Deletion of Histone Deacetylase 3 reveals critical roles in S-phase progression and DNA damage control (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 11.
Abstract
Histone deacetylases (HDAC) are enzymes that modify key residues in histones to regulate chromatin architecture, and play a vital role in cell survival, cell cycle progression, and tumorigenesis. To understand the function of Hdac3, a critical component of the N-CoR/SMRT repression complex, a conditional allele of Hdac3 was engineered. Cre-recombinase-mediated inactivation of Hdac3 led to a delay in cell cycle progression, cell-cycle dependent DNA damage, and apoptosis in mouse embryonic fibroblasts (MEFs). While no overt defects in mitosis were observed in _Hdac3_−/− MEFs, including normal H3Ser10 phosphorylation, DNA damage was observed in _Hdac3_−/−interphase cells, which appears to be associated with defective DNA double strand break repair. Moreover, we noted that _Hdac3_−/− MEFs were protected from DNA damage when quiescent, which may provide a mechanistic basis for the action of histone deacetylase inhibitors on cycling tumor cells.
Introduction
HDACs are divided into different classes based on sequence conservation. Class I HDACs include HDACs 1, 2, 3, and 8, which are homologous to yeast RPD3 and deacetylate histones and non-histone proteins in vitro and in vivo (Bernstein et al., 2000; Hubbert et al., 2002; Jenuwein and Allis, 2001; Taunton et al., 1996; Zhang et al., 2007). Class II HDACs consist of HDACs 4, 5, 6, 7, and 9 and are more similar to the yeast Hda1 enzyme. While these factors mediate transcriptional repression of many DNA binding transcription factors, the class IIa HDACs (4, 5, 7 and 9) have little enzymatic activity when acetylated histone peptides were used as substrates (Chang et al., 2004; Gallinari et al., 2007; Gregoire et al., 2007; Lahm et al., 2007; Zhang et al., 2002), but can associate with HDAC3 (Fischle et al., 2002). The third class includes the yeast Sir2 protein and its mammalian homologues that are involved in transcriptional silencing and whose activity is dependent on NAD (Gray and Ekstrom, 2001; Khochbin and Kao, 2001). HATs and HDACs are not only involved in acetylation and deacetylation of histones, but also play a role in cell survival and cell cycle progression (Gallinari et al., 2007; Linggi et al., 2005).
Several forms of leukemia are caused by the inappropriate recruitment of HDACs by mutant transcription factors resulting from chromosomal translocations (Altucci et al., 2005). For example, the chimeric proteins formed between RUNX1 and members of Myeloid Translocation Gene family (MTG also known as ETO) as a result of the t(8;21) and the t(16;21) act as repressors through the recruitment of the nuclear hormone co-repressors N-CoR/SMRT (Amann et al., 2001; Gelmetti et al., 1998; Lutterbach et al., 1998; Wang et al., 1998). HDAC3 forms a stable complex with N-CoR and SMRT (Li et al., 2000; Wen et al., 2000), but the t(8;21) can also associate with HDAC3 independently of N-CoR (Amann et al., 2001).
A wide variety of histone deacetylase inhibitors (HDI) targeted toward class I and class II HDACs are in Phase I and Phase II clinical trials and are used in therapy against both solid and hematological tumors. These inhibitors include short aliphatic acids such as butyric acid, hydroxamic acids such as Trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA, known clinically as Vorinostat), and cyclic peptides such as depsipeptide (Bolden et al., 2006; Marks, 2007; Marks and Breslow, 2007). HDIs can trigger cellular differentiation and/or the induction of apoptosis in cell lines derived from leukemia or carcinomas (Bolden et al., 2006; Insinga et al., 2005; Marks, 2007; Marks and Breslow, 2007). However, the mechanism by which HDIs selectively target tumor cells is still not completely understood (Marks, 2007). Therefore, it is important to elucidate the function of individual HDACs to not only understand the downstream effects of these compounds, but to also design better and/or more specific HDIs that are more effective in cancer therapy.
Given the vital role of HDAC3 in normal cells and in the generation and treatment of various cancers, a conditional deletion of Hdac3 was engineered in mice. The deletion of Hdac3 in the germ line yielded very early embryonic lethality. Therefore, mouse embryonic-derived fibroblasts (MEFs) containing the Hdac3 conditional allele were used to examine the function of Hdac3 in cell viability and in cell cycle control. Consistent with RNAi-mediated knock-down studies and inactivation of Hdac3 in transformed cell lines (Li et al., 2006; Takami and Nakayama, 2000; Wilson et al., 2006), removal of Hdac3 in MEFs led to apoptosis. However, rather than mitotic catastrophe, the apoptosis was preceded by DNA damage that correlated with inefficient repair of DNA double strand breaks.
Results
Creation of _Hdac3_-deficient mice
To understand the biological functions of Hdac3 in normal development and in cell cycle control, mice lacking Hdac3 were created. Because of the reported cell lethal phenotype, Exon 7 of Hdac3 was flanked by LoxP sites and a G418 resistance gene (Neo) was inserted between Exon 7 and Exon 8 to engineer a Cre-recombinase-dependent allele (Fig. S1A). Mice harboring the targeted allele were crossed to EIIA-Cre transgenic mice to obtain progeny with either a “floxed” allele or with a null allele. No _Hdac3_-null animals were obtained from 17 litters of mice derived from the intercross of heterozygous mice, indicating embryonic lethality (Mendelian ratio of 1:2.2:0 of Wt:Het:Null). Timed matings failed to detect null embryos at E12.5, E11.5 or E9.5 indicating that the defect was prior to E9.5 (e.g., crosses of heterozygous mice at E9.5 also yielded ratios of 1:2.2:0 of Wt:Het:Null). Thus, Hdac3 is required for early embryonic development, which is consistent with the reported cellular lethality in cancer cell lines lacking HDAC3 (Wilson et al., 2006).
To probe the cellular basis of the requirement of Hdac3, MEFs were isolated from E13.5–14.5 embryos obtained from a cross between Hdac3+/− and Hdac3FL/FL mice. The MEFs obtained from Hdac3FL/+ or Hdac3FL/− embryos were then infected with recombinant Adenovirus expressing the Cre recombinase (Ad-Cre) to delete Exon 7 yielding Hdac3+/− and _Hdac3_−/− MEFs, respectively. PCR analysis of the genomic DNA isolated from Hdac3FL/+ and Hdac3FL/− MEFs following a 24 hr Ad-Cre infection showed the excision of the floxed Exon 7 (Fig. S1B). Western blot analysis with whole cell extracts prepared from Hdac3FL/+ or Hdac3FL/− MEFs showed nearly complete depletion of Hdac3 in Hdac3FL/− MEFs after Ad-Cre infection (Fig. S1C). Furthermore, immunofluoresence analysis demonstrated the lack of Hdac3 in Hdac3FL/− MEFs after Cre expression (Fig. S1D).
