Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories (original) (raw)
. Author manuscript; available in PMC: 2008 Jul 3.
Published in final edited form as: Mol Psychiatry. 2007 Jan 30;12(7):656–670. doi: 10.1038/sj.mp.4001957
Abstract
Brain-derived neurotrophic factor (BDNF) is known to play a critical role in the synaptic plasticity underlying the acquisition and/or consolidation of certain forms of memory. Additionally, a role has been suggested for neurotrophin function within the hippocampus in protection from anxiety and depressive disorders. Understanding the function of this important gene in adult animals has been limited however, because standard knockouts are confounded by gene effects during development. There are no BDNF receptor-specific pharmacological agents, and infusions of neuropeptides or antibodies have other significant limitations. In these studies, we injected a lentivirus expressing Cre recombinase bilaterally into the dorsal hippocampus in adult mice floxed at the BDNF locus to facilitate the site-specific deletion of the BDNF gene in adult animals. Significant decreases in BDNF mRNA expression are demonstrated in the hippocampi of lenti-Cre-infected animals compared with control lenti-GFP-infected animals. Behaviorally, there were no significant effects of BDNF deletion on locomotion or baseline anxiety measured with startle. In contrast, hippocampal-specific BDNF deletions impair novel object recognition and spatial learning as demonstrated with the Morris water maze. Although there were no effects on the acquisition or expression fear, animals with BDNF deletions show significantly reduced extinction of conditioned fear as measured both with fear-potentiated startle and freezing. These data suggest that the cognitive deficits and impairment in extinction of aversive memory found in depression and anxiety disorders may be directly related to decreased hippocampal BDNF.
Keywords: fear, brain-derived neurotrophic factor, anxiety, depression, hippocampus, Cre-Lox
Introduction
Brain-derived neurotrophic factor (BDNF) undoubtedly plays a role in development, trophic support and neural plasticity within the mammalian hippocampus.1–4 Correlational studies show that BDNF is increased in its expression following hippocampal-dependent tasks.3,5,6 Genetic manipulation studies suggest that animals with decreased levels of BDNF or its receptor, TrkB, are deficient in behavioral tasks thought to be hippocampally dependent.4,7 Furthermore, mice with inducible deletions of BDNF that do not involve the entire brain, but that are forebrain-specific, including cortex, hippocampus and amygdala also display deficits in some of these behaviors.8–10 However, all of these studies are limited, in that none of them is designed to examine specifically the effects of BDNF deletion only within the hippocampus of adults while sparing BDNF in other forebrain regions. As an alternative approach, rat studies have used antisense RNA to knockdown expression and TrkB ‘receptor bodies’ to ‘soak up’ extracellular BDNF within the hippocampus.6,11,12 However, these studies have other limitations, and there are no pharmacological-specific antagonists for the BDNF TrkB receptor. Thus, even with behaviors that are well accepted to be hippocampus-dependent tasks, definitive behavioral experiments proving a role for hippocampus-specific BDNF expression are lacking.
There are a number of behaviors that are thought to be at least partially hippocampus-dependent, which have been studied primarily with lesion or inactivation experiments. Spatial learning, as indicated with the Morris water maze (MWM) and object recognition memory are two of the most robust measures of hippocampal functioning.13–15 Additionally, context-dependent fear learning has also been shown to be hippocampus-dependent, although negative studies have also been reported.16–18 A developing literature suggests that the hippocampus also may be involved in the extinction of fear,19 which is defined as the reduction of fear following unreinforced exposure to the fearful stimulus.20 In this study, we specifically examine the role of BDNF expression using mice in which the BDNF locus is flanked by loxP sites,21 in conjunction with dorsal hippocampus-specific injections of a lentiviral vector that constitutively expresses the Cre recombinase protein. This allows for the examination of hippocampus-specific BDNF within adult animals in a variety of hippocampally dependent tasks. We find expected deficits in spatial and object recognition tasks in animals with reduced hippocampal BDNF. We also report a novel deficit in extinction, but not acquisition, of conditioned fear in these animals. These data suggest that animals with reduced BDNF may have normal fear learning, but diminished extinction of fear.
A variety of data now exists in which decreased BDNF is associated with animal models of anxiety and depression, and which is reversed by antidepressant treatment.8,10,22,23 Furthermore, diminished BDNF in the hippocampus is thought to possibly be related to the decreased hippocampal volume found in patients with major depressive disorder and posttraumatic stress disorder (PTSD).24–28 These disorders have also been hypothesized to result in part from a deficit in extinction of fear.29–32 Our data combined with these previous studies suggest that there may be a preservation of aversion learning combined with a deficit in extinction of fear and stress-related memories in such patients that may contribute to maintenance of psychopathology.
Materials
Conditional mutant mice
Homozygous BDNF-floxed mice were originally obtained from Jackson Labs (Bdnftm3Jae/J; Bar Harbor, ME, USA), and bred within our animal facility. Generated by Jaenisch and co-workers,21 these mice possessed loxP sites both upstream and downstream of exon 5 of the BDNF gene. This strain was originated and maintained on a mixed B6, 129S4, BALB/c background and did not display any gross physical or behavioral abnormalities. The floxed-stop lacZ reporter mice (129S-Gt(ROSA)26Sortm1Sor/J)33 were also obtained from Jackson Labs. Mice were housed in groups of four in plastic cages (30 × 20 × 16 cm) on corn dust bedding with ad libitum access to food and water. They were maintained on a 12/12-h light/dark cycle at 24°C. Experiments were approved by the Emory Institutional Protocol Approval Committee in accordance with Yerkes Primate Research Center Regulations.
Experiments were conducted on male transgenic mice between 4 and 10 weeks of age and weighing 20–30 g. All mice were evaluated on the following order of behavioral tests 10–14 days after surgery: (1) novel object recognition, (2) MWM, (3) baseline activity, (4) baseline startle, (5) fear conditioning and (6) extinction of fear. This provided an order of testing in which the least stressful preceded the more stressful, minimizing any confounds of order effects. All animals were sacrificed for histological evaluation 3–5 days after behavioral testing. A total of 32 mice were injected for the study, with a final number of 14 LV-GFP mice and 13 LV-Cre mice used for all behavioral and histological analyses.
Lentiviral constructs and virus production
Viral vectors were derived from the HIV-based lentivirus backbone pLV-CMV-GFP-U3Nhe (as described previously,34,35 a generous gift of Inder Verma, Salk Institute), which allows for virally mediated expression of green fluorescent protein (GFP) driven by a cytomegalovirus (CMV) promoter. We created a Cre-recombinase expressing viral vector (LV-Cre) by replacing the GFP coding sequence in pLV-CMV-GFP-U3Nhe with the coding sequence for Cre-recombinase (coding sequence was replaced as a BamH1/Sal1 fragment). Notably, the coding sequence for Cre included a 5′ nuclear localization sequence (translated sequence: MAPKKKRKV), which is known to enhance the efficiency of Cre-mediated recombination. Viral production procedures are described in detail previously.36,37 In brief, active viral particles were produced by co-transfecting these lentiviral packaging constructs with plasmids coding for delta8.9 and VSV-G into HEK-293T cells. The packaged, unconcentrated virus was collected over a period of 5 days post-transfection, and then concentrated using ultracentrifugation and resuspension in sterile PBS/1% BSA. The resulting titer was assessed in HEK-293T cells, and the observed titer of the GFP-and Cre-expressing viruses used here ranged from 1× 108 to 1 × 109 infectious particles per ml.
For pLV-CMV-GFP-U3Nhe control vector (hereafter referred to as LV-GFP), assessment of viral titer was achieved through serial dilution of concentrated virus, followed by infection of HEK-293T cells with diluted virus. GFP-expressing HEK-293T cells were then counted with fluorescence microscopy to determine the number of infectious particles present per ml of concentrated virus.
