RNA interference guides histone modification during the S phase of chromosomal replication (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 8.
Published in final edited form as: Curr Biol. 2008 Apr 8;18(7):490–495. doi: 10.1016/j.cub.2008.03.016
Summary
Background
Heterochromatin is chromosomal material that remains condensed throughout the cell division cycle and silences genes nearby. It is found in almost all eukaryotes, and although discovered (in plants) almost 100 years ago, the mechanism by which heterochromatin is inherited has remained obscure. Heterochromatic silencing and histone H3 lysine-9 methylation (H3K9me2) depend, paradoxically, on heterochromatic transcription and RNA interference (RNAi).
Results
Here we show that heterochromatin protein 1 in fission yeast (Swi6) is lost via phosphorylation of H3 serine-10 (H3S10) during mitosis, allowing heterochromatic transcripts to transiently accumulate in S phase. Rapid processing of these transcripts into small interfering RNA promotes restoration of H3K9me2 and Swi6 after replication when cohesin is recruited. We also show that RNAi in fission yeast is inhibited at high temperatures, providing a plausible mechanism for epigenetic phenomena that depend on replication and temperature, such as vernalization in plants and position effect variegation in animals.
Conclusions
These results explain how “silent” heterochromatin can be transcribed, and lead to a model for epigenetic inheritance during replication.
Introduction
The centromeres of S.pombe are composed of a kinetochore binding region flanked by innermost repeats and then by outermost dh and dg repeats (Fig. 1). RNAi triggers heterochromatin assembly and is required for its’ maintenance during growth [1, 2]. Reporter genes inserted into those repeats are silenced, but loss of RNAi pathway components Dicer (_dcr1_−), Argonaute (_ago1_−) or RNA dependent RNA polymerase (_rdp1_−) leads to loss of silencing and a decrease of H3K9me2 [1]. In addition, RNAi mutants exhibit chromosome segregation and cohesion defects [2, 3]. RNAi is involved in both transcriptional and post-transcriptional silencing of centromeric repeats: the forward strand is silenced transcriptionally via Swi6, while the reverse strand is strongly transcribed, but silenced posttranscriptionally [1, 4]. The reverse transcript is cleaved or “sliced” by the RNase-H-like activity of Ago1 leading to recruitment of the RDRC complex which generates double stranded RNAs [5, 6]. Dicer processes dsRNA into siRNA[7, 8], which is loaded onto Ago1 [9] and incorporated into the RITS complex, which associates with chromatin through Chp1 binding to H3K9me2 [10]. Ago1 slicing is thought to recruit the Rik1 complex, which includes the histone methyltransferase Clr4, as well as Clr8/Dos1, Clr7/Dos2 and Pcu4 [11–13]. Methylation of H3K9 by Clr4 results in recruitment of Swi6 and silencing of the forward strand, which accumulates, at 5–10 fold lower levels than the reverse strand [1].
Figure 1. Heterochromatic repeats and replication origins near centromere 2.
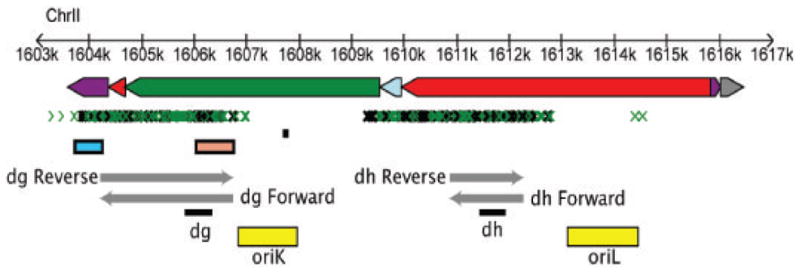
Repeats are oriented with telomere to the left and centromere to the right. Non coding features are depicted as dg, dh and imr repeats (green, red and purple rectangles, respectively) along with chromosomal coordinates. Small RNA appear as small green [40] and black [8] arrows. Centromeric promoters [4] are represented by blue (reverse) and sepia (forward) rectangles while transcripts from dg [4] and dh [1] are shown as grey arrows. Origins of replication [18] are depicted by yellow rectangles: oriK is the strongest and contains an ars-binding protein (Abp1) binding site consensus sequence (black). Black rectangles under dh and dg transcripts indicate regions used for PCR amplification (p30 and p33 probes, respectively).
