The mouse and human Ah receptor differ in recognition of LXXLL motifs (original) (raw)
. Author manuscript; available in PMC: 2009 Mar 15.
Published in final edited form as: Arch Biochem Biophys. 2008 Jan 26;471(2):215–223. doi: 10.1016/j.abb.2008.01.014
Abstract
The aryl hydrocarbon receptor (AhR) is a ligand inducible transcription factor that exhibits interspecies differences, with the human and mouse AhR C-terminal transactivation domain sharing only 58% amino acid sequence identity. The AhR has a transactivation domain comprised of proline/serine/threonine-rich, glutamine-rich and acidic amino acid subdomains. A truncated mAhR and hAhR containing only the acidic subdomain displayed widely differing transactivation potentials. Whether the glutamine-rich subdomain of the mouse AhR and the human AhR differentially recruit LXXLL-motif coactivators was investigated. Transiently expressed GAL4 DNA binding domain (GAL4DBD)-LXXLL-motif fusion proteins were used to map the critical LXXLL binding sequence of the hAhR to amino acid residues 663-688. Several LXXLL-motif GAL4DBD fusion proteins dramatically differed in their ability to influence the transactivation potential of the mAhR and hAhR. These findings suggest that the human and mouse AhR may display differential recruitment of coactivators and hence may exhibit divergent regulation of target genes.
Keywords: Ah receptor, LXXLL, coactivators, TCDD, glutamine-rich, dioxin
Introduction
The Aryl hydrocarbon receptor (AhR1) is a ligand activated transcription factor. The AhR has a somewhat promiscuous ligand binding site and thus binds a structurally diverse group of compounds [1]. AhR ligands include polycyclic aromatic hydrocarbons, halogenated polycyclic aromatic hydrocarbons, as well as atypical ligands such as carbaryl and myristicin [1]. 2,3,7,8-tetrachlorodibenzo-_p_-dioxin (TCDD) is considered a prototypical high affinity ligand for the AhR, which mediates most of the toxic and carcinogenic affects of TCDD exposure. Exposing rodents to TCDD results in a number of adverse biological effects, including thymic atrophy, immunotoxicity, tumor promotion, teratogenicity, reproductive toxicity, hepatotoxicity, chloracne, and birth defects; while in humans the hallmark of TCDD exposure is chloracne [2]. Studies in Ahr null mice demonstrate that the AhR plays a physiological role in liver, cardiac vasculature development and ovarian follicle maturation [3–5]. A human AhR “knock-in” mouse model has been generated to explore whether the human AhR can mediate the same spectrum of toxicity as the mouse AhR. Studies revealed that TCDD-mediated toxicity differed between the hAhR and murine AhRd mice, suggesting that indeed the human AhR may differ in its ability to regulate gene expression [6].
In its inactive state the AhR exists as part of a stable multiprotein complex comprised of two molecules of heat shock protein 90 (Hsp90), immunophilin-like AhR interacting protein (AIP, also known as XAP2 and ARA9) and p23. Hsp90 interaction with the human AhR is less stable and requires the presence of molybdate in buffers to stabilize the receptor/Hsp90 complex [7, 8]. The interaction of XAP2 with the AhR/Hsp90 complex appears to differentially affect the mouse and human AhR. For example, XAP2 remains bound to the hAhR upon translocation into the nucleus, while XAP2 must dissociate from the mAhR prior to translocation [9]. Upon ligand binding the receptor translocates to the nucleus and heterodimerizes with the Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT), apparently through ARNT mediated displacement of Hsp90 [10, 11]. The AhR/ARNT heterodimer then binds to dioxin responsive elements (DREs) and activates gene expression through recruitment of coactivator complexes and subsequent histone modifications. The AhR has a C-terminal transactivation domain (TAD) consisting of proline/serine/threonine-rich (P/S/T), glutamine (Q-rich) and acidic subdomains, each of which function independently and exhibit varying levels of activation [12–14]. The AhR has been shown to directly regulate a number of genes involved in phase I and II xenobiotic metabolism, such as CYP1A1, CYP1A2, CYP1B1, glutathione S-transferase Ya, and UDP-glucuronsyltransferase 1A1 [15]. The list of genes directly regulated by the AhR continues to grow, with direct target genes being identified with diverse biological activities, such as epiregulin and slug [16, 17]. The AhR has also been shown to modulate gene expression through non-classical mechanisms such as recruitment to estrogen receptor (ER) bound to its cognate response element, as well as the ability to inhibit NFkB activity [18, 19].
Coactivators are central components involved in gene regulation and overall transcriptional activity. A number of coactivators have been previously shown to interact with the human and mouse AhR transactivation domain (TAD), including the steroid receptor coactivator 1 (SRC-1), nuclear coactivator 2 (NcoA2, SRC-2), p300/CBP co-integrator protein (p/CIP), GAC63 and CREB binding protein (CBP) [20–22]. Interestingly, the co-regulator RIP140 has been shown to both enhance and repress AhR transcription, dependent on the level of RIP140 expression [23]. To date, the spectrum of coactivators that have been shown to be recruited to the AhR is similar to what has been observed with the ER despite having divergent TAD motifs.