A novel reporter plasmid (CX1-LEL-DR) was constructed to assess Cre-recombinase activity in vitro, and to allow for the titering of concentrated LV-Cre virus in vitro. This reporter plasmid was based on the pBS-CX1-LEL plasmid,38 in which a ubiquitously expressing CX1-promoter (a hybrid of the _β_-actin promoter and enhancer elements of the CMV promoter) was placed upstream of the coding sequence for GFP. To allow for Cre-mediated excision of the GFP, the coding sequence for GFP (including stop codon) was flanked by LoxP sites (‘floxed’). In the reporter plasmid used in this study (CX1-LEL-DR), the coding sequence for a red fluorescent protein (DsRed2, Invitrogen Inc., Carlsbad, CA, USA) was placed downstream of the coding sequence for GFP. In the absence of Cre expression, this plasmid only expressed GFP, as the DsRed coding sequence was placed downstream of the GFP stop codon. If the CX1-LEL-DR reporter plasmid was expressed in the presence of a Cre-expressing plasmid or virus, the GFP coding sequence (along with its stop codon) was excised, allowing for CX1-driven DsRed expression. In this way, the presence of red fluorescence in CX1-LEL-DR-transfected HEK-293T cells could be used as a marker for virally mediated Cre-recombinase expression in vitro, allowing for assessment of viral titer.
Novel-object recognition apparatus
The object recognition apparatus consisted of an open box (44 × 44 × 8 cm) made of white polyvinyl chloride (PVC). The apparatus was placed in a sound-isolated testing room, illuminated by normal housing lights. Four different objects made of a combination of plastic, metal, and rubber were employed in this task. Each object was similar in size (approximately 7 cm height and 6 cm diameter), and the weight of the objects ensured that they could not be displaced by mice. These objects were selected on the basis of previous observations that demonstrated a lack of preferential exploration for one object over the other. Animals training and testing behaviors were filmed on a video camera mounted over the training arena.
Hidden-platform Morris Water Maze
MWM consisted of a circular pool made of white PVC plastic with 45-cm-high walls and a diameter of 200 cm. The maze was filled to a depth of 26 cm with 25°C water-rendered opaque nontoxic latex paint. A small clear Plexiglas escape platform was placed at a fixed position in the center of one quadrant and was hidden 1 cm beneath the water surface. The MWM was placed in a sound-isolated testing room, illuminated by normal housing lights and surrounded by a number of fixed extramaze cues.
Fear-conditioning apparatus
Mice were given fear-conditioning in standard rodent modular test chambers (ENV-008-VP; Med Associates Inc. Georgia, VT, USA) with an inside area of 30.5 cm (L) × 24.1 cm (W) × 21.0 cm (H). The tone conditioned stimulus (CS) was a 30 s, 70-dB SPL, 12-kHz tone delivered through a high-frequency speaker (Motorola, Model 948, Schaumburg, IL, USA) attached to side of each chamber. The unconditioned stimulus (US) was scrambled footshock delivered to a removable grid floor that consisted of 36 stainless-steel rods (3.2 mm) placed 7.9 mm apart. Footshock intensity was 0.4 mA verified by using a 0.5 kΩ resistor across the bars of the shock grids and measuring the voltage drop between the bars to calculate the constant current. Stimuli presentations were controlled by an IBM PC-compatible computer using MED-PC IV, software interfaced to chambers. Conditioned freezing responses were recorded with video cameras mounted in front of each conditioning apparatus.
Startle apparatus
Fear-potentiated startle (FPS) was measured in eight identical startle response systems (SR-LAB, SDI, San Diego, CA, USA). Each system consisted of a nonrestrictive Plexiglas cylinder, 5.5 cm in diameter and 13 cm long, mounted on a Plexiglas platform, which was located in a ventilated, sound-attenuated chamber. Cylinder movements were sampled each millisecond (ms) by a piezoelectric accelerometer mounted under each platform. Startle amplitude was defined as the peak accelerometer voltage that occurred during the first 100 ms after the onset of the startle stimulus. The output sensitivity of all response systems was calibrated to be nearly identical (SR-LAB Startle Calibration System, San Diego, CA, USA). Startle stimuli were presented through a high-frequency speaker located 15 cm above the chambers. The tone CS was generated by a Tektronix function generator audio oscillator (Model CFG253, Beaverton, OR, USA) and was delivered through a high-frequency speaker (Motorola, Model 948) located 13 cm above each cylinder. The chamber ventilation fans produced a 55 dB white-noise background, which was present during fear testing sessions. Sound intensities were measured by an audiometer (Radio Shack, Ft. Worth, TX, USA, #33-2055). Stimuli presentation and data acquisition were controlled, digitized and stored by an interfacing IBM PC-compatible computer using SR-LAB software.
Procedures
Surgery and histology
Before surgery, mice were anesthetized by i.p. (intraperitoneal) injections of ketamine–metomidine (ketamine, 80 mg/kg; metomidine, 1.0 mg/kg), then mounted in a stereotaxic apparatus (Stoelting Instruments, Wood Dale, IL, USA). Small holes were drilled in the skull above the injection site, and a 30-gauge Hamilton 0.5 _μ_l microsyringe (model #75) was lowered to the following coordinates (two hippocampal sites on each side) from bregma based on the mouse brain atlas of Paxinos and Franklin:39 anteroposterior, −1.7; mediolateral, ±0.8 and ±1.8; dorsoventral, −2.2. The microsyringe was lowered to appropriate coordinates and left in place 4 min before and 4 min after each injection. For each injection site, a total a volume of 1.0 _μ_l LV-CRE or LV-GFP was administered at a rate of 0.12 _μ_l/min. After surgery, the incision was closed with cyanoacrylate glue. Mice were given post-surgical i.p. injection of antisedan (4.0 mg/kg; metomidine reversing agent) and placed on a heated pad. After recovery from anesthesia, mice were given a narcotic analgesic (buprenorphine, 0.05 mg/kg, s.c.) and returned to home cages for 14 days of recovery before testing. Body weight, eating and drinking were monitored daily.
After behavioral testing, mice were anesthetized with isoflurane and the brains were rapidly dissected and frozen on crushed dry ice. Coronal sections (20 _μ_m) of brains were cut on a cryostat (Leica; Nussloch, Germany) at −20°C, mounted on gelatin-coated slides, and stored at −80°C until processed for in situ hybridization histochemistry. For each brain, sections were placed on five consecutive sets of slides. Two sets of slides from each brain were used for in situ hybridization analyses of Cre recombinase and BDNF mRNA (see below). A third slide from each mouse was stained with cresyl violet (Nissl stain).
In situ hybridization
In situ hybridization was performed to examine the expression of mRNA as described previously.40,41 Briefly, hybridization riboprobes were prepared from linearized clones using appropriate RNA polymerase and radioactive [35S]-UTP (1250 Ci/mmol, 12.5 mCi/ml, NEN, Boston, MA, USA) in the polymerase reaction. Radiolabeled antisense RNA strands were base hydrolyzed to average lengths of 100–300 bp and isolated using a Sephadex spin columns (Roche Quick Spin G-25 G-50, Indianapolis, IN, USA). Probes were diluted to a concentration of 20–50 000 cpm/_μ_l in hybridization buffer, and sections were incubated overnight in humid chambers at 52°C with 75 _μ_l/slide of diluted probe covered with Parafilm. Slides were then stringently washed, air-dried and opposed to Biomax MR autoradiography film (Eastman Kodak Co., Rochester, NY, USA) for 1–5 days.
For image analyses, Biomax MR film was scanned using an Epson 3700 scanner (3000 dpi) and was saved in JPEG format with compression quality 10, at a size of 32 000 × 18 000 pixels (~580 MB). For each section, hybridization density was determined for the regions of interest (ROI), as well as an adjacent background area that lacked hybridization (e.g., dorsal thalamus). Normalized density levels were assessed using the luminosity histogram feature of Adobe Photoshop at a level of exposure that produced linear densities with 14C standards.40,42
Histological analyses
Of the 32 mice that received LV injections (LV-Cre = 16 and LV-GFP = 16), three mice died during surgery or recovery period. One mouse from each group was also excluded from statistical analysis owing to lack of observable LV infection. As expected, in the remaining LV-GFP mice (n = 14) an evaluation of brain sections for Cre-recombinase expression by way of in situ hybridization revealed no Cre mRNA signals. In contrast, prominent Cre hybridization signals were evident in LV-Cre animals (n = 13). For all histological analyses, successive brain sections spanning the dorsal hippocampus were distributed across five different slide sets, and one set was used to evaluate either Cre, BDNF or cresyl violet Nissl stain. In all LV-Cre animals, infection was evident in each hippocampus subregion subject to analyses (i.e. CA1, CA3 and DG). No infection was noted in the subiculum or thalamus. Overall, LV-Cre expression extended from rostral areas of the dorsal hippocampus at the level of the mediodorsal thalamus (AP, −1.4 mm) to the anterior portion of the pretectal nucleus of the thalamus (AP, −2.2 mm). In brain sections at central levels of the injection sites, the estimated percentage of infection for all hippocampus subregions was 53%, with means of 41% in CA1, 57% in CA3 and 61% in the DG (Figure 2d).