RNAi-mediated silencing plays a significant role in the function of the centromere due to the recruitment of Swi6 and the consequent retention of cohesin during the G2 phase of the cell cycle, when sister chromatids must align with the spindle before segregation [14]. It has been reported that _ago1_− and _dcr1_− mutants have delayed cytokinesis, and hence decreased growth rate. G1, S and M checkpoints are also affected [15]. Dicer knockouts result in cell death and premature chromosome segregation in vertebrate cells [16].
In S. pombe heterochromatic centromeric outer repeats are replicated early, just before the beginning of S phase via origins of replication [17, 18] that lie between the dh and dg transcription units (Fig. 1). In order to measure heterochromatin transcription and modification during the cell cycle we synchronized wild type cells. This can be achieved by first arresting them in the cell cycle, and then releasing to progress through each phase at more or less the same time. Cells can be arrested by using drugs or by using temperature sensitive mutants in cell cycle progression. We have found that RNAi occurs specifically in the S phase of the cell cycle, but is inhibited at high temperatures such as those used to arrest cell cycle mutants. Our results have implications for epigenetic phenomena in animals and plants, many of which also occur in dividing cells and are sensitive to temperature.
Results
RNAi is inhibited at elevated temperatures
In order to investigate RNAi during the cell cycle we arrested mutant cells at different points in the cell cycle when grown at the restrictive temperature, namely cdc25-22 (G2 arrest), cdc10_–_129 (G1/S phase arrest) and nda3-KM11 (M phase arrest). Synchronization was followed using cell cycle marker genes expressed in G2, M and S phase, and was most complete in cdc25-22. We found that small RNA accumulated at low levels at the restrictive temperature, and gradually increased through the M, S, and G2 phases reaching a peak at the end of the second S phase (Fig. 2). This pattern of accumulation is not cell cycle regulated, but rather increases gradually after release. For example, levels of siRNA were much higher from 180–210 minutes than from 0–30 minutes, although both were in G2, the predominant phase in unsynchronized cells. When nda3-KM11 mutant cells were arrested in mitosis at cold-temperatures, we observed the opposite pattern, with a steady decline through S, G2 and subsequent S phases (data not shown). We therefore suspected that RNA interference in S. pombe is suppressed at high temperatures, but enhanced at low temperatures, confounding any cell cycle regulation. In unsynchronized wild type cells, we found that centromeric transcripts were sharply elevated at 36C (Fig. 2A) consistent with the loss of centromeric silencing at high temperatures previously reported [19]. We therefore used alternative means of synchronizing cells to study RNAi.
Figure 2. Quantitative analysis of temperature effect on RNAi.

A Unsynchronized with type cells were grown at four different temperatures; 23C, 27C, 32C and 36C. Total RNA from cells grown at each temperature was analyzed by RT-PCR using primers from the dh heterochromatic repeat. The signal was quantified using actin (act1). RNAi mediated silencing is lost at 36 degrees, confounding the use of temperature sensitive cell cycle mutants to study RNAi.
B Synchronization of cdc25-22 temperature sensitive mutant cells was achieved by incubation at 36C, release and growth at 26C. Synchronization was analyzed with cell cycle specific genes; dashed black-cig2 (early S), blue-hhf2 (S) and brown-psu1 (G2). siRNA levels, shown below, increase as the cells are released and grown at permissive temperature (26C).
RNAi occurs in S phase
We synchronized cells by incubation in hydroxyurea (HU), which depletes dNTP pools preventing cells from completing DNA synthesis in S phase. Cells were released when septation was complete. Measurement of the septation index and expression of 4 different cell cycle specific genes indicated the cells went through one full cycle and one partial cycle of synchronous division (Fig. 3). Fractions were collected every 10 minutes and analyzed for expression of strand specific dh and dg repeats, small interfering RNAs, and histone H3 modification (Fig. 3).
Figure 3. Cell cycle analysis of centromeric transcripts, small RNA and histone modifications in WT cells.
