Protein Phosphatases Decrease Sarcoplasmic Reticulum Calcium Content by Stimulating Calcium Release in Cardiac Myocytes (original) (raw)
Abstract
Phosphorylation/dephosphorylation of Ca2+ transport proteins by cellular kinases and phosphatases plays an important role in regulation of cardiac excitation−contraction coupling; furthermore abnormal protein kinase and phosphatase activities have been implicated in heart failure. However, the precise mechanisms of action of these enzymes on intracellular Ca2+ handling in normal and diseased hearts remains poorly understood. We have investigated the effects of protein phosphatases PP1 and PP2A on spontaneous Ca2+ sparks and SR Ca2+ load in myocytes permeabilized with saponin. Exposure of myocytes to PP1 or PP2A caused a dramatic increase in frequency of Ca2+ sparks followed by a nearly complete disappearance of events. These effects were accompanied by depletion of the SR Ca2+ stores, as determined by application of caffeine. These changes in Ca2+ release and SR Ca2+ load could be prevented by the inhibitors of PP1 and PP2A phosphatase activities okadaic acid and calyculin A. At the single channel level, PP1 increased the open probability of RyRs incorporated into lipid bilayers. PP1-medited RyR dephosphorylation in our permeabilized myocytes preparations was confirmed biochemically by quantitative immunoblotting using a phosphospecific anti-RyR antibody. Our results suggest that increased intracellular phosphatase activity stimulates RyR-mediated SR Ca2+ release leading to depleted SR Ca2+ stores in cardiac myocytes.
In heart muscle cells, the process of excitation-contraction (EC) coupling is mediated by Ca2+ influx through sarcolemmal L-type Ca2+ channels activating Ca2+ release channels (ryanodine receptors, RyRs) in the sarcoplasmic reticulum (SR). Once activated, the RyR channels allow Ca2+ to be released from the SR into the cytosol to induce contraction. This mechanism is known as Ca2+-induced calcium release (CICR) (Fabiato, 1985; Bers, 2002). During relaxation, most of the Ca2+ is resequestered into the SR by the Ca2+-ATPase. The amount of Ca2+ released and the force of contraction depend on the magnitude of the Ca2+ trigger signal, the functional state of the RyRs and the amount of Ca2+ stored in the SR.
Reversible phosphorylation of proteins composing the EC coupling machinery plays an important role in regulation of cardiac contractility (Bers, 2002). Thus, during stimulation of the β-adrenergic pathway, phosphorylation of several target proteins, including the L-type Ca2+ channels, RyRs and phospholamban, by protein kinase A (PKA) leads to an overall increase in SR Ca2+ release and contractile force in heart cells (Callewaert et al. 1988, Spurgeon et al. 1990; Hussain & Orchard, 1997; Zhou et al. 1999; Song et al. 2001; Viatchenko-Karpinski & Gyorke, 2001). PKA-dependent phosphorylation of the L-type Ca2+ channels increases the Ca2+ current (_I_Ca), increasing both the Ca2+ trigger for SR Ca2+ release and the SR Ca2+ content (Callewaert et al. 1988; Hussain & Orchard, 1997; DelPrincipe et al. 2001). Phosphorylation of phospholamban (PLB) relieves the tonic inhibition dephosphorylated PLB exerts on the SR Ca2+-ATPase (SERCA) resulting in enhanced SR Ca2+ accumulation and enlarged Ca2+ release (Kranias et al. 1985; Simmermann & Jones, 1998). With regard to the RyR, despite clear demonstration of phosphorylation of the channel in biochemical studies (Takasago et al. 1989; Yoshida et al. 1992), the consequences of this reaction to channel function have not been clearly defined. RyR phosphorylation by PKA and Ca2+-calmodulin-dependent protein kinase (CaMKII) has been reported to increase RyR activity in lipid bilayers (Hain et al. 1995; Marx et al. 2000; Uehara et al. 2002). Moreover, it has been reported that in heart failure (HF), hyperphosphorylation of RyR causes the release of FK-506 binding protein (FKBP12.6) from the RyR, rendering the channel excessively leaky for Ca2+ (Marx et al. 2000). However, other studies have reported no functional effects (Li et al. 2002) or even found phosphorylation to reduce RyR channel steady-state open probability (Valdivia et al. 1995; Lokuta et al. 1995).
The action of protein kinases is opposed by dephosphorylating phosphatases. Three types of protein phosphatases (PPs), referred to as PP1, PP2A and PP2B (calcineurin), have been shown to influence cardiac performance (Neumann et al. 1993; Rusnak & Mertz, 2000). Overall, according to most studies phosphatases appear to downregulate SR Ca2+ release and contractile performance (Neumann et al. 1993; duBell et al. 1996, 2002; Carr et al. 2002; Santana et al. 2002). Furthermore, PP1 and PP2A activities appear to be increased in heart failure (Neumann, 2002; Carr et al. 2002). However, again the precise mode of action of these enzymes on intracellular Ca2+ handling in normal and diseased hearts remains poorly understood. In the present study, we have investigated the effects of protein phosphatases PP1 and PP2A on local Ca2+ release events, Ca2+ sparks, in cardiac cells. Our results show that phosphatases activate RyR-mediated SR Ca2+ release leading to depletion of SR Ca2+ stores. These results provide novel insights into the mechanisms and potential role of protein phosphorylation/dephosphorylation in regulation of Ca2+ signalling in normal and diseased hearts.