Many protein-protein interactions involving coactivators, co-repressors and nuclear receptors are mediated by LXXLL motifs [24], which are short α-helical sequences, consisting of conserved leucine residues flanked by non-conserved residues. Coactivators often feature multiple LXXLL motifs in different nuclear receptor (NR) boxes capable of interacting with various receptors. A number of coactivators including pCIP, TIF-1α, GRIP1, and SRC-1, have been shown to bind to the AF2 domains of nuclear receptors through their LXXLL motifs [25]. LXXLL motifs are a conserved feature of the coactivator receptor interacting domain; a region which facilitates ligand-dependent direct interactions with nuclear receptors [24]. A variety of experimental approaches, including site-directed mutagenesis and peptide competition experiments, have provided strong evidence for the requirement of these LXXLL motifs in the interactions of coactivators with ligand-activated receptors [24, 26, 27]. Additionally, other studies have used phage-displayed peptide libraries to demonstrate that LXXLL peptides interacted only with agonist-bound ER [28]. Interestingly, LXXLL motifs appear to be part of a receptor specific code whereby different nuclear receptors preferentially bind distinct NR boxes of the receptor interacting domain [29]. The interaction of ER and other receptors may also be influenced by the sequences flanking these LXXLL motifs [27]. Therefore, different transcription factors may interact with different regions of a coactivator, or show preferential binding or affinity to specific subsets of coactivators, based on the sequences of the non-conserved LXXLL motif residues [24, 26, 30]. Receptor ligands have also been shown to direct NR box binding selectivity. For example, troglitazone-bound peroxisome proliferator activated receptor γ preferred NR box ii over box i, while the opposite was observed in indomethacin-treated cells [31]. Recently, the ability of ligands to modulate the specificity of coactivator recruitment to a hAhR-VP16 chimeric protein was tested in mammalian two-hybrid assay [32]. Indeed, the type of ligand bound to the AhR can have a profound effect on the specificity of coactivator recruitment. The LXXLL motifs of SRC-1 have been shown to directly interact with the Q-rich region of the hAhR and are required for SRC-1-mediated enhanced AhR transcriptional activity [33]. We therefore decided to investigate whether divergent TAD structure of the mAhR and hAhR results in the differential recruitment of coactivators. Using a DRE-driven cell-based reporter assay, we have found that the acidic subdomain is sufficient to mediate mAhR transcriptional activity. In contrast, the acidic domain was insufficient to mediate hAhR transcriptional activity. The mouse and human AhR displayed differences in transcriptional activity in response to increasing amounts of GAL4-LXXLL motif fusion proteins in DRE-driven cell-based reporter assays. The LXXLL interacting domain of the hAhR was mapped to amino-acids 663-688, a region previously shown to be indispensable for hAhR receptor transcriptional activity [34]. This evidence suggests that the lack of TAD conservation between the mouse and human AhR may result in distinct molecular function and differential regulation of gene expression in response to ligand activation.
Materials and Methods
Plasmids
The pMsx vectors capable of mediating expression of LXXLL motif peptide-GAL4DNA-binding domain (GAL4DBD) fusion proteins were obtained from Dr. Donald P. McDonnell (Duke University) [27]. The isolated subdomains of hAhR transactivation domain (TAD) were transiently expressed using the following vectors; pEF1/V5-HisA-SP163-GST/NLS, pEF1/V5-HisA-SP163-GST-acidic-hAhR/NLS, pEF1/V5-HisA-SP163-GST-Qrich-hAhR/NLS and pEF1/V5-HisA-SP163-GST-P/S/T-hAhR/NLS, generated as described previously [35]. pEF1/V5-HisA-SP163-GST-Qrich-hAhR/NLS plasmid was used to generate deletion mutants, GST-Q663 (Δ664-713), GST-Q688 (Δ689-713), and GST-Q663-688 (Δ606-662) using the loop-out mutagenesis approach using QuikChange site-directed mutagenesis kit (Stratagene). The luciferase reporter vector pGUDLUC6.1 was a generous gift from Dr. Michael Denison (University of CA, Davis) and the pFR2-Luc reporter plasmid was purchased from Stratagene. pDJM/βgal obtained from Dr. M. W. McBurney consists of a β-galactosidase reporter gene under the control of the murine _pgk_-1 (phosphoglycerate kinase 1) promoter, which is constitutively active in mammalian cells [36]. The pSG5-SRC-1a vector was a gift from Dr. Malcolm Parker [37]. The plasmid pcDNA3-βmAhR was used as a template for generation of plasmids for expression of mAhR deletion mutants, stop codons at appropriate sites were introduced by site-directed mutagenesis and a QuikChange site-directed mutagenesis kit following manufacturer’s instructions. The pEF-V5-hAHR deletion constructs were generated previously [34]. The mAhR and hAhR Q-rich GAL4DBD constructs were generated by PCR amplification of mouse and hAhR cDNA encompassing amino acid sequence 665-690 or 663-688, using the following primer pairs 5′-CCGGAATTCAATTGTCCACAGCAAGAC-3′, and 5′-CCCAAGCTTTTATTTGTAGGGGAACTCTTG-3′ or 5′-CCCAAGCTTTTATTTGTAGGGAAATTCCTG-3′ and CCGGAATTCAACTGTCCCCAGCAG-3′ for the human and mouse AhR, respectively. The PCR generated products and pM GAL4DBD plasmid (BD Biosciences, San Jose CA.) were each digested with HindIII and EcoR1 restriction enzymes and ligated together.