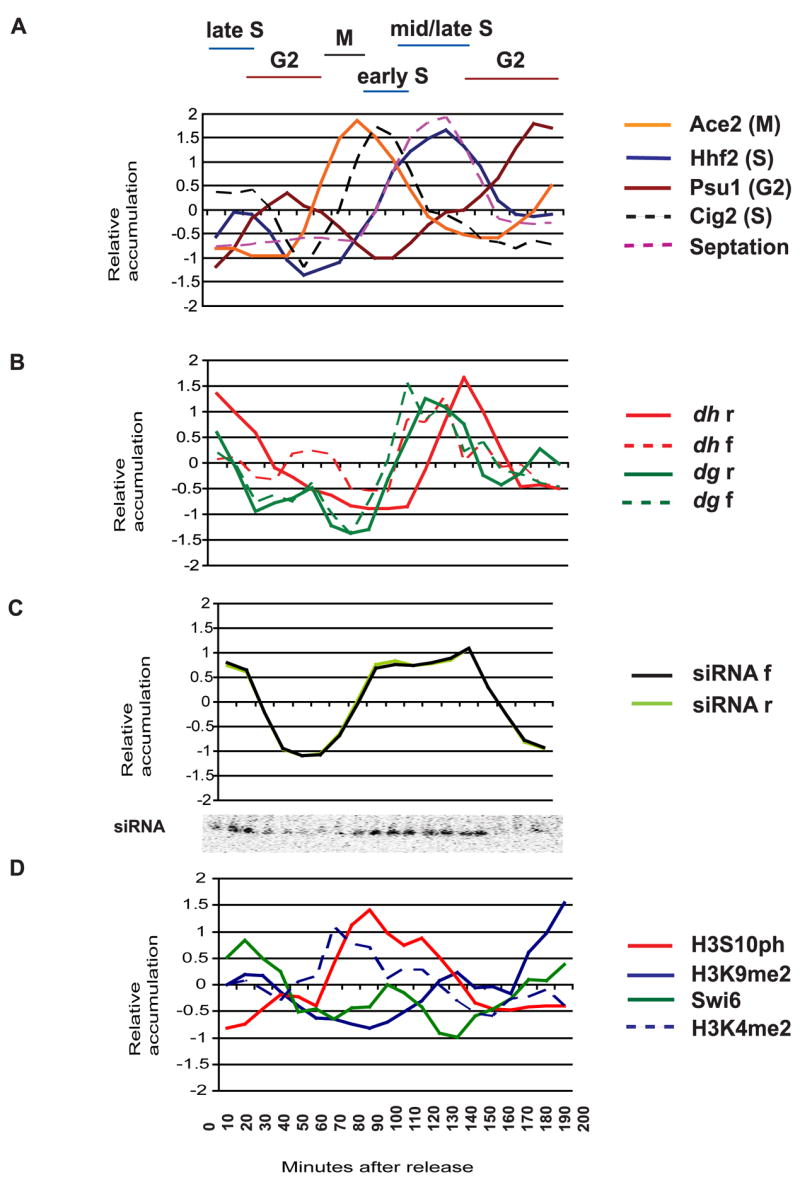
A Synchronization of wild type S.pombe cells with hydroxyurea (HU) was followed by quantitative RT-PCR of cell cycle genes; blue- hhf2 (S phase), dashed black-cig2 (early S phase), brown- psu1 (G2), orange- ace2 (M) and by measuring the septation index, which peaks during S phase (dashed pink).
B Quantitative PCR analysis of strand specific dh (red) and dg (green) transcripts. In both cell cycles, both dh and dg forward transcripts (dashed lines) appear before reverse transcripts (solid lines). Reverse transcripts are much more abundant than forward transcripts, but have been normalized for comparison (Experimental Procedures).
C Forward (black) and reverse (lime) strands of siRNAs appear throughout S phase, as shown on the small RNA Northern Blot (inset). The two strands accumulate in parallel with each other.
D Chromatin immunoprecipitation of H3S10ph (red), H3K9me2 (blue), Swi6 (green) and H3K4me2 (dashed blue) associated with the dh repeat was performed on samples taken between 0 and 210 minutes following HU release. H3K9me2 is highest in G2. Swi6 (green) is lost in M and again in S phase when H3S10 is phosphorylated (red). Swi6 levels return to normal in G2.