METHODS
Confocal microscopy
Single ventricular myocytes were obtained from adult male Sprague-Dawley rat hearts by enzymatic dissociation (Lukyanenko & Gyorke, 1999). Rats were anaesthetized with nembutal and killed by exsanguination. The protocol for animal use was reviewed and approved by the Institutional Animal Care and Use Committee and complied with US Public Health Service policy on humane care and use of laboratory animals.
The standard extracellular Tyrode solution contained (mM): 140 NaCl, 5.4 KCl, 0.5 MgCl2, 1 CaCl2, 10 Hepes, 0.25 NaH2PO4, 5.6 glucose (pH 7.3). The cells were permeabilized by exposure to saponin (0.01 % for 45-60 s; Lukyanenko & Gyorke, 1999). The permeabilization solution contained (mM): 120 potassium aspartate, 20 KCl, 3 MgATP, 10 phosphocreatine, 5 U ml−1 creatine phosphokinase, 0.5 EGTA (pCa 7) and 20 Hepes (pH 7.2). The control experimental solution contained (mM): 120 potassium aspartate, 20 KCl, 3 MgATP, 10 phosphocreatine, 5 U ml−1 creatine phosphokinase, 0.03 Fluo-3 potassium salt, 0.5 EGTA (pCa 7) and 20 Hepes (pH 7.2).
All experiments were performed at room temperature (21-23 °C). Spontaneous Ca2+ sparks, were recorded with a Bio-Rad Laser Scanning Confocal system (MRC-1024ES, Bio-Rad Laboratories, Hercules, CA, USA) using an Olympus × 60 1.4 N.A. objective as described previously (Lukyanenko et al. 2001). Fluo-3 was excited by light at 488 nm (25 mW argon laser, intensity attenuated to 0.3 %), and the fluorescence was acquired at wavelengths of > 515 nm in the line scan mode at rate of 2 or 6 ms per scan. Ca2+ sparks were quantified using a detection and analysis computer algorithm implemented in IDL (Research Systems Inc., Boulder CO, USA) (Lukyanenko et al. 2001). Changes in the total SR Ca2+ content under different experimental conditions were assessed from the peak amplitude of the caffeine-induced Ca2+ transients considering the amount (0.5 mM) and dissociation constant (_K_D ≈ 163 nM) of the dominant Ca2+ buffer in the external solution, EGTA, as described previously (Lukyanenko et al. 2000).
Lipid bilayer experiments
Heavy SR microsomes were isolated by differential centrifugation from the ventricles of dog hearts (Gyorke & Gyorke, 1998). SR vesicles prepared from canine ventricles are the most commonly used preparation for studying cardiac RyR channels. Dogs were anaesthetized with nembutal and killed by exsanguination with procedures approved by the Institutional Animal Care and Use Committee. The three dog hearts used in our experiments were available from dogs killed for other studies. Single RyRs were reconstituted by fusing SR microsomes into planar lipid bilayers and single channel currents recorded as described previously (Gyorke & Gyorke, 1998). Channel incorporation was performed in solutions containing (mM): 350 CsCH3SO3, 0.02 CaCl2, 20 Hepes (pH 7.4), on the cytosolic (cis) side of the bilayer, and 20 CsCH3SO3, 0.02 CaCl2, 20 Hepes (pH 7.4) on the lumenal (trans) side of the bilayer. The experimental solutions contained (mM): 350 CsCH3SO3, 3 MgATP, 0.6 MgCl2, 3 dibromo-BAPTA ([Ca2+]free ≈ 5 μmol l−1), 20 Hepes (pH 7.4) (cis), and 20 CsCH3SO3, 0.02 CaCl2, 20 Hepes (pH 7.4) (trans). Single channel currents were recorded at room temperature (21-23 °C) with an Axopatch 200A (Axon Instruments, Union City, CA, USA) patch-clamp amplifier. Data were digitized at 5-10 kHz and filtered at 2 kHz. Acquisition and analysis of data were performed using pCLAMP 6.01 software (Axon Instruments).
Dephosphorylation of RyR by PP1
Saponin-permeabilized myocytes were treated with PP1 (2 U ml−1 or 10 U ml−1, 5 min, 23 °C) under conditions similar to those used for our Ca2+ spark measurements. To determine the maximal levels of RyR phosphorylation, PKA (10 U ml−1) and okadaic acid (10 μM) were added to some of the permeabilized cells (10 min, 23 °C). Reactions were terminated by pelleting cells and adding solubilization buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 15 mM MgCl2, 1 % Nonidet P40, 0.3 % Triton X-100, 0.5 % sodium deoxycholate, 1 mM dithiothreitol, protease inhibitor cocktail, 10 mM _N_-ethylmaleimide, pH 7.8). Levels of phosphorylated RyR in our control, phosphatase- and PKA/okadaic acid-treated samples were determined by immunoblot analysis using a RyR phosphoepitope-specific antibody (Rodriguez et al. 2003). Twenty micrograms of cell lysate proteins was subjected to 14 % SDS-PAGE, blotted onto polyvinylidene difluoride (PVDF) membranes (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and probed with an antibody against phosphorylated RyR (1:2500, P-S2809, Badrilla, West Yorkville, UK). Blots were developed with Super Signal West Pico (Pierce Biotechnology, Inc., Rockford, IL, USA) and quantified using a Visage 2000 Blot Scanning and Analysis system (BioImage Systems, Inc., Jackson, MI, USA).