Figure 2.

Reduction in BDNF mRNA in dorsal hippocampus with no changes in baseline activity or startle. (a) Qualitative figure showing BDNF in situ hybridization of dorsal hippocampus following a sham injection (top, −Cre) or following LV-Cre injection (bottom, + Cre). (b) Nissle-stained sections (from different experiment than (a) from hippocampi that have been infected with LV-GFP or LV-Cre. We found no discernable morphological changes with either viral infection compared to control. (c) Relative mRNA expression in dentate gyrus (DG), and CA1 and CA3 regions and the average of all regions (Avg) of dorsal hippocampus in LV-Cre (Cre)- or LV-GFP (GFP)-infected mice. (d) The estimated percentage of total area of each hippocampal region infected within the examined sections. (e) Baseline activity (%time moving) within the SR-LAB behavior box in mice that have been infected bilaterally with LV-Cre or LV-GFP. (f) Baseline acoustic startle reflex (startle amplitude) in mice that have been infected bilaterally with LV-Cre or LV-GFP (*P < 0.001).
To estimate area of LV-Cre infection, the total area of each hippocampal subregion (in pixels) was first outlined by freehand tool of Adobe Photoshop with reference to Nissl-stained section from the same mouse and the atlas of Paxinos and Franklin.39 Next, gray area measurements, which reflected areas of Cre mRNA expression, were delimited within hippocampal subregion. Area of infection were calculated by dividing total gray areas by total subregion area and multiplied by 100. All data were expressed as mean ± s.d.
The expression level of BDNF was likewise evaluated by first outlining hippocampal subregions. Within each region, the levels of BDNF were quantified using the luminosity histogram feature of Adobe Photoshop on film in which time of exposure resulted in a distribution of gray scale intensity values ranging from 0 to 256. Measures of gray scale intensity from each hippocampal subregion were subtracted from background levels and used as measures of BDNF mRNA levels.
Novel-object recognition testing
The object recognition experiment was carried out according to the method of Ennaceur et al.43,44 with minor modifications. On each of 2 days before testing, each animal was placed into the open box and allowed to explore for 10 min in the absence of objects to allow for habituation of handling and open box environment. One day following habituation, animals were given a sample session in which two objects were placed in the back corner of the box, 10 cm from the side wall. Each session began with the mouse being placed in the maze facing the wall at the opposite end from the objects and continued for 5 min, at which point the mouse was promptly removed and returned to the housing colony. The sample objects on this session were identical. Twenty-four hours after the sample session, mice were returned to the testing room for choice session, which was identical to the sample session except that the objects were replaced by one identical copy and a novel object. The role objects as sample or novel as well as the location of the novel object during sessions were counterbalanced between mice and across sessions. Before each session, the open box and objects were cleaned with a solution of 50% alcohol and allowed to dry thoroughly. Exploration of an object was defined as strictly on the basis of active exploration in which mice had to be touching the object with at least their nose. Videotape analyses of total time spent exploring each of the objects were obtained using video recordings and an observer blind to treatment conditions. Measurement of the time spent exploring each object during the choice session was expressed as a percentage of the total exploration time in seconds. When compared to the novel object, significantly less time spent exploring the same object (previously explored) is considered as a measure of retention in this task.44 Statistical analyses were performed using a within–between design ANOVA with object (same, novel) as the repeated factor and treatment (CRE, GFP) as the between factor. A habituation index was also calculated as the difference in time spent exploring the two objects during the sample session minus the time spent exploring the object during choice session. For between-group comparison of the habituation index, we used a one-factor ANOVA design.
Hidden-platform MWM testing
The acquisition phase consisted of five consecutive training days (sessions) with four trials per day, starting at four different positions in a semirandom order. On each trial, mice were placed in a starting location facing the pool wall and allowed to swim until finding a submerged platform or until a maximum of 120 s. On finding the platform, mice remained on the platform for 30 s before removal to the retaining cage. If an animal did not reach the platform within 120 s, it was placed on the platform where it had to remain for 30 s before being returned to its retaining cage. Mice remained in retaining cage for 60 s before the start of the next trial (i.e. 60 s inter-trial interval) or the return to home cage at end of training session. Two days after the acquisition phase, each animal was given a probe session. During this session, the platform was removed from the maze and animals were allowed to swim freely for 120 s. For each acquisition session, a mean latency was calculated for each mouse by averaging the latency to reach the platform across all four session trials. For the probe session, the percent time spent in the target quadrant and the number of platform location crossings was calculated. Mean latency to platform during acquisition was analyzed with two-way ANOVA. Time spent in the target quadrant and number of platform crossings were analyzed with independent _t_-tests.
Fear-conditioning session
Two days after object recognition testing, mice were trained for fear-conditioning. During training, each mouse was placed into the fear-conditioning apparatus and after 4 min presented with 5 CS–US pairings at an inter-trial interval of 3 min. Each paring consisted of a 30-s tone CS, which co-terminated with a 1-s 0.4 mA, footshock US. Mice were left in chambers for an additional 1 min after the last CS–US pairing.
Conditioned freezing and fear-potentiated startle tests
One day after training, freezing behavior was examined in the same context in which training took place and was used to test for conditioned freezing to the context and CS. This test was similar to the conditioning session with the exception that no footshock US was delivered after CS presentations. Conditioned freezing responses during training and testing were analyzed using video recordings by a rater blind to the treatment conditions. Freezing was characterized by a crouching posture with an absence of visible movement except that due to breathing.45,46 Behaviors were sampled for 2 s during the inter-trial interval (at 60 and 40 s before CS onset) and during the CS presentation (at 10 and 20 s after CS onset).
One day after the conditioned freezing test, fear was assessed in a different context using the FPS paradigm.47 For this test, each mouse was placed in the cylinder and 5 min later presented with four startle stimuli at each of three different startle stimulus intensities (95, 105 and 115 dB). After these initial trials, mice were presented with four additional startle-alone trials and four tone + startle trials at each of the three startle stimulus intensities. On tone + startle test trials, the startle stimulus was presented 29 s after the onset of the tone cue. All trials were presented in a pseudorandom order with the constraint that each trial type occurred only once in each consecutive six trial block. The inter-trial interval (defined as the interval between startle stimuli) was 1 min. In the FPS paradigm, fear is inferred by the presence of an augmented startle response in the presence of a CS previously paired with shock. To measure CS-induced changes in startle, mean startle amplitudes were computed for the trials in the presence of the CS (tone + startle) and the trials in the absence of the light (startle-alone). A percentage of FPS was computed for each mouse by dividing the difference between these two trial types by the mean startle amplitude on startle-alone trials: %FPS = (difference/startle-alone) × 100.
Freezing as measured during the extinction trials was obtained from the same sessions as the FPS sessions. For a period of 5 s in the middle point of the 29 s tone cue, the automated SR-LAB system gathered data every millisecond to obtain activity measures (see Jones et al.48 for full details). Owing to within-session extinction, we only used the activity scores from the first one-half of each testing session in these analyses. Percentage reduction in freezing during tone cue presentation was used as a measure of extinction and was computed as %reduction = [(%freezing at pretest − %freezing at posttest]/%freezing at pretest) × 100. This serves simply as a measure of relative decrease in freezing across the extinction of fear between animals. For all statistical tests, two-tailed analyses with P < 0.05 is considered the cutoff for significance, unless stated otherwise.
Results
Lenti-Cre production and function
The lentivirus containing Cre recombinase was initially tested in vitro by examining its efficiency in removing DNA flanked by loxP sites from a transiently transfected plasmid vector containing a CMV constitutive promoter followed by a floxed GFP and a polyA site. This was followed by an additional DsRed2 coding region and polyA site. Cells transfected with this plasmid express GFP, with no detectable red fluorescent leakage, until Cre recombinase is expressed, at which point they remove green fluorescence and express red fluorescence constitutively. Figure 1a demonstrates the use of this ‘stop-light’ expression system for examining functional Cre titer in vitro. All viruses used for subsequent in vivo experiments were first determined to have a titer of 108–109 infectious units/milliliter.
Figure 1.
