Distinct Methylation of the Interferon γ (IFN-γ) and Interleukin 3 (IL-3) Genes in Newly Activated Primary CD8+ T Lymphocytes: Regional IFN-γ Promoter Demethylation and mRNA Expression Are Heritable in CD44highCD8+ T Cells (original) (raw)
Abstract
Differential genomic DNA methylation has the potential to influence the development of T cell cytokine production profiles. Therefore, we have conducted a clonal analysis of interferon (IFN)-γ and interleukin (IL)-3 gene methylation and messenger (m)RNA expression in primary CD8+ T cells during the early stages of activation, growth, and cytokine expression. Despite similar distributions and densities of CpG methylation sites, the IFN-γ and IL-3 promoters exhibited differential demethylation in the same T cell clone, and heterogeneity between clones. Methylation patterns and mRNA levels were correlated for both genes, but demethylation of the IFN-γ promoter was widespread across >300 basepairs in clones expressing high levels of IFN-γ mRNA, whereas demethylation of the IL-3 promoter was confined to specific CpG sites in the same clones. Conversely, the majority of clones expressing low or undetectable levels of IFN-γ mRNA exhibited symmetrical methylation of four to six of the IFN-γ promoter CpG sites. Genomic DNA methylation also has the potential to influence the maintenance or stability of T cell cytokine production profiles. Therefore, we also tested the heritability of IFN-γ gene methylation and mRNA expression in families of clones derived from resting CD44lowCD8+ T cells or from previously activated CD44highCD8+ T cells. The patterns of IFN-γ gene demethylation and mRNA expression were faithfully inherited in all clones derived from CD44high cells, but variable in clones derived from CD44low cells. Overall, these findings suggest that differential genomic DNA methylation, including differences among cytokine genes, among individual T cells, and among T cells with different activation histories, is an important feature of cytokine gene expression in primary T cells.
Keywords: interferon, interleukin, CD8+ T cell, DNA methylation, gene expression
Differential cytokine gene expression in activated T lymphocytes plays an important part in the diversification of T cell cytokine production patterns and polarization towards type 1, type 2, and other profiles (1–4). The underlying molecular mechanisms are likely to operate combinatorially, involving a number of pretranscriptional, transcriptional, and posttranscriptional levels of control with multiple interacting factors at each level (5–7).
At the pretranscriptional/transcriptional level, the involvement of genomic DNA methylation in the regulation of differential cytokine gene expression has not yet been widely investigated. In other systems, methylation of the cytosine residue in CpG dinucleotides can be a negative regulatory influence on the expression of various inducible tissue-specific genes (8–10). Off-on effects occur when CpG methylation influences chromatin conformation and accessibility (11, 12). In addition, quantitative influences on gene expression are possible since methylation can differentially inhibit the binding of individual transcription factors, directly via the protruding methyl group or indirectly via methyl-CpG–binding proteins (13–15). Importantly, the methylation status of CpG sites can be heritable, thus contributing to epigenetic regulation of gene expression (16).
Only a few studies have examined the role of DNA methylation in cytokine gene expression in leukocytes. Inhibition of DNA methylation has been used to establish constitutive IL-2 secretion in EL-4 thymoma clones (17). Reduced CpG methylation in the TNF-α promoter has been described in myeloid leukemia cells and differentiated monocyte cell lines expressing TNF-α mRNA (18, 19). Differences in IFN-γ expression among long-term T cell clones, T cell lines, and primary human T cell populations have been found to be associated with differential CpG methylation in this gene (20–22). The latter important findings are supported by data on methylation-mediated alterations in transcription factor–binding to the proximal IFN-γ promoter element (23). Whether heritable methylation patterns in the IFN-γ gene can account for the stability of augmented IFN-γ expression in memory/effector T cells, for example, has not yet been tested.
Until recently, analysis of genomic DNA methylation has generally required relatively large amounts of DNA for methylation-dependent restriction endonuclease digestion followed by Southern blotting– or semi-quantitative PCR– based evaluation of restriction patterns. These approaches have been largely superseded by the development of a PCR- and DNA sequencing–based method for positive display of methylated cytosine residues: bisulfite genomic sequencing (24, 25). This method allows evaluation of the methylation status of all cytosine residues on both DNA strands, regardless of their restriction enzyme recognition context. In addition, PCR amplification of bisulfite-modified DNA from small numbers of cells allows clonal analyses (26).
In this investigation, we have applied bisulfite genomic sequencing to analyze the methylation patterns of the mouse IFN-γ and IL-3 genes during development of short-term CD8+ T cell clones from normal animals. We have also coupled the methylation studies to quantitative competitive PCR (QCPCR)1 analyses of IFN-γ and IL-3 mRNA expression. To our knowledge, this is the first report of this combination of analyses for any cytokine gene in any type of leukocyte. The bisulfite sequencing results extend the previous findings for one CpG site in the IFN-γ promoter to cover all of the other IFN-γ promoter methylation sites, and provide entirely new information for all of the methylation sites in the IL-3 promoter.
The results show that, despite similar distributions and densities of CpG methylation sites, the IFN-γ and IL-3 promoters exhibit differential demethylation in the same T cell clone. The coupled methylation QCPCR results show that demethylation of the IFN-γ and IL-3 genes is intimately associated with differential expression of these two cytokines in activated CD8+ T cell clones. The pattern of IFN-γ promoter demethylation is widespread and compatible with regulatory mechanisms based on altered chromatin conformation. In contrast, the pattern of IL-3 promoter demethylation is more site specific and focussed on sequence elements and transcription factor–binding sites additional to those currently studied. Thus, differential methylation patterns among cytokine genes and among primary T cell clones suggest that DNA methylation may be one of the mechanisms that underpins the generation of diversity in T cell cytokine profiles during primary immune responses. Furthermore, we show that both IFN-γ gene methylation patterns and mRNA expression levels are heritable in the subclones derived from CD44highCD8+ (recently activated or memory/effector) cells, but variable among the offspring derived from CD44lowCD8+ (resting or naive) cells. This indicates that heritable gene methylation patterns correlate with CD44 and IFN-γ expression and thus have the potential to serve as molecular markers of memory/effector T cells. Together, these results suggest a role for gene methylation in establishing and/or maintaining differential cytokine gene expression patterns in primary T cells.