Cell cultures and transient transfections
COS-1 cells were routinely grown in αMEM (Sigma), supplemented with 10% fetal bovine serum (Hyclone), 100 units of pencillin/ml and 100 μg streptomycin/ml. Cultures were maintained under standard conditions in a 37°C incubator at 5% CO2, 95% air and media was changed after every 48 h. For transient transfection, cells were seeded in 6-well tissue culture plates 24 h prior to transfection, and were transfected using LipofectAMINE Plus reagent (Invitrogen, Carlsbad, CA), according to manufacturer’s instructions for reporter gene assays. In the LXXLL motif-containing GAL4DBD fused peptide library reporter assays, cells were transfected with 1.5 μg of total DNA, consisting of 200 ng of the DRE-driven luciferase reporter plasmid pGUDLUC 6.1 and the pDJM/βGAL constitutively expressed β-galactosidase, 10 ng of pCI-hAhR or pcDNA3-mAhR, along with variable amounts of pM-GAL4DBD-LXXLL or pM constructs. After 24 h, transfected cells were treated with 10 nM TCDD for 8 h prior to lysis. In reporter assays involving 5X GAL4-Luc reporter (pFR) driven transactivation, cells were transiently transfected with a total of 1.5 μg of DNA comprised of 200 ng of pDJM/βGAL control, 300 ng of pFR reporter, 500 ng of D11 or D14 or D43 or 293 plasmid selected from the LXXLL-peptide library, and 500 ng of either GST, GST-acidic, or GST-Q-rich or GST-P/S/T-rich plasmid or GST-Q-rich deletion-mutants (GST-Q663, GST-Q688 and GST-Q663-688). Cells were harvested 24 h after transfection and assayed for luciferase activity normalized to GST activity. In the reporter assay involving truncated mAhR and hAhR; COS-1 cells were transiently transfected with 200 ng of receptor, 100 ng of DRE-driven luciferase reporter, and 200 ng of DJM-β-galactosidase using LipofectAMINE plus (Invitrogen, Carlsbad, CA). Cells were transfected and treated 24 h later with 10 nM TCDD for 6 h, and the cell free lysate assayed for luciferase activity. For reporter assays transiently expressing mouse or hAhRQ-rich-GAL4DBD fusion proteins, COS-1 cells were transfected in 6-well plates with 2 μg of pM-GAL4DBD-mAhRQ-rich(663-688) or pM-GAL4DBD, along with increasing amounts of pSG5-SRC-1a, 100 ng of constitutively expressed pDJM/β-gal plasmid and 400 ng of pFR2-5XGAL4 vector. Luciferase activity was measured 24 h after transfection and normalized to β-galactosidase activity.
Statistical analysis
Mouse or hAhRQ-rich-GAL4DBD reporter assays were done in triplicate and statistical analysis was performed using one-way ANOVA using GraphPad Prism (GraphPad Software Inc. San Diego, CA).
Immunoblotting
Transiently transfected COS-1 cells were lysed in MENG buffer (25 mM MOPS, 2mM EDTA, 0.02% NaN3, 10% glycerol pH 7.4) containing 1% NP-40 + protease inhibitors (Sigma) and centrifuged at 10,000 g for 120 min. The supernatants (50 μg protein) were separated on 8% SDS-Tricine polyacrylamide gels. The proteins were transferred to PVDF membrane and the AhR detected with monoclonal antibody RPT1 (Affinity BioReagents). GAL4DBD fusion proteins were detected using anti-GAL4DBD antibody (sc-577, Santa Cruz Biotechnology). Primary antibodies were visualized with I125-secondary antibodies (GE Healthcare) and autoradiography.
Results
Transcriptional activity of the TAD subdomains of the mouse and human AhR
Since the mouse and human AhR TAD primary protein structures share limited homology we decided to investigate whether the role of the TAD subdomains in receptor transcriptional activity differ for each receptor. COS-1 cells were transiently transfected with P/S/T, Q-rich or acidic domain deletion mutants of the mouse and human AhR in a DRE-driven reporter assay. As shown previously, deletion of the Q-rich subdomain (hAhR-599) of the human receptor completely blocks both TCDD-induced and constitutive hAhR transcriptional activity (Fig. 1) [34]. For both receptors, the Q-rich subdomain was shown to be important for AhR transcriptional activity, as expected. However, deletion of the mAhR’s Q-rich subdomain (mAhR-593) reduced, but did not entirely abrogate, constitutive or inducible mAhR activity. This suggests that the acidic subdomain of the mAhR may also contribute to overall mAhR-mediated transcriptional activity, in contrast to this domain in the hAhR.
Figure 1. Role of TAD subdomains in mouse and human AhR transactivation potential.
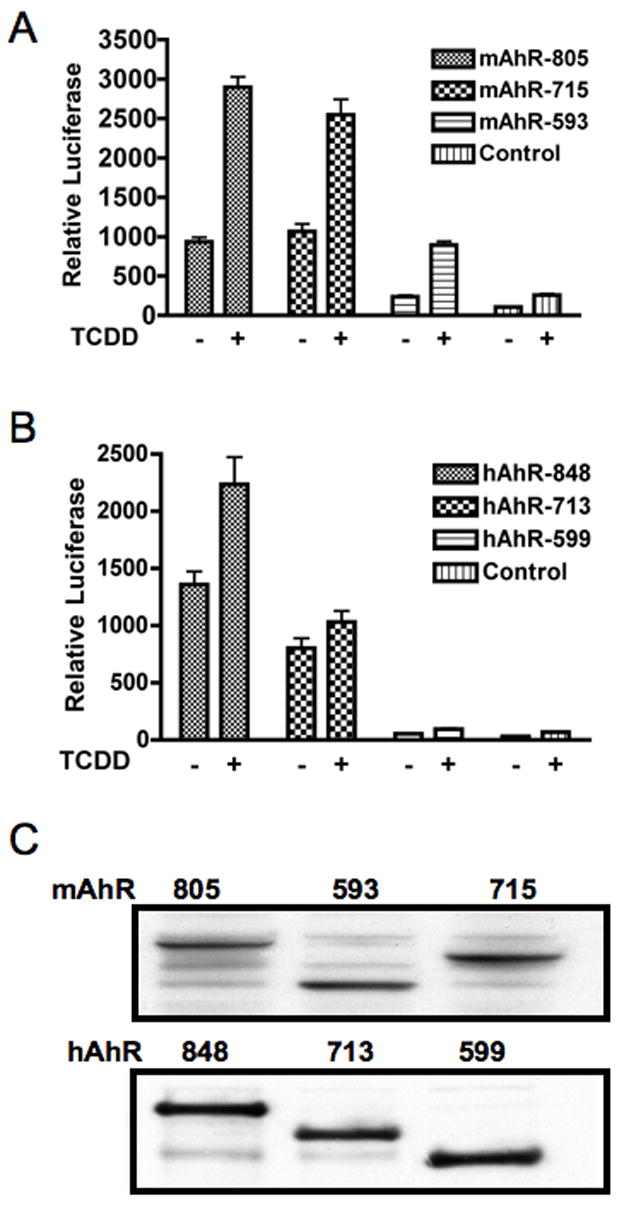
TAD subdomain deletion mutants of the mouse and human AhR were transiently transfected into COS-1 cells and assayed for their transcriptional activity in response to TCDD activation. The 713/715 receptor mutants have the P/S/T region deleted, while the 593/599 receptor mutants have the P/S/T- and Q-rich domains deleted.