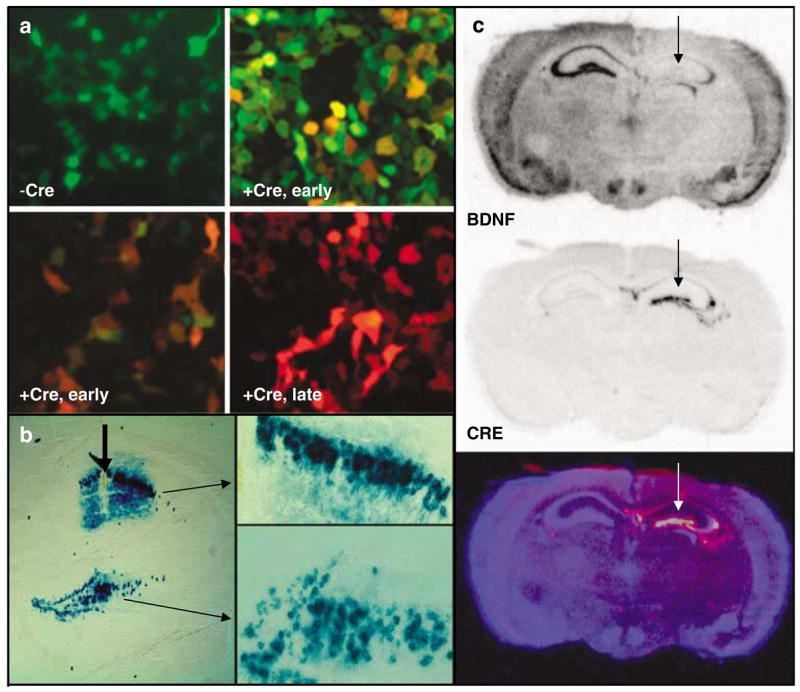
In vitro and in vivo validation of LV-Cre virus. (a) HEK-293 cells transiently transfected with the green-red fluorescent cre-reporter plasmid, pLoxPGFP-DsRed and visualized 2 days later (−Cre), or infected with LV-Cre and visualized 1 day later (+ Cre, early (top), green filter; + Cre, early (bottom), red filter), or visualized 2 days later (+ Cre, late). (b) The floxed-stop lacZ reporter mice(129S-Gt(ROSA)26Sortm1Sor/J)33 were injected within dorsal hippocampus, and sections were processed for lacZ histochemistry with x-gal 14 days later to visualize Cre-dependent recombination in vivo. These demonstrate × 10 and × 20 magnification of LV-Cre-infected cells within CA1 and DG, illustrating the density and intact morphology of LV-Cre-infected hippocampal neurons. (c) BDNF (top) and Cre-recombinase (middle) mRNA expression visualized with in situ hybridization 2 weeks after LV-Cre infection into Bdnftm3Jae/J floxed mice.21 The bottom figure represents a pseudocolor overlay of the two in situ sections.
The first test of in vivo function with this virus was performed using the floxed-stop lacZ Rosa26 animals that have been described previously.33 Mice were injected unilaterally into striatum, hippocampus or amygdala with 1–2 _μ_l of virus and allowed to recover for 14 days. Animals were then killed and brain slices were processed for lacZ immunohistochemistry. Figure 1b demonstrates the efficient ability of LV-Cre to remove the floxed-stop sequences upstream of lacZ in the Rosa mice in a CA1 and DG region of the dorsal hippocampus, allowing for strong production of LacZ with no leakage in areas not infected with virus.
For the behavioral experiments in this study, BDNF within the hippocampus was deleted by performing bilateral injections of LV-Cre into the dorsal hippocampus of mice that are floxed at BDNF exon 5.21 Initial verification of this approach was performed with unilateral injections followed 14 days later with in situ hybridization for the Cre-recombinase gene, or the BDNF gene. Figure 1c demonstrates BDNF mRNA expression, which is specifically missing only within the dorsal hippocampus where the Cre is expressed (Figure 1c, top), strong and stable expression of Cre within the dorsal hippocampus (Figure 1c, middle), and an overlay picture in which the converse expression of the two genes is clear (Figure 1c, bottom). These data as well as Nissl-stained sections (not shown) demonstrate intact cellular and regional morphology at the site of injection, with no clear evidence of cell death or degradation at the time points examined.
Specificity and efficiency of BDNF deletions within the dorsal hippocampus
Both qualitative and quantitative analyses of mRNA expression levels reveal that the deletion of BDNF in the mice used in this study was largely limited to the hippocampus and statistically significant. Figure 2a demonstrates qualitatively (in conjunction with Figure 1c) the decrease in BDNF expression within the dorsal hippocampus following LV-Cre infection. Qualitative assessment shows that no other brain regions had noticeably different levels of BDNF expression. Figure 2b demonstrates in two different Nissl-stained sections infected with lentivirus that we found no discernable histological abnormalities in dorsal hippocampi from animals infected with either LV-GFP or LV-Cre. We also examined the levels of BDNF in control, LV-GFP-infected mice and experimental, LV-Cre-infected mice. We found a significant hippocampal region × lentivirus-type interaction with ANOVA suggesting that Cre-infected hippocampal regions expressed less BDNF mRNA than GFP-infected regions (F(2.50) = 7.9; P < 0.001). Figure 2c shows that significantly less BDNF was expressed in LV-Cre as compared with LV-GFP-infected brains in the dentate gyrus (DG) t(25) = 4.04, P < 0.001; CA1: t(25) = 3.89, P < 0.001; and CA3 t(25) = 4.78; P < 0.001) subregions of the dorsal hippocampus. With reference to GFP control animals, the relative expression level of remaining BDNF within the dorsal hippocampus in LV-Cre-infected animals were 35, 30 and 45% for DG, CA1 and CA3, respectively. Regarding overall level of infection within the dorsal hippocampus, the Cre in situ hybridization results suggest that the estimated percentage of infected area for all hippocampus subregions was 53%, with means of 41% in CA1, 57% in CA3 and 61% in the DG (Figure 2d).
No significant effect of BDNF deletion on locomotion or baseline startle
If the lenti-Cre virus injections into dorsal hippocampus did not detrimentally affect baseline function, we should find no difference between lenti-Cre- and lenti-GFP-infected animals on a variety of baseline measures of behavior. Thus, we first examined baseline locomotion and startle within the SR-LAB boxes. We found no differences in baseline locomotor activity (measured as %time moving in SR box) between mice (Figure 2e: LV-Cre: 75%, LV-GFP: 79%, P > 0.5). We also found no differences between baseline acoustic startle response between mice (Figure 2f: LV-Cre: 257, LV-GFP: 274, P > 0.5).
BDNF deletion within dorsal hippocampus impairs spatial learning in Morris water maze
Before fear learning, the 27 analyzed animals (LV-GFP, N = 14; LV-Cre, N = 13) were tested for their spatial learning ability using the MWM with five training sessions spaced over 5 separate days. After five days of MWM training, an overall ANOVA revealed a significant Group × Session interaction, F(4.100) = 4.01, P < 0.01, indicating differences between Cre and GFP groups in the pattern of MWM acquisition (Figure 3a). Simple effects at each level of group indicated a significant session effect for LV-GFP animals, _F_(4.52) = 14.36, _P_ < 0.001, but not for LV-Cre animals, _F_(4,52) = 1.96, _P_ > 0.05. Post hoc tests showed that the mean escape latency values for the CRE-injected group were significantly higher than the GFP group in sessions 2, 4 and 5, t(25) > 2.20, P < 0.05. For the probe phase of the MWM, both groups showed statistically similar latencies to the target quadrant; however, GFP mice displayed a greater number of target platform crossings than CRE mice (Figure 3b, Cre: 2.7 crossings, GFP: 5.5 crossings, t(25) = 3.8, P < 0.001). Additional analyses of the amount spent in the target quadrant during the probe test also demonstrates that the LV-GFP-infected animal spent significantly more time in the target quadrant as compared with the adjacent or opposite quadrants (F(2.50) = 3.48, P < 0.05), whereas the LV-Cre-infected animal did not (Figure 3c).
Figure 3.

