] [Google Scholar]
- Urbatsch, I.L., G.A. Tyndall, G. Tombline, and A.E. Senior. 2003. P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J. Biol. Chem. 278:23171–23179. [DOI] [PubMed] [Google Scholar]
- Venglarik, C.J., B.D. Schultz, R.A. Frizzell, and R.J. Bridges. 1994. ATP alters current fluctuations of cystic fibrosis transmembrane conductance regulator: evidence for a three-state activation mechanism. J. Gen. Physiol. 104:123–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon, G., S.V. Albers, B.W. Dijkstra, A.J. Driessen, and A.M. Thunnissen. 2003. a. Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations. J. Mol. Biol. 330:343–358. [DOI] [PubMed] [Google Scholar]
- Verdon, G., S.V. Albers, N. van Oosterwijk, B.W. Dijkstra, A.J. Driessen, and A.M. Thunnissen. 2003. b. Formation of the productive ATP-Mg2+-bound dimer of GlcV, an ABC-ATPase from Sulfolobus solfataricus. J. Mol. Biol. 334:255–267. [DOI] [PubMed] [Google Scholar]
- Vergani, P., A.C. Nairn, and D.C. Gadsby. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J.E., M. Saraste, M.J. Runswick, and N.J. Gay. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, D.J., T.V. Strong, M.K. Mansoura, D.L. Wood, S.S. Smith, F.S. Collins, and D.C. Dawson. 1997. CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. 273:L127–L133. [DOI] [PubMed] [Google Scholar]
- Winter, M.C., and M.J. Welsh. 1997. Stimulation of CFTR activity by its phosphorylated R domain. Nature. 389:294–296. [DOI] [PubMed] [Google Scholar]
- Winter, M.C., D.N. Sheppard, M.R. Carson, and M.J. Welsh. 1994. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys. J. 66:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, Y.R., S. Blecker, O. Martsinkevich, L. Millen, P.J. Thomas, and J.F. Hunt. 2001. The crystal structure of the MJ0796 ATP-binding cassette. Implications for the structural consequences of ATP hydrolysis in the active site of an ABC transporter. J. Biol. Chem. 276:32313–32321. [DOI] [PubMed] [Google Scholar]