LXXLL-peptide library differently modulates mouse and human AhR mediated transcriptional activity
The peptide sequence; LXXLL, has previously been shown to be an important core motif involved in cofactor binding to ligand-activated receptors [24, 30]. In order to investigate whether there are possible differences in coactivator recruitment between the human and mouse AhR, we compared the ability of a library of LXXLL motif-GAL4DBD fusion proteins to alter the transcriptional activity of the mAhR and hAhR in cell-based reporter assays. The library of LXXLL motif containing GAL4DBD proteins was originally used by Chang et al. [27], and they have been classified into categories, depending on the sequences flanking the conserved LXXLL motif. The Class I peptides contain a conserved serine at the −2 position and a positively charged residue arginine at the −1 position. Class II peptides have a proline occupying the −2 position and a hydrophobic leucine residue directly preceding the LXXLL motif. Two of the three peptides in class II also contain a charged histidine at the −3 position. Class III peptides share a conserved serine or threonine at the −2 position, followed by a hydrophobic leucine or isoleucine at the −1 position (Fig. 2). Different classes of LXXLL peptides were transiently co-transfected with mouse or human AhR and DRE-driven luciferase reporter in COS-1 cells. COS-1 cells express a very low level of AhR and thus serve as an appropriate model system to examine transiently expressed receptor. Table 1 shows the LXXLL library peptides screened for their potential effect on mAhR or hAhR receptor transcriptional activity in these transfection experiments. This initial screen indicated there were significant differences in the ability of certain LXXLL fusion proteins to alter AhR transcriptional activity in a species-dependent manner. Seven LXXLL-GAL4DBD fusion proteins were selected for further analysis; increasing amounts of these fusion proteins were expressed and they differentially modulated mouse and human AhR transcriptional activity (Fig. 3). The seven-peptide sequences selected also represent different classes of LXXLL peptides. The representative LXXLL-peptides corresponding to the LXXLL motifs of SRC-1 enhanced both mouse and human AhR transactivation (Table 1); this result is consistent with the previous studies that establish that SRC-1 can interact with the hAhR [20]. Interestingly the LXXLL-peptide #293, which has been previously shown to specifically inhibit ERβ transcriptional activity [27], and also inhibited hAhR and mAhR activity here (Table 1). Other classes of LXXLL peptides, such as the Class I peptides D2 or D11 and the Class III peptides D22 or D43, enhanced mouse AhR activity but repressed hAhR transcriptional activity (Table 1 and Fig 3). Both mAhR and hAhR-mediated transactivation potential was repressed by co-expression of Class II peptides C33 or D14. However, hAhR-dependent transactivation was repressed more than that of the mAhR (Table 1 and Fig 3). The differential ability of different classes of LXXLL peptides to alter mouse versus human AhR transcriptional activity suggests that coactivator or co-repressor recruitment may differ between these receptors.
Figure 2. LXXLL motif-containing GAL4DBD peptide library.

LXXLL-peptides are classified into three different classes based on sequences flanking the conserved LXXLL motif. Peptide # 293 was generated from a random peptide library and has been shown to bind to activated ERβ [27]. Three distinct LXXLL motifs found in the GRIP-1 and SRC-1 coactivators are also shown. This figure has been adapted from a published figure [29].
Table 1.
Differential effect of LXXLL-motif fused GAL4DBD-peptides on mouse and human AhR-mediated transactivation potential
| LXXLL Peptides | mAhR | hAhR |
|---|---|---|
| D2 | + | − |
| D11 | + | − |
| D30 | + | + |
| C33 | − | − |
| D14 | − | − |
| F4 | NC | NC |
| F6 | NC | NC |
| D15 | − | − |
| D22 | + | NC |
| D40 | NC | NC |
| D43 | NC | − |
| D48 | − | − |
| SRC1 | + | + |
| GRIP | + | + |
| 293 | − | − |
Figure 3. Mouse and human AhR exhibit differences in mediating transactivation of a DRE-driven reporter gene when co-expressed with LXXLL-peptides.