Morris water maze and object recognition are impaired in hippocampal BDNF KO animals. (a) Acquisition of the location of the hidden platform, measured as the average latency to find the platform over several sessions of training, each separated by a day. LV-Cre-infected animals demonstrated significantly slower acquisition compared with LV-GFP-infected animals. (b) Following training, mice were tested on a probe trial in the absence of a platform. Number of crossings of the target location demonstrates that mice with LV-Cre injections made significantly fewer crossings over the target area than did LV-GFP controls. (c) Percent of time spent in the target, opposite or adjacent (average of both) quadrant during the probe test for each group of animals. (d) Percent of time spent exploring the new vs old object during the test day for novel object recognition. LV-GFP-infected animals spent significantly more time exploring the novel compared to the previously habituated object. The LV-Cre-infected animals did not differentiate between the two. (e) Scatter plot demonstrating a significant positive correlation t between animals performance on the MWM (number of target quadrant crossings) vs Novel Object Recognition (%time with novel object). LV-Cre-infected animals are represented with a diamond (N = 13), and LV-GFP animals with a square (N = 14). This correlation suggests that the manipulation of the dorsal hippocampus affected each spatial learning paradigm in a similar way (*P < 0.01; **P < 0.001).
BDNF deletion impairs novel object recognition task
These same mice (LV-GFP, N = 14; LV-Cre, N = 13) were tested within the novel object recognition task as a separate measure both of spatial learning and novelty response. Mice were first exposed to a single object, later re-exposed to that object or a novel object. Amount of time spent exploring the previous (old) object vs the novel object was measured. A within–between design ANOVA with object (same, novel) as the repeated factor and group (LV-Cre and LV-GFP) as the between factor revealed a significant Object × Group interaction during the choice session, F(1.25) = 5.53, P < 0.03. LV-GFP-infected animals spent significantly more time exploring the novel object compared with the old object indicative of novel object recognition (Figure 3d, paired _t_-test, _t_(13) = 3.19, _P_ < 0.01). This is compared with LV-Cre-infected animals that displayed no recognition, as indicated by non-discriminant exploration of objects (_t_(12) = .68, _P_ > 0.5). Additionally, when percent of time exploring novel object is compared between the LV-GFP and LV-Cre groups, the LV-GFP group spends significantly more time with the Novel Object (60 vs 47%; t(25) = 2.4; P < 0.05). No group differences in object preference were observed during the initial sample session, _t_(25) = 1.3, _P_ > 0.1, suggesting that the effect seen in the object recognition task is not due to non-specific object preference.
The MWM and Novel Object Recognition tasks measure some similar aspects of spatial learning, but they also measure distinctly different aspects of hippocampal function as well. We examined whether there was a correlation between animal performance on these two different tests as a way of investigating if the BDNF hippocampal deletion effects on the two tests were similar. Linear regression analysis suggests that there is a significant correlation across the groups between performance on the MWM (number of target crossings during test phase) and Novel Object (%time with novel object) (Figure 3e, F(1.26) = 11, P < 0.005; R = 0.55, _r_2 = 0.30).
No significant effect of BDNF deletion on fear-conditioning
We next examined whether BDNF deletion within the dorsal hippocampus would affect acquisition or expression of conditioned fear. Fear training to an auditory cue was performed in a Med Associates conditioning chamber and fear testing was performed in both the same context (for contextual and cue fear measures) and a separate context (for cue-dependent measures of FPS). Before training trials, neither LV-Cre (N = 13) nor LV-GFP (N = 14) animals displayed any freezing activity in the conditioning chambers, suggesting no gross differences in motor activity between groups, t(25) = 1.76, P > 0.05. During the conditioning phase of the session, freezing scores similarly increased from the first to the last CS presentation in the two groups as shown in Figure 4a. Thus, the ANOVA showed a significant main effect of trial (F(4,100) = 65.60, P < 0.001), but no group × trial interaction (_F_(4,100) = 0.76, _P_ > 0.5).
Figure 4.

Conditioned fear is not altered in mice with dorsal hippocampus BDNF deletions. (a) Acquisition of cue-conditioned fear, as measured with freezing after the onset of the auditory CS, during the conditioned fear acquisition session. There was no difference in acquisition of fear in animals receiving bilateral hippocampal injections of LV-Cre or LV-GFP. (b) Animals were tested within the same context in which training occurred for the presence of contextual fear as measured with freezing. There was no difference between the groups on level of contextual fear. (c) When animals were tested following acquisition in a different context, there was no difference in cue-conditioned fear as measured with FPS. (d) To examine explicitly the animals’ ability to discriminate the conditioned cue within a novel context, the level of acoustic startle reflex was measured during CS+ and CS− presentations of the acoustic startle stimulus. There is very tight overlap in the groups’ abilities to discriminate the fear-conditioned auditory cue (*P < 0.01).
When tested 24 h after conditioning, both CRE and GFP groups displayed similar levels of contextual freezing and tone CS freezing. As shown in Figure 4b, a direct comparison between freezing scores before conditioning and during re-exposure to context revealed only a significant session effect (F(1.25) = 15.2, P < 0.001), indicating both groups displayed contextual fear as measure by freezing. No differences were observed between groups (_P_’s > 0.05).
Twenty-four hours after the conditioned freezing test, mice were tested for FPS in a different context. In the absence of the CS, both groups showed similar levels of responses on noise-alone trials, t(25) = 0.32, P > 0.5, indicating no differences in baseline startle responses. Like conditioned freezing, CRE and GFP groups displayed similar percent levels of FPS in the presence of the tone CS (Figure 4c). Thus, statistical analysis failed to reveal any significant group effect on this variable, t(25) = 0.02, P > 0.5. Note that both groups of animals were able to discriminate exquisitely between the fearful cue and non-fearful cue during the FPS test (Figure 4d, P > 0.5). Together, these data suggest that the level of BDNF deletion within the dorsal hippocampus achieved in this study produced no deficits in the acquisition, consolidation or expression of cued or contextual fear as measured with freezing or FPS.
BDNF deletion impairs extinction of conditioned fear
Following cued fear acquisition, mice were repeatedly tested over 4 days with the auditory cue within the SR-LAB apparatus for analysis of initial cued fear, as measured with FPS, and extinction of fear over several sessions of unreinforced conditioned stimuli. Although there were no differences between initial levels of conditioned fear, there were significant differences between the levels of FPS at the last extinction session (Figure 5a, t(25) = 3.3, P < 0.005). Additionally, the LV-GFP infected-animals (_N_ = 14) demonstrated significant extinction of fear, but the LV-Cre animals (_N_ = 13) did not (LV-GFP: pre = 64%, post = −13%, _t_(13) = 3.5, _P_ < 0.005; LV-Cre: pre = 63%, post = 31%, _t_(12) = 1.9, _P_ > 0.05). Note that mice frequently display an unconditioned inhibition of acoustic startle, so that %FPS can exhibit a non-zero baseline as seen in Figure 5a.48,49 When the separate post-extinction tests (sessions 2–4) were analyzed together (Figure 5b), we found a significant group effect for LV-GFP vs LV-Cre on %FPS (F(1.25) = 6.0, P < 0.05).
Figure 5.

Extinction of conditioned fear is impaired in mice with dorsal hippocampus BDNF deletions. (a) Percent FPS is graphed for the first post-fear training test (pre-extinction) vs the last test (post-extinction). Mice infected with LV-GFP demonstrate significant decreases in their level of conditioned fear as measured with %FPS compared with LV-Cre-infected mice. (b) Impaired extinction of fear, measured with %FPS, is stable across multiple testing sessions. (c) Percent freezing is graphed for the first post-fear training test (pre-extinction) vs the last test (post-extinction). The percent reduction in freezing from pre- to post-extinction is significantly greater for the LV-GFP group than the LV-Cre group. (d) Impaired extinction of fear measured with freezing demonstrated across testing sessions (*P < 0.05; **P < 0.005).
Automated measures of conditioned freezing were also gathered during the same repeated testing trials that were used to gather the FPS data. In the 30 s before the startle stimulus, level of activity was obtained as described in the methods allowing for the automated measure of %freezing during each CS presentation. To compare directly the extinction between animals, we measured relative reduction in freezing (as calculated in method section) from pre- to post-extinction. We found that both groups of animals had a significant reduction in %freezing between the first and last test trial (Figure 5c and d, Cre: t(12) = 2.5, P < 0.05; GFP: _t_(13) = 5.3, _P_ < 0.001). When percentage reduction of freezing was examined between the first and last test, we found that the GFP group showed significantly greater extinction than did the Cre group (Figure 5c, _t_(25) = 2.0, _P_ < 0.05). On the basis of these results from Figure 5a–c, a one-tailed test was utilized to demonstrate a significant difference in level of freezing between the infected groups for the final two freezing sessions (Figure 5d, _F_(1.25) > 3.0, P < 0.05).