Materials
PP1 and PKA catalytic subunits were purchased from Sigma (St Louis, MO, USA). PP2A was from Promega Corp. (Madison, WI, USA). Okadaic acid and calyculin A were from Alomone Labs (Jerusalem, Israel). Fluo-3 potassium salt was from TefLabs (Austin, TX, USA). Dibromo-BAPTA was from Molecular Probes (Eugene, OR, USA).
Statistics
Data were expressed as means ± s.e.m. Comparisons were performed by using Student's paired _t_-test, and significance was defined at P < 0.05.
RESULTS
Effects of PP1 and PP2A on Ca2+ sparks and SR Ca2+ content
To investigate the effects of phosphatases on properties of SR Ca2+ release, we carried out experiments in myocytes permeabilized with saponin (Lukyanenko & Gyorke, 1999). Figure 1_A_ shows images of Ca2+ sparks from a permeabilized myocyte before and at different times (1 and 5 min) after addition of 2 U ml−1 PP1 to the bathing solution. On its addition, PP1 first produced a dramatic increase (≈5-fold) in Ca2+ spark frequency. This effect was followed by a nearly complete disappearance of events (78 % inhibition of spark frequency). Pooled data on the impact of PP1 at two different concentrations (0.5 and 2 U ml−1) on spark frequency during the initial and late phases are presented in Fig. 1_B_. At higher doses (5 and 10 U ml−1) PP1 caused an even larger initial potentiation of Ca2+ sparks; however, quantification of these effects in terms of changes in Ca2+ spark parameters was complicated due to elevated background [Ca2+]i and overlap of individual events (not shown). Thus PP1 caused an early transient potentiation of Ca2+ spark frequency followed by a delayed inhibition of event occurrence.
Figure 1. Effects of PP1 on properties of Ca2+ sparks and SR Ca2+ content in rat permeabilized myocytes.
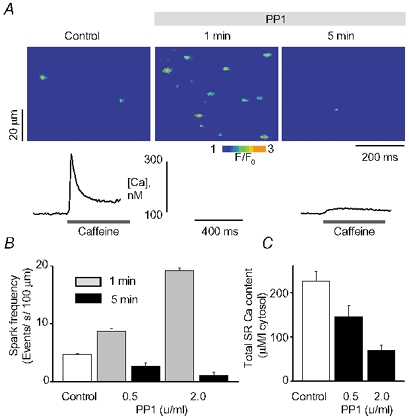
A, spontaneous Ca2+ spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 2 U ml−1 PP1. Traces below the images are Ca2+ transients induced by application of 10 mm caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP1 in the same cell. The Ca2+ transients were elicited by a whole bath application of 10 mm caffeine. B, averaged spark frequency at early (1 min) and late (5 min) times following the addition of either 0.5 or 2 U ml−1 of PP1 to the bathing solution. C, averaged SR Ca2+ content for 0.5 or 2 U ml−1 of PP1 measured before and 5 min after exposure to the enzyme. Data are presented as means ± s.e.m. of 6 experiments in different cells.
In addition to affecting Ca2+ spark frequency in a biphasic manner, PP1 produced similar biphasic effects on the magnitude and spatio-temporal characteristics of Ca2+ sparks (Table 1). Specifically, during the potentiatory phase (1 min after addition of the enzyme), PP1 significantly increased the amplitude, rise-time, duration and width of Ca2+ sparks; during the inhibitory phase (5 min after addition of the enzyme), all these parameters were significantly suppressed by PP1.
Table 1.
Effects of phosphatases on spatio-temporal properties of Ca2+ sparks in permeabilized ventricular myocytes
| δ_F/F_0 | Rise time (ms) | Duration (ms)† | Width (μm)† | n | |
|---|---|---|---|---|---|
| Control | 1.23 ± 0.02 | 6.2 ± 0.1 | 11.8 ± 0.1 | 1.94 ± 0.002 | 520 |
| PP1 1 min | 1.34 ± 0.02* | 7.4 ± 0.2* | 15.3 ± 0.2* | 2.13 ± 0.03* | 399 |
| PP1 5 min | 0.82 ± 0.05 | 5.9 ± 0.4 | 11.9 ± 0.3 | 1.96 ± 0.07 | 47 |
| PP2A 1 min | 1.37 ± 0.02* | 7.6 ± 0.2* | 15.5 ± 0.1* | 2.17 ± 0.03* | 474 |
| PP2A 5 min | 0.78 ± 0.06 | 5.7 ± 0.4 | 11.5 ± 0.4 | 1.92 ± 0.08 | 52 |
One possible reason for the observed biphasic effects of PP1 on Ca2+ sparks is that the SR Ca2+ content of myocytes was changed by this enzyme. To examine the loading status of the SR in the course of these experiments, we used applications of caffeine (10 mM). Traces below the images illustrate caffeine-induced Ca2+ transients measured from the same myocyte before and 5 min after addition of PP1. Changes in the total SR Ca2+ content were assessed from the peak amplitude of the caffeine-induced Ca2+ transients by taking into account binding of Ca2+ to EGTA in the bathing solution (Lukyanenko et al. 2000). Based on these assessments, the SR Ca2+ content decreased by 35 % or 69 % following the exposure of myocytes to either 0.5 or 2 U ml−1 PP1, respectively (Fig. 1_C_).