Transcripts from both strands of the dg and dh repeats accumulated predominantly in S phase, with the forward strand transcript appearing first, but at much lower levels than the reverse strand[1, 4]. The forward transcripts initiate closer to the origins of replication (Fig. 1). dh and dg reverse strand heterochromatic transcripts are processed co-transcriptionally into 21–24 nucleotide long siRNA via RdRP and Dicer[5, 20, 21], and we found that these siRNA accumulate in S phase, detected on Northern blots in the first and second cell cycles (Fig. 3). Origins of replication fire under hydroxyurea arrest [22], but replication forks fail to elongate, so that siRNA accumulation follows origin firing.
Histone modification
The levels of modified histones associated with dh heterochromatic repeats throughout the cell cycle were investigated by Chromatin Immunoprecipitation (ChIP) (Fig. 3). Synchronized WT cells were fixed in formaldehyde and incubated with various antibodies. Quantification was achieved using input DNA from whole cell extract (WCE) and control primers, via quantitative PCR (Experimental Procedures). H3K9me2 levels associated with heterochromatic repeats decreased during the cell cycle, reaching a minimum in early S phase, precisely when the repeats have replicated (Fig. 3). This may reflect distribution of parental modified histones on each of the two daughter chromatids immediately after replication [23]. Histone H3 serine-10 (H3S10) phosphorylation peaked in mitosis, as expected [24]. H3S10ph prevents binding of the heterochromatin protein-1 (HP-1) homolog Swi6 to H3K9me2 via the “phospho-methyl switch” [25]. As H3S10ph increased, levels of Swi6 fell, first to a local minimum in M phase, and then to an absolute minimum in S phase. Each minimum corresponded to a peak of H3S10ph, and Swi6 levels only returned to normal in G2, once H3S10ph was lost (Fig. 3). Swi6 levels were thus inversely related to H3S10ph, and the septation index.
Swi6 is required after S phase for retention of cohesin in heterochromatin and ensures proper chromosome segregation [14]. It is also required for transcriptional silencing [1] so that loss during the cell cycle might allow accumulation of transcripts from dh and dg heterochromatic repeats. In agreement with this model, forward transcripts accumulated weakly during M phase, and much more strongly during S phase. The more abundant reverse transcripts[1], followed the same pattern, and accumulated predominantly in S phase (Fig. 3). H3K4me2 is a robust mark of active transcription and was associated with dh repeats when Swi6 was lost (Fig. 3). This pattern of transcripts accounts for S phase specific accumulation of siRNA (Fig. 3), as dsRNA is generated from reverse transcripts by Rdp1 and processed into siRNAs[1, 4]. It neatly accounts for the paradoxical transcription of heterochromatic repeats, as heterochromatic marks are reduced transiently during replication.
In a very recent study of cdc25-22 mutant cells, reverse dh pre-siRNA transcript accumulation was found to be unchanged throughout the cell cycle, while siRNA steadily increased [26] (Supplementary Figures 1 and 10, respectively), in close agreement with our study of these mutants (Fig. 2). However, forward transcripts and PolII were found to be associated with dh repeats during S phase, along with Ago1 [26]. The authors conclude that heterochromatic repeats can still be transcribed in G2 on the reverse strand, despite being associated with silent heterochromatic marks. Presumably this inconsistency reflects inhibition of RNAi at high temperatures, calling these results into question. However, most of the cell cycle changes observed in cdc25-22 cells following arrest and release [26] can be reconciled with those in HU treated cells (see below), except that our measurements were taken at much higher resolution allowing M, G1 and S phase to be distinguished using cell cycle markers (Fig. 3).
Dcr1, Rdp1 and Ago1 themselves are not cell cycle regulated [27], although lack of _dcr1_− and _ago1_− has been linked to cytokinesis defects [15]. siRNA, H3K9me2 and Swi6 are lost or reduced in RNAi mutants, while centromeric transcripts accumulate to high levels [1]. We performed cell cycle analysis of centromeric transcripts in _dcr1_− mutants (Supplemental Fig. 1). Septation was delayed in _dcr1_− as previously reported[15]. dh forward transcripts were elevated in G2 (Supplemental Fig. 1), consistent with loss of transcriptional silencing, and in agreement with expression levels in _clr4_− cells [26]. However, reverse transcripts still peaked in S phase, at much higher levels than in WT, consistent with loss of post-transcriptional silencing as well [1].