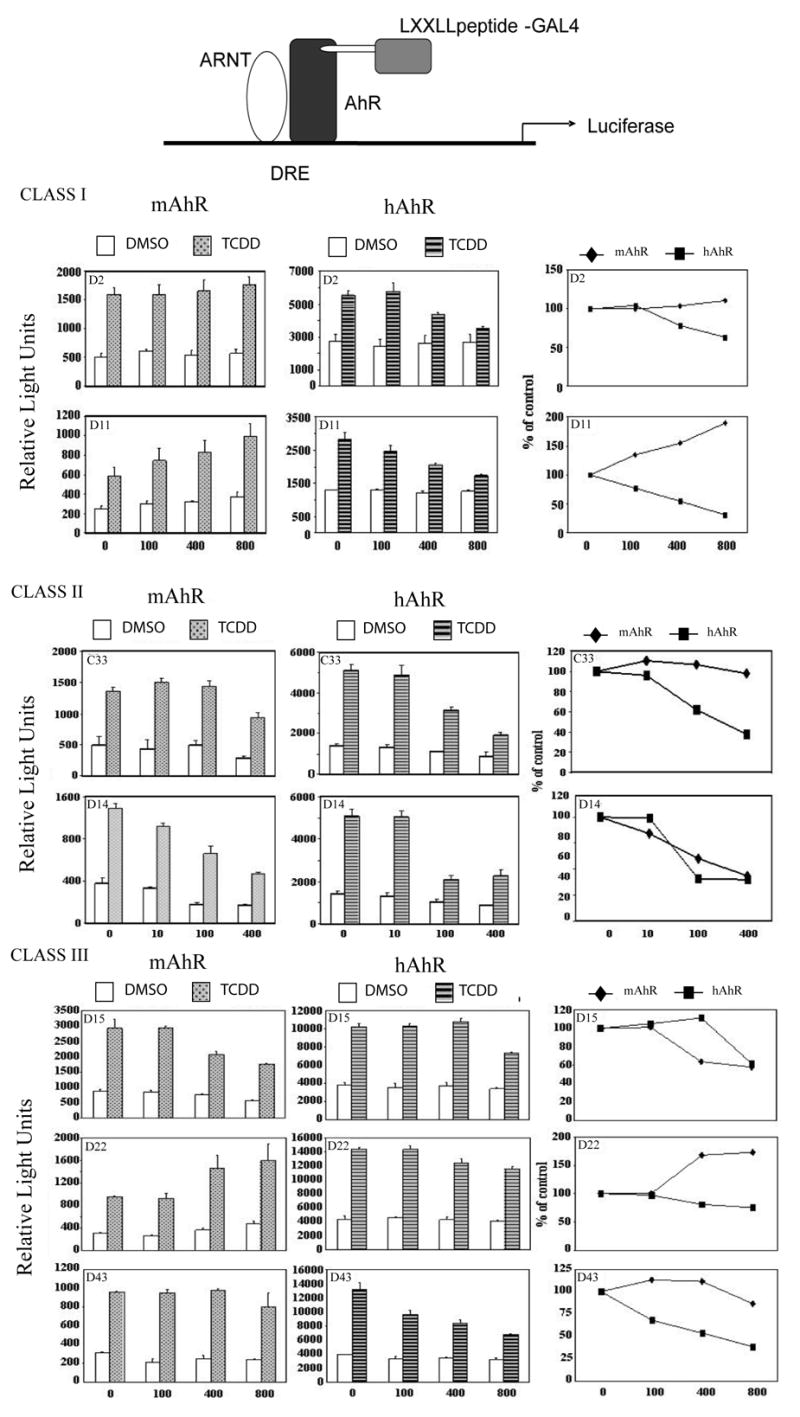
COS-1 cells were co-transfected in 6-well plates with 10 ng of human or mouse AhR expressing construct, 200 ng of DRE-driven luciferase reporter plasmid pGUDLUC 6.1, 200 ng of internal control pDJM/βgal and variable amounts of LXXLL motif-containing peptides or control plasmid pM. Cells were treated for 8 h with 10 nM TCDD or carrier solvent. 24 h after transfection, luciferase activity was measured and normalized to β-galactosidase activity. The far right panel illustrates the affect of each class of LXXLL peptide on the percent reporter activity relative to control.
Isolated subdomains of the hAhR TAD modulate LXXLL-mediated transcriptional activity
We wanted to identify the subdomains of the AhR TAD that were capable of modulating LXXLL-peptide-GAL4 transcriptional activity. This was accomplished using the GST fusion proteins; GST-Q-rich, GST-P/S/T-rich and GST-acidic co-expressed with four different LXXLL-GAL4 fusion proteins, which exhibited an ability to modulate the transcriptional activity of the mouse and human AhR were selected. D11, D14, D43 and #293 plasmids were co-transfected with pFR-Luc reporter plasmid (5XGAL4-Luc) in the presence or absence of GST, GST-Q-rich or GST-P/S/T-rich or GST-acidic subdomains expressing constructs of the hAhR (Fig 4). Co-transfection of the LXXLL-peptides D11, D14, D43, and #293 plasmids in the presence of GST-Q-rich or GST-P/S/T subdomain fusion protein induced a marked increase in reporter activity (Fig 4B). Also no change in reporter expression in the presence of the GST-Acidic subdomain was observed. These findings suggest that both the P/S/T-rich and Q-rich subdomains may be involved in LXXLL-dependent coactivator recruitment by the activated hAhR.
Figure 4. Modulation of GAL4DBD-LXXLL-peptide fusion protein transcriptional activity by specific subdomains of hAhR TAD. (A).
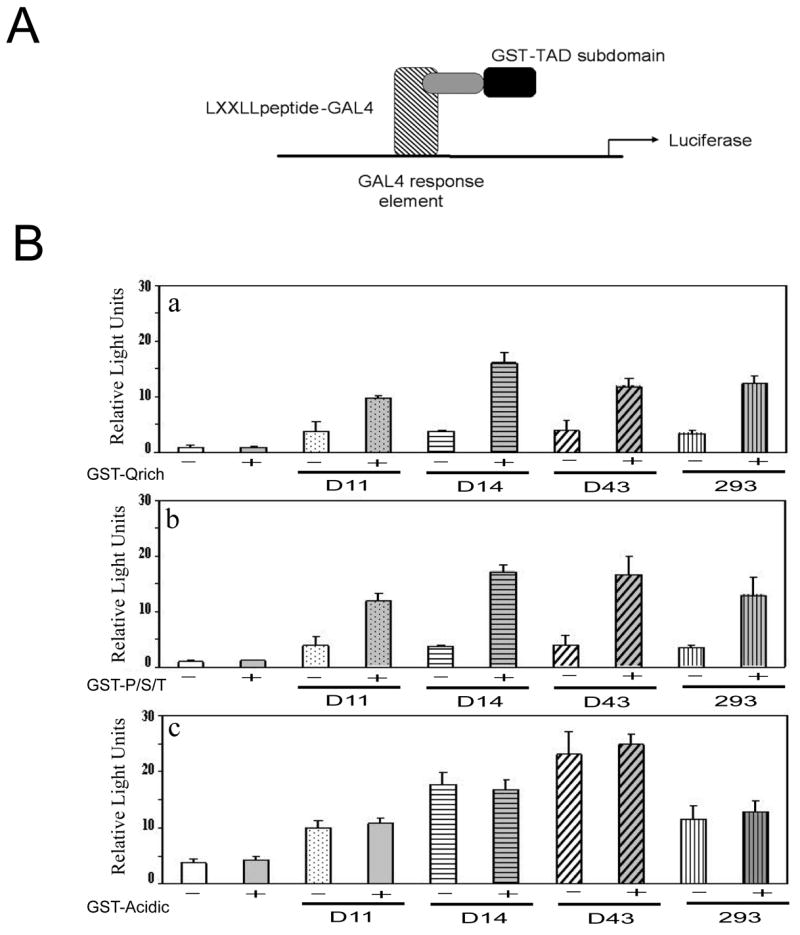
Schematic model of transcriptional assay utilized in B. (B) COS-1 cells were transfected with 300 ng of 5XGAL4Luc-driven luciferase reporter, pFR, 200 ng of internal control pDJM/βgal and 500 ng of either D11 or D14 or D43 or #293 in presence (shaded bars) or absence (clear bars) of 500 ng GST or GST-Q-rich (AA 600–712), GST-P/S/T-rich (AA 713–848) GST-acidic (AA 500–599) domains. Luciferase activity in the presence of GST was equivalent to background reporter activity without GST expression and was subtracted from the data presented. Luciferase activity was measured and normalized to β-galactosidase activity 24 h after transfection.
