Structure of the Main Protease from a Global Infectious Human Coronavirus, HCoV-HKU1 (original) (raw)
Abstract
The newly emergent human coronavirus HKU1 (HCoV-HKU1) was first identified in Hong Kong in 2005. Infection by HCoV-HKU1 occurs worldwide and causes syndromes such as the common cold, bronchitis, and pneumonia. The CoV main protease (Mpro), which is a key enzyme in viral replication via the proteolytic processing of the replicase polyproteins, has been recognized as an attractive target for rational drug design. In this study, we report the structure of HCoV-HKU1 Mpro in complex with a Michael acceptor, inhibitor N3. The structure of HCoV-HKU1 provides a high-quality model for group 2A CoVs, which are distinct from group 2B CoVs such as severe acute respiratory syndrome CoV. The structure, together with activity assays, supports the relative conservation at the P1 position that was discovered by sequencing the HCoV-HKU1 genome. Combined with structural data from other CoV Mpros, the HCoV-HKU1 Mpro structure reported here provides insights into both substrate preference and the design of antivirals targeting CoVs.
Coronaviruses (CoVs) are positive-strand RNA viruses that have been identified as the main etiologic agents responsible for a vast number of enteric, gastric, and respiratory syndromes of both humans and animals (14-17, 19, 21, 23, 26, 30, 31, 34, 45). CoVs can be divided into three groups: group 1 (including human CoV 229E [HCoV 229E] and transmissible gastric enteritis virus [TGEV]), group 2 (including HCoV-OC43, murine hepatitis virus [MHV], and bovine CoV [BCoV), and group 3 (including avian infectious bronchitis virus [IBV]). Shortly after the emergence of severe acute respiratory syndrome CoV (SARS-CoV) in 2003, group 2 CoVs were further divided into two subgroups, termed 2A and 2B (46). The classical group 2 viruses constitute subgroup 2A, while the newly emergent SARS-CoV and its animal counterparts (37) form subgroup 2B. Group 1 and group 2 CoVs have more impact on human health than group 3, since group 3 CoVs (such as avian IBV) can only infect avian species. Following the outbreak of SARS, group 2 CoVs have continued to attract greater attention for two reasons. First, they consist of human viruses (SARS-CoV and HCoV-OC43) as well as several important animal viruses (MHV and BCoV) that serve as useful models for CoV-host interactions. Second, group 2 CoVs are reported to have crossed the animal-to-human species barrier in two instances: one bat-to-human transmission in group 2B (27, 37) and one transmission event in group 2A CoVs, in which BCoV led to the emergence of HCoV-OC43 (36).
Group 2A HCoVs were less widely studied prior to the global SARS epidemic in 2003. However, they are closely associated with a wide range of acute or chronic respiratory syndromes (3, 4, 7-9, 11, 12, 15, 20, 22, 35, 39, 40, 47). In the wake of the SARS outbreak, several novel HCoVs have been discovered, one of which is HCoV-HKU1 (9, 39). HCoV-HKU1 has achieved global distribution since it was first identified in 2005: infections were first characterized in Hong Kong (26), followed by the identification of several strains of the virus in Korea (9), Europe (5, 17), Australia (31), and North America (14). In contrast to the lethal SARS-CoV, infection by HCoV-HKU1 usually leads to self-limiting syndromes affecting the lower respiratory tract. Nevertheless, the consequences could be more severe in patients with a compromised or immature immune system, such as asthma sufferers or newborn infants (24). Genome sequencing has confirmed that the HCoV-HKU1 virus belongs to CoV group 2A and shares high sequence homology with MHV and BCoV (39).
The functional components of the CoV replication machinery are released via posttranslational cleavage by two or three proteases. These proteases were first designated the papain-like protease (PLP) and 3C-like protease (3CL) for their respective sequence homology to the papain and rhinovirus 3C proteases. The 3CL protease also is commonly known as the main protease (Mpro) because of the major role it plays in the proteolytic pathway, which makes it the most attractive pharmacological target for anti-CoV drug design. CoV Mpros have been intensively studied, and crystal structures have been determined for the Mpros from the following CoVs: HCoV strain 229E (HCoV-229E) (2), porcine TGEV (1), avian IBV (41), and SARS-CoV (44). These structures are representative of group 1 (HCoV-229E and TGEV), group 2B (SARS-CoV), and group 3 (IBV) CoVs. However, no structure of the Mpro from a group 2A CoV (MHV, HCoV-HKU1, and HCoV-OC43) has been determined to date. The absence of structural data presents a major obstacle for structure-aided drug optimization targeting group 2A CoVs.
The Mpros from different CoV groups are homologous in both sequence and main-chain architecture. They share a similar substrate binding sequence, with a requirement for glutamine at the P1 position and a strong preference for leucine/methionine at P2. Based on this information, broad-spectrum lead compounds (43) with micromolar K i values have been designed that target CoV Mpros. However, structural data for the Mpros from classical group 2A CoVs still are not available, posing a problem for further optimization.
Although CoV Mpros exhibit absolute specificity for glutamine in the P1 position, recent research (38) has shown that the Mpro from HCoV-HKU1 may possess an unusual substrate preference at P1 site quite different from that of other CoV Mpros. Here, we report the structure of HCoV-HKU1 Mpro, which serves as a model for group 2A CoVs in complex with a synthetic peptidomimetic inhibitor, N3. The structure and subsequent enzyme activity assays help to resolve the issue of the relative conservation at the P1 position based on genome sequencing. Moreover, this complex structure provides further structural data for rational drug design against HCoVs.