In summary, these data suggest that although there does not appear to be a direct effect of BDNF deletion on acquisition or expression of conditioned fear, there is an impairment of extinction of conditioned fear measured with both FPS and freezing.
Relationship between hippocampal BDNF expression and behavioral measures
To examine further the relationship between the deficits found in the above behavioral tests and BDNF gene deletion, we performed linear regression analyzing level of BDNF mRNA expression and behavioral outcome measures. The level of BDNF varies across animals, as can be seen in Supplementary Figure 1, such that there is a range of expression even within control animals that received LV-GFP virus. As a gene with a dynamic level of expression (e.g., Rattiner et al.42), BDNF gene expression can vary somewhat within a similar group of animals limiting the power of regression analyses. One possible reason for the variation in these experiments was that the animals were killed from their home cage after all other behavioral tests were completed. Thus, the recent behavioral activity of the animal before perfusion was not controlled. Despite this within-group variation, as shown in Supplementary Figure 1 and Figure 2c, the mean levels of BDNF expression between the LV-GFP and LV-Cre groups are quite different, supporting the use of group comparisons as used in the previous analyses.
We next examined correlations between behavior and the level of BDNF mRNA expression within DG. We found a weak, but significant, correlation between BDNF level and latency to find the platform on the MWM during the final acquisition test day (day 5, Figure 3a) animals with the shortest latency (and thus the most robust spatial memory) having the most BDNF expression (R = −0.43, _r_2 = 0.18, F(1.26) = 5.5, P < 0.05). Examination of the novel object test revealed that animals that spent more time examining the novel object also expressed more BDNF within the dorsal hippocampus (R = 0.42, _r_2 = 0.18, F(1,26) = 5.3, P < 0.05). Consistent with these results, %FPS following extinction (test 4, Figure 5b) was significantly inversely correlated with BDNF expression, such that those with more BDNF expression in DG demonstrated the largest reduction in fear (Supplementary Figure 1, R = −0.48, _r_2 = 0.23, F(1.26) = 7.7, P < 0.01). Additionally, extinction as measured with conditioned freezing (percentage reduction of freezing from Figure 5c) was weakly, but significantly, correlated with BDNF expression in DG (R = 0.37, _r_2 = 0.14, F(1.26) = 4.1, P = 0.05) and in CA1 (R = 0.42, _r_2 = 0.18, F(1.26) = 5.4, P < 0.05). Together, these data suggest that the extinction deficits may be correlated with the level of BDNF deletion within the dorsal hippocampus.
As a final test, we asked if the performance of the animals within the groups was related to the extinction of fear. We found that latency to find the platform on the MWM during the final acquisition test day (day 5, Figure 3a) was weakly, but significantly, correlated with level of fear on the final day of testing for FPS following extinction (test 4, Figure 5b) as shown in Supplementary Figure 2, R = 0.44, _r_2 = 0.19, F(1.26) = 6.2, P < 0.05. This suggests that animals with deficits in spatial memory following dorsal hippocampal BDNF deletion also have deficits in extinction of fear.
Discussion
Although BDNF has been implicated in numerous hippocampus-dependent functions including learning and memory, stress response, and depression and anxiety, definitive studies have not yet demonstrated that loss of endogenous BDNF within the hippocampus in adults is causal relative to the behaviors being tested. In these experiments, we combined lentiviral-mediated expression of Cre recombinase with floxed BDNF transgenic mice to examine specifically the behavioral effects of hippocampus-dependent BDNF deletion in adults. We found that spatial and object recognition learning is impaired as predicted, but that there was not a predicted effect on contextual fear learning. Interestingly, we found a novel effect of hippocampal BDNF deletion on the extinction of aversive memory, which we believe has significant impact for understanding the neurotrophic model of stress-related disorders.
Hippocampal BDNF and spatial learning
The hidden platform water maze is a well-accepted measure of hippocampus-dependent spatial learning in mice.13,14 Lesioning the hippocampus results in significantly impaired learning as indicated by delays in time to locate the hidden platform with repeated testing.50,51 Mice heterozygous for developmental BDNF deletions were found to have a deficit in water maze learning.4 This is also consistent with heterozygous BDNF knockout mice demonstrating radial arm maze deficits.6 Gorski et al.9 examined the hidden platform water maze with inducible BDNF deletions using the Emx1 promoter driving Cre recombinase in the same floxed BDNF background as used in our experiments. They found significant differences in escape latencies and swim distances during acquisition as well as during probe trials between forebrain-restricted (Emx1 expressing regions) BDNF knockouts and controls. The Emx1 promoter drives expression throughout cortical areas as well as hippocampus and amygdala developmentally. In our studies, with dorsal hippocampus-specific deletions induced virally during adulthood, we found similar deficits in water maze acquisition and testing. Together these data support the concept that BDNF expression in adults around the time of learning is required for normal acquisition and expression of water maze learning, and that these previously reported effects are not due to developmental alterations and are unlikely to be secondary to long-term BDNF effects on hippocampal neuronal survival or morphology.
Hippocampal BDNF and novel object recognition
A role for the hippocampus in object recognition memory has previously been suggested, although depending on the task different results have been found.52,53 Hammond et al.54 found that lidocaine inactivation of dorsal hippocampus did not interfere with acquisition of object recognition, but interfered with 24 h retention of object memory. More complex behavioral tasks have suggested, based on lesion data, that the hippocampal deficits in these types of tasks are primarily a function of deficits in spatial object recognition as opposed to deficits in individual object recognition per se,55 or involving a deficiency in relationship changes among objects.56 Our results show that BDNF deletions within the dorsal hippocampus reduces the amount of time spent with the novel compared to the habituated object, and that BDNF levels within the hippocampus across treatment groups correlate with performance in the novel object test. Additionally, consistent with the putative role of the hippocampus in the spatial components of object recognition,55 we found a significant correlation between performance on the novel object test and the water maze test. Together, these data suggest that BDNF expression in adult hippocampus is involved in the encoding or consolidation of some component of object recognition memory.
Fear-conditioning
We were somewhat surprised to find no obvious deficit in contextual fear learning. The hippocampus does not appear to be involved in Pavlovian-cued fear-conditioning as demonstrated in a variety of lesion and inactivation studies.57 A role for the hippocampus in contextual fear-conditioning appears to be more complicated, however. Initial studies, principally those using freezing as a paradigm, suggested that an intact hippocampus is required for contextual fear learning (cf. Anagnostaras et al.,17 Sanders et al.58). However, there have also been studies employing similar lesion approaches with FPS as a measure of fear in which no effect on contextual fear was found.18,59 Studies examining the role of the dorsal hippocampus in the retrieval or expression of context-dependent fear may help to clarify the likely complex role of the hippocampus in this form of learning.60–62 With regard to the role of BDNF in contextual fear learning, it has been reported that a heterozygous developmental knockout is impaired with contextual fear learning.63 Inducible BDNF knockouts using a forebrain-specific CamKII promoter have reduced contextual fear learning as well.10 However, these studies are contrasted with a different forebrain-specific knockout of BDNF, using the Emx1 promoter, which did not find contextual fear learning deficits.9 It is notable that the Emx1 and CamKII promoters have different levels of expression across hippocampus, amygdala, cortex and other forebrain regions, which may partially account for these different findings. Furthermore, the heterozygous knockout used by Liu et al.63 is knocked down throughout the brain early in embryonic development. Our data suggest that localized partial deletion of BDNF within the adult hippocampus does not result in deficits in contextual or cued fear learning, at least using our specific training parameters.
Having deletions of BDNF limited to only the dorsal hippocampus and only during a time-limited period in adulthood may also lead to a number of differences compared with the developmental and spatially broader BDNF deletions studied earlier. It is likely that normal hippocampal functioning may be required for the contextual discrimination that occurs with contextual fear learning, but the neuroplasticity underlying this learning may not actually occur within dorsal hippocampus. Developmental knockouts of BDNF could easily lead to subtle alterations in hippocampal functioning or wiring that may not be noticeable at the gross cellular morphology level, but could nonetheless alter normal functioning. In contrast, the time- and spatially-limited BDNF knockout in this study likely only affects functional BDNF needed for neuroplasticity during the time of the study. Thus, we suspect that the more specific functional deletion of BDNF in this study provides a more accurate assessment for a lack of BDNF effect within dorsal hippocampus during contextual fear learning.