Materials and Methods
Mouse Lymphocyte Preparation.
Specific pathogen–free female C57BL/6 mice (Animal Resources Centre, Murdoch, Western Australia) were used when 6–12 wk old. CD8+ T cells were isolated from pooled brachial, axillary, cervical, inguinal, para-aortic, and mesenteric LNs as previously described (27). In brief, LN cells were stained with PE-conjugated rat anti–mouse CD4 mAb (GK1.5), FITC-conjugated rat anti–mouse CD8 mAb (53.6-7), and 0.5 μg/ml propidium iodide. In some experiments, the staining with anti-CD4 mAb was substituted with biotinylated anti-CD44 mAb (IM7.8.1) followed by PE-conjugated streptavidin. Fluorescence-activated cell sorting was conducted using a FACS®Vantage flow cytometer with Lysis II software (Becton Dickinson, Sunnyvale, CA). Viable CD8+ cells were positively sorted and, upon reanalysis, were >97% pure in all experiments. In some experiments, the CD44low and CD44high subsets of the CD8+ population were also separated by sorting for the lowest or highest 15%, respectively, of the staining distribution. On a four-decade scale, the reanalyzed sorted CD44low cells were contained within the first decade with a mean fluorescence intensity of 4.6, whereas the sorted CD44high cells were within the third decade with a mean intensity of 192. For clonal cultures, single CD8+ T cells were then seeded directly into the wells of prepared (see below) 60-well Terasaki tissue culture plates (Nunc, Roskilde, Denmark) using the automated cell deposition unit of the flow cytometer.
Accessory Cell–free CD8+ T Lymphocyte Culture.
Terasaki and 96-well flat-bottomed tissue culture plates were coated with a mixture of hamster anti–mouse CD3ε mAb (145-2C11) at 10 μg/ml, rat anti–mouse LFA-1 mAb (I21/7.7) at 5 μg/ml, and rat anti–mouse CD8 mAb (53.6-7) at 10 μg/ml diluted in PBS. In the presence of IL-2, this mAb combination activates CD8+ T cells in a TCR-dependent manner without accessory cells, and yields cloning frequencies of 75% for CD8+ cells from LNs of normal mice (27, 28). After 12–16 h at 37°C, the coated plates were washed once with PBS and twice with modified DMEM supplemented with 216 mg/liter l-glutamine, 5 × 10−5 M 2-ME, antibiotics, and 10% heat-inactivated FCS (CSL Ltd., Parkville, Victoria, Australia). Bulk T cell cultures were initiated at 104cells/well in 200 μl of supplemented DMEM with 10% FCS, 600 IU/ml human _r_IL-2 (Cetus Corp., Emeryville, CA), and varying doses of fresh 5-azacytidine (see below). For clonal cultures, after automated deposition of single T cells into Terasaki plates, additional medium was placed in each well to attain final concentrations of 15% FCS and 600 IU/ml IL-2. Subcloning was performed by micromanipulation (27, 29) after 4 d of stimulation and the parent clones and subclones were harvested 2–3 d after micromanipulation.
Secreted Cytokine Assays.
Supernatants harvested from bulk CD8+ T cell cultures after varying periods of stimulation were assayed for IL-3 by FDC-P1 reporter cell assay as previously described in detail (27). Appropriate controls demonstrated that the presence of 5-azacytidine did not interfere with this bioassay at the dilutions where titration end points were determined (data not shown). Supernatant IFN-γ levels were quantitated using ELISA kits (Endogen Inc., Cambridge, MA).
Genomic DNA and Cytoplasmic RNA Extraction and cDNA Synthesis.
Nucleic acids were extracted using a combination of published methods (30–32). After supernatant removal, T cell clones were resuspended in 11 μl of lysis buffer (0.2 × PBS, 20 μg/ml oligo-dT [Boehringer Mannheim, Castle Hill, New South Wales, Australia], 1% NP-40, 4 mM dithiothrietol, 4 U/ml RNasin [Promega Corp., Madison, WI], 100 μg/ml tRNA [_Boehringer Mannheim_]) and snap frozen at −70°C. Nuclei were pelleted and the supernatants were removed for cDNA synthesis while the pellets were processed for DNA extraction. For cDNA synthesis, supernatants were heated to 70°C for 5 min before addition of 14 μl of cDNA buffer (36 mM (NH4)2SO4, 134 mM Tris HCl [pH 9], 0.018% Tween, 8.6 mM MgCl2, 0.7 mM dNTPs, 1.4 mM dithiothrietol, 0.7 U/ml RNasin, and 0.14 U/ml AMV reverse transcriptase [_Promega Corp._]) and incubation at 42°C for 2 h. For genomic DNA isolation, nuclei were resuspended in 100 μl of GIT buffer (4 M guanidinium isothiocyanate, 25 mM sodium acetate, 0.84% 2-ME, 10 μg/ml glycogen [_Boehringer Mannheim_], and 5 μg/ml λ DNA [_Promega Corp._]), and the DNA was precipitated with isopropanol. After centrifugation at 14,000 g for 30 min at 4°C, the precipitates were washed twice with 75% ethanol, air-dried and resuspended in 20 μl of water.
Bisulfite Modification of Genomic DNA.