Swi6 and the phospho-methyl switch
siRNA accumulation guides modification of H3K9me2[1], which increases steadily during S phase and peaks in G2, closely following the G2 cell cycle marker Psu1 (Fig. 3). One prediction is that mutants in Histone H3 S10 phosphorylation should lose H3K9me2 after each round of replication. Substitution of alanine for serine-10 in histone H3 is viable in S.pombe and prevents phosphorylation [28]. Consistent with our model, H3K9me2 accumulation is lost in these strains along with centromeric silencing [28]. However, alanine substitution and/or loss of H3K9me2 also reduces binding of Swi6 (Supplemental. Fig. 2) [28]. We therefore reduced levels of serine-10 phosphorylation transiently as well, so as not to lose H3K9me2. Histone H3S10 is phosphorylated by aurora kinase (Ark1), which has many other substrates and is essential for growth. We used an nmt1::ark1+ “shut off” plasmid to reduce Ark1 levels in an Δ_ark1_ deletion strain, which undergoes cell cycle arrest [29]. H3S10ph levels decreased upon repression of nmt1::ark1+ (Supplemental Fig. 3) while levels of Swi6 were elevated in arrested cells, without loss or gain of H3K9me2. In contrast, _swi6_− mutants gain H3K9me2 [25]. Taken together, these genetic experiments strongly support our model.
Discussion
Inheritance of modified histones after replication
Heterochromatin is defined by its inheritance from interphase to interphase [30] and represents an intriguing example of propagation of epigenetic material. Our results suggest a mechanism for epigenetic inheritance of heterochromatin during chromosomal replication (Fig. 4). Replication results in transmission of genetic information (DNA) into each daughter cell, but nucleosomes are disrupted as the replication fork proceeds [31], resulting in the loss of modified histones as fresh histones are deposited onto replicated DNA [32]. H3S10 phosphorylation during mitosis leads to loss of Swi6 and transcription of heterochromatic repeats. Double stranded RNA is quickly processed into siRNA leading to restoration of H3K9me2 at the end of S phase. H3K9me2 may even “spread” across the replication fork from unreplicated to replicated DNA, via transcription of the reverse strand and processing by RNAi [5]. Thus, we can view epigenetic inheritance as a cyclic event that is based on inheritance of modified histones during replication, guided by RNAi. Interestingly, heterochromatic silencing in budding yeast also requires passage through S phase, though neither DNA replication nor RNAi are thought to be involved [33]. Thus fission and budding yeasts seem to have evolved distinct mechanisms to propagate silent chromatin after replication.
Figure 4. A model for cell cycle regulation of heterochromatic RNA interference and histone modification.

Cells in G2 have high levels of H3K9me2 and Swi6 associated with heterochromatic repeats, and retain cohesin. Phosphorylation of H3S10 in mitosis results in loss of Swi6, and transcription of the repeats in early S phase. K9me2 levels fall during replication, but are restored in S phase by RNA interference of the transcripts, which guides K9 methylation, recruiting Swi6.
In multi-cellular organisms, if heterochromatic siRNA only arise in S phase, then they would only accumulate in dividing cells or in cells undergoing endoreduplication. Indeed, genes involved in replication are also required for RNAi-mediated transgene silencing in Arabidopsis and in fission yeast [34]. S phase specificity has major implications for epigenetic phenomena that depend on RNAi, such as transposon silencing, and for phenomena that depend on non-coding RNA, such as X inactivation and imprinting. Occasional failure to maintain heterochromatic siRNA during replication could contribute to variegated patterns of expression, which are hallmarks of these epigenetic phenomena. Further, paramutation in maize and position effect variegation in Drosophila, exhibit temperature sensitivity and are regulated, at least in part, by RNAi [35, 36]. Our results suggest that siRNA in each case should accumulate in dividing cells. They also provide an attractive explanation for vernalization in Arabidopsis: The process by which FLOWERING LOCUS C (FLC) is silenced during long periods of cold, resulting in a cellular memory of winter and flowering the following Spring [12]. Permanent silencing only occurs if cold periods are experienced by dividing cells [37] and requires, among other things, histone methylation [30] and heterochromatic RNAi [38]. Thus replication-dependent and temperature-sensitive RNA-mediated silencing may contribute to environmental response, as well as to the epigenetic inheritance of heterochromatin.