MATERIALS AND METHODS
cDNA and plasmid.
The cDNA encoding HCoV-HKU1 Mpro was kindly provided by K. Y. Yuen from the Department of Microbiology, Hong Kong University, Hong Kong Special Administrative Region, China. BamHI and XhoI restriction sites were attached to the 5′ and 3′ ends separately by PCR, and the PCR product first was inserted into the pMD-18T vector (Takara). The DNA of interest then was cleaved from the T vector and subcloned into a glutathione _S_-transferase-tagged expression vector, pGEX-4T-1. The validity of the whole procedure was confirmed by DNA sequencing.
Protein expression and purification.
The plasmid was first transformed into the commercial Escherichia coli strain BL21(DE3) Rosetta (Invitrogen). After incubation at 37°C overnight on an Amp+ algae Luria-Bertani (LB) plate, fresh transformants were inoculated into 5 ml LB medium in the presence of 100 μg/ml ampicillin. After growth for 12 h, the incubation system was scaled up to 1 liter LB medium with the same concentration of antibiotics in a 2-liter flask, and the solution was shaken vigorously at 37°C until the optical density at 600 nm reached 0.6. Cells were induced by 0.5 mM isopropyl-β-d-thiogalactopyranoside (Sigma) at 16°C overnight.
Cell pellets were harvested by centrifugation, resuspended in 40 ml phosphate-buffered saline buffer with 2 mM dithiothreitol and 7 mM β-mercaptoethanol, and sonicated on ice for 25 min. The supernatant was collected after the centrifugation of the sonicant at 15,000 rpm for 40 min.
Affinity purification was achieved by letting the supernatant flow through 2 ml glutathione _S_-transferase affinity medium twice. On-column digestion lasted for 16 h at 4°C with thrombin (New England BioLabs), and the protein of interest was harvested and concentrated to 30 mg/ml. The N3 inhibitor then was added to a final molar ratio of 1:1 and incubated at 4°C overnight. Finally, the HCoV-HKU1 Mpro-inhibitor complex was purified by gel filtration using a Superdex 200 (10/30) column (GE Healthcare). The protein concentration was adjusted to 20 mg/ml for crystallization trials.
Crystallization and structure determination.
Crystals of HCoV-HKU1 Mpro were grown in 0.1 M imidazole, pH 6.0, and 0.6 M sodium acetate by the hanging drop vapor diffusion method. Synchrotron X-ray diffraction data were collected on beamline BL-5A of the Photon Factory (Tsukuba, Japan) and processed to 2.5-Å resolution, using HKL2000 (29) for data indexing and scaling. Molecular replacement using the SARS Mpro structure (Protein Data Bank entry 2AMQ; 48% identity) as a template was performed with PHASER (32). The manual rebuilding of the structure was performed using Coot (13), and the structure was refined using REFMAC in the CCP4 suite (10). Final modification was carried out using CNS (6). The volume of the S1 cavity was calculated using VOIDOO (25).
Enzyme activity assays.
Substrates and analogs were designed through three rounds of affinity optimization (42) by substrate mimicry and from a library of substrate analogs. The substrate and analogs were synthesized by Dawei Ma from the Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China.
The strategy employed for enzyme activity assays of HCoV-HKU1 Mpro has been described previously (43). Activity assays for HCoV-HKU1 Mpro against the CoV consensus substrate and the HCoV-HKU1-specific substrate followed a similar protocol, which is described briefly below. The consensus substrate and HCoV-HKU1-specific substrates were fluorescent compounds with the sequences MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 and MCA-PRLHCTTN-Lys(Dnp)-Lys-NH2, respectively (greater than 95% purity; GL Biotech Shanghai Ltd., Shanghai, China). A P1 single-mutant substrate also was synthesized with sequence MCA-AVLHSGFR-Lys(Dnp)-Lys-NH2.
The excitation and emission wavelengths of the fluorescent substrates were 320 and 405 nm, respectively. A buffer consisting of 50 mM Tris-HCl (pH 7.3) and 1 mM EDTA was used for enzyme activity assays at 30°C. The reaction was initiated by adding protease (final concentration, 2 μM) to a solution containing different final concentrations of the substrate (3.2 to 40 μM). Strict kinetic parameters for the inhibition assay were determined according to the previously reported protocol (43). All results from enzyme activity assays were calculated using data based on at least three independent parallel experiments.
Coordinate accession number.
Coordinates and structure factors for the HKU1 Mpro in complex with inhibitor N3 have been deposited in the Protein Data Bank under entry ID 3D23.
RESULTS
Structural overview.
Four protein molecules (denoted A, B, C, and D) occupy one asymmetric unit, with one N3 molecule per protomer. Two of the protomers form a typical homodimer, while the remaining two protomers dimerize with their adjacent symmetry-related counterparts (Fig. 1a). Each protomer exhibits a three-domain (I to III) architecture that is common to other CoV Mpro structures (1, 2, 42, 44): domains I and II have chymotrypsin-like folds, and domain III displays a globular α-helical cluster that is unique to CoV Mpro. The catalytic site, including the Cys-His dyad, and the relatively shallow substrate binding pocket of HCoV-HKU1 Mpro are located in the cleft between domains I and II. The substrate-binding pocket features two deeply buried sites (P1 and P2) and several sites with different levels of solvent exposure (P3, P4, and P5) (Fig. 1b). X-ray data collection and refinement statistics are summarized in Table 1.