Mapping the region of the Q-rich subdomain of hAhR that modulates LXXLL-motif-mediated transcriptional activity
Previous studies have shown that the Q-rich domain, and more precisely, the region between 663-688 of the hAhR, is necessary and sufficient for AhR transactivation [34]. Furthermore, a leucine residue 678 was required for hAhR-mediated transactivation of a DRE-driven reporter gene construct. Also the Q-rich region can directly bind to SRC-1 and RIP140 [33, 34]. To expand upon these results, we decided to test whether hAhR amino acid sequence 663-688 was directly involved in LXXLL motif recruitment, with the ultimate goal being to determine if this region of the TAD differs in a specific-dependent manner. Three GST fusion proteins were co-expressed, GST-Q600-688, GST-Q600-663, and GST-Q663-688 in a one mammalian hybrid GAL4-driven luciferase assay with D11, D14, D43 or #293 selected LXXLL motif-GAL4DBD fusion proteins (Fig 5). Co-expression of GST-Q600-688 and peptides D11, D14, D43 or #293 resulted in enhanced luciferase expression (2–3 fold) but no increase in reporter expression was seen with GST-Q600-663 and the same peptides. This data suggests peptide residues 663-688 are sufficient for recruitment and binding of the LXXLL motif peptides.
Figure 5. Modulation of GAL4DBD-LXXLL-peptide fusion protein transcriptional activity by segments of the Q-rich subdomain of hAhR TAD.
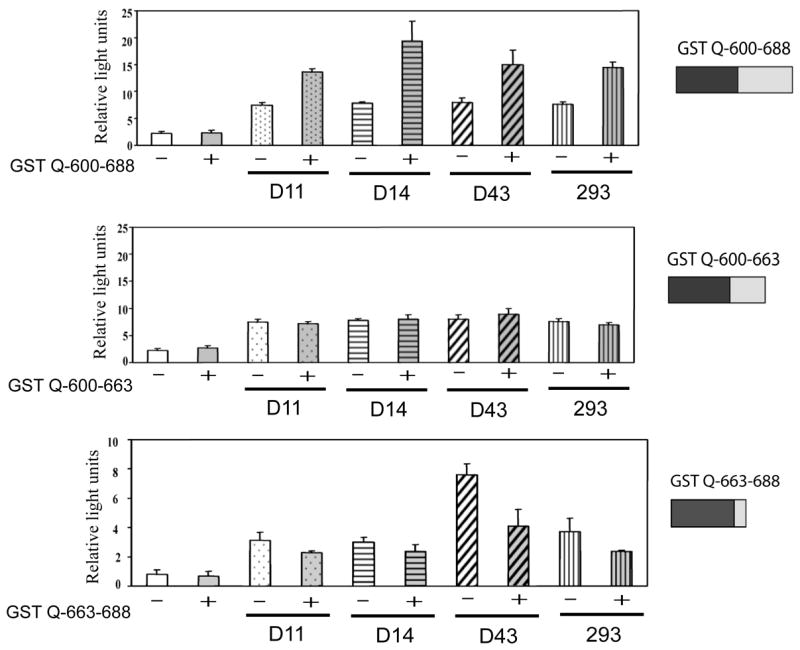
COS-1 cells were co-transfected with 1.5 μg of total DNA. The cells were transfected with 300 ng pFR, 200 ng of internal control pDJM/βgal, 500 ng of either D11 or D14 or D43 or 293 in presence or absence of 500 ng pEF1/GST-Q600-688, pEF1/GST-Q600-663 or pEF1/GST-Q663-688.
Coactivators modulate mouse and human AhR Q-rich-mediated transcriptional activity
The core sequence of the Q-rich region of the mouse and human AhR are somewhat divergent as revealed in figure 6A, conserved residues are underlined. The human and mouse GAL4DBD-Q-rich region (663-688) were co-expressed with increasing amounts of SRC-1, previously shown to directly interact with the hAhR [21, 33], along with a pFR reporter construct. Transient expression of SRC-1 along with the mouse and human Q-rich (663-688) GAL4DBD fusion proteins significantly enhanced the expression of the GAL4 reporter construct, suggesting that both receptors may utilize their Q-rich subdomain for interaction with LXXLL motifs in the NR boxes of coactivators (Fig 6B). However, no significant difference in SRC-1-mediated enhanced transactivation potential was observed between the mouse and human Q663-688 region.
Figure 6. Mouse and human AhR Q-rich subdomain region 663-688 transcriptional activity is enhanced by SRC-1 expression. (A).