By 90 hr after infection with Ad-Cre when Hdac3 is completely depleted in the cells, we noted the initiation of cell death in _Hdac3_−/− MEFs, and by 120 hr post infection the percentage of cells dying increased to 20–30% of the cells in the culture (data not shown). Hoechst staining of DNA indicated that the dying cells displayed nuclear blebbing, a characteristic marker of apoptosis (Fig. S1E) and annexin V staining confirmed that this was apoptosis (data not shown).
In addition to the Ad-Cre system, Hdac3FL/FL and Hdac3+/− mice were cross-bred with transgenic mice expressing a tamoxifen responsive Cre-estrogen receptor fusion protein (Cre-ER) to yield Hdac3FL/−/Cre-ER+, or Hdac3FL/+/Cre-ER+ mice (Hayashi and McMahon, 2002). When MEFs were isolated from these mice and were treated with tamoxifen, the loss of Hdac3 was also accompanied by apoptosis (Fig. S2). In both systems, at 60–90 hr post infection, there was little apoptosis and even at 120 hr the majority of cells were still viable allowing us to investigate the effect of loss of Hdac3 on cell cycle regulation.
Changes in histone modifications and gene expression upon the loss of Hdac3.
Embryonic stem cells derived from _Hdac1_-null mice showed an increase in the acetylation of histones H3 and H4, and a decrease in H3 methylation (Lagger et al., 2002). Therefore, we used western analysis of nuclear extracts prepared from Ad-Cre infected Hdac3FL/+ or Hdac3FL/− MEFs to examine alterations in histone modifications following the loss of Hdac3. Hdac3 mRNA or protein is long lived, as we were still able to detect some Hdac3 64 hr after infection (Fig. 1, upper panel). The acetylation levels of histone H4 K5, K12 and K16 were modestly increased between 48 and 64 hr following Ad-Cre infection in _Hdac3_−/− MEFs (Fig. 1). Acetylation of these sites is typically associated with the deposition of histones onto newly synthesized DNA (Benson et al., 2006; Mello and Almouzni, 2001), suggesting a role for Hdac3 in S-phase. There was also a modest increase in histone H3 K9/K14 acetylation with a concomitant decrease in H3K9 trimethylation (Fig. 1).
Figure 1. Loss of Hdac3 alters histone acetylation.
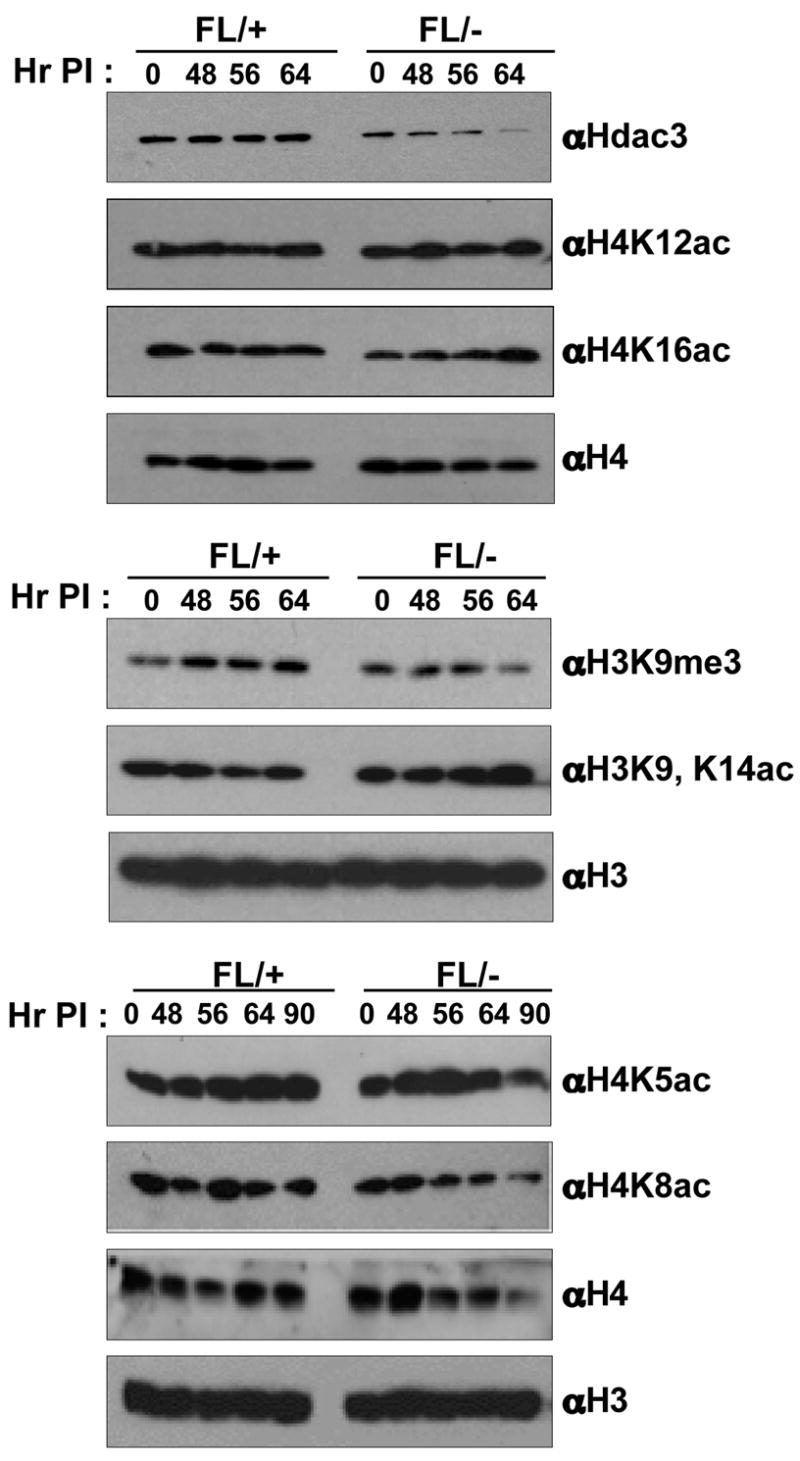
Western blot analysis of nuclear extracts prepared from Ad-Cre infected Hdac3FL/+ and Hdac3FL/− MEFs for changes in post-translational modifications of histone H3 and H4. Two independent western analyses were done using two different isolates of MEFs. A representative blot from one analysis is shown. The levels of H3 and H4 served as a loading control. The numbers above each lane indicate the time in hr after the initiation of Ad-Cre infection (Hr PI).