As shown in Fig. 2 and Table 1, qualitatively similar results were obtained with phosphatase PP2A. Similar to the effects of PP1, PP2A (5 U ml−1) produced a transient increase in Ca2+ spark frequency (≈4-fold) followed by a depression of event occurrence and decreased SR Ca2+ content (by 82 % and 65 %, respectively). Also similar to the action of PP1, PP2A increased the amplitude and spatio-temporal spread (i.e. rise-time, duration and width) of Ca2+ sparks at 1 min and suppressed the same parameters at 5 min of exposure to the enzyme (Table 1). Together, these results suggest that phosphatases enhance spark-mediated SR Ca2+ release, leading to decreased SR Ca2+ content.
Figure 2. Effects of PP2A on properties of Ca2+ sparks and SR Ca2+ content in rat permeabilized myocytes.
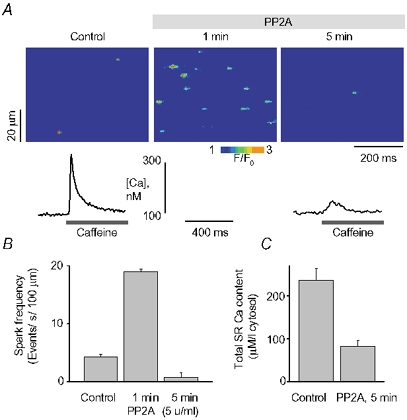
A, spontaneous Ca2+ spark images recorded under reference conditions, and 1 or 5 min after exposure of the cell to 5 U ml−1 PP2A. Traces below the images are Ca2+ transients induced by application of 10 mm caffeine immediately following the acquisition of sparks before (3 min) and after (5 min) application of PP2A in the same cell. B and C, averaged spark frequency (B) and SR Ca2+ content (C) for the same conditions as in A. Data are presented as means ± s.e.m. of 6 experiments in different cells.
Preventive effects of calyculin A and okadaic acid
We examined the effects of PP1/PP2A inhibitors okadaic acid and calyculin A on Ca2+ sparks. Okadaic acid shows a degree of selectivity for PP2A, whereas calyculin A inhibits both phosphatases with similar potencies (Ishihara et al. 1989). Sensitivity of PP1 and PP2A to these inhibitors is related to the homologous catalytic domains shared by these enzymes (Denu et al. 1996). In these experiments, myocytes were exposed to calyculin A (1 μM) or okadaic acid (5 μM) for up to 5 min, after which PP1 or PP2A was added to the medium. As shown in Fig. 3, calyculin by itself produced no significant changes in Ca2+ sparks. However, in the continuous presence of this compound, PP1 failed to produce the characteristic changes in Ca2+ spark frequency and SR Ca2+ content. Similar results were obtained with sequential additions of okadaic acid and PP2A (Fig. 4). These results confirm that the effects of phosphatases on Ca2+ sparks and SR Ca2+ load involve catalytic activities of these enzymes. In addition, since these phosphatase inhibitors produced no detectable effects on Ca2+ sparks, our results suggest that the background phosphatase activity is relatively low in the permeabilized rat myocyte preparation.
Figure 3. Prevention of PP1 effects on Ca2+ release by calyculin A.
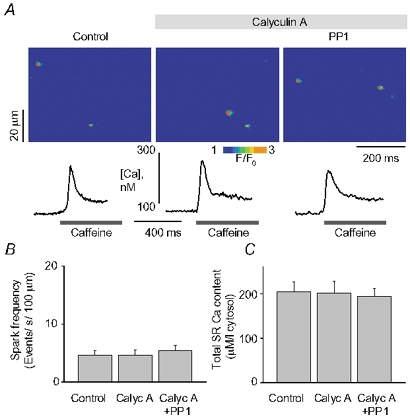
A, images of sparks along with traces of caffeine-induced Ca2+ transient recorded under reference conditions, after exposure of the cell to 1 μm calyculin A and 5 min after 2 U ml−1 of PP1 was added to the bathing solution. The Ca2+ transients were elicited by a whole bath application of 10 mm caffeine. B and C, averaged spark frequency (B) and SR Ca2+ content (C) for the same conditions as in A. Data are presented as means ± s.e.m. of 6 experiments in different cells.
Figure 4. Prevention of PP2A effects on Ca2+ release by okadaic acid.
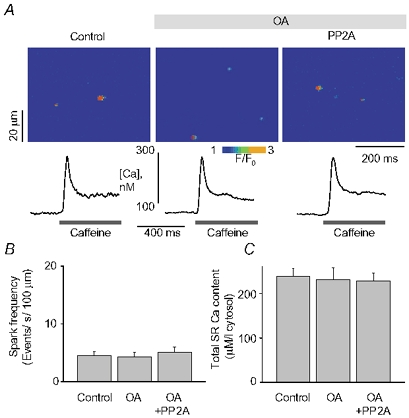
A and B, images of sparks along with traces of caffeine-induced Ca2+ transient recorded under reference conditions, after exposure of the cell to 5 μm of okadaic acid (OA), and 5 min after 5 U ml−1 of PP2A was added to the bathing solution. The Ca2+ transients were elicited by a whole bath application of 10 mm caffeine. B and C, averaged spark frequency (B) and SR Ca2+ content (C) for the same conditions as in A. Data are presented as means ± s.e.m. of 6 experiments in different cells.