FIG. 1.

(a) Structural overview of four protomers (A, green; B, cyan; C, magenta; and D, yellow) in one asymmetric unit, represented as cartoons. N3 inhibitors are shown as blue sticks. (b) Structural overview of the enzyme-inhibitor complex of one monomer unit. The main chain of the enzyme is represented as blue cartoons, and the synthetic inhibitor is shown as yellow sticks. The three domains are labeled.
TABLE 1.
X-ray data-processing and refinement statistics
| Statistic | Value for the HCoV-HKU1 Mpro-N3 complexe |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.0 |
| Resolution limit (Å) | 50.0-2.48 (2.62-2.48) |
| Space group | P41 |
| Cell parameters | |
| a (Å) | 91.770 |
| b (Å) | 91.770 |
| c (Å) | 187.914 |
| β (°) | 90 |
| Total no. of reflections | 510,408 |
| No. of unique reflections | 108,501 |
| Completeness (%) | 99.1 (98.64) |
| Redundancy | 5.0 (4.9) |
| _R_mergea | 0.11 |
| Sigma cutoff | 0 |
| I/σ(I) | 16 (5) |
| Refinement | |
| Resolution range (Å) | 50.0-2.5 |
| _R_workb (%) | 22.9 |
| _R_free (%) | 28.5 |
| rmsdc from ideal geometry | |
| Bonds (Å) | 0.012 |
| Angles (°) | 1.59 |
| Avg B factor (Å2) | |
| Protein | 38.7 |
| Small molecule | 43.7 |
| Ramachandran plotd | |
| Favored (%) | 86.8 |
| Allowed (%) | 12.2 |
| Generously allowed (%) | 0.9 |
| Disallowed (%) | 0.1 |
Michael acceptor and catalytic dyad.
Clear and continuous electron density was observed between the reactive backbone carbon atom of the N3 substrate and the Sγ atom of Cys145 in the inhibitor-bound HCoV-HKU1 Mpro structure. We conclude that this reaction can be categorized as an electrophilic addition mediated by a Michael acceptor, obeying the K i − _k_3 kinetics, where K i is the dissociation constant and _k_3 is the turnover number, according to the following scheme:
 |
(1) |
|---|
As the covalently bound inhibitor is a mimic of the real peptide substrate, it is possible to model the transition state by treating the enzyme-inhibitor complex structure as a snapshot of the catalytic dyad, and hence to predict parameters of the K m − _k_cat kinetics, according to the following scheme:
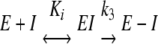 |
(2) |
|---|
This catalytic dyad involves residues His41 and Cys145, and the intermediate state might be stabilized by the oxyanion hole (28) formed by the backbone amides of the oxyanion loop from Phe140 to Cys145 (Fig. 2a). The oxyanion hole is crucial to the stabilization of the intermediate state, so the formation of the oxyanion hole has a significant influence on _k_cat. As discussed for the inhibitor design targeting human rhinovirus 3C proteases (28), the correct organization of this oxyanion loop also is essential to the _k_3 step for mechanism-based suicide inhibitors.
FIG. 2.

(a) Details of the interaction between the P1 side chain and the defined oxyanion loop, shown in stereo representation. Side chains are shown as sticks, and the crucial hydrogen bond between His163 and the substrate side chain is shown by a cyan dashed line. (b) Details of the substrate-binding pocket. The inhibitor is shown in the following color scheme: C, white; O, red; and N, blue. The crucial residues of the enzyme are shown in the following color scheme: C, cyan; O, red; N, blue; and S, yellow. Hydrogen bonds are shown as red dashed lines.
Surrounding the Sγ atom of Cys145, we observe well-defined amides from the loop from residues 142 to 145. Similarly to human rhinovirus 3C proteases (28), these amide dipoles construct a tetrahedral oxyanion hole. From native and complex structures of SARS-CoV Mpro, the correct orientation of these backbone amides is triggered and maintained by substrate binding, in particular by the binding of the P1 residue and interaction between the N finger and the substrate (2, 44), in which the backbone carbonyl of Leu141 is hydrogen bonded to the side chain oxygen of Ser144. The correct position of Leu141 is maintained by a hydrogen bond between the carbonyl group of Phe140 and the amide group of the P1 side chain and by hydrophobic stacking between His163 and Phe140. Although the analysis of the HCoV-HKU1 Mpro structure in complex with N3 (Fig. 2a) shows that the P1 side chain exerts no direct influence on the residues forming the oxyanion hole, its side chain oxygen atom forms a strong hydrogen bond (2.6 Å) with His163 and helps to strengthen the stacking interaction with Phe140. Furthermore, the nitrogen atom of the P1 side chain also forms a hydrogen bond (3.1 Å) with the backbone of Phe140, thus helping to maintain the oxyanion loop (Phe140-Cys145) in its proper conformation. For the above reasons, we conclude that the P1 side chain is important for the network of interactions stabilizing the oxyanion hole.
The S1 pocket has a smaller size to accommodate P1 histidine.