Given the links between Hdac3 and transcriptional repression, we used oligonucleotide micro arrays to examine RNA isolated from control and Cre recombinase expressing MEFs to define the changes in gene expression upon inactivation of Hdac3. Initially, we compared RNA from Ad-Cre infected Hdac3FL/+ and Hdac3FL/− MEFs, but found that some of the gene expression changes were due to variation between the initial MEF isolates (data not shown). Therefore, we extended the analysis to H_dac3FL_/− MEFs expressing Cre-ER. When the gene expression of ethanol-treated (vehicle) and tamoxifen-treated MEFs was compared at 72 hr post-induction, 83 genes were induced and 111 genes were down-regulated at least 1.5-fold upon inactivation of Hdac3. Approximately, 48 genes were up-regulated in both Ad-Cre and tamoxifen-treated MEFs at the 72 hr time point (Fig. S3).
The majority of the affected genes were associated with signal transduction and metabolism (Fig. 2A). Real time qRT-PCR analysis confirmed the microarray results and showed very good correlation with the microarray data with a 2–3 fold induction of the cholesterol or fatty acid synthesis regulating genes (Fig. 2B). Many of these genes were also induced when Hdac3 was inactivated in the liver (Vanden-Eynden et al., unpublished data). Overall, few of the up-regulated genes at the 72hr time point in Ad-Cre and tamoxifen-treated MEFs are associated with apoptosis or with cell cycle control (Fig. 2A). Thus, these metabolic regulatory genes may be “core” Hdac3-regulated genes, and alterations in gene expression are unlikely to trigger apoptosis. Examination of histone acetylation up-stream of the transcriptional start site of Cyp51, Sqe, and Hmgcs1 indicated that the changes in gene expression correlated with increased acetylation of histone H3K9, K14 (Fig. 2C).
Figure 2. Inactivation of Hdac3 affects gene expression.

(A) Classification of genes that were up- regulated at least 1.5-fold in both tamoxifen-treated Hdac3FL/− ERCre+ and in Ad-Cre treated Hdac3FL/−MEF at the 72hr time point. The data were analyzed using Gene Spring software to obtain the gene list and then these genes were categorized based on their different biological functions such as development (Dev.), signal transduction (Signal Trans.), cell cycle, and metabolism, using the Panther Gene Classification software. (B) Total RNA was isolated 72 hr after addition of tamoxifen to Cre-ER containing MEFs and real time PCR (Q-PCR) analysis was performed to validate the genes that were up regulated in both Ad-Cre and tamoxifen (64hr and 72hr) treated MEFs. Real time PCR was done in duplicate with the RNA isolate that was used for the microarray and with a different isolate of RNA (biological replicate) obtained from tamoxifen- and ethanol-treated Hdac3FL/−_/ERCre+_ MEFs. The values represent the average fold-increase ± S.E. of duplicate samples. (C) MEFs (Hdac3 FL/+ and Hdac3 FL/−) were infected with Ad-Cre and ChIP analysis was performed 50hr post infection and the data was quantified by real-time PCR analysis. The level of acetylated histone H3 obtained in uninfected and infected samples was normalized to the total histone H3 occupancy. The fold-increase in the acetylated H3 level was then calculated for the Ad-Cre infected Hdac3FL/− and Hdac3FL/+ MEFs after normalization to the values obtained from the uninfected cells as the control. The relative fold-increase in Ad-Cre infected Hdac3FL/− MEFs, denoted as _Hdac3_−/− in the figure, was calculated by normalizing the values to those obtained from infected Hdac3FL/+ MEFs denoted as Con. For Cyp51, primers 2 Kbp upstream of the transcriptional start site, at the transcriptional start site, and in Exon 5 were used. The data shown are the average fold-increase ± S.E. obtained from two different MEF preparations, each done in duplicate.
_Hdac3_−/− MEFs progress through mitosis
RNAi-mediated knockdown of Hdac3 in human colon cancer cell lines or HeLa cells caused an accumulation of cells in the G2 and/or M phase (Wilson et al., 2006), loss of phosphorylation of histone H3Ser10 during metaphase, and mitotic catastrophe (Li et al., 2006). Therefore, we tested whether inactivation of Hdac3 has a similar phenotype in primary cells. Surprisingly, immunofluorescence analysis indicated that there was no significant difference in the levels of histone H3Ser10 phosphorylation between Hdac3FL/− and _Hdac3_−/− MEFs in either the Ad-Cre or Cre-ER expressing cells (Fig. 3A and Fig. S4). In western blot analysis, there was only a slight decrease in the global levels of H3Ser10 phosphorylation (data not shown). In addition, there was no evidence of problems transiting mitosis, and the small reduction of H3Ser10 phosphorylation was likely due to the fact that _Hdac3_−/− cells have 2-fold fewer cells in metaphase when compared to Hdac3FL/− MEFs (2.1 % vs. 1.2%). Thus, _Hdac3_-null MEFs are impaired in cell cycle progression prior to metaphase.
Figure 3. Examination of H3Ser10 phosphorylation in _Hdac3_−/− MEFs and restoration of H3Ser10 phosphorylation by caffeine treatment in _Hdac3_−/− NIH 3T3 cells.
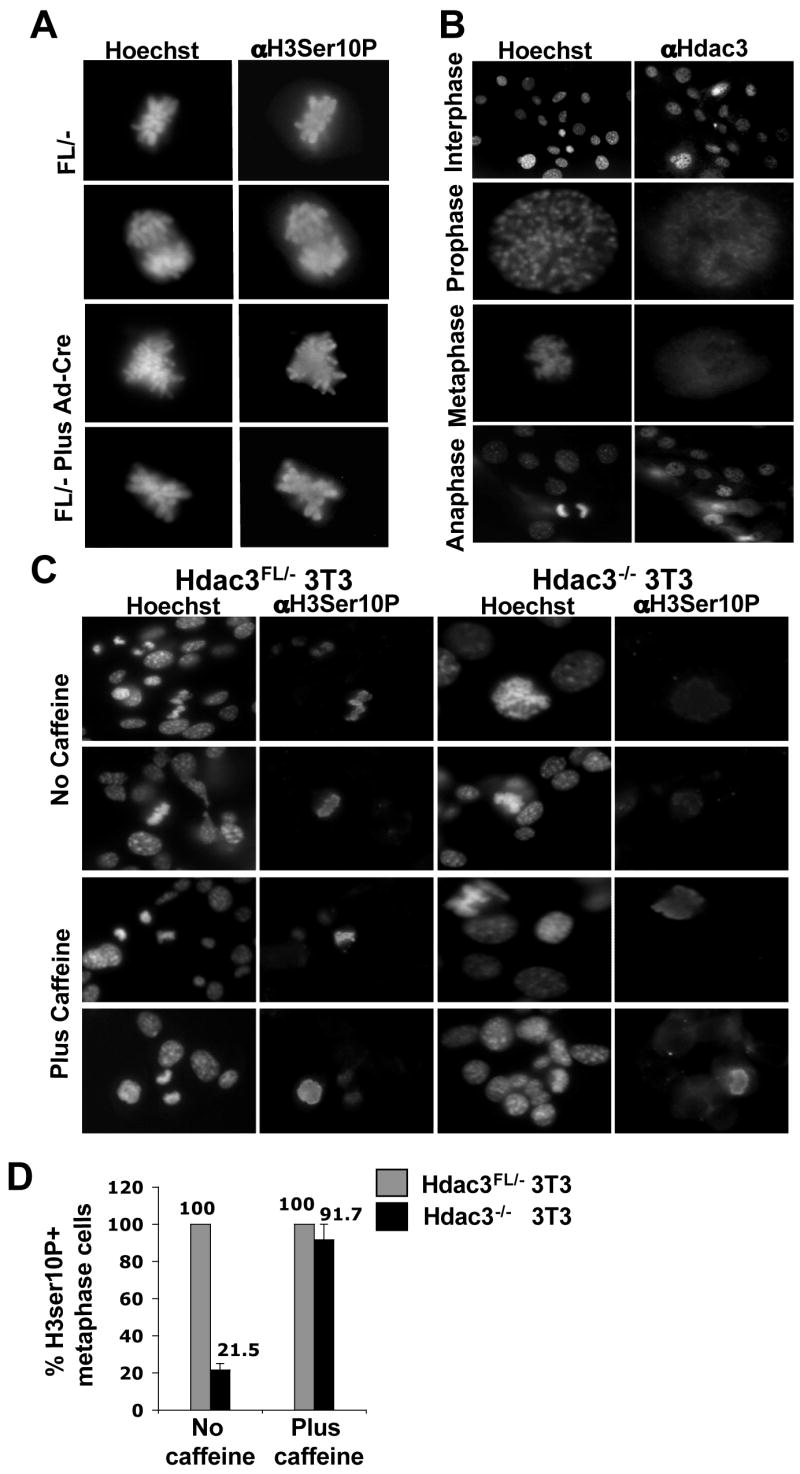