Preventive effects of ryanodine
We used ryanodine to test whether the observed effects of PP1 and PP2A on Ca2+ release are indeed mediated by the RyR Ca2+ release channel. At high concentrations ryanodine inhibits RyR channel openings (Coronado et al. 1994). Exposure of cells to 100 μM ryanodine completely eliminated Ca2+ sparks (not shown). Application of either PP1 or PP2A produced no measurable changes in fluorescence under these conditions. Similar results were obtained in a total of nine cells (5 and 4 cells for PP1 and PP2A, respectively). PP1 and PP2A also failed to produce any changes in Ca2+ release in myocytes pretreated with another RyR channel blocker, ruthenium red (10 μM). Similarly, phosphatases induced no Ca2+ release events in myocytes in which the SR Ca2+ content was depleted by thapsigargin (10 μM, for 5 min).
Effects of PP1 on single RyRs
To assess the effects of phosphatases on the activity of RyR channels more directly, we carried out experiments with single RyRs incorporated into lipid bilayers. We fused heavy cardiac SR microsomes into lipid bilayers and then recorded single RyR channel activity using Cs+ as the charge carrier in the presence of cytosolic Mg2+ and ATP. [Ca2+] on the cytosolic (cis) side of the channel was buffered at 5 μM with dibromo-BAPTA. Addition of PP1 (10 U ml−1) to the cytosolic side of the channel resulted in a significant increase in open channel probability (_P_o) in 5 out of 11 channels (Fig. 5). The mean _P_o in these channels increased about 4-fold (0.04 ± 0.02 and 0.17 ± 0.05 before and after addition of PP1, respectively). In the other six channels PP1 produced no significant changes in _P_o. The heterogeneity of responses to PP1 may reflect variations in the phosphorylation state of individual RyRs. Clearly, only channels that are phosphorylated at certain sites can respond to addition of PP1. To test the hypothesis that the observed heterogeneity of channel responses to PP1 was due to differences in the initial phosphorylation status of the RyRs, we carried out additional experiments in which RyRs were treated with PKA before their exposure to PP1. In these experiments, RyRs were incorporated into the lipid bilayers in the presence of PKA (10 U ml−1) and exposed to this phosphorylating agent for at least 5 min prior to replacing PKA by PP1 in the cis solution. Consistent with our hypothesis, under these conditions, most channels (3 out 4) showed increased _P_o on application of PP1 (0.03 ± 0.01 and 0.15 ± 0.06 before and after addition of PP1, respectively). These results suggest that PP1 can act directly on RyR or a closely associated regulatory protein.
Figure 5. Effects of PP1 on activity of single RyR channels.

Representative single channel recordings performed in symmetrical Cs+ under control conditions and after addition (2 min) of 10 U ml−1 PP1 to the cis side of the channel. The cis chamber contained 5 μm Ca2+ and 3 mm MgATP. Channel openings are upward. Holding potential was +40 mV.
PP1-mediated RyR dephosphorylation
The cardiac RyR is phosphorylated at Ser-2809 (in the rabbit sequence) by both PKA and CAMKII (Witcher et al. 1991; Marx et al. 2000). Although additional phosphorylation sites may exist on the RyR (Rodriguez et al. 2003), Ser-2809 is believed to be the only site that is phosphorylated by PKA, and RyR hyperphosphorylation at this site has been reported in heart failure (Marx et al. 2000). To test whether indeed phosphatases dephosphorylated the RyR in our permeabilized myocyte experiments we performed quantitative immunoblotting using an antibody that specifically recognizes the phosphorylated form of the RyR at Ser-2809 (Rodriguez et al. 2003). As shown in Fig. 6, myocytes exhibited a significant level of phosphorylation under baseline conditions. Maximal phosphorylation was 201 % of control. When exposed to 2 U ml−1 PP1, RyR phosphorylation was 58 % of the control basal condition. Exposing to a higher PP1 concentration (10 U ml−1) further reduced RyR phosphorylation to 22 % of control. Thus, consistent with the results of our functional measurements, PP1 decreased RyR phosphorylation in cardiac myocytes.
Figure 6. PP1-dependent dephosphorylation of RyR in permeabilized myocytes.
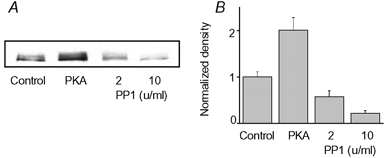
A, representative immunoblots detecting phosphorylated RyR (at Ser-2809) in saponin-permeabilized myocytes under baseline conditions (1st lane), conditions of maximal phsosphorylation (10 U ml−1 PKA and 10 μm okadaic acid, lane 2) and after exposure to 2 or 10 U ml−1 PP1 (lanes 3 and 4, respectively). B, pooled results (means ± s.e.m.) normalized to control (n = 5).