Given its crucial role in the catalytic process, glutamine outperforms other residues as the signature of the Mpro substrate at the P1 position. In addition to this advantage, the side chain of glutamine in the P1 position suitably fits with residues forming the S1 subsite via Van der Waals interactions (Fig. 2b). From the HCoV-HKU1 genome sequence, 11 out of 12 Mpro recognition sites have Gln at the P1 position. In our structure, the N3 molecule has a lactam ring as an analog to the glutamine residue (the cross-linking between the Cγ and N atoms helps to select the stretching conformation from the ensemble of rotamers and better occupy the binding cleft). In HCoV-HKU1 Mpro structures, the lactam ring protrudes into the S1 pocket via a hydrogen bond to the imidazole ring NH of His162 at a distance of 2.6 Å. However, unlike the SARS-CoV Mpro structure in complex with N3, the NH of the HKU1-N3 lactam ring fails to recruit a water molecule to satisfy a second S1 hydrogen bond. Instead, the N-terminal Oγ atom might provide a weak electronegative interaction to stabilize the NH atom; the interaction likely is stronger due to the presence of redundant residues as a cloning artifact, hindering the N-terminal Ser from coming any closer to the NH of P1 side chain.
Nevertheless, compared with the Mpros from other CoV groups, the structure of the HCoV-HKU1 Mpro has an S1 pocket with a relatively smaller volume of ∼18.1 Å3. In contrast, the volume of the S1 pocket of TGEV Mpro is ∼19.1 Å3, that of of IBV Mpro is ∼21.7 Å3, and that of SARS-CoV Mpro is ∼19.5 Å3. The reduced size of the S1 pocket might be caused by the position of the loop Leu167-Cys171, which is bent upward by about 90°. As a result, the smaller S1 pocket might tolerate mutation to short-chain residues at the P1 position, in which case a weakened oxyanion hole is to be expected. Novel substrate specificity already has been found in the HCoV-HKU1 genome, in which the Mpro recognition site between the helicase and exonuclease utilizes histidine instead of glutamine at the P1 position. Mimicking proteolysis in the cell, enzyme activity assays using a synthetic fluorogenic substrate confirm the existence of such a cleavage event in vitro and exhibit novel enzymatic properties not seen with the consensus substrate (Table 2).
TABLE 2.
Activity assay of HCoV-HKU1
| Substratea | K m (μM) | _k_cat |
|---|---|---|
| C | 83.2 ± 13.3 | 1.1 ± 0.12 |
| H | 22.3 ± 5.2 | 0.09 ± 0.013 |
| SMC | 265.3 ± 21.5 | 0.03 ± 0.001 |
Enzyme activity assays indicate that the affinity for a substrate containing a single mutation at the P1 position decreases to 30% of the affinity for the native consensus substrate, which can be attributed to the loss of a hydrogen bond resulting from the mutation of glutamine to histidine. The scissile velocity decreases to 3%, which is to be expected, since the histidine residue lacks an oxygen atom that is required to form a strong hydrogen bond and support the intermediate oxyanion hole. However, when determining the influence of P2-P5 variance in the HCoV-HKU1-specific substrate, we observed an unusual fourfold elevation in the K m compared to that of the consensus substrate and a minor rescue of the scissile velocity (a threefold elevation of the single-mutant substrate). This could be explained by the contribution of the non-P1 residues in the HCoV-HKU1-specific substrate, since activity assays for the single-mutant substrate imply that mutation at the P1 position has a detrimental effect not only on the substrate binding affinity but also on the substrate scissile velocity.
The S2 pocket presents group-specific features but no group-specific substrate preferences.
The P2 side chain of the ligand protrudes into the S2 pocket via interactions with the hydrophobic side chains of Met25, Pro52, and Tyr54 (Fig. 2B). The lid of the pocket is covered by a short 310 helical region from Ser45-Asn51. To compare the diversity of the S2 pockets of all three CoV groups, the backbones of Mpro complex structures from all groups were superimposed (Fig. 3): for group 1, TGEV Mpro in complex with the inhibitor N1, an ancestor of N3; for group 3, IBV Mpro in complex with N3; and for group 2B, SARS-CoV in complex with inhibitor N3. We observed three modes of secondary structure: the 310 helix (HCoV-HKU1 and SARS-CoV), a loose loop (IBV), and a tight loop (TGEV). Interestingly, the clustering of the secondary structure correlates with the temporary classification of CoVs. We then explored the natural recognition sequences to examine whether the group-specific features could result in different substrate specificities at the P2 site (Table 3). After summarizing the P2 residue type in the protease recognition sites of the HKU1 PP1ab genome, we observe that Mpros prefer a hydrophobic residue at this position, which is also the case for SARS-CoV, IBV, and TGEV. Although there are a few exceptions, such as asparagine or valine residues, leucine/methionine are the most abundant. This is consistent with the observation from our structure that the hydrophobic P2 side chain extends into the deep S2 site without clashing with the Van der Waals surface of the pocket (Fig. 2b). Therefore, on the one hand, considering the flexibility of S2 pocket as well as the residual space after occupation by the P2 residue, the optimal choice for leucine or methionine might be related to the size of the S2 pocket. On the other hand, the similar preferences on S2 sites among group 1, 2a, 2b, and 3 CoV Mpros does challenge the efficacy of designing group-specific inhibitors by altering only P2 moieties.
FIG. 3.