(A) Immunofluorescence analysis of mock infected or Ad-Cre infected Hdac3FL/−MEFs using anti-phospho-H3Ser10 at 90hr post-infection. Cells were stained with Hoechst 33258 to visualize the nucleus. (B) Immunofluorescence analysis using anti-Hdac3 shows the localization of Hdac3 with chromatin in interphase and prophase stages of the cell cycle. The wild type MEFs were counterstained with Hoechst 33258 to visualize the DNA. (C) Caffeine restores histone H3Ser10 phosphorylation in _Hdac3_−/−NIH 3T3 cells. Immunofluoresence staining of untreated and caffeine-treated cells using anti-phospho H3Ser10 on Hdac3FL/− and _Hdac3_−/− NIH 3T3 cells. Cells were treated with 2mM caffeine for 30 min prior to fixation. Hoechst staining of the cells was performed to visualize the nucleus. Two independent experiments were performed and the images shown are from a representative experiment. (D) Quantification of Phospho-H3Ser10 positive metaphase cells in Hdac3FL/− and _Hdac3_−/− NIH 3T3 cells in the absence and presence of caffeine. The values represent average percentage ± S.E. obtained from two independent experiments.
Given the normal phosphorylation of H3Ser10 phosphorylation, we examined Hdac3 association with chromatin in various stages of the cell cycle in wild type MEFs. Hdac3 was associated with chromatin in the interphase and prophase stages, but not in the metaphase or anaphase stages of the cell cycle, which is consistent with a role of Hdac3 prior to metaphase (Fig. 3B). To further investigate changes in chromatin, we examined the levels and/or the cellular localization of Heterochromatin Protein-1 (HP1α) (Bartova et al., 2005; Taddei et al., 2001) and Orc2. Orc2 is a subunit of the origin recognition complex, which is tightly associated with HP1α in the G1 and S-phases and is associated with centromeres during late S, G2 and M phases (Prasanth et al., 2004). Western blot analysis revealed no change in the levels of HP1α upon inactivation of Hdac3 (Fig. S5A) and immunofluoresence analysis showed that both HP1α and Orc2 localized properly (Fig. S5B, S5C). In addition, FACS analysis after Propidium Iodide staining did not uncover dramatic alterations in cell cycle progression (Fig. S5D).
_Hdac3_-null mitotic phenotypes are associated with the establishment of cell lines
To further examine the distinction between the phenotypes observed upon siRNA-mediated knockdown of Hdac3 in cell lines versus our conditional deletion of Hdac3 in primary cells, we established immortalized cell lines from Hdac3FL/− MEFs following the NIH 3T3 regimen (Todaro and Green, 1963). The immortalized NIH 3T3 Hdac3FL/− cells were then infected with Ad-Cre to excise the floxed Exon 7 of Hdac3. Although the majority of the Hdac3FL/− NIH 3T3 cells were not viable after Ad-Cre infection, we were able to establish an _Hdac3_−/− NIH 3T3 cell line. Western blot analysis of these cells confirmed the loss of Hdac3 without a compensatory increase in either Hdac1 or Hdac2 (data not shown). In concordance with previous studies of the role of HDAC3 in transformed cell lines, we found that the removal of Hdac3 in immortalized cells caused a decrease in phosphorylated histone H3Ser10 (Fig. 3C and Fig. 3D, upper panels) and fewer cells in metaphase (data not shown). Thus, the apparent _Hdac3_-null M phase defects required the immortalization process.
A decrease in mitotic cells is characteristic of an active DNA damage checkpoint (Bartek et al., 2004). Therefore, to test whether the cell cycle response of the _Hdac3_−/− NIH3T3 cells was due to an active ATM/ATR-dependent DNA damage response, we added the checkpoint inhibitor caffeine to the culture medium (Cortez, 2003). Caffeine restored histone H3Ser10 phosphorylation in metaphase chromosomes of the _Hdac3_−/− cells (Fig. 3C and D) and also restored the number of _Hdac3_−/− NIH 3T3 cells progressing to metaphase to the levels observed in control cells (data not shown). Therefore, the metaphase defects observed are either due to a DNA damage response or due to replicative stress, or both, and Hdac3 is not required for Aurora B kinase activity in primary cells or in the presence of caffeine.
Loss of Hdac3 decreases S-phase cells and causes a cell cycle delay
The analysis of NIH 3T3 cells and the effects of caffeine (Fig. 3, S4), suggested that the _Hdac3_-null phenotype might be traced back to defects in the S-phase. Therefore, BrdU pulse-chase analysis was used to more stringently examine the transit of _Hdac3_-null MEFs through S-phase. After Ad-Cre infection, Hdac3+/− and _Hdac3_−/− MEFs were labeled with BrdU for 45 min, and the BrdU labeled cells were “chased” through the cell cycle using flow cytometry. MEFs that lack Hdac3 had ~2-fold fewer BrdU-positive cells, suggesting that Hdac3 plays a vital role in the cycling of the cells (Fig. 4A). Treatment of the cells with Z-VAD-FMK (a general caspase inhibitor) impaired apoptosis, but it did not increase BrdU incorporation of the _Hdac3_−/− MEFs showing that the inefficient BrdU incorporation in these cells was not due to apoptosis (data not shown). As the BrdU label was chased throughout the cell cycle, the BrdU positive cells were delayed in clearing S-phase (Fig. 4B, 4C, compare 2 hr time points) and there was nearly a 2 hr delay in cells moving from S phase, through G2/M, and re-entering G1 (Fig. 4B, 4C, compare the 6 hr time points). When only the BrdU positive cells were analyzed, the delayed progression of the cells through S-phase was more evident (Fig. 4C).