Genomic DNA was bisulfite-treated using a method optimized for small cell numbers (26). In brief, extracted genomic DNA was sheared by pipetting and then denatured in 0.3 N NaOH for 20 min at 75°C. Fresh 4.8 M sodium metabisulfite (pH 5.0) was prepared by adding 4.55 g of Na2S2O5 and 0.4 ml of 10 N NaOH to 8.2 ml H2O and mixing gently. To each 22-μl sample of denatured genomic DNA, 250 μl of 4.8 M Na2S2O5, 14 μl of fresh 10 mM hydroquinone, and paraffin oil were added and the samples were incubated at 55°C, shielded from light, for 4 h. Modified DNA was then purified using Geneclean® kits (BIO 101, La Jolla, CA), and desulfonated in 0.3N NaOH at 37°C for 20 min. Desulfonated DNA was precipitated with ammonium acetate and ethanol, pelleted, washed with 70% ethanol, and resuspended in 20 μl H2O.
PCR and Sequencing of Bisulfite-modified Genomic DNA.
Primers flanking CpG sites in the mouse IFN-γ and IL-3 promoters and specific for either the coding or noncoding strands of the bisulfite-modified genomic DNA (Table 1) were designed using the OLIGOTM program (Bresatec, Thebarton, South Australia) and the following criteria in addition to those previously reported (25): (a) % G + C > 25%; (b) homopolymer strings <9 bases; (_c_) predicted C to T conversions >25% with a high density of conversions at the 3′ primer end; (d) >50°C predicted annealing temperature; and (e) no false priming within the predicted PCR product. Careful primer design was the most important requirement for specific amplification of bisulfite-modified DNA. Phosphodiester oligodeoxynucleotide primers were obtained commercially (GIBCO BRL, Gaithersburg, MD).
Table 1.
PCR and Sequencing Primers for Bisulfite-modified Genomic DNA
| Primer | Sequence (5′ to 3′) | Position/strand* | Use |
|---|---|---|---|
| MIFNGBIS1 | ggtgtgaagtaaaagtgtttttagagaattttat | −262/coding | 1st and 2nd round PCR, sequencing |
| MIFNGBIS2 | tagagaattttataagaatggtataggtgggtat | −241/coding | 2nd round PCR, sequencing |
| MIFNGBIS3 | ccataaaaaaaaactacaaaaccaaaatacaata | +159/coding | 2nd round PCR, sequencing |
| MIFNGBIS4 | caataacaaccaaaaacaaccataaaaaaaaact | +179/coding | 1st round PCR |
| MIFNGBIS5 | ataaaattatagttgtaatgt | −149/coding | Sequencing |
| MIFNGBIS6 | cttctatctcaaatcaaaaaatc | +77/coding | Sequencing |
| MIFNGBIS7 | gttagaaatagttatgaggaagagttgtaaagtt | +170/noncoding | 1st and 2nd round PCR, sequencing |
| MIFNGBIS8 | taggaggagaagtttagaatttttgttttaagtt | +96/noncoding | 2nd round PCR, sequencing |
| MIFNGBIS9 | acaatttccaacccccaccccaaataatataaaa | −287/noncoding | 2nd round PCR, sequencing |
| MIFNGBIS10 | acaaaaactccctatactatactctataaataaa | −360/noncoding | 1st round PCR |
| MIL3BIS1 | ggctgtatagttagggttaagtttgtgtaagg | −337/coding | 1st round PCR |
| MIL3BIS2 | gatggtaggatgagattttattgtatagaaagt | −306/coding | 2nd round PCR, sequencing |
| MIL3BIS3 | acctacaccaaaccaacctcaaccaattactc | +228/coding | 2nd round PCR, sequencing |
| MIL3BIS4 | cataaaactcactaacttaaaacacctattaaaac | +265/coding | 1st round PCR |
| MIL3BIS5 | aagaagatggtttttgaataa | −203/coding | Sequencing |
| MIL3BIS6 | aacatcaaaaacaaaaacaac | +88/coding | Sequencing |
| MIL3BIS7 | tacataaaaaacccaaatactcaaaaccaaac | −284/noncoding | 1st and 2nd round PCR, sequencing |
| MIL3BIS8 | aataacctttaaataaacaatctttcttcccata | −198/noncoding | 2nd round PCR, sequencing |
| MIL3BIS9 | gttattgattgaagtttggagttttaggtggaa | +122/noncoding | 2nd round PCR, sequencing |
| MIL3BIS10 | ggttaattttagttagttatttattgggagtttt | +217/noncoding | 1st round PCR |
25 μl PCR reactions were assembled using 0.3 U/tube Red Hot polymerase (Advanced Biotechnologies, Leatherhead, Surrey, UK) with the reaction buffer supplied by the manufacturer (1.5 mM MgCl2, 200 μM dNTPs, 100 ng of each PCR primer, 1–4 μl bisulfite-modified DNA, and 50 μl paraffin oil). At least 20% of each set of reactions were negative control tubes lacking template DNA. The reactions were amplified in an Omnigene thermal cycler (Hybaid, Teddington, Middlesex, UK) using a touchdown strategy (decreasing annealing temperatures from 59 to 52°C in 1°C increments with two cycles per increment, and a final annealing temperature of 50°C for 30 cycles). This strategy minimizes early false priming and facilitates specific amplification by low annealing temperature primers during later stages of the program (26, 33). A nested or semi-nested approach was then applied wherein 1 μl of the first-round PCR reactions was reamplified in fresh 25-μl second-round reactions using internal primers (Table 1) with a repeat of the touchdown strategy. First-round negative controls were also reamplified in the second-round to exclude contamination. Second-round PCR reactions were subjected to agarose electrophoresis and the products were harvested and purified using QIAquickTM kits (Qiagen, Hilden, Germany).
Purified PCR products were sequenced directly using the PCR and internal sequencing primers (Table 1) with dye terminator cycle sequencing reagents (Applied Biosystems, Burwood, Victoria, Australia) and analysis on a model 377 ABI automated DNA sequencer. Both strands of each PCR product were sequenced using a total of 2–4 different primers (Table 1), thus verifying the methylation status of each cytosine residue in each genomic DNA strand 2–4 times.