DISCUSSION
In the present study, we have investigated the impact of physiologically relevant exogenous protein phosphatases PP1 and PP2A on RyR-mediated SR Ca2+ release (measured as Ca2+ sparks) in permeabilized heart cells. Our principal finding is that phosphatases stimulated RyR channels leading to depleted SR Ca2+ stores. These results have important ramifications for understanding the mechanisms and role of protein phosphorylation/dephosphorylation in modulation of Ca2+ handling in normal and diseased heart.
Modulation of SR Ca2+ release by protein phosphorylation/dephophorylation
Several lines of experimental evidence suggest that the observed stimulation of Ca2+ sparks by PP1 and PP2A involves a dephosphorylation reaction promoting openings of the RyR channel. The evidence includes: (1) application of PP1 and PP2A produced transient increases in the magnitude and frequency of sparks followed by depletion of SR Ca2+ stores, as observed previously with known stimulators of RyR channels, such as caffeine (Lukyanenko et al. 2001); (2) changes in sparks could be prevented by calyculin A and okadaic acid, potent inhibitors of PP1 and PP2A catalytic activities; (3) phosphatase application induced no Ca2+ release when RyR channels were inhibited by the specific blockers of these channels, ryanodine (100 μM) or ruthenium red, thereby excluding the possibility that the effects of phosphatases are mediated by other Ca2+ efflux pathways; (4) PP1 increased the open probability of single RyRs incorporated into lipid bilayers; and (5) RyR was phosphorylated to a considerable degree (at Ser-2809) under baseline conditions and exposure to PP1 drastically reduced RyR phosphorylation in permeabilized myocyte settings.
Our finding that exogenous phosphatases activate Ca2+ sparks appears paradoxical given the large body of evidence demonstrating that increasing activities of cellular protein kinases augment SR Ca2+ release (Callewaert et al. 1988; Spurgeon et al. 1990; Hussain & Orchard, 1997; Zhou et al. 1999; Song et al. 2001; Viatchenko-Karpinski & Gyorke, 2001). Protein phosphorylation and dephosphorylation are opposing processes that would be expected to produce opposite functional effects. To reconcile this apparent paradox it is important to consider the transient vs. sustained nature of these potentiatory influences as well as the fact that at least two Ca2+ transport systems (i.e. RyR and the SERCA-PLB complex) are involved in these phenomena. Thus, PP1 and PP2A produce only transient activation of Ca2+ sparks because opening of RyRs leads to unloading of the SR (e.g. Lukyanenko et al. 2001). Dephosphorylation of PLB also may contribute to the reduced SR Ca2+ accumulation by decreasing the Ca2+ transport efficiency of SERCA in the presence of phosphatases (Kranias et al. 1988; MacDougall et al. 1991; Steenaart et al. 1992). In support of this possibility, the kinetics of decay of Ca2+ sparks were slowed by PP1 and PP2A (Table 1). Thus, the long-term depression of SR Ca2+ releasing function by phosphatases is likely to result from enhanced Ca2+ leak via RyRs and reduced Ca2+ uptake by SERCA.
On the other hand, enhanced protein kinase activity causes a maintained potentiation of both global and local SR Ca2+ release signals without a decrease in the SR Ca2+ content (Zhou et al. 1999; Song et al. 2001; Viatchenko-Karpinski & Gyorke, 2001; DelPrincipe et al. 2001; Li et al. 2002). It appears these effects are predominantly due to phosphorylation of PLB augmenting SERCA-mediated Ca2+ uptake (Li et al. 2002). Since protein dephosphorylation clearly resulted in increased functional activity of the Ca2+ release channel, our results imply that a reverse, phosphorylation reaction should reduce RyR activity. If indeed such effects take place, why do they not manifest in inhibition of Ca2+ sparks? One possibility is that enhanced Ca2+ uptake by SERCA masks or overcomes the effects phosphorylation may have on RyRs. In addition, the potential inhibitory influence of protein phosphorylation on RyR activity in myocytes could be countered by feedback mechanisms involving changes in luminal Ca2+ (Trafford et al. 2002; Gyorke et al. 2002). In particular, reduced open probability of RyRs would be expected to lead to increased Ca2+ accumulation in the SR; increased intra-SR [Ca2+] in turn would increase activity of RyRs at their luminal Ca2+ regulatory sites as demonstrated for the RyR channel inhibitor tetracaine (Gyorke et al. 1997; Overend et al. 1997). Thus potentiation of SERCA combined with the intrinsic capacity of the release mechanism to self-regulate could explain at least in part why PKA-mediated protein phoshorylation results in maintained potentiation of Ca2+ sparks despite a potential initial decrease in RyR activity.
It is also conceivable that both phosphorylation and dephosphorylation stimulate the RyR by acting at different phosphorylation sites. Recent evidence suggest that the RyR is phopshorylated on at least five different phosphorylation sites (one site is phosphorylated by PKA and CAMKII and four additional sites are phosphorylated by CAMKII; Rodriguez et al. 2003). If phosphorylation at different sites, or sets of sites, were to affect the RyR differently, this could explain how phosphorylating and dephosphorylating agents could both lead to enhanced channel activity.