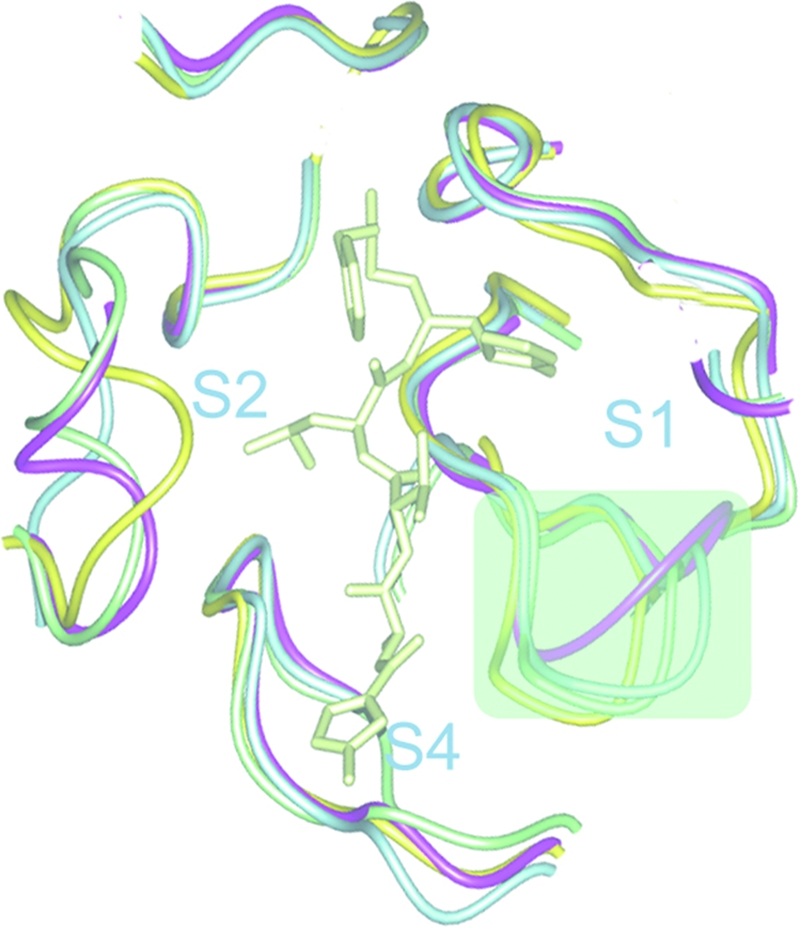
S1 and S2 binding sites of HCoV-HKU1 Mpro main chains of four Mpro structures are superimposed and displayed in the neighborhood of the substrate-binding site. The S1 and S2 binding sites are highlighted by light green shadows. The main chains are represented in worm forms. Different colors are used to represent the strain of CoV. Lemon, synthetic compound; magenta, HCoV-HKU1; light green, SARS-CoV; light blue, TGEV; and yellow, avian IBV.
TABLE 3.
P2 residues in different CoV genomes
| Mpro cleavage site no. | P2 residue for: | |||
|---|---|---|---|---|
| SARS-CoV | HCoV-HKU1 | IBV | TGEV | |
| 1 | L | L | L | L |
| 2 | F | L | L | N |
| 3 | V | I | M | V |
| 4 | L | L | L | L |
| 5 | L | M | L | L |
| 6 | L | L | L | L |
| 7 | M | V | V | M |
| 8 | L | M | L | L |
| 9 | L | L | L | L |
| 10 | L | L | L | L |
| 11 | L | M | L | L |
The P3 position.
Two out of four protomers in one asymmetric unit exhibit a solvent-exposed P3 side chain, which may interact weakly with the edge of the substrate-binding cleft via Van der Waals forces (Fig. 2b). To assess whether P3 side chain variation can influence the potency of inhibition, we synthesized a small library of six inhibitors (see Table S1 in the supplemental material) to assay their affinity by the second-order reaction coefficients. The results are summarized in Fig. S1 in the supplemental material. It appears unlikely that the preference can be attributed exclusively to weak interactions. When investigating other possible P3-related interactions found in our HCoV-HKU1 Mpro structure, we scrutinized and evaluated the mainly hydrophobic structural interaction (Fig. 4a) between molecules B and D (Fig. 4b). From the crystal packing, we observed a crystallographic contact close to the P3 position (see Fig. S3 in the supplemental material) that we suspect might affect this property of the P3 residue. To examine the physiological relevance, we conducted dynamic light-scattering experiments to check for higher-molecular-weight states that should be expected in solution if this interaction is related to one of the stable physiological states. However, dynamic light-scattering experiments did not identify a tetramer or higher-molecular-weight complex in aqueous solution (see Fig. S2 in the supplemental material). Thus, the substrate selectivity at the P3 position may be attributed to other weak factors, such as solvent-side chain interactions and Van der Waals interactions with the substrate binding cleft, rather than to a strong and direct interaction, which is more likely to be influenced by crystal contacts.
FIG. 4.

(a) Overview of the P3 pocket. The inhibitor resides between the interface of molecules B and D. A cyan surface model is shown covering protomers B and D. The inhibitor is shown in magenta. (b) P3 interaction site of substrate in detail. Neighboring residues within 4 Å of the S3 site are colored green. An Fo-Fc map contoured at 1.5σ around the inhibitor is displayed in cyan. For the inhibitor, C atoms are colored yellow, N atoms are colored blue, and O atoms are colored red. The protein carbon atoms are colored gray. Neighboring main chains are displayed as white ribbons.
DISCUSSION
The HKU1 structure is a suitable model for group 2A CoV Mpros.