Quantitative Competitive Reverse Transcriptase PCR Assay of Cytokine mRNA Levels.
Competitor plasmids bearing small deletions in the IFN-γ or IL-3 cDNAs were constructed as described elsewhere. For IFN-γ, the competitor lacks 82 bp around an introduced XhoI site compared with the wild-type sequence. The IL-3 competitor lacks 77 bp compared with the wild-type sequence. Construction integrity was verified by DNA sequencing. For QCPCR standards, the competitor plasmids were adjusted to equimolar amounts, serially diluted in 10 mM Tris HCl (pH 8.0), 0.1 mM EDTA, and 1 μg/ml tRNA, and stored in aliquots at −70°C.
25-μl QCPCR reactions comprising 0.3 U Red Hot polymerase with the supplied reaction buffer, 2 mM MgCl2, 200 μM dNTPs, 10 μM of each PCR primer, competitor plasmid dilutions, T cell cDNA, and paraffin oil were assembled in 96-well plates (Becton Dickinson). Separate amplifications for IFN-γ or IL-3 were conducted using the primer pairs MIFNGIN5′ and MIFNGIN3′ or MIL3IN5′ and MIL3IN3′ (34), respectively, with each 3′ primer biotinylated. Reactions were cycled using one cycle of 94°C for 4 min then 60°C for 1 min and 72°C for 1.5 min, followed by 39 cycles of 94°C for 0.5 min then 60°C for 1 min and 72°C for 1.5 min. Each cDNA was tested against fivefold competitor dilutions, ranging over seven (IFN-γ) or five (IL-3) orders of magnitude. To account for RNA extraction and cDNA synthesis variations, separate determinations of the stimulus-resistant T cell–specific CD3ε mRNA (29, 35, 36) were used to correct the cytokine mRNA levels measured. Using these methods, the correlation between clone size (30–300 cells) and CD3ε mRNA levels is routinely high (r >0.75).
Aliquots of QCPCR reactions were separated by electrophoresis to verify appropriate product sizes and estimate titration equivalence points. Additional aliquots were tested by PCR-ELISA (37) for hybridization with oligonucleotide probes specific for the exogenous competitor products or the endogenous intact cytokine cDNA products. In brief, the biotinylated PCR products were diluted in PBS/0.2% Tween 20 (PBST) then bound to streptavidin-coated plates. The bound products were denatured with 50 mM NaOH/2 mM EDTA for 2 min, then incubated with 100 ng/ml FITC-labeled oligonucleotide probe diluted in 6× SSC, 20% formamide, and 1 μg/ml denatured fish sperm DNA for 16 h at 42°C. After four washes with PBST, bound probes were detected with an alkaline phosphatase–conjugated antifluorescein antibody (Boehringer Mannheim) followed by para-nitrophenol colorimetric substrate development and spectrophotometric ELISA read-out. The sensitivity of the QCPCR assay for IFN-γ and IL-3 mRNA was ∼100–500 input competitor molecules by agarose electrophoresis and ∼20–100 input molecules by PCR-ELISA. Amplification rates for the competitor and endogenous PCR products were equivalent, semilogarithmic titration data were linear over >3 orders of magnitude, and probe specificity was high with negligible background or cross-reactivity (data not shown). Accordingly, the competition equivalence points of the exogenous competitor– and endogenous cDNA–derived PCR products were virtually identical when determined by agarose electrophoresis and PCR-ELISA. The equivalence point reproducibility among assays of positive control samples was high, with standard deviations <15% of the means (data not shown).
Results
Potential Methylation Sites in the Mouse IFN-γ and IL-3 Promoters.
Sequence analysis revealed several, mostly clustered, CpG methylation sites in the mouse IFN-γ and IL-3 promoters within ∼300 bp of the transcription start site (Fig. 1). Despite the low overall sequence conservation, 1–4 CpG sites ∼40–60 bases upstream of the transcription start site, were present in both genes of all species. Several of the CpG sites overlapped with the binding sites of transcription factors shown to be involved in the regulation of human IFN-γ or IL-3 promoter activity. The binding of some of these factors, including the cyclic AMP response element binding protein (CREB) and ETS members, has been shown to be directly affected by cytosine methylation (13,23, 38). Of all these CpG sites in these two genes, only the −53 site in the IFN-γ promoter has hitherto been characterized for its methylation status in T cells (21, 22).
Figure 1.