Extinction deficit with hippocampal BDNF deletions
Recent studies by Maren and co-workers19 have demonstrated that muscimol inactivation of dorsal hippocampus reduced the rate of extinction and prevented the context dependency of extinction. Previous studies by this group have suggested that muscimol into dorsal hippocampus produces selective impairment in the context specificity of extinction.64 Other studies demonstrated that neurotoxic lesions of dorsal hippocampus abolished reinstatement of extinction, which is thought to depend on existing context-US associations.65 Similarly, mice with a congenitally reduced hippocampal commissure were shown to have impaired extinction of contextual fear.66 A series of studies by Izquierdo and co-workers67–71 suggests that extinction of inhibitory avoidance learning is blocked with a variety of kinase, NMDA, mRNA and protein synthesis inhibitors into the dorsal hippocampus. However, the specific role of protein synthesis within the hippocampus for extinction of contextual fear has been questioned.72,73 Recent data also suggest that structural plasticity involving actin rearrangement within dorsal hippocampus is required for extinction of context conditioned fear.72 Finally, imaging data suggest that the hippocampus may be involved in extinction of fear in humans.74,75
The role of BDNF within the hippocampus on extinction of fear has not been evaluated previously. Our data suggest that hippocampal expression of BDNF is required for the neural plasticity underlying the acquisition or consolidation of extinction memories. This is combined with recent data suggesting that amygdala-dependent TrkB activation is required for the consolidation of extinction of fear.76 Future studies should examine whether this hippocampal BDNF dependency of extinction is related only to the context specificity of the extinction memory as suggested by the data described above, or whether there is a more general effect of hippocampal neural plasticity on fear inhibition.
Hippocampal BDNF and psychopathology
A growing literature suggests that neurotrophic factors, particularly BDNF, within the hippocampus are required for normal emotional function and that alterations in hippocampal neurotrophic function may underlie stress-related mood disorders (for review see Duman and Monteggia8). Several different models of depression, including chronic immobilization stress,77 social isolation,78 social defeat79 and corticosterone overexpression,80 lead to decrease in hippocampal BDNF. Similarly, BDNF appears to increase in the hippocampus following treatment with a variety of antidepressants22,81,82 and electroconvulsive therapy.22 Additionally, some have suggested that the reduced hippocampal volume repeatedly found in depressed patients and those with PTSD,24–28 which appears to be reversed with antidepressant treatment,83 may be in part owing to chronically low levels of BDNF within the hippocampus.8
Two important pieces of information have been missing from the neurotrophic hypothesis of depression, however. First, there has been a lack of definitive support showing depression-like symptoms in animals with induced decreases in BDNF. Second, how the hippocampus neural circuitry leads to behavioral depression-like phenotypes has been difficult to understand. Recently, mice with an inducible BDNF knockout using the CamKII promoter were indistinguishable from controls in the forced swim test but displayed a reduced response to antidepressants.10 Our study demonstrates that hippocampus-specific deletions of BDNF have deficits in certain forms of cognitive functioning. Working memory, cognitive and executive function is classically impaired in humans with major depressive episodes.84,85
Regarding depression-like phenotypes, a deficit in extinction of aversive memories potentially offers a new way to understand and study the affective component of depression. Our data suggest that mice with specific deletions of hippocampal BDNF show decreased extinction and that BDNF levels correlate with level of extinction across the study groups. These data are consistent with data from the Maren lab that extinction of fear is partially dependent upon the hippocampus.19,64 Although it has been rarely studied, there is recent evidence that animal models of depression, such as congenital learned helplessness, are associated with deficits in extinction of aversive memory.86 Similarly, PTSD, which is highly co-morbid with depression, has been proposed based on a variety of data, to be due in part to a deficit in extinction of fear.29–32 A recently published study also demonstrates that there may be spatial memory deficits, for example copying figures and other similar neurological soft signs, in twin subjects with PTSD compared to their identical twin without PTSD,87 consistent with the correlations seen here between spatial memory performance in the water maze and extinction of fear.
Our data suggest that BDNF deletions within the hippocampus will be consistent with the persistence of fear or stress-related memories, possibly owing to the loss of neural plasticity necessary for new extinction memories to be formed. Among the most salient, yet poorly understood, symptoms of depression is the mood bias toward negative emotions, essentially the ‘depressed affect’, that is distinct from many of the easier to study cognitive and neurovegetative signs of depression. Impaired extinction of aversive memories as a result of decreased hippocampal BDNF may partially explain this set of mood symptoms and offers new avenues for understanding emotion regulation in patients with mood and anxiety disorders.
Supplementary Material
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
Acknowledgments
Support was provided by NIH (MH069884, KR and MH070218, JPC), NARSAD (KR), NIH/NCRR base grant (P51RR000165) to Yerkes National Primates Research Center and the Center for Behavioral Neuroscience (NSF agreement IBN-987675). We would also like to acknowledge C Todd French, MS, for excellent technical support.
References
- 1.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 2.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- 4.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 5.Kesslak JP, So V, Choi J, Cotman CW, Gomez-Pinilla F. Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav Neurosci. 1998;112:1012–1019. doi: 10.1037//0735-7044.112.4.1012. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J Neurosci. 2000;20:7116–7121. doi: 10.1523/JNEUROSCI.20-18-07116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, et al. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 8.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- 10.Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gomez-Pinilla F. Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience. 2004;123:429–440. doi: 10.1016/j.neuroscience.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris RG, Garrud P, Rawlins JN, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- 14.Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- 15.Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, et al. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 16.Wiltgen BJ, Sanders MJ, Anagnostaras SG, Sage JR, Fanselow MS. Context fear learning in the absence of the hippocampus. J Neurosci. 2006;26:5484–5491. doi: 10.1523/JNEUROSCI.2685-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anagnostaras SG, Gale GD, Fanselow MS. Hippocampus and contextual fear-conditioning: recent controversies and advances. Hippocampus. 2001;11:8–17. doi: 10.1002/1098-1063(2001)11:1<8::AID-HIPO1015>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 18.Gewirtz JC, McNish KA, Davis M. Is the hippocampus necessary for contextual fear-conditioning? Behav Brain Res. 2000;110:83–95. doi: 10.1016/s0166-4328(99)00187-4. [DOI] [PubMed] [Google Scholar]
- 19.Corcoran KA, Desmond TJ, Frey KA, Maren S. Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci. 2005;25:8978–8987. doi: 10.1523/JNEUROSCI.2246-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- 21.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 22.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 24.Bremner JD. Functional neuroanatomical correlates of traumatic stress revisited 7 years later, this time with data. Psychopharmacol Bull. 2003;37:6–25. [PubMed] [Google Scholar]
- 25.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 26.Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry. 1995;152:973–981. doi: 10.1176/ajp.152.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bremner JD. Neuroimaging studies in post-traumatic stress disorder. Curr Psychiatry Rep. 2002;4:254–263. doi: 10.1007/s11920-996-0044-9. [DOI] [PubMed] [Google Scholar]
- 28.Fossati P, Radtchenko A, Boyer P. Neuroplasticity: from MRI to depressive symptoms. Eur Neuropsychopharmacol. 2004;14:S503–S510. doi: 10.1016/j.euroneuro.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Gillespie CF, Ressler KJ. Emotional learning and glutamate: translational perspectives. CNS Spectr. 2005;10:831–839. doi: 10.1017/s1092852900010439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann NY Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- 31.Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, et al. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol Med. 2005;35:791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 34.Tiscornia G, Singer O, Ikawa M, Verma IM. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA. 2003;100:1844–1848. doi: 10.1073/pnas.0437912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeifer A, Brandon EP, Kootstra N, Gage FH, Verma IM. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc Natl Acad Sci USA. 2001;98:11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 39.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2. Academic Press; San Diego: 2001. [Google Scholar]