The crystal structure of Mpro from HCoV-HKU1 is the first to be determined from a group 2A Mpro. Since members of the group 2A CoVs share particularly high sequence identity (4) (Fig. 5a), nonconservative changes occur mainly in flexible regions of the HCoV-HKU1 Mpro structure, including domain linkages and the molecular surface. Notable variations from residues 46 to 71 in group 2A sequences are located in or nearby the S2 pocket, which might infer properties relating to enzyme activity. However, since even greater differences between the different groups of CoVs exhibit no particular enzyme-specific preferences in the S2 pocket, the relatively small variations here may be unlikely to challenge the consensus substrate preference among CoVs at the P2 position. The residue ranges 20 to 40, 140 to 160, and 187 to 189, as well as residue 166, are highly conserved and are involved in the formation of the S-1 and S1 pockets, together with the walls of the binding pocket of the P3 side chain. Thus, it is reasonable to conclude that the HCoV-HKU1 Mpro structure is a suitable model for the study of group 2A CoVs, both in terms of enzyme activity and inhibitor design.
FIG. 5.

(a) Sequence alignment of a typical Mpro from CoV group 2A exhibits high homology. The alignment was performed with ClustalW (33), and the final figure was generated with ESPript1.0 (18). White letters with red backgrounds refer to identical residues, red letters with white backgrounds refer to conservative variation, and black letters with white backgrounds refer to nonconservative mutations. (b) Three-dimensional representation of nonconserved mutations in group 2A CoV Mpros mapped onto the HCoV-HKU1 Mpro structure. Identical residues are colored white, conserved mutations are colored yellow, and nonconserved mutations are colored red. The inhibitor is colored blue.
Michael acceptor inhibitors interact with CoV Mpro in a similar manner.
As Michael acceptor suicide inhibitors, N3 and its derivatives cocrystallize with the CoV Mpro in a similar manner (Fig. 6). The backbones of the peptidomimetic compounds align antiparallel to the β-strands, constituting the binding cleft. The P1 and P2 residues fit into the S1 and S2 pockets, respectively, and have a major contribution to substrate preference: glutamine at P1 and leucine/methionine at P2. In our future optimization of Mpro inhibitors, we think that the glutamine (or its analog) might be worth keeping in the P1 position, while it would be reasonable to conduct a comparison of leucine to methionine for the evaluation of the P2 residue. Aside from these deeply buried side chains, the solvent-exposed P3 provides no straightforward information for the substrate-enzyme interaction, though the variation at this position shows an obvious impact on inhibition. Alternatively, we might employ random screening for further optimization at the P3 position.
FIG. 6.
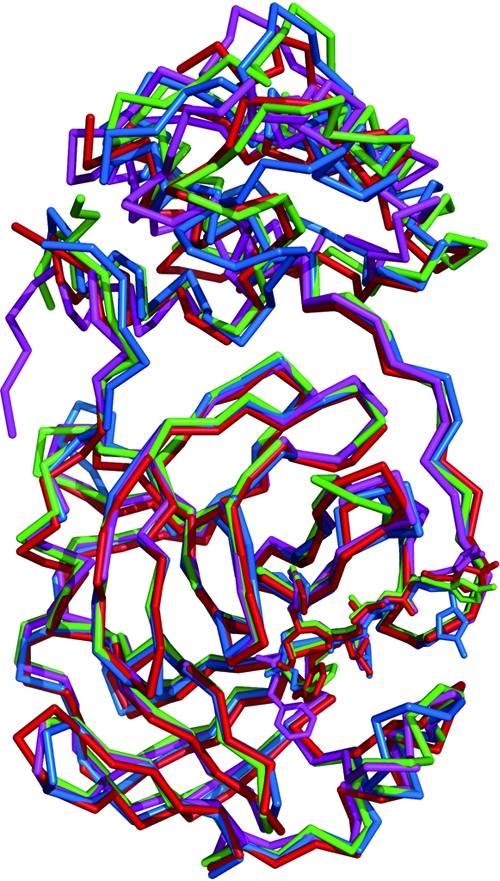
Superposition of representative CoV Mpro in complex with Michael acceptor-based inhibitors of group 1 (TGEV; blue), group 2A (HCoV-HKU1; green), group 2B (SARS-CoV; magenta), and group 3 (IBV; red). The color of each inhibitor is consistent with that of its host.
Conclusions.
Structural data now are available for CoV Mpro-inhibitor complexes from all CoV groups, including the two subgroups of the group 2 CoVs. Moreover, these structures provide further confirmation for the efficacy of wide-spectrum inhibitors at atomic resolution. From enzyme activity assays, we succeeded in identifying the atypical substrate specificity of HCoV-HKU1 Mpro with higher affinity (K m) and lower reactivity (_k_cat) than those of the consensus CoV Mpro substrate. We attributed these properties to the contribution of non-P1 residues and the distortion of the oxyanion hole. Although the S2 pockets from different groups share group-specific features, an investigation of the natural recognition sequences does not find different residue-type specificity at the P2 site.
Considering the high identity shared by group 2A CoVs, these structural features of HCoV-HKU1 Mpro, together with corresponding enzyme activity assays, will help to profile HCoV-HKU1 and other newly emerging etiologic agents from this group of CoVs.
Supplementary Material
[Supplemental material]
Acknowledgments
We thank K. Y. Yuen for providing the cDNA of HCoV-HKU1 Mpro, as well as Zhiyong Lou and Xiaohang Tong for technical assistance and data collection for HCoV-HKU1 Mpro in complex with N3.
This work was supported by Project 973 of the Ministry of Science and Technology of China (grant numbers 2006CB806503 and 2007CB914301), the National Natural Science Foundation of China (grant numbers 30221003 and 30730022), the Sino-German Center [grant number GZ236(202/9)], the Sino-European Project on SARS Diagnostics and Antivirals (SEPSDA) of the European Commission (grant number 003831), and the Tsinghua University Ph.D. student innovation fund.