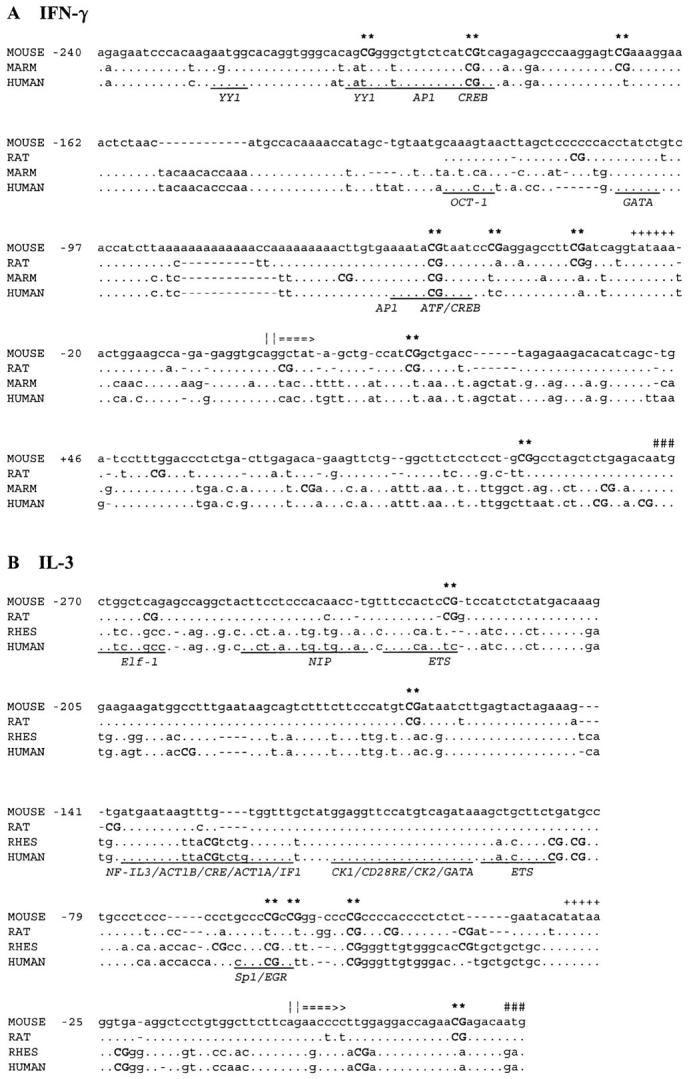
Homology of CpG sites in the IFN-γ (A) and IL-3 (B) genes in different species. The mouse sequence is numbered relative to the transcription start site (||===>). Marmoset (MARM), human, rat, and/or rhesus monkey (RHES) sequences are also shown. The mouse sequence is the reference for the alignments, determined using the FASTA program, with identical residues (.) and gaps (–) indicated. CpG sites (upper case and bold, and asterisked for the mouse sites), TATA motifs (++++), and ATG start codons (###) are also indicated. Potential transcription factor–binding sites defined for the human sequence are underscored and annotated with the sequence element and/or transcription factor acronyms.
Inhibition of DNA Methylation Increases CD8+ T Cell IFN-γ and IL-3 Secretion.
The cytosine methylation inhibitor 5-azacytidine has previously been shown to increase the expression of IFN-γ in long-term T cell clones and lines (21, 39). Given the similar pattern of potential methylation sites in the mouse IFN-γ and IL-3 promoters (Fig. 1), we tested the effect of this inhibitor on IFN-γ and IL-3 expression in primary mouse CD8+ T cells activated in vitro in a TCR-dependent, accessory cell–independent, solid-phase mAb system. Treatment with 5-azacytidine increased IFN-γ expression up to 25-fold and IL-3 expression up to 14-fold in a dose-dependent manner, with different dose optima for the two cytokines (Fig. 2). Within the dose range shown, T cell proliferation was unaffected but 5-azacytidine doses >8 μM resulted in inhibition of proliferation and cell death and consequently cytokine levels below those of untreated cultures (data not shown). Increased cytokine secretion in the 5-azacytidine–treated cultures was maximal on day 2 (IFN-γ) or 3 (IL-3), with trends towards plateau levels at day 3 or 4, respectively (Fig. 2). These kinetics represent an acceleration of the expression of IFN-γ and IL-3 in this culture system (27, 28). Thus, these results, in support of the sequence analysis above, suggested a potential role for DNA methylation in regulation of expression of both the IFN-γ and IL-3 genes in primary mouse CD8+ T cells.
Figure 2.


5-azacytidine treatment increases IFN-γ and IL-3 secretion by primary mouse CD8+ T lymphocytes. CD8+ T cells from normal C57BL/6 mouse LNs were activated in low density in vitro cultures using solid-phase mAb against CD3ε, CD8, and LFA-1 in the presence of IL-2 and varying concentrations of 5-azacytidine for the indicated periods of time. Cumulative secreted cytokine levels were quantitated by ELISA (A, IFN-γ) or reporter cell bioassay (B, IL-3). The results shown are representative of three independent experiments.
Bisulfite Genomic DNA Sequencing of the Mouse IFN-γ and IL-3 Promoters.
To study comprehensively the potential cytosine methylation sites in the mouse IFN-γ and IL-3 promoters, we used bisulfite genomic DNA sequencing to display the methylation status of all of the cytosines on both DNA strands of the promoter regions analyzed (Figs. 1 and3). In both genes, cytosines in a CpG sequence context were virtually the only nonmodified (i.e., methylated) cytosines detected. Methylation at CpNpG sites (40) was encountered at <1% of all potential CpNpG sites and in <5% of all samples. Consequently, >99% of non-CpG cytosines exhibited cytosine to thymidine conversion, thus internally controlling for the efficiency of the bisulfite modification and for PCR and sequencing artifacts. Repeated bisulfite modifications, PCR, and direct PCR product sequencing of control mixes of methylated and nonmethylated molecules, similar to experiments reported previously (25, 41), confirmed that this method reproducibly displays the predominant methylation pattern of a heterogeneous population of molecules (data not shown). Coincident clear cytosine and thymidine peaks (Fig. 3), indicative of possible mixed or transitional CpG methylation status, were seen at a frequency of <13% of the sites examined in the panel of clones (see below). The methylation of CpG bps was usually symmetrical with identical patterns on the coding and noncoding DNA strands in 80% of the sites tested. This result, derived from independent PCR reactions on separate aliquots of modified genomic DNA from each clone, also affirmed the reproducibility of the analyses. Symmetrical methylation of all of the predicted CpG sites in the coding and noncoding strands of the IFN-γ and IL-3 promoters was observed in the control mouse FDC-P1 myelomonocytic cell line that does not express IFN-γ or IL-3 (42, 43), in bulk populations of normal nonstimulated spleen cells, and in nonstimulated LN CD8+ T cells (Fig. 4 and data not shown).
Figure 3.