- 40.Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear-conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chhatwal JP, Myers KM, Ressler KJ, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- 43.Ennaceur A, Neave N, Aggleton JP. Neurotoxic lesions of the perirhinal cortex do not mimic the behavioural effects of fornix transection in the rat. Behav Brain Res. 1996;80:9–25. doi: 10.1016/0166-4328(96)00006-x. [DOI] [PubMed] [Google Scholar]
- 44.Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- 45.Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- 46.Blanchard RJ, Blanchard DC. Passive and active reactions to fear-eliciting stimuli. J Comp Physiol Psychol. 1969;68:129–135. doi: 10.1037/h0027676. [DOI] [PubMed] [Google Scholar]
- 47.Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 48.Jones SV, Heldt SA, Davis M, Ressler KJ. Olfactory-mediated fear-conditioning in mice: simultaneous measurements of fear-potentiated startle and freezing. Behav Neurosci. 2005;119:329–335. doi: 10.1037/0735-7044.119.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heldt SA, Ressler KJ. Lesions of the habenula produce stress- and dopamine-dependent alterations in prepulse inhibition and locomotion. Brain Res. 2006;1073–1074:229–239. doi: 10.1016/j.brainres.2005.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 51.Logue SF, Paylor R, Wehner JM. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- 52.Aggleton JP, Blindt HS, Rawlins JN. Effects of amygdaloid and amygdaloid-hippocampal lesions on object recognition and spatial working memory in rats. Behav Neurosci. 1989;103:962–974. doi: 10.1037//0735-7044.103.5.962. [DOI] [PubMed] [Google Scholar]
- 53.Cassaday HJ, Rawlins JN. The hippocampus, objects, and their contexts. Behav Neurosci. 1997;111:1228–1244. doi: 10.1037//0735-7044.111.6.1228. [DOI] [PubMed] [Google Scholar]
- 54.Hammond RS, Tull LE, Stackman RW. On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem. 2004;82:26–34. doi: 10.1016/j.nlm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 55.Lee I, Hunsaker MR, Kesner RP. The role of hippocampal subregions in detecting spatial novelty. Behav Neurosci. 2005;119:145–153. doi: 10.1037/0735-7044.119.1.145. [DOI] [PubMed] [Google Scholar]
- 56.Moses SN, Cole C, Driscoll I, Ryan JD. Differential contributions of hippocampus, amygdala and perirhinal cortex to recognition of novel objects, contextual stimuli and stimulus relationships. Brain Res Bull. 2005;67:62–76. doi: 10.1016/j.brainresbull.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 57.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear-conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 58.Sanders MJ, Wiltgen BJ, Fanselow MS. The place of the hippocampus in fear-conditioning. Eur J Pharmacol. 2003;463:217–223. doi: 10.1016/s0014-2999(03)01283-4. [DOI] [PubMed] [Google Scholar]
- 59.McNish KA, Gewirtz JC, Davis M. Evidence of contextual fear after lesions of the hippocampus: a disruption of freezing but not fear-potentiated startle. J Neurosci. 1997;17:9353–9360. doi: 10.1523/JNEUROSCI.17-23-09353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frankland PW, Cestari V, Filipkowski RK, McDonald RJ, Silva AJ. The dorsal hippocampus is essential for context discrimination but not for contextual conditioning. Behav Neurosci. 1998;112:863–874. doi: 10.1037//0735-7044.112.4.863. [DOI] [PubMed] [Google Scholar]
- 61.Maren S, Aharonov G, Fanselow MS. Neurotoxic lesions of the dorsal hippocampus and Pavlovian fear-conditioning in rats. Behav Brain Res. 1997;88:261–274. doi: 10.1016/s0166-4328(97)00088-0. [DOI] [PubMed] [Google Scholar]
- 62.Maren S, Holt W. The hippocampus and contextual memory retrieval in Pavlovian conditioning. Behav Brain Res. 2000;110:97–108. doi: 10.1016/s0166-4328(99)00188-6. [DOI] [PubMed] [Google Scholar]
- 63.Liu IY, Lyons WE, Mamounas LA, Thompson RF. Brain-derived neurotrophic factor plays a critical role in contextual fear-conditioning. J Neurosci. 2004;24:7958–7963. doi: 10.1523/JNEUROSCI.1948-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frohardt RJ, Guarraci FA, Bouton ME. The effects of neurotoxic hippocampal lesions on two effects of context after fear extinction. Behav Neurosci. 2000;114:227–240. doi: 10.1037//0735-7044.114.2.227. [DOI] [PubMed] [Google Scholar]
- 66.Schimanski LA, Wahlsten D, Nguyen PV. Selective modification of short-term hippocampal synaptic plasticity and impaired memory extinction in mice with a congenitally reduced hippocampal commissure. J Neurosci. 2002;22:8277–8286. doi: 10.1523/JNEUROSCI.22-18-08277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cammarota M, Bevilaqua LR, Kerr D, Medina JH, Izquierdo I. Inhibition of mRNA and protein synthesis in the CA1 region of the dorsal hippocampus blocks reinstallment of an extinguished conditioned fear response. J Neurosci. 2003;23:737–741. doi: 10.1523/JNEUROSCI.23-03-00737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK and CaMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- 69.Vianna MR, Igaz LM, Coitinho AS, Medina JH, Izquierdo I. Memory extinction requires gene expression in rat hippocampus. Neurobiol Learn Mem. 2003;79:199–203. doi: 10.1016/s1074-7427(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 70.Vianna MR, Szapiro G, McGaugh JL, Medina JH, Izquierdo I. Retrieval of memory for fear-motivated training initiates extinction requiring protein synthesis in the rat hippocampus. Proc Natl Acad Sci USA. 2001;98:12251–12254. doi: 10.1073/pnas.211433298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bevilaqua LR, da Silva WN, Medina JH, Izquierdo I, Cammarota M. Extinction and reacquisition of a fear-motivated memory require activity of the Src family of tyrosine kinases in the CA1 region of the hippocampus. Pharmacol Biochem Behav. 2005;81:139–145. doi: 10.1016/j.pbb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 72.Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21:5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Knight DC, Smith CN, Cheng DT, Stein EA, Helmstetter FJ. Amygdala and hippocampal activity during acquisition and extinction of human fear-conditioning. Cogn Affect Behav Neurosci. 2004;4:317–325. doi: 10.3758/cabn.4.3.317. [DOI] [PubMed] [Google Scholar]
- 75.LaBar KS, Phelps EA. Reinstatement of conditioned fear in humans is context dependent and impaired in amnesia. Behav Neurosci. 2005;119:677–686. doi: 10.1037/0735-7044.119.3.677. [DOI] [PubMed] [Google Scholar]
- 76.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, et al. Brain-derived neurotrophic factor mRNA down-regulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121:847–853. doi: 10.1016/s0306-4522(03)00564-5. [DOI] [PubMed] [Google Scholar]
- 79.Pizarro JM, Lumley LA, Medina W, Robison CL, Chang WE, Alagappan A, et al. Acute social defeat reduces neurotrophin expression in brain cortical and subcortical areas in mice. Brain Res. 2004;1025:10–20. doi: 10.1016/j.brainres.2004.06.085. [DOI] [PubMed] [Google Scholar]
- 80.Benninghoff J, Schmitt A, Mossner R, Lesch KP. When cells become depressed: focus on neural stem cells in novel treatment strategies against depression. J Neural Transm. 2002;109:947–962. doi: 10.1007/s007020200078. [DOI] [PubMed] [Google Scholar]
- 81.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Hoomissen JD, Chambliss HO, Holmes PV, Dishman RK. Effects of chronic exercise and imipramine on mRNA for BDNF after olfactory bulbectomy in rat. Brain Res. 2003;974:228–235. doi: 10.1016/s0006-8993(03)02584-8. [DOI] [PubMed] [Google Scholar]
- 83.Vermetten E, Vythilingam M, Southwick SM, Charney DS, Bremner JD. Long-term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol Psychiatry. 2003;54:693–702. doi: 10.1016/s0006-3223(03)00634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rose EJ, Ebmeier KP. Pattern of impaired working memory during major depression. J Affect Disord. 2006;90:149–161. doi: 10.1016/j.jad.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 85.Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology. 2004;29:1765–1781. doi: 10.1038/sj.npp.1300506. [DOI] [PubMed] [Google Scholar]
- 86.Shumake J, Barrett D, Gonzalez-Lima F. Behavioral characteristics of rats predisposed to learned helplessness: reduced reward sensitivity, increased novelty seeking, and persistent fear memories. Behav Brain Res. 2005;164:222–230. doi: 10.1016/j.bbr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 87.Gurvits TV, Metzger LJ, Lasko NB, Cannistraro PA, Tarhan AS, Gilbertson MW, et al. Subtle neurologic compromise as a vulnerability factor for combat-related posttraumatic stress disorder: results of a twin study. Arch Gen Psychiatry. 2006;63:571–576. doi: 10.1001/archpsyc.63.5.571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)