Experimental Procedures
Fission yeast strains
_ago1_−, _dcr1_−, and _rdp1_− null alleles with G418 resistant cassettes were generated as described previously [1]. cdc25-22, nda3-KM11 and cdc10_–_129 temperature sensitive mutants were obtained from M. Yanagida. The mutants were grown at permissive temperatures (26C for _cdc25_-22 and cdc10_–_129, 33C for nda3-KM11). Ark1+ function was ablated by growth in thiamine as described [29].
Hydroxyurea synchronization and analysis of dh centromeric transcripts
Log phase wild type S.pombe cells were treated with 15mM hudroxyurea (Sigma) for 4.5 hours. The cells were washed twice and grown for 5 hours. Total RNA was prepared using hot phenol [5]. Synchronization efficiency was estimated by septation index and PCR analysis of cell cycle specific genes; Hhf2 (S phase), Ace2 (M) and Psu1 (late G2) of cDNA samples taken at 10 minute intervals.
Strand specific analysis of dh and dg centromeric transcripts
Following a hot phenol RNA extraction, each sample was treated with DNAseI and reverse transcribed with either forward or reverse dh (p30) or dg (p33) primers along with either forward or reverse actin (act1) primers to obtain strand specific cDNA (SuperScript III Reverse Transcriptase; Invitrogen). The strand specific samples were amplified using SYBR Green qPCR Universal Kit (Invitrogen) with either dh (p30) or dg (p33) primers and control actin (act1) primers. The samples were analyzed with Opticon Program. In order to standardize all three technical replicates, the mean of all values for a given profile was subtracted from each individual value for that profile. The difference was divided by the standard deviation of the entire profile, giving a value similar to a Z score. A moving average was calculated for every 3 consecutive samples and plotted.
Cdc25-22 temperature sensitive mutant analysis
The cdc25-22 cells were grown at 26C and shifted to 36C for 4 hours to induce arrest. The cells were then released back to permissive temperature (26C) and grown for 4,5 hours. The small RNA blots were probed with dh (p30) forward and reverse probe, as well as U6. The blots were quantified by dividing dh probe signal by U6 signal.
Temperature effect analysis
Unsynchronized wild type S.pombe cells were grown at four different temperatures; 23C, 27C, 32C and 36C. RNA was isolated and RT-PCR was performed using dh (p30) and actin (act1) primers. Gel scans were used for quantification. The enrichment was calculated by dividing dh signal by actin signal.
Small RNA quantification
Small interfering RNAs were isolated from total RNA using Polyethylene glycol precipitation. Samples were resuspended in formamide and run on 15% urea denaturing polyacrylamide gel. The blots were transferred onto a nitrocellulose membrane and incubated at 42C with both sense and antisense α32P-UTP labeled riboprobe specific for dh centromeric repeats (p30), which was generated using T3 and T7 vitro transcription Maxiscript (Ambion) and hydrolyzed with sodium bicarbonate. The blot was probed with a U6 snRNA con end labeled with γ32P-ATP and Polynucleotide Kinase (NEB). Washing was with 2×SSC 2% SDS at 50C. Blots were exposed onto phosphoimager screens for 1–3 days and quantified. Enrichment was calculated by dividing siRNA signal by U6 control signal.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as previously described [39]. HU synchronized fractions of wild type S.pombe cells were fixed in 1% formaldehyde and the chromatin was purified, sonicated and incubated overnight with either of the antibodies: anti-H3K9me2 (Upstate), anti- H3K4me2 (Upstate), anti-phospho-H3 Ser10 (Upstate) or rabbit polyclonal sera against Swi6 (Abcam). DNA was extracted using phenol and Ethanol-precipitated. For quantification, ChIP samples were amplified using multiplex PCR in the presence of α-32P-dCTP, run on 5% polyacrylamide gel, dried and exposed to a phosphoimager screen. Quantification was obtained using actin as a measurement of background (with the exception of H3S10ph, which was quantified without normalization to actin, because actin association with H3S10ph is also cell-cycle regulated) and comparing the intensity of the test and actin bands in the ChIP sample with those in DNA purified from Whole Cell Extract (WCE). The enrichment was calculated as (target band IP/normalization band IP)/(target band WCE/normalization band WCE). In Fig. 3, ChIP results were analyzed by quantitative PCR in 3 replicates. The following formula was used: 2^-(input _dh_-input actin)/2^-(wce _dh_-wce actin), except for H3S10ph, for which the formula 2^-(input dh)/2^-(wce dh), was used. This is because the euchromatic actin gene also associates with H3S10ph. The profiles were then standardized as described above and the moving avarage of all three replicates was plotted on the graph.