Figure 4. Deletion of Hdac3 results in a cell cycle delay.

(A) Flow cytometry analysis of BrdU incorporation versus DNA content performed for Hdac3FL/+ and Hdac3FL/− MEFs at 90 hr following Ad-Cre infection. The experiment was done two times and a representative profile is shown as BrdU incorporation versus propidium iodide staining. (B) Ad-Cre infected Hdac3FL/+ and Hdac3FL/− MEFs were incubated with 10 μM BrdU for 45min, washed, and cultured in the absence of BrdU for the indicated times. BrdU/PI flow cytometry analysis of a representative experiment is shown. (C) Histogram representation of BrdU+ cells from (B) progressing through the cell cycle over a 10 hr time course.
DNA damage and S-phase checkpoint activation in _Hdac3_−/− MEFs
The decreased BrdU incorporation in _Hdac3_−/− cells and the ability of caffeine to bypass the defects in NIH 3T3 cells lacking Hdac3, led us to hypothesize that the S-phase defect in _Hdac3_−/− MEFs is a consequence of DNA damage. Phosphorylated H2AX (known as γH2AX) forms distinct nuclear foci and a 1:1 correspondence exists between the number of H2AX foci and the number of DSBs (Fernandez-Capetillo and Nussenzweig, 2004). γH2AX foci were counted in 100–150 cells in three independent experiments and classified into groups of cells with 0, 1–10, 10–20 and greater than 20 foci. At 72 hr (Fig. S6) and at 90 hr after Ad-Cre infection (Fig. 5A), time points well before or at the beginning of when cell death was observed (any apoptotic nuclei were excluded), there were increased numbers of cells with greater than 20 foci in _Hdac3_−/−MEFs when compared to the Hdac3+/− MEFs (Fig. 5A and Fig. 5B, p = 0.02). Similar results were obtained with tamoxifen-treated Hdac3FL/−/Cre-ER+ MEFs in which tamoxifen was added to the culture media for 48 hr and the cells were allowed to recover from exposure to Cre prior to the assessment of DNA damage (Fig. S9, DNA damage in MEFs cultured in 10% serum). It is notable that Adenovirus-mediated expression of the Cre recombinase in control cells did not cause significant DNA damage (data not shown, but see Fig. 6B).
Figure 5. Inactivation of Hdac3 causes DNA damage.

(A) Immunofluoresence analysis of γH2AX 90 hr after Ad-Cre infection of Hdac3FL/+ and Hdac3FL/− MEFs. The experiment was done three times and images from a representative experiment are shown. (B) To obtain quantitative data of the H2AX foci distribution, the number of foci in 100–150 cells were manually counted and the percentage of cells containing 0, 1–10, 10–20, and greater than 20 foci per cell was calculated. Data represent the average percentage ± S.E. of three independent experiments. Student’s t-test demonstrated that the percentage of cells with greater than 20 foci in Hdac3FL/− MEFs is statistically significant with a p value of 0.017. (C) Detection of KAP1 phosphorylation at Ser824 in Ad-Cre infected Hdac3FL/+ and Hdac3FL/− MEFs was performed by immunofluoresence. The data shown are representative of two experiments performed with two different isolates of MEF. (D) The percentage of the infected Hdac3FL/+ and Hdac3FL/− cells that are positive for KAP1 phosphorylation was determined after counting approximately 100 cells in each experiment. The data represent the average percentage ± S.E. of 2 independent experiments. (E) Western analysis for phospho MCM2 (S108) and for p53 levels of Hdac3FL/+ and Hdac3FL/− MEFs infected with Ad-Cre. Extracts prepared from doxorubicin (2μg/ml)- treated NIH 3T3 cells were used as a positive control. The level of total histone H3 was used as a loading control. (F) MEFs (Hdac3FL/+) were treated either with increasing doses of SAHA (0.5–4μM) or with DMSO for 24hr. Immunofluoresence analysis of γH2AX in treated MEFs was performed and quantification of the γH2AX foci was calculated as the average percentage of cells with 20 or more foci in two different MEF isolates, and approximately 50 cells were counted in each isolate. The each point on the graph is the mean of the percentages ± S.E. obtained with two different MEF isolates.
Figure 6. DNA damage in _Hdac3_−/− MEFs is cell cycle dependent.

(A) MEFs (Hdac3FL/+ and Hdac3FL/−) were mock infected or infected with Ad-Cre for 3 hr and 45 hr later the cells were cultured in media containing 0.5% serum for 60 hr prior to immunofluorescence using anti-γH2AX or p-KAP1. (B) Quantification of the γH2AX foci or p-Kap1 positive cells in serum starved Hdac3FL/+, Hdac3FL/−, Hdac3+/−, and _Hdac3_−/− MEFs. Shown are the averages of 2 independent experiments with the standard error. Student’s t-test indicated that the differences in cell with 20 γH2AX foci were significant (p = 0.007).
To further corroborate the presence of DNA damage following the loss of Hdac3, we examined additional markers of the DNA damage response. KAP-1 (KRAB-associated protein) is phosphorylated on Serine 824 following DNA damage, which allows chromatin relaxation (Ziv et al., 2006). A robust induction of Kap1 phosphorylation was observed in 10% of the _Hdac3_−/− cells, whereas fewer cells that exhibited only a weak signal for Kap1 phosphorylation were seen in Hdac3+/− MEFs (Fig. 5C and Fig. 5D; Fig. S9). In addition, Minichromosome Maintenance 2 (MCM2), a replication protein and a direct substrate for ATM and ATR (Cortez et al., 2004; Yoo et al., 2004) was hyperphosphorylated in _Hdac3_−/− MEFs (Fig. 5E). Finally, western analysis of the p53 tumor suppressor, a key downstream mediator of the DNA damage response (Kurz and Lees-Miller, 2004; Prives and Hall, 1999), showed a slightly higher level of total p53 in _Hdac3_−/− MEFs compared to the Hdac3+/− MEFs (Fig. 5E).