Footnotes
▿
Published ahead of print on 18 June 2008.
REFERENCES
- 1.Anand, K., G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr, and R. Hilgenfeld. 2002. Structure of CoV main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 213213-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand, K., J. Ziebuhr, P. Wadhwani, J. R. Mesters, and R. Hilgenfeld. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 3001763-1767. [DOI] [PubMed] [Google Scholar]
- 3.Arden, K. E., M. D. Nissen, T. P. Sloots, and I. M. Mackay. 2005. New human coronavirus, HCoV-NL63, associated with severe lower respiratory tract disease in Australia. J. Med. Virol. 75455-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bastien, N., K. Anderson, L. Hart, P. Van Caeseele, K. Brandt, D. Milley, T. Hatchette, E. C. Weiss, and Y. Li. 2005. Human coronavirus NL63 infection in Canada. J. Infect. Dis. 191503-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosis, S., S. Esposito, H. G. Niesters, E. Tremolati, S. Pas, N. Principi, and A. D. Osterhaus. 2007. Coronavirus HKU1 in an Italian pre-term infant with bronchiolitis. J. Clin. Virol. 38251-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brünger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54905-921. [DOI] [PubMed] [Google Scholar]
- 7.Chang, L. Y., B. L. Chiang, C. L. Kao, M. H. Wu, P. J. Chen, B. Berkhout, H. C. Yang, and L. M. Huang. 2006. Lack of association between infection with a novel human coronavirus (HCoV), HCoV-NH, and Kawasaki disease in Taiwan. J. Infect. Dis. 193283-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiu, S. S., K. H. Chan, K. W. Chu, S. W. Kwan, Y. Guan, L. L. Poon, and J. S. Peiris. 2005. Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin. Infect. Dis. 401721-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi, E. H., H. J. Lee, S. J. Kim, B. W. Eun, N. H. Kim, J. A. Lee, J. H. Lee, E. K. Song, S. H. Kim, J. Y. Park, and J. Y. Sung. 2006. The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000-2005. Clin. Infect. Dis. 43585-592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50760-763. [DOI] [PubMed] [Google Scholar]
- 11.Dominguez, S. R., M. S. Anderson, M. P. Glode, C. C. Robinson, and K. V. Holmes. 2006. Blinded case-control study of the relationship between human coronavirus NL63 and Kawasaki syndrome. J. Infect. Dis. 1941697-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebihara, T., R. Endo, X. Ma, N. Ishiguro, and H. Kikuta. 2005. Detection of human coronavirus NL63 in young children with bronchiolitis. J. Med. Virol. 75463-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 602126-2132. [DOI] [PubMed] [Google Scholar]
- 14.Esper, F., C. Weibel, D. Ferguson, M. L. Landry, and J. S. Kahn. 2006. Coronavirus HKU1 infection in the United States. Emerg. Infect. Dis. 12775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esposito, S., S. Bosis, H. G. Niesters, E. Tremolati, E. Begliatti, A. Rognoni, C. Tagliabue, N. Principi, and A. D. Osterhaus. 2006. Impact of human coronavirus infections in otherwise healthy children who attended an emergency department. J. Med. Virol. 781609-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garbino, J., S. Crespo, J. D. Aubert, T. Rochat, B. Ninet, C. Deffernez, W. Wunderli, J. C. Pache, P. M. Soccal, and L. Kaiser. 2006. A prospective hospital-based study of the clinical impact of non-severe acute respiratory syndrome (non-SARS)-related human coronavirus infection. Clin. Infect. Dis. 431009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerna, G., E. Percivalle, A. Sarasini, G. Campanini, A. Piralla, F. Rovida, E. Genini, A. Marchi, and F. Baldanti. 2007. Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J. Clin. Virol. 38244-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gouet, P., E. Courcelle, D. I. Stuart, and F. Metoz. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15305-308. [DOI] [PubMed] [Google Scholar]
- 19.Granzow, H., U. Meyer, P. Solisch, E. Lange, and D. Fichtner. 1981. Morphology of coronaviruses—electron microscopic demonstration by negative contrast technic of the transmissible gastroenteritis virus of swine. Arch. Exp. Veterinarmed. 35177-186. [PubMed] [Google Scholar]
- 20.Han, T. H., J. Y. Chung, S. W. Kim, and E. S. Hwang. 2007. Human coronavirus-NL63 infections in Korean children, 2004-2006. J. Clin. Virol. 3827-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hök, K. 1990. Demonstration of feline corona virus (FCV) antigen in organs of cats suspected of feline infectious peritonitis (FIP) disease. APMIS 98659-664. [DOI] [PubMed] [Google Scholar]
- 22.Huang, I. C., B. J. Bosch, W. Li, M. Farzan, P. M. Rottier, and H. Choe. 2006. SARS-CoV, but not HCoV-NL63, utilizes cathepsins to infect cells: viral entry. Adv. Exp. Med. Biol. 581335-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ignatov, G., L. Vasileva, and K. Kharalambiev. 1979. Purification of coronaviruses from contaminated cattle feces. Vet. Med. Nauki 1623-27. [PubMed] [Google Scholar]
- 24.Kistler, A., P. C. Avila, S. Rouskin, D. Wang, T. Ward, S. Yagi, D. Schnurr, D. Ganem, J. L. DeRisi, and H. A. Boushey. 2007. Pan-viral screening of respiratory tract infections in adults with and without asthma reveals unexpected human coronavirus and human rhinovirus diversity. J. Infect. Dis. 196817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleywegt, G. J., and T. A. Jones. 1994. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50178-185. [DOI] [PubMed] [Google Scholar]