Analysis of ark1 mutant
The enrichment was calculated as (target band IP/normalization band IP)/(target band WCE/normalization band WCE)
Supplementary Material
01
Acknowledgments
We thank Jim Hicks, Bruce Stillman and Janet Leatherwood for helpful advice. We are grateful to Robin Allshire, Janni Petersen and Mitsuhiro Yanagida for providing mutant strains. This work is in partial completion of the requirements for a PhD (AK). MZ was supported by a postdoctoral fellowship from the Spanish Ministry of Science and Education. Research in the author’s laboratory is supported by grants from the National Science Foundation Plant Genome Program DBI-0421651 and the NIH (R01GM076396).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science (New York, NY) 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 2.Hall IM, Noma K, Grewal SI. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:193–198. doi: 10.1073/pnas.232688099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, Allshire RC. RNA interference is required for normal centromere function in fission yeast. Chromosome Res. 2003;11:137–146. doi: 10.1023/a:1022815931524. [DOI] [PubMed] [Google Scholar]
- 4.Djupedal I, Portoso M, Spahr H, Bonilla C, Gustafsson CM, Allshire RC, Ekwall K. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes & development. 2005;19:2301–2306. doi: 10.1101/gad.344205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irvine DV, Zaratiegui M, Tolia NH, Goto DB, Chitwood DH, Vaughn MW, Joshua-Tor L, Martienssen RA. Argonaute slicing is required for heterochromatic silencing and spreading. Science (New York, NY) 2006;313:1134–1137. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- 6.Colmenares SU, Buker SM, Buhler M, Dlakic M, Moazed D. Coupling of double-stranded RNA synthesis and siRNA generation in fission yeast RNAi. Molecular cell. 2007;27:449–461. doi: 10.1016/j.molcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 8.Reinhart BJ, Bartel DP. Small RNAs correspond to centromere heterochromatic repeats. Science (New York, NY) 2002;297:1831. doi: 10.1126/science.1077183. [DOI] [PubMed] [Google Scholar]
- 9.Buker SM, Iida T, Buhler M, Villen J, Gygi SP, Nakayama J, Moazed D. Two different Argonaute complexes are required for siRNA generation and heterochromatin assembly in fission yeast. Nature structural & molecular biology. 2007;14:200–207. doi: 10.1038/nsmb1211. [DOI] [PubMed] [Google Scholar]
- 10.Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science (New York, NY) 2004;303:672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horn PJ, Bastie JN, Peterson CL. A Rik1-associated, cullin-dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes & development. 2005;19:1705–1714. doi: 10.1101/gad.1328005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amasino RM. Vernalization and flowering time. Current opinion in biotechnology. 2005;16:154–158. doi: 10.1016/j.copbio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Hansen KR, Ibarra PT, Thon G. Evolutionary-conserved telomere-linked helicase genes of fission yeast are repressed by silencing factors, RNAi components and the telomere-binding protein Taz1. Nucleic acids research. 2006;34:78–88. doi: 10.1093/nar/gkj415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 15.Carmichael JB, Provost P, Ekwall K, Hobman TC. ago1 and dcr1, two core components of the RNA interference pathway, functionally diverge from rdp1 in regulating cell cycle events in Schizosaccharomyces pombe. Molecular biology of the cell. 2004;15:1425–1435. doi: 10.1091/mbc.E03-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nature cell biology. 2004;6:784–791. doi: 10.1038/ncb1155. [DOI] [PubMed] [Google Scholar]
- 17.Kim SM, Huberman JA. Regulation of replication timing in fission yeast. The EMBO journal. 2001;20:6115–6126. doi: 10.1093/emboj/20.21.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JG, Caddle MS, Bulboaca GH, Wohlgemuth JG, Baum M, Clarke L, Calos MP. Replication of centromere II of Schizosaccharomyces pombe. Molecular and cellular biology. 1995;15:5165–5172. doi: 10.1128/mcb.15.9.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allshire RC, Javerzat JP, Redhead NJ, Cranston G. Position effect variegation at fission yeast centromeres. Cell. 1994;76:157–169. doi: 10.1016/0092-8674(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 20.Buhler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 21.Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science (New York, NY) 2005;309:467–469. doi: 10.1126/science.1114955. [DOI] [PubMed] [Google Scholar]