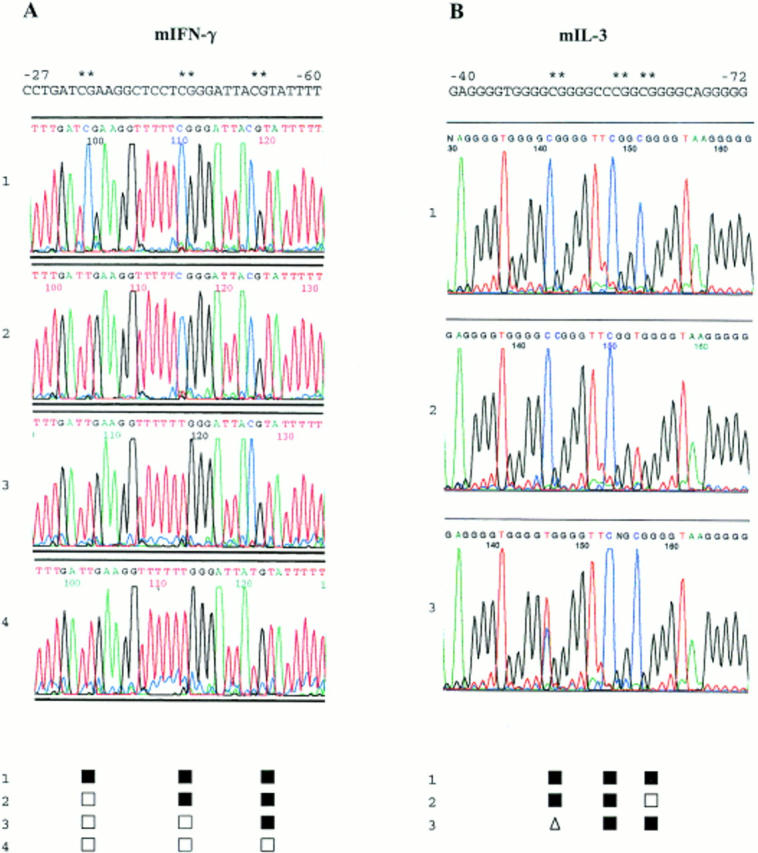
Primary bisulfite genomic DNA sequencing data. Nuclear DNA from clonal cultures of primary mouse CD8+ T cells was purified, bisulfite modified, amplified, and sequenced directly using dye terminator chemistry and automated fluorescent sequence analysis. Sequence results for the noncoding strands of the IFN-γ promoter between bases −27 and −60 in four different clones are shown in the left panel. Sequence results for the noncoding strands of the IL-3 promoter between bases −40 and −72 in three different clones are shown in the right panel. Methylated cytosines are displayed by retained blue cytosine peaks while nonmethylated cytosines are converted by bisulfite modification and PCR to red thymidine peaks. These peaks were scored for each clone and are shown below the chromatograms by the following symbols: ▪, methylated; □, demethylated. The presence of clear coincident cytosine and thymidine peaks, e.g., at position −52 in the third IL-3 sequence, was scored: ▵, partially methylated.
Figure 4.

Differential methylation of the IFN-γ and IL-3 genes in CD8+ T cell clones and independence of clone size. CpG sites in the two promoters are depicted schematically and numbered as described in Fig.1. Asterisks above the −53 site in the IFN-γ promoter indicate the only CpG site hitherto characterized in T cells. Methylation of the coding (upper line of symbols) and noncoding (lower line of symbols) strands for the IFN-γ and IL-3 promoters are shown for the control cell line FDCP1 and 16 CD8+ T cell clones ranging in size from 35 to >256 cells. Methylation patterns were scored as described in Fig. 3.
Differential Methylation of the IFN-γ and IL-3 Promoters in Activated CD8+ T Cells.
Single CD8+ T cells were cultured with anti-CD3ε, anti-CD8, and anti–LFA-1 mAbs in the presence of IL-2 for 4–5 d, until clones of 32–256 cells had formed. The methylation status of the IFN-γ and IL-3 promoters in individual clones was then examined by bisulfite genomic sequencing. In both the IFN-γ and IL-3 promoters, clonal heterogeneity was seen with >30 distinct patterns recorded in the panel of 40 clones (Fig. 4 and data not shown). The methylation patterns in the two genes were not related to each other or to clone size, which is an approximation of division number. In the IFN-γ promoter, the patterns ranged from the two extremes of methylation of nearly all sites on the coding and noncoding strands (e.g., Fig. 4, clone 1414), to demethylation of all 14 of these cytosines (e.g., Fig. 4, clone 1386). Contrastingly, in the IL-3 promoter, demethylation of all sites was never seen. Instead, site-specific differences predominated, ranging from high-frequency demethylation of one or both of the coding or noncoding strand cytosines at the −164 CpG site, to low-frequency demethylation of both cytosines at the −52 site. In both the IFN-γ and IL-3 genes, demethylation of the CpG site immediately downstream of the transcription start site was infrequent. It was concluded that there was differential methylation of these two cytokine genes in the same T cell clone, and that the methylation patterns of the two genes were not closely related to clone size. Therefore, these results were consistent with a clonally variable, division-independent demethylation process, affecting these two cytokine genes differentially.
Quantitation of Early IFN-γ and IL-3 mRNA Expression in Clonal Cultures of Activated Primary CD8+ T cells.
To disclose links between promoter methylation patterns and transcriptional activity, the bisulfite genomic methylation analysis above was coupled to QCPCR measurements of cytokine mRNA levels. In this way, parallel steady state genomic DNA and cytoplasmic mRNA data were derived for each clone at the time of cell lysis.
The QCPCR results for the panel of clones showed that, by 4-5 d of activation, approximately half of the clones had detectable levels of IFN-γ and/or IL-3 mRNA that ranged over at least two (IL-3) or four (IFN-γ) orders of magnitude (Fig. 5). The frequency of IFN-γ mRNA expression after 4–5 d was higher than that of IL-3 mRNA at these early times. In comparison, clones in the 4–32 cell stage analyzed after 2–3 d of stimulation expressed IFN-γ mRNA at a lower frequency, and IL-3 mRNA expression was usually undetectable (data not shown). Correlations between mRNA levels and clone size (Fig. 5, A and B) were not significant (r <0.5). However, the probability of detection of IFN-γ or IL-3 mRNA was significantly higher in clones larger than 256 cells, and clones with detectable IL-3 mRNA almost always coexpressed IFN-γ mRNA (Fig. 5 C).