Comparison with previous studies
Our results are consistent with the reports of others regarding the effects of phosphatases on SR Ca2+ release and EC coupling. Thus, the observed depletion of the SR Ca2+ stores by PP1 and PP2 could account, at least in part, for the depressed cardiac performance in transgenic mice overexpressing the PP1 catalytic subunit and in mice with ablation of the PP1 inhibitor, l-1 (Carr et al. 2002). Our data are also in general agreement with positive inotropic influences of phosphatase inhibitors okadaic acid and calyculin A in cardiac myocytes (Neumann et al. 1993; duBell et al. 2002). duBell et al. (1996) reported that intracellular dialysis of rat ventricular myocytes with either PP1 or PP2A decreased the magnitude of steady-state intracellular Ca2+ transients without producing measurable changes in SR Ca2+ content (assessed by caffeine application). Although, the long-term depression of SR Ca2+ release is consistent with our results, it is not clear how such a depression could occur without changes in the SR Ca2+ load. Because the SR Ca2+ content at steady-state is determined by a balance between SR Ca2+ uptake and Ca2+ leak through RyRs, any changes in RyR gating must lead to altered SR Ca2+ content unless the uptake is also affected (Trafford et al. 2002). The uptake rate was reported not to change in this study based on measurements of kinetics of decay of the Ca2+ transients. A possible explanation for these results is that the caffeine protocol used for detection of SR Ca2+ load may not have been sufficiently sensitive to detect changes in load caused by enhanced leak through RyRs. Consistent with this possibility, in our hands PP1 produced significant decreases in the magnitude of both electrically evoked and caffeine-induced Ca2+ transients in patch-clamped cardiac myocytes at doses higher (>10 U ml−1) than those used by duBell et al. (1996) under otherwise similar conditions (not shown).
At the single channel level, studies of protein kinase and phosphatase effects on RyRs have yielded conflicting results. Hain et al. (1995) have reported that phosphorylation by PKA and CAMKII increases the steady-state activity of single RyR2 channels in lipid bilayers. They also showed that dephosphorylation by potato acid phosphatase or PP1 reversed the potentiatory effects of protein kinases. On the other hand, Valdivia and collaborators (Lokuta et al. 1995; Valdivia et al. 1995) showed that phosphorylation by PKA or CAMKII reduced, whereas dephosphorylation by acid phosphatase increased, steady-state activity of RyRs. Our results are in line with the latter reports. The reasons for these discrepancies are not known but they could reflect differences in SR membrane isolation procedures. RyRs in vivo are thought to be associated with endogenous protein kinases and phosphatases (Marx et al. 2000). In addition, the RyR Ca2+ release channel complex contains other regulatory proteins, such as FKBP12.6, that could modulate the effects of phosphorylation on RyR activity. Thus, the functional state of RyR channels and their responsiveness to exogenous protein kinases and phosphatases in lipid bilayer studies may vary with the degree of dissociation of RyRs. This underscores the importance of analysis of protein phosphorylation/dephosphorylation effects on the Ca2+ release mechanism in a relatively intact physiological environment.
Role of altered RyR phosphorylation in heart failure
Marx et al. (2000) have proposed that enhanced levels of circulating catecholamines lead to increased phosphorylation of RyR in heart failure. Based on biochemical observations as well as on studying properties of single RyRs incorporated into artificial lipid bilayers, these investigators have hypothesized that hyperphosphorylation of RyRs contributes to pathogenesis of heart failure by making the channel excessively leaky due to dissociation of FKBP12.6 from the channel. We show that the mode of modulation of RyRs by phosphatases does not support this hypothesis as dephosphorylation caused activation instead of inhibition of activity of RyR channels in a relatively intact setting. Interestingly, our results provide the basis for a different possibility in which dephophosphorylation of RyR rather than its phosphorylation causes depletion of SR Ca2+ stores by stimulating RyRs in failing hearts. It has been reported that PP1 and PP2 activities are increased in heart failure (Huang et al. 1999; Neumann et al. 1997; Neuman, 2002). Furthermore, overexpression of PP1 or ablation of the endogenous PP1 inhibitor, l-1, results in depressed contractile performance and heart failure (Carr et al. 2002). Our finding that PP1 causes depletion of SR Ca2+ stores by activating RyRs could account for, or contribute to, these results.
Acknowledgments
This work was supported by grants from the American Heart Association and the National Institutes of Health (HL03739).