The human and mouse AhR have divergent amino acid sequences within their Q-rich subdomains, amino acid residues 663-688 or 665-690, respectively. (B) The human and mouse GAL4DBD-AhR-Q-rich fusion proteins display enhanced transcriptional activity upon co-expression of the coactivator SRC-1. (C) Protein blot analysis demonstrating expression of mouse and human Q-richGAL4DBD fusion proteins in COS-1 cells. COS-1 cells were transfected in 6-well plates with 2μg of GAL4DBD fused to human, mouse AhR Q-rich (663-688) subdomains or GAL4DBD pM control plasmid, along with increasing amounts (20–40 ng) of the coactivators SRC-1 (100 ng) of constitutively expressed pDJM/βgal plasmid and 400 ng of pFR2 5XGAL4 driven luciferase reporter. Luciferase activity was measured 24 h after transfection and normalized to β-galactocidase activity, (*) represents significant differences (P< 0.05) between control (0ng) and SRC-1 transfected samples (20 and 40ng of SRC-1 plasmid).
Discussion
The mAhR has a ~10-fold higher affinity for TCDD than the hAhR, yet interspecies comparisons of the mouse and human AhR have mainly focused on reduced ligand binding affinity as a defining component of hAhR activity and related gene transactivation [38, 39]. Consequently other traits that define hAhR activity such as coactivator recruitment have not been thoroughly investigated and incorporated into studies aimed at comparing the mAhR and hAhR. There is 86% amino acid identity in the N-terminal regions and only 58% identity in the C-terminal regions between mAhR and hAhR. Interestingly, many of the proline resides in the TAD are not conserved. Here we have provided evidence that this lack of conservation between the mouse and human AhR TADs might actually result in differences in LXXLL motif recognition. Using GAL4 one-hybrid assays we were able to map the region that functionally interacts with LXXLL motifs to residues 663-688 of Q-rich subdomain of the human AhR TAD (Fig 5). Although, we have been unable to demonstrate direct interaction between GST 663-688 and LXXLL peptides in vitro (data not shown). This region was also necessary to mediate transcription of a hAhR mutants with progressive deletion from the carboxyl end [34]. Interestingly this region of both receptors is not completely conserved as shown in Fig 6A, which suggests that the structural differences between each receptor may actually contribute to the contrasting interactions of each receptor with LXXLL-motif peptides. The isolated Q-rich regions (663-688) of both the mouse and human receptor, however, did not show differences in their interaction with SRC-1 (Fig 6B) whereas the full-length mAhR and hAhR did show differential interaction with LXXLL peptides (Fig 3). This would suggest that receptor-coactivator interaction is complex and may also be influenced by other factors such as AhR protein folding and other non-LXXLL-motif interactions.
The TAD subdomains of each receptor revealed dissimilar roles in transactivation potential for the acidic domain in the mouse and human AhR. In this study we demonstrated that the mouse AhR acidic subdomain was independently able to activate reporter gene expression, in agreement with an earlier study [40]. Also the acidic domain of the hAhR has been shown to be insufficient to mediate receptor driven transcriptional activity [34]. However these results were surprising considering that the hAhR and mAhR acidic subdomains share significant homology, in particular the critical regions within the mouse acidic subdomain previously mapped, amino acid residues 539-546 and 563-570 are conserved [41]. However, a comparison of amino acid residues between 515-530 in the mouse and human AhR reveals two proline residues present in the mouse receptor are absent in the hAhR. This may suggest that the tertiary structure of the mAhR versus hAhR acidic subdomain may differ. These results imply that the hAhR TAD directs coactivator binding to the Q-rich region whereas the mAhR may utilize both the acidic and Q-rich TAD subdomains for coactivator recruitment and gene activation. In addition, the acidic domain was previously shown not to bind SRC-1 and data presented here failed to demonstrate an effect of the acidic domain on LXXLL-GAL4DBD-mediated transcriptional activity [33]. Whether the human and mouse P/S/T subdomains differ in LXXLL motif recruitment remains to be explored. However, we have established that GST-P/S/T fusion protein can modulate LXXLL-GAL4DBD transcriptional activity, suggesting that the P/S/T can interact with LXXLL motifs. Perhaps the number of regions each receptor utilizes in coactivator binding may actually influence the relative stability of the mouse and human AhR/coactivator interactions as well. Whether the relative stability of AhR/coactivator interactions will result in differences between human and mouse AhR signaling has not yet been evaluated.
The non-conserved residues in LXXLL motifs have been shown to be important in the selective recognition of proteins by functionally different nuclear receptors [27]. In this study, a number of different classes of LXXLL peptides differentially enhanced human and mouse AhR transcriptional activity, suggesting that each receptor may differentially interact with coactivators. Although coactivators share functional overlap, they can display distinct functional characteristics. For example, microinjection of expression plasmids for SRC-1 or NCoA-2, but not pCIP, were able to rescue retinoic acid receptor-dependent activation in SRC-1 immunodepleted cells, suggesting a non-redundant role of members of the SRC-1 coactivator family [42]. Therefore, the differential recruitment of LXXLL motif-containing coactivators by the mouse and human AhR could lead to differentially regulate gene expression through selective recruitment of coactivators. Future studies utilizing mice that express the hAhR can be utilized to directly test this hypothesis.
Acknowledgments
We thank Dr. Donald P. McDonnell for the LXXLL motif peptide fusion protein expressing plasmid library. We also thank Marcia H. Perdew for editorial review of this manuscript. This study was funded by Dow Chemical Company and NIH grants ES04869 and ES011834.