The cell death and DNA damage associated with the inactivation of Hdac3 suggested that this enzyme is a key target of HDIs that are being tested against various types of cancers in clinical trials. Therefore, we examined whether SAHA or Trichostatin A, widely used HDIs that target the class I HDACs (Gallinari et al., 2007), also cause DNA damage similar to the loss of Hdac3. MEFs were cultured in the presence of 5 μM or 10 μM TSA or 0.5 to 4 μM doses of SAHA for 24 hr and DNA damage was assessed using anti-γH2AX for immunofluorescence (Fig. 5F and Fig. S7). Although TSA is rapidly turned over in vivo and is therefore not an effective drug, it caused widespread DNA damage in MEFs (Fig. S7). Likewise, while higher levels of SAHA were required to induce DNA damage in 40% of the cells (10 μM, data not shown), at 0.5 to 1.0 μM, which are therapeutic levels commonly achieved in patients (Duvic et al., 2007; O’Connor et al., 2006), 15–25% of the cells displayed γH2AX foci and 4–7% contained greater than 20 γH2AX foci (Fig. 5F, Fig. S7, and data not shown).
Given that high levels of SAHA caused even greater DNA damage than deletion of Hdac3, we used siRNAs to impair the expression of Hdac1 and Hdac2 to explore whether these enzymes also contribute to HDI-mediated DNA damage. Although MEFs do not transfect well, immunofluoresence analysis showed that siRNA directed towards either Hdac1 or Hdac2 caused a dramatic reduction in the levels of these proteins in 70% of the cells. The loss of these Hdacs correlated with an increase in the percentage of phospho-Kap1 containing cells and it appeared that loss of Hdac2 caused more DNA damage as compared to Hdac1 (Fig. S8A). Therefore, we extended the analysis of Hdac2 siRNA treated cells using anti-γH2AX, and cells lacking Hdac2 expression were more prone to DNA damage (Fig. S8B). Thus, the DNA damage caused by HDIs may be a cumulative effect of inhibiting multiple class I Hdacs.
Next, we tested whether the observed DNA damage required cell cycle progression. Hdac3FL/+ and Hdac3FL/− MEFs were serum-starved for 60 hr following Ad-Cre infection and γH2AX levels were measured using immunofluoresence. Labeling of the serum-starved cells with anti-BrdU-FITC confirmed that only 1–2% of the cells were in the S phase following serum starvation (data not shown). Taking the cells out of the cell cycle decreased the percentage of _Hdac3_−/− cells with greater than 20 γH2AX foci, as well as the percentage of cells that were positive for KAP1 phosphorylation (Fig. 6A and 6B). Similar results were found with tamoxifen-treated Hdac3FL/−/Cre-ER+ MEFs or when serum starved cells were treated with SAHA (Fig. S9 and Fig. S10). In addition, immunofluoresence analysis of _Hdac3_−/− MEFs labeled with BrdU revealed the presence of γH2AX foci in a large number of cells that were BrdU+ (Fig. S11). However, some cells containing γH2AX, were not BrdU positive (data not shown), suggesting that if the DNA damage occurred during S phase, it was not efficiently repaired.
Depletion of Hdac3 causes inefficient repair of IR-induced NDA Damage in MEFs
To examine DNA repair, MEFs were serum starved to curtail the naturally occurring DNA damage in _Hdac3_−/− MEFs, and then exposed to 1 Gy of ionizing radiation (IR). γH2AX foci and p-Kap1 positive cells were counted at 0 hr, 2 hr and 24 hr post-irradiation. _Hdac3_−/− MEFs displayed 3-fold more cells with greater than 20 γH2AX foci immediately after IR treatment, when compared to the Hdac3+/− cells, indicating that _Hdac3_−/− MEFs are more sensitive to IR (Fig. 7A and Fig. 7B). 2 hr after irradiation, the percentage of control cells with 10–20 γH2AX foci had decreased by 2-fold with a simultaneous increase in the number of cells with 1–10 foci (Fig. 7A and Fig. 7B). Similarly, the number of p-Kap1 negative cells increased to 30% 2 hr after IR treatment (Fig. 7C and 7D). By 24 hr post-irradiation, the vast majority of the control cells contained less than 10 γH2AX foci and showed no KAP1 phosphorylation (Fig. 7A–D). By contrast, 2 hr after damage over half of the _Hdac3_−/− MEFs contained more than 10 γH2AX foci and 88% of the cells retained p-KAP1 (Fig. 7A–D). 24 hr post-irradiation, roughly 10% of the null cells still displayed markers of a DNA damage response (Fig. 7A–D). Similar results were observed using single cell gel electrophoresis (“comet” assays) of tamoxifen-treated Hdac3FL/−/Cre-ER+ MEFs 4hr after 1 Gy of ionizing radiation (Fig. 7E and Fig. S12). Thus, inactivation of Hdac3 sensitized MEFs to IR.
Figure 7. Inactivation of Hdac3 sensitizes MEFs to irradiation.

(A) Hdac3FL/+ and Hdac3FL/− MEFs infected with Ad-Cre were serum starved for 60 hr and irradiated with 1Gy of IR and γH2AX immunofluoresence was performed at 0 hr, 2 hr, and 24 hr following irradiation. (B) Quantitative analysis of γH2AX foci distribution following irradiation. (C) Immunofluoresence analysis of infected and serum starved Hdac3FL/+ and Hdac3FL/− MEFs at 0, 2, and 24 hr following 1Gy irradiation using anti-phospho-KAP-1. (D) Quantitative analysis of the data shown in C. The experiment was performed using two different preparations of MEFs and representative data are shown. (E) Quantification of the “comet” assays shown in Fig. S12. The DNA damage is expressed as the “tail moment” at the time of IR treatment (upper panel) and 4 hr later (lower panel). Tail moments of at least 25 comets in each treatment were calculated using the Comet Score software and the percentage of cells with different categories of tail moments was calculated. The experiment was done with two different MEF preparations and representative data from one experiment is shown.
Discussion
Inactivation of Hdac3 causes DNA damage and impedes DNA repair
Hdac3 is a key component of N-CoR/SMRT/TBL1 repression complex and may contribute to the action of the class IIa HDACs (Fischle et al., 2002; Guenther et al., 2001; Wen et al., 2000; Yoon et al., 2003). Using conditional inactivation of Hdac3, we have dissected the cell cycle requirements for this enzyme in both primary and immortalized cell lines. Using MEFs with a genetic deletion of Hdac3, we found that the inactivation of this key regulator of chromatin structure triggers apoptosis. Apoptosis was preceded by changes in the epigenetic program of the cells including altered histone H3 K9/K14 methylation, which demonstrates how Hdac3 can indirectly affect chromatin-modifying programs. However, no evidence of “mitotic catastrophe” or impaired phosphorylation of histone H3Ser10 was observed. By contrast, immortalized NIH 3T3 cells lacking Hdac3 did show a decrease in histone H3 Ser10 phosphorylation, but this appeared to be due to an altered response to replicative stress or to DNA damage, as when the ATM/ATR checkpoint was bypassed using caffeine, H3Ser10 phosphorylation was restored (Fig. 3). The deletion of Hdac3 in primary cells demonstrated that the inactivation of Hdac3 affects S-phase progression (Fig. 4), and causes DNA damage (Fig. 5), perhaps by disrupting DNA repair (Fig. 7), which likely contributes to the observed apoptosis.