- 26.Lau, S. K., P. C. Woo, C. C. Yip, H. Tse, H. W. Tsoi, V. C. Cheng, P. Lee, B. S. Tang, C. H. Cheung, R. A. Lee, L. Y. So, Y. L. Lau, K. H. Chan, and K. Y. Yuen. 2006. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 442063-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, W., Z. Shi, M. Yu, W. Ren, C. Smith, J. H. Epstein, H. Wang, G. Crameri, Z. Hu, H. Zhang, J. Zhang, J. McEachern, H. Field, P. Daszak, B. T. Eaton, S. Zhang, and L. F. Wang. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310676-679. [DOI] [PubMed] [Google Scholar]
- 28.Matthews, D. A., P. S. Dragovich, S. E. Webber, S. A. Fuhrman, A. K. Patick, L. S. Zalman, T. F. Hendrickson, R. A. Love, T. J. Prins, J. T. Marakovits, R. Zhou, J. Tikhe, C. E. Ford, J. W. Meador, R. A. Ferre, E. L. Brown, S. L. Binford, M. A. Brothers, D. M. DeLisle, and S. T. Worland. 1999. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 9611000-11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode, p. 307-326. In C. W. Carter, Jr., and R. M. Sweet (ed.), Macromolecular crystallography, part A, vol. 276. Academic Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 30.Pyrc, K., B. Berkhout, and L. van der Hoek. 2007. The novel human coronaviruses NL63 and HKU1. J. Virol. 813051-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sloots, T. P., P. McErlean, D. J. Speicher, K. E. Arden, M. D. Nissen, and I. M. Mackay. 2006. Evidence of human coronavirus HKU1 and human bocavirus in Australian children. J. Clin. Virol. 3599-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storoni, L. C., A. J. McCoy, and R. J. Read. 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60432-438. [DOI] [PubMed] [Google Scholar]
- 33.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vabret, A., J. Dina, S. Gouarin, J. Petitjean, S. Corbet, and F. Freymuth. 2006. Detection of the new human coronavirus HKU1: a report of 6 cases. Clin. Infect. Dis. 42634-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Hoek, L., K. Pyrc, and B. Berkhout. 2006. Human coronavirus NL63, a new respiratory virus. FEMS Microbiol. Rev. 30760-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vijgen, L., E. Keyaerts, P. Lemey, P. Maes, K. Van Reeth, H. Nauwynck, M. Pensaert, and M. Van Ranst. 2006. Evolutionary history of the closely related group 2 coronaviruses: porcine hemagglutinating encephalomyelitis virus, bovine coronavirus, and human coronavirus OC43. J. Virol. 807270-7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, L. F., Z. Shi, S. Zhang, H. Field, P. Daszak, and B. T. Eaton. 2006. Review of bats and SARS. Emerg. Infect. Dis. 121834-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo, P. C., Y. Huang, S. K. Lau, H. W. Tsoi, and K. Y. Yuen. 2005. In silico analysis of ORF1ab in coronavirus HKU1 genome reveals a unique putative cleavage site of coronavirus HKU1 3C-like protease. Microbiol. Immunol. 49899-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo, P. C., S. K. Lau, C. M. Chu, K. H. Chan, H. W. Tsoi, Y. Huang, B. H. Wong, R. W. Poon, J. J. Cai, W. K. Luk, L. L. Poon, S. S. Wong, Y. Guan, J. S. Peiris, and K. Y. Yuen. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79884-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu, P. S., L. Y. Chang, B. Berkhout, L. van der Hoek, C. Y. Lu, C. L. Kao, P. I. Lee, P. L. Shao, C. Y. Lee, F. Y. Huang, and L. M. Huang. 2008. Clinical manifestations of human coronavirus NL63 infection in children in Taiwan. Eur. J. Pediatr. 16775-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue, X., H. Yu, H. Yang, F. Xue, Z. Wu, W. Shen, J. Li, Z. Zhou, Y. Ding, Q. Zhao, X. C. Zhang, M. Liao, M. Bartlam, and Z. Rao. 2008. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 822515-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang, H., M. Bartlam, and Z. Rao. 2006. Drug design targeting the main protease, the Achilles' heel of coronaviruses. Curr. Pharm. Des. 124573-4590. [DOI] [PubMed] [Google Scholar]
- 43.Yang, H., W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei, J. Ziebuhr, R. Hilgenfeld, K. Y. Yuen, L. Wong, G. Gao, S. Chen, Z. Chen, D. Ma, M. Bartlam, and Z. Rao. 2005. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang, H., M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G. F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld, and Z. Rao. 2003. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 10013190-13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zakstel'skaia, L., and A. V. Sheboldov. 1971. New group of viruses: corona viruses. Vestn. Akad. Med. Nauk SSSR. 2664-69. [PubMed] [Google Scholar]
- 46.Zheng, W. X., L. L. Chen, H. Y. Ou, F. Gao, and C. T. Zhang. 2005. Coronavirus phylogeny based on a geometric approach. Mol. Phylogenet. Evol. 36224-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu, R. N., Y. Qian, L. Q. Zhao, J. Deng, F. Wang, and B. Liao. 2006. Human coronavirus-NL63 was detected in specimens from children with acute respiratory infection in Beijing, China. Zhonghua Er Ke Za Zhi 44202-205. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental material]