- 22.Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. DNA replication origins fire stochastically in fission yeast. Molecular biology of the cell. 2006;17:308–316. doi: 10.1091/mbc.E05-07-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129:813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- 24.Dormann HL, Tseng BS, Allis CD, Funabiki H, Fischle W. Dynamic regulation of effector protein binding to histone modifications: the biology of HP1 switching. Cell cycle (Georgetown, Tex. 2006;5:2842–2851. doi: 10.4161/cc.5.24.3540. [DOI] [PubMed] [Google Scholar]
- 25.Yamada T, Fischle W, Sugiyama T, Allis CD, Grewal SI. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Molecular cell. 2005;20:173–185. doi: 10.1016/j.molcel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 27.Rustici G, Mata J, Kivinen K, Lio P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bahler J. Periodic gene expression program of the fission yeast cell cycle. Nature genetics. 2004;36:809–817. doi: 10.1038/ng1377. [DOI] [PubMed] [Google Scholar]
- 28.Mellone BG, Ball L, Suka N, Grunstein MR, Partridge JF, Allshire RC. Centromere silencing and function in fission yeast is governed by the amino terminus of histone H3. Curr Biol. 2003;13:1748–1757. doi: 10.1016/j.cub.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 29.Petersen J, Hagan IM. S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr Biol. 2003;13:590–597. doi: 10.1016/s0960-9822(03)00205-7. [DOI] [PubMed] [Google Scholar]
- 30.Bastow R, Mylne JS, Lister C, Lippman Z, Martienssen RA, Dean C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature. 2004;427:164–167. doi: 10.1038/nature02269. [DOI] [PubMed] [Google Scholar]
- 31.Gruss C, Wu J, Koller T, Sogo JM. Disruption of the nucleosomes at the replication fork. The EMBO journal. 1993;12:4533–4545. doi: 10.1002/j.1460-2075.1993.tb06142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santoro R, De Lucia F. Many players, one goal: how chromatin states are inherited during cell division. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2005;83:332–343. doi: 10.1139/o05-021. [DOI] [PubMed] [Google Scholar]
- 33.Kirchmaier AL, Rine J. DNA replication-independent silencing in S. cerevisiae. Science (New York, NY) 2001;291:646–650. doi: 10.1126/science.291.5504.646. [DOI] [PubMed] [Google Scholar]
- 34.Vaughn MW, Tanurdzic M, Martienssen R. Replication, repair, and reactivation. Developmental cell. 2005;9:724–725. doi: 10.1016/j.devcel.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Chandler VL. Paramutation: from maize to mice. Cell. 2007;128:641–645. doi: 10.1016/j.cell.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Zaratiegui M, Irvine DV, Martienssen RA. Noncoding RNAs and gene silencing. Cell. 2007;128:763–776. doi: 10.1016/j.cell.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 37.Finnegan EJ, Dennis ES. Vernalization-induced trimethylation of histone H3 lysine 27 at FLC is not maintained in mitotically quiescent cells. Curr Biol. 2007;17:1978–1983. doi: 10.1016/j.cub.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 38.Swiezewski S, Crevillen P, Liu F, Ecker JR, Jerzmanowski A, Dean C. Small RNA-mediated chromatin silencing directed to the 3′ region of the Arabidopsis gene encoding the developmental regulator, FLC. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3633–3638. doi: 10.1073/pnas.0611459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li F, Goto DB, Zaratiegui M, Tang X, Martienssen R, Cande WZ. Two novel proteins, dos1 and dos2, interact with rik1 to regulate heterochromatic RNA interference and histone modification. Curr Biol. 2005;15:1448–1457. doi: 10.1016/j.cub.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 40.Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nature genetics. 2005;37:809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01