Defective DNA double strand break repair could account for most of the phenotypes observed in _Hdac3_-null cells. Inefficient repair would cause the DNA damage that naturally occurs during DNA replication to persist, thus triggering the intra-S-phase checkpoint and impairing transit through G2 (Fig. 4). Replication stress and S-phase associated DNA damage, engage the ATR-dependent checkpoint, which is bypassed by caffeine (Bartek et al., 2004; Byun et al., 2005; Lupardus et al., 2002) and may be the underlying reason for the decrease in BrdU incorporation and impaired cycling of _Hdac3_−/− cells. These repair defects may be exacerbated by hyperacetylation of histones (Fig. 1), which may affect chromatin assembly as newly synthesized histones H3 and H4 are acetylated and must be deacetylated after deposition on to newly replicated DNA (Benson et al., 2006); (Smith and Stillman, 1989; Verreault et al., 1996). The failure to deposit newly synthesized histones or to remove acetyl residues from the histones following deposition may lead to defects in the chromatin assembly, which in turn can lead to accumulation of DNA damage.
Therapeutic implications of inactivating Hdac3
Pharmacological inhibitors of HDACs are in phase I and phase II clinical trials for the treatment of various cancers, yet the mode of action of these class I HDAC inhibitors (HDIs) is not fully understood (Bolden et al., 2006). As the catalytic component of the N-CoR/SMRT repression complex and the mediator of class IIa HDAC activities (Fischle et al., 2002; Guenther et al., 2001; Wen et al., 2000; Yoon et al., 2003), Hdac3 is likely a key target of therapeutic HDIs, but Hdac2 may also be an important target (Fig. S8). While pulses of drugs such as SAHA yield only transient inhibition of the class I enzymes, at levels achieved in patients treated with SAHA (Duvic et al., 2007; O’Connor et al., 2006), DNA damage was observed in cycling cells (Fig. S10). This result is consistent with the observation that treatment of leukemic cells with TSA caused DNA damage and triggered apoptosis (Gaymes et al., 2006). Thus, DNA damage appears to be a contributing mechanism of action for these compounds. Given that the DNA damage correlates with inefficient DNA repair (Fig. 7), this work provides a molecular mechanism for the observations that HDIs cooperate with ionizing radiation and DNA damaging agents to kill cancer cells (Camphausen and Tofilon, 2007; Karagiannis and El-Osta, 2006).
It is also notable that the distinction between the requirement for Hdac3 in primary cells and in immortalized cells (Figs. 3, 5) may provide a mechanism for the observed therapeutic window in the action of these HDIs. That is, the lack of cell cycle checkpoints in tumor cells may cooperate with the acquired DNA lesions to trigger mitotic catastrophe and apoptosis, whereas transient inhibition of HDACs with HDIs may allow normal cells to repair the damage (Fig. 6). The therapeutic window is even wider considering that _Hdac3_-null and SAHA-treated cells were protected from the DNA damage when quiescent, indicating why histone deacetylase inhibitors preferentially affect cycling cells (e.g., tumor cells). Thus, specific targeting of Hdac3 may have very important therapeutic benefits in the treatment of cancers.
Materials and Methods
Preparation of MEFs, NIH 3T3 cells, and their culture conditions
The generation of conditional _Hdac3-_null mice is described in the supplementary methods section. MEFs were isolated from E13.5–14.5 embryos and cultured in the Dubecco’s modified Eagle’s medium (DMEM) (Cellgro) supplemented with 10% FBS (Geminin Bio-products), 50μg/ml Penicillin-Streptomycin, 2mM L-Glutamine, and 1% non-essential amino acids (Cellgro). To arrest the cell cycle, cells infected with Ad-Cre were cultured in media containing 0.5% FBS for 60 hr prior to analysis. For adenovirus infection, MEFs were incubated with Ad-Cre (Ad5-CMV-Cre obtained from the Vector Development Lab, Baylor Univ., 2 × 105 particles/cell) for 3 hr. For tamoxifen treatment, MEFs were treated with 1μM tamoxifen (Sigma) and fresh tamoxifen containing media was added to the cells after 48hr. MEFs were immortalized into 3T3 cells according to the standard NIH 3T3 protocol (Todaro and Green, 1963) and cultured in DMEM media containing 10% bovine calf serum.
Protein analyses
Nuclei were prepared from MEFs and resuspended in RIPA buffer and subjected to 12% SDS-PAGE analysis. The antibodies against γH2AX and Phospho MCM2 (Ser108) were as described (Lovejoy et al., 2006). Anti-histone and anti-HP1α antibodies were obtained from Upstate Cell signaling. The antibodies for Hdac3 (ab-16047), GAPDH (clone 6C5, ab8245), HDAC2 (Y461), and HDAC1 (ab19845) were purchased from Abcam. The antibodies for p53 (FL-393), HDAC1 (H-51), and HDAC2 (C-19) were purchased from Santa Cruz Biotechnology. Phospho KAP1 (S824) (A300–767A) antibody was purchased from Bethyl Laboratories and Orc2 antibody (551179 clone: 920-4-41) was obtained from BD biosciences. Anti-BrdU-mouse IgG-Alexa 488 (A21303) was obtained from Molecular Probes.
Cell cycle analysis and Immunofluoresence microscopy
For BrdU-pulse chase analysis, cells were labeled with 10 μM BrdU for 45 min and were washed with complete media to remove BrdU and fresh MEF media was added to the cells. Cells were then fixed in ice-cold ethanol at various time points (0 hr, 2 hr, 4 hr, 6 hr, 8 hr and 10 hr) and were processed for the BrdU-PI FACS analysis as described (Strom et al., 2000).
Cells grown on coverslips were washed with PBS and fixed in 3.7% formalin for 10 min. The cells were then permeabilized with 0.5% Triton-X-100 in PBS for 3–5 min at room temperature. Cells were blocked with 2.5ml of 10% normal goat serum (Sigma) for 30 min and stained with the primary antibody for 1hr at room temperature. Cells were then incubated with the secondary antibody (anti-mouse or anti-rabbit IgG, Alexa 488/546) at a 1:600 dilution for 45 min at RT, and counterstained with Hoechst 33258 (Sigma, 1:1000) to visualize the nuclei. Images were captured using a Zeiss Axiophot microscope with a 40X lens and analyzed using Metamorph software.
Microarray and real-time quantitative PCR
Total RNA was isolated from MEFs that were treated with either AdCre, or with ethanol (solvent for tamoxifen), or with tamoxifen using the Perfect Pure Tissue Kit (5 Prime). Three separate preparations of MEFs were used for biological replicates so as to control for variation in gene expression in different isolates of MEFs Genes that were altered in their expression by more than 1.5 fold, as determined using the Student’s t-test, in these MEF isolates prior to Cre-mediated recombination were ignored. Microarray analysis was performed with the ABI Expression system in the Vanderbilt University Microarray Core. For the quantitative PCR in real time, 3 μg of total RNA was transcribed into cDNA using the Mo-MLV reverse transcriptase and 2μl of the diluted cDNA was used for the quantitative real time PCR analysis with the iQ Sybr Green Supermix (Bio-Rad) in an iCycler (Bio-Rad). The expression level of the gene of interest was calculated by normalizing the expression values of the samples to the levels of β-actin.