Figure 5.
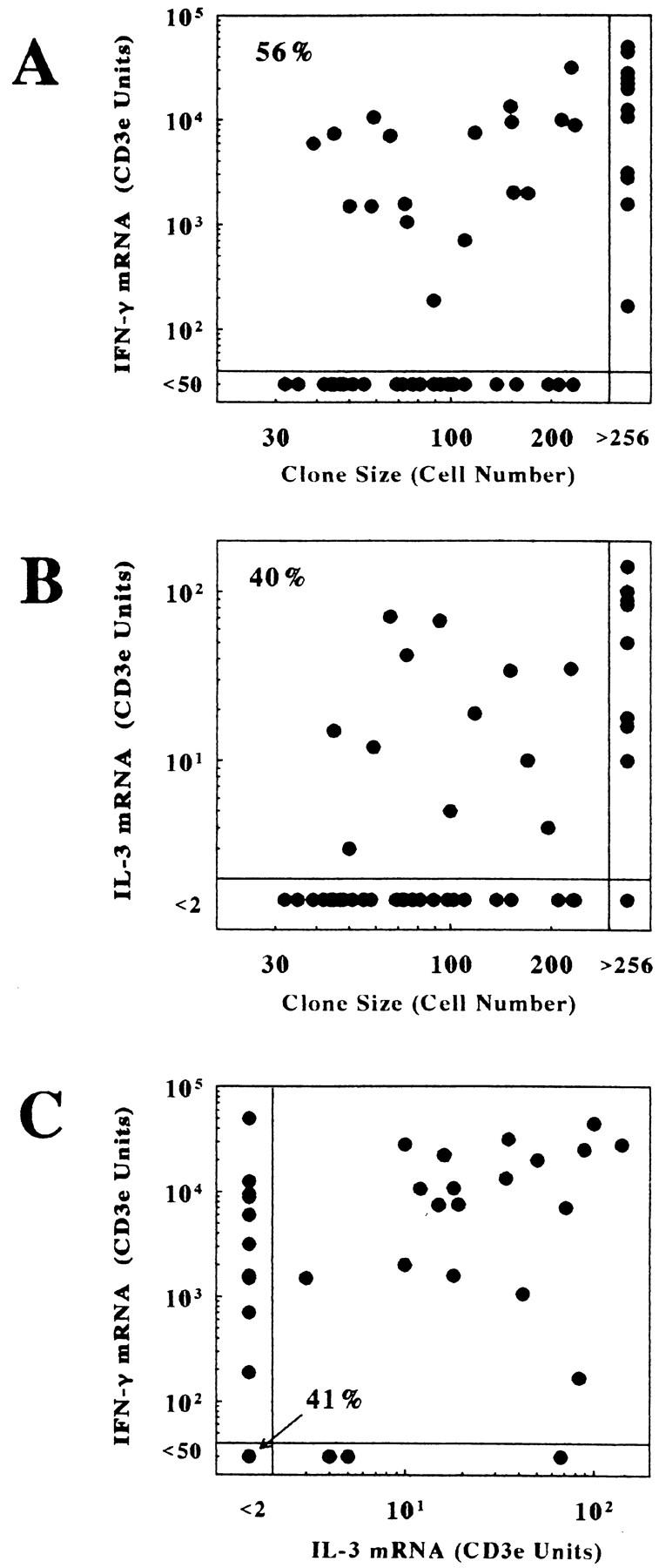
Quantitation of IFN-γ and IL-3 mRNA levels in a panel of CD8+ clones by competitive PCR after 4–5 d of stimulation. Levels of mRNA were determined by QCPCR and corrected for CD3ε mRNA levels as described in Materials and Methods. A shows relative IFN-γ mRNA levels in relation to clone size. B shows relative IL-3 mRNA levels in relation to clone size in the same set of clones. C shows the relationship between IFN-γ and IL-3 expression levels for each clone in the panel. The results for 53 clones are shown.
These data concurred with many of the previous reported findings on the kinetics and relative levels of IFN-γ and IL-3 protein expression by primary mouse CD8+ T cell clones (27, 28, 44, 45), and supported the choice of this time frame for study of primary CD8+ T cells during an early phase of in vitro development, when about half the clones had initiated cytokine mRNA expression.
Regional Demethylation of the IFN-γ Promoter Is Associated with High-Level IFN-γ mRNA Expression in Activated CD8+T Cells.
Comparing the methylation and mRNA data for individual clones revealed a striking overall association between demethylation of the IFN-γ promoter and expression of mRNA (Fig. 6). Clones lacking detectable levels of IFN-γ mRNA exhibited relatively dense methylation of 8–13 of the CpG cytosines examined on the two DNA strands of the IFN-γ promoter. In contrast, the majority of IFN-γ mRNA-positive clones exhibited multiple and sometimes uninterrupted demethylation of all CpG sites in the IFN-γ promoter between positions −210 and +1. A small number of clones (e.g., clones 1417, 1427, and 1408, Fig. 6) expressed relatively low levels of IFN-γ mRNA and retained symmetrical or hemi-methylation of most of the CpG sites.
Figure 6.