REFERENCES
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Clemann L, Morad M. Epinephrine enhances Ca2+ current regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proc Natl Acad Sci U S A. 1988;85:2009–2013. doi: 10.1073/pnas.85.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AN, Schmidt AG, Suzuki Y, Del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, Depaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM. Structure and function of ryanodine receptors. Am J Physiol. 1994;266:C1485–1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- DelPrincipe F, Egger M, Pignier C, Niggli E. Enhanced E-C coupling efficiency after beta-stimulation of cardiac myocytes. Biophys J. 2001;80:64a. [Google Scholar]
- Denu JM, Stuckey JA, Saper MA, Dixon JE. Form and function in protein dephosphorylation. Cell. 1996;87:361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- duBell WH, Gigena MS, Guatimosim S, Long X, Lederer WJ, Rogers TB. Effects of PP1/PP2A inhibitor calyculin A on the E-C coupling cascade in murine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;282:H38–48. doi: 10.1152/ajpheart.00536.2001. [DOI] [PubMed] [Google Scholar]
- duBell WH, Lederer WJ, Rogers TB. Dynamic modulation of excitation-contraction coupling by protein phosphatases in rat ventricular myocytes. J Physiol. 1996;493:793–800. doi: 10.1113/jphysiol.1996.sp021423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci. 2002;7:d1454–d1463. doi: 10.2741/A852. [DOI] [PubMed] [Google Scholar]
- Gyorke I, Lukyanenko V, Gyorke S. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500:297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Huang B, Wang S, Qin D, Boutjdir M, El-Sherif N. Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of beta-adrenergic pathway, G(i) protein, phosphodiesterase, and phosphatases. Circ Res. 1999;85:848–855. doi: 10.1161/01.res.85.9.848. [DOI] [PubMed] [Google Scholar]
- Hussain M, Orchard CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. J Physiol. 1997;505:385–402. doi: 10.1111/j.1469-7793.1997.385bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, Hartshorne M. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- Kranias EG, Garvey JL, Srivastava RD, Solaro RJ. Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem J. 1985;226:113–121. doi: 10.1042/bj2260113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranias EG, Gupta RC, Jakab G, Kim HW, Steenaart NA, Rapundalo ST. The role of protein kinases and protein phosphatases in the regulation of cardiac sarcoplasmic reticulum function. Mol Cell Biochem. 1988;82:37–44. doi: 10.1007/BF00242513. [DOI] [PubMed] [Google Scholar]
- Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487:609–622. doi: 10.1113/jphysiol.1995.sp020904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol. 1999;521:575–585. doi: 10.1111/j.1469-7793.1999.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V, Viatchenko-Karpinski S, Smirnov A, Wiesner TF, Gyorke S. Dynamic regulation of sarcoplasmic reticulum Ca2+ content and release by luminal Ca2+-sensitive leak in rat ventricular myocytes. Biophys J. 2001;81:785–798. doi: 10.1016/S0006-3495(01)75741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDaugall LK, Jones LR, Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur J Biochem. 1991;196:725–734. doi: 10.1111/j.1432-1033.1991.tb15871.x. [DOI] [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12. 6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Neumann J. Altered phosphatase activity in heart failure, influence on Ca2+ movement. Basic Res Cardiol. 2002;97:I91–I95. doi: 10.1007/s003950200036. [DOI] [PubMed] [Google Scholar]
- Neumann J, Boknik P, Herzig S, Schmitz W, Scholz H, Gupta RC, Watanabe AM. Evidence for physiological functions of protein phosphatases in the heart: evaluation with okadaic acid. Am J Physiol. 1993;265:H257–266. doi: 10.1152/ajpheart.1993.265.1.H257. [DOI] [PubMed] [Google Scholar]
- Neumann J, Eschenhagen T, Jones LR, Linck B, Schmitz W, Scholz H, Zimmermann N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol. 1997;29:265–272. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- Overend CL, Eisner DA, O'Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. J Physiol. 1997;502:471–479. doi: 10.1111/j.1469-7793.1997.471bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine-2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003 doi: 10.1074/jbc.C301180200. (in press) [DOI] [PubMed] [Google Scholar]
- Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol. 2002;544:57–69. doi: 10.1113/jphysiol.2002.020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman HK, Jones LR. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. beta2-Adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. Am J Physiol. 1990;258:H574–586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- Steenaart NA, Ganim JR, Di Salvo J, Kranias EG. The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch Biochem Biophys. 1992;293:17–24. doi: 10.1016/0003-9861(92)90359-5. [DOI] [PubMed] [Google Scholar]
- Takasago T, Imagawa T, Shigekawa M. Phosphorylation of the cardiac ryanodine receptor by cAMP-dependent protein kinase. J Biochem. 1989;106:872–877. doi: 10.1093/oxfordjournals.jbchem.a122945. [DOI] [PubMed] [Google Scholar]
- Trafford AW, Diaz ME, O'Neill SC, Eisner DA. Integrative analysis of calcium signalling in cardiac muscle. Front Biosci. 2002;7:d843–d852. doi: 10.2741/trafford. [DOI] [PubMed] [Google Scholar]
- Uehara A, Yasukochi M, Mejia-Alvarez R, Fill M, Imanaga I. Gating kinetics and ligand sensitivity modified by phosphorylation of cardiac ryanodine receptors. Pflugers Arch. 2002;444:202–212. doi: 10.1007/s00424-002-0791-3. [DOI] [PubMed] [Google Scholar]
- Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatchenko-Karpinski S, Gyorke S. Modulation of the Ca2+-induced Ca2+ release cascade by beta-adrenergic stimulation in rat ventricular myocytes. J Physiol. 2001;533:837–348. doi: 10.1111/j.1469-7793.2001.t01-1-00837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem. 1991;266:11144–11152. [PubMed] [Google Scholar]
- Yoshida A, Takahashi M, Imagawa T, Shigekawa M, Takisawa H, Nakamura T. Phosphorylation of ryanodine receptors in rat myocytes during beta-adrenergic stimulation. J Biochem. 1992;111:186–190. doi: 10.1093/oxfordjournals.jbchem.a123735. [DOI] [PubMed] [Google Scholar]
- Zhou YY, Song LS, Lakatta EG, Xiao RP, Cheng H. Constitutive beta2-adrenergic signaling enhances sarcoplasmic reticulum Ca2+ cycling to augment contraction in mouse heart. J Physiol. 1999;521:351–361. doi: 10.1111/j.1469-7793.1999.00351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]