Footnotes
1
Abbreviations used: AhR, aryl hydrocarbon receptor; TCDD, 2,3,7,8-tetrachlorodibenzo-_p_-dioxin; ARNT, AhR nuclear translocator; DRE, dioxin responsive element; TAD, transactivation domain; P/S/T, proline/serine/threonine; SRC-1, steroid receptor coactivator 1; NR, nuclear receptor; ER, estrogen receptor; GST, glutathione-S-transferase
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Denison MS, Nagy SR. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 2.Poland A, Knutson JC. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt JV, Su GHT, Reddy JK, Simon MC, Bradfield CA. Proc Natl Acad Sci. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott BD, Schmid JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Toxicol Appl Pharmacol. 1999;155:62–70. doi: 10.1006/taap.1998.8601. [DOI] [PubMed] [Google Scholar]
- 5.Lund AK, et al. Toxicol Appl Pharmacol. 2003;193:177–187. doi: 10.1016/j.taap.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Moriguchi T, Motohashi H, Hosoya T, Nakajima O, Takahashi S, Ohsako S, Aoki Y, Nishimura N, Tohyama C, Fujii-Kuriyama Y, Yamamoto M. Proc Natl Acad Sci. 2003;100:5652–5657. doi: 10.1073/pnas.1037886100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, Perdew GH, Bradfield CA. J Biol Chem. 1997;272:8581–8593. doi: 10.1074/jbc.272.13.8581. [DOI] [PubMed] [Google Scholar]
- 8.Manchester DK, Gordon SK, Golas CL, Roberts EA, Okey AB. Cancer Res. 1987;47:4861–4868. [PubMed] [Google Scholar]
- 9.Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Biochemistry. 2004;43:700–709. doi: 10.1021/bi035827v. [DOI] [PubMed] [Google Scholar]
- 10.Perdew GH. Arch Biochem Biophys. 1991;291:284–290. doi: 10.1016/0003-9861(91)90136-7. [DOI] [PubMed] [Google Scholar]
- 11.McGuire J, Whitelaw ML, Pongratz I, Gustafsson JA, Poellinger L. Mol Cell Biol. 1994;14:2438–2446. doi: 10.1128/mcb.14.4.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain S, Dolwick KM, Scmidt JV, Bradfield CA. J Biol Chem. 1994;269:31518–31524. [PubMed] [Google Scholar]
- 13.Sogawa K, Iwabuchi K, Abe H, Fuji-Kuriyama AY. J Cancer Res Clin Oncol. 1995;121:612–620. doi: 10.1007/BF01197779. [DOI] [PubMed] [Google Scholar]
- 14.Rowlands JC, McEwan IJ, Gustafsson J. Mol Pharmacol. 1996;50:538–548. [PubMed] [Google Scholar]
- 15.Kohle C, Bock KW. Biochem Pharmacol. 2007;73:1853–1862. doi: 10.1016/j.bcp.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Patel RD, Kim DJ, Peters JM, Perdew GH. Toxicol Sci. 2006;89:75–82. doi: 10.1093/toxsci/kfi344. [DOI] [PubMed] [Google Scholar]
- 17.Ikuta T, Kawajiri K. Exp Cell Res. 2006;312:3585–3594. doi: 10.1016/j.yexcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 19.Ruby CE, Leid M, Kerkvliet N. Mol Pharmacol. 2002;62:722–728. doi: 10.1124/mol.62.3.722. [DOI] [PubMed] [Google Scholar]
- 20.Hankinson O. Arch Biochem Biophys. 2005;433:379–386. doi: 10.1016/j.abb.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 21.Beischlag TV, Wang S, Rose DW, Torchia J, Reisz-Porszasz S, Muhammad K, Nelson WE, Probst MR, Rosenfeld MGHO. Mol Cell Biol. 2002;22:4319–4333. doi: 10.1128/MCB.22.12.4319-4333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YH, Beischlag TV, Kim JH, Perdew GH, Stallcup MR. J Biol Chem. 2006;281:12242–12247. doi: 10.1074/jbc.M512537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar MB, Tarpey RW, Perdew GH. J Biol Chem. 1999;274:22155–22164. doi: 10.1074/jbc.274.32.22155. [DOI] [PubMed] [Google Scholar]
- 24.Heery DM, et al. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 25.Ding XF, Anderson CM, Ma H, Hong H, Uht RM, Kushner PJ, Stallcup MR. Mol Endocrinol. 1998;12:303–313. doi: 10.1210/mend.12.2.0065. [DOI] [PubMed] [Google Scholar]
- 26.Litterst CM, Phitzner E. J Biol Chem. 2002;277:36052–36060. doi: 10.1074/jbc.M203556200. [DOI] [PubMed] [Google Scholar]
- 27.Chang CY, et al. Mol Cell Biol. 1999;19:8226–8239. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, Hamilton PT, Fowlkes DMMDP. Science. 1999;285:744. doi: 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- 29.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker D, et al. Mol Cell Biol. 1999;19:5601–5607. doi: 10.1128/mcb.19.8.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG. Genes Dev. 1998;12 doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S, Rowlands C, Safe S. Toxicol Appl Pharmacol. 2007 doi: 10.1016/j.taap.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar MB, Perdew GH. Gene Expr. 1999;8:273–286. [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar MB, Ramadoss P, Reen RK, Vanden Heuvel JP, Perdew GH. J Biol Chem. 2001;276:42302–42310. doi: 10.1074/jbc.M104798200. [DOI] [PubMed] [Google Scholar]
- 35.Reen RK, Cadwallader A, Perdew GH. Arch Biochem Biophys. 2002;408:93–102. doi: 10.1016/s0003-9861(02)00518-0. [DOI] [PubMed] [Google Scholar]
- 36.Adra CN, Boer PH, McBurney MW. Gene. 1987;60:65–74. doi: 10.1016/0378-1119(87)90214-9. [DOI] [PubMed] [Google Scholar]
- 37.Kalkhoven E, Valentine JE, Heery DM, Parker MG. EMBO J. 1998;17:232–243. doi: 10.1093/emboj/17.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramadoss P, Perdew GH. Mol Pharmacol. 2004;66:129–136. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- 39.Harper PA, Golas CL, Okey AB. Cancer Res. 1988;48:2388–2395. [PubMed] [Google Scholar]
- 40.Ko HP, Okino ST, Ma Q, Whitlock JP., Jr Mol Cell Biol. 1997;17:3497–3507. doi: 10.1128/mcb.17.7.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones LC, Whitlock JP., Jr J Biol Chem. 2001;276:25037–25042. doi: 10.1074/jbc.M102910200. [DOI] [PubMed] [Google Scholar]
- 42.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]