Chromatin Immunoprecipitation analysis
MEFs (1×106) were crossed linked with 37% formaldehyde for 10 min at 37°C, lysate was prepared with lysis buffer (50 mM Hepes (pH 7.8), 20 mM KCl, 3 mM MgCl2, 0.1% Nonidet P-40, and a mixture of protease inhibitors), and the lysate was sonicated with 8 10-sec pulses at 4 watts with a 1–2 minute refractory period in between the sonications, to reduce the chromatin fragments to ~ 500bp. Clarified lysates were precleared with Protein A-agarose (Santa Cruz Biotechnology Inc.) for 30 min and immunoprecipitated with either anti-acetyl-Histone H3 or anti-Histone H3 antibody. Following incubation with protein A-agarose, immune complexes were washed once with low salt, high salt, and LiCl immune complex wash buffer and eluted with 400 μl elution buffer (1%SDS, 0.1M NaHCO3). Cross links were reversed with 5M NaCl at 65°C, DNA was recovered by ethanol precipitation, the mixture treated with RNase and proteinase K digestion, and the DNA was purified using the PCR purification kit (Qiagen), and used for real time PCR.
Supplementary Material
01
Acknowledgments
We thank all the members of Hiebert lab for helpful discussions and advice. We also thank Mahesh Chandrasekharan, Sandra Zinkel, Courtney Lovejoy, Jeremy Myers, Yelena Janumyan, and Xin Xu for their valuable suggestions and reagents. We also thank the Vanderbilt Imaging Core and Vanderbilt Microarray Core for services and support. This work was supported by National Institutes of Health grants (R01-CA64140 and RO1-CA77274) and core services performed through Vanderbilt University Medical Center’s Digestive Disease Research grant NIDDK P30DK58404 and the Vanderbilt-Ingram Cancer Center support grant NCI P30-CA68485.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altucci L, Clarke N, Nebbioso A, Scognamiglio A, Gronemeyer H. Acute myeloid leukemia: therapeutic impact of epigenetic drugs. Int J Biochem Cell Biol. 2005;37:1752–1762. doi: 10.1016/j.biocel.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, Downing JR, Meyers S, Hiebert SW. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–6483. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Bartova E, Pachernik J, Harnicarova A, Kovarik A, Kovarikova M, Hofmanova J, Skalnikova M, Kozubek M, Kozubek S. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J Cell Sci. 2005;118:5035–5046. doi: 10.1242/jcs.02621. [DOI] [PubMed] [Google Scholar]
- Benson LJ, Gu Y, Yakovleva T, Tong K, Barrows C, Strack CL, Cook RG, Mizzen CA, Annunziato AT. Modifications of H3 and H4 during chromatin replication, nucleosome assembly, and histone exchange. J Biol Chem. 2006;281:9287–9296. doi: 10.1074/jbc.M512956200. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Tong JK, Schreiber SL. Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci U S A. 2000;97:13708–13713. doi: 10.1073/pnas.250477697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25:4051–4056. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem. 2003;278:37139–37145. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Nussenzweig A. Linking histone deacetylation with the repair of DNA breaks. Proc Natl Acad Sci U S A. 2004;101:1427–1428. doi: 10.1073/pnas.0307342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkuhler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- Gaymes TJ, Padua RA, Pla M, Orr S, Omidvar N, Chomienne C, Mufti GJ, Rassool FV. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res. 2006;4:563–573. doi: 10.1158/1541-7786.MCR-06-0111. [DOI] [PubMed] [Google Scholar]
- Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SG, Ekstrom TJ. The human histone deacetylase family. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- Gregoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol. 2007;27:1280–1295. doi: 10.1128/MCB.00882-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Insinga A, Minucci S, Pelicci PG. Mechanisms of selective anticancer action of histone deacetylase inhibitors. Cell Cycle. 2005:741–743. doi: 10.4161/cc.4.6.1717. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Karagiannis TC, El-Osta A. Modulation of cellular radiation responses by histone deacetylase inhibitors. Oncogene. 2006;25:3885–3893. doi: 10.1038/sj.onc.1209417. [DOI] [PubMed] [Google Scholar]
- Khochbin S, Kao HY. Histone deacetylase complexes: functional entities or molecular reservoirs. FEBS Lett. 2001;494:141–144. doi: 10.1016/s0014-5793(01)02327-4. [DOI] [PubMed] [Google Scholar]
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. Embo J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfi A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. Embo J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, Phelan C, Lazar MA. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linggi BE, Brandt SJ, Sun ZW, Hiebert SW. Translating the histone code into leukemia. J Cell Biochem. 2005;96:938–950. doi: 10.1002/jcb.20604. [DOI] [PubMed] [Google Scholar]
- Lovejoy CA, Lock K, Yenamandra A, Cortez D. DDB1 maintains genome integrity through regulation of Cdt1. Mol Cell Biol. 2006;26:7977–7990. doi: 10.1128/MCB.00819-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, Cimprich KA. A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 2002;16:2327–2332. doi: 10.1101/gad.1013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Mello JA, Almouzni G. The ins and outs of nucleosome assembly. Curr Opin Genet Dev. 2001;11:136–141. doi: 10.1016/s0959-437x(00)00170-2. [DOI] [PubMed] [Google Scholar]
- O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. Embo J. 2004;23:2651–2663. doi: 10.1038/sj.emboj.7600255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Smith S, Stillman B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell. 1989;58:15–25. doi: 10.1016/0092-8674(89)90398-x. [DOI] [PubMed] [Google Scholar]
- Strom DK, Nip J, Westendorf JJ, Linggi B, Lutterbach B, Downing JR, Lenny N, Hiebert SW. Expression of the AML-1 oncogene shortens the G(1) phase of the cell cycle. J Biol Chem. 2000;275:3438–3445. doi: 10.1074/jbc.275.5.3438. [DOI] [PubMed] [Google Scholar]
- Taddei A, Maison C, Roche D, Almouzni G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001;3:114–120. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem. 2000;275:16191–16201. doi: 10.1074/jbc.M908066199. [DOI] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A. 1998;95:10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YD, Perissi V, Staszewski LM, Yang WM, Krones A, Glass CK, Rosenfeld MG, Seto E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci U S A. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- Yoo HY, Shevchenko A, Shevchenko A, Dunphy WG. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem. 2004;279:53353–53364. doi: 10.1074/jbc.M408026200. [DOI] [PubMed] [Google Scholar]
- Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. Embo J. 2003;22:1336–1346. doi: 10.1093/emboj/cdg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, et al. HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01