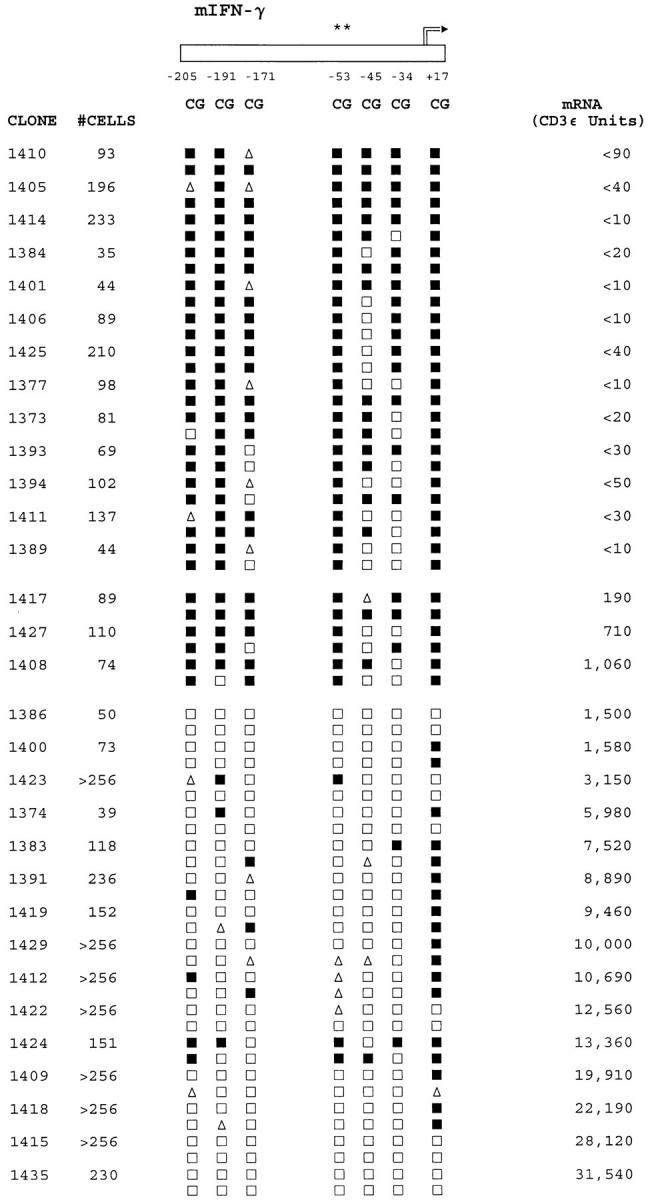
Regional IFN-γ promoter methylation patterns are related to IFN-γ mRNA expression levels in CD8+ T cells. Clone sizes and methylation patterns are shown as described in the legend to Fig. 4. Levels of mRNA are also shown as described in the legend to Fig. 5. The results depicted are representative of a panel of 40 clones for which complete DNA and mRNA data were obtained. Clones with undetectable mRNA levels are ranked by methylation pattern, whereas mRNA-positive clones are ranked by mRNA level.
Although these promoter-wide or regional demethylation patterns were pronounced, clone- and site-specific differences across the IFN-γ promoter were also evident. For the panel as a whole, symmetrical or hemi-demethylation of the −45 and −34 CpG sites occurred with the highest frequency, whereas the site at position +17 exhibited the lowest frequency of demethylation, yet the status of these sites was not related to IFN-γ mRNA levels. In contrast, the −205, −191, and −53 CpG sites were symmetrically methylated in most IFN-γ mRNA-negative clones, and their demethylation was closely correlated with expression of >103 U of IFN-γ mRNA. As shown in Fig. 1 A, these three CpG sites are adjacent to conserved activator protein 1 (AP-1) and activating transcription factor (ATF)/CREB transcription factor–binding sites.
Site-specific Demethylation of the IL-3 Promoter Is Associated with IL-3 mRNA Expression in Activated CD8+ T Cells.
In the same panel of CD8+ T cell clones, the patterns of DNA methylation and mRNA expression for IL-3 were distinctly different from IFN-γ. Although clones lacking detectable levels of IL-3 mRNA again exhibited relatively dense methylation of 8–12 of the CpG cytosines examined on both strands of the IL-3 promoter from bases −270 to +30, widespread demethylation of multiple CpG sites across the IL-3 promoter was never seen (Fig. 7). Among the IL-3 mRNA-negative clones, all of the CpG sites except position −164 exhibited high-frequency methylation. In the IL-3 mRNA-positive clones, symmetrical demethylation of the −164 CpG site was frequent (10 out of 12 clones, or 83%) and was linked to a slightly higher frequency of symmetrical or hemi-demethylation of the −62 CpG site. However, symmetrical demethylation of the −164 CpG site in the IL-3 promoter also occurred in a small proportion (6 out of 18, or 33%) of IL-3 mRNA-negative clones. As shown in Fig. 1 B, the −164 CpG site abuts a GATA transcription factor–binding site that is not conserved in primates, whereas the −62 site overlaps with a partially conserved Sp1-binding site.
Figure 7.

Site-specific IL-3 promoter methylation patterns are related to IL-3 mRNA expression levels in CD8+ T cells. Clone sizes, methylation patterns, and mRNA levels for the panel of clones are shown as described in the legend to Fig. 6.
Heritability of IFN-γ Promoter Methylation Patterns in CD44 Subsets of CD8+ T Cells.
We have previously presented evidence for the inheritance of cytokine production patterns in families of subclones of CD4+ and CD8+ T cells from normal mice (27, 29). Therefore, we tested whether the IFN-γ gene methylation patterns and levels of mRNA expression described above were heritable characteristics of CD8+ T cell clones, and whether this would vary for T cells at different stages of in vivo differentiation. To this end, LN cells from normal mice were sorted into CD8+CD44high or CD8+CD44low subpopulations to enrich for previously activated or resting T cells, respectively (46– 48). Single cell cultures were initiated as described above and, after 4 d, the resultant parent clones were subcloned by micromanipulation. Parent and progeny subclones were harvested 2–3 d later, thus allowing clonal expansion for two to six divisions under the stimulation conditions used.
Fig. 8 shows that, in families derived from CD44highCD8+ T cells, most of the CpG sites in the IFN-γ promoter were demethylated in all of the parent clones and their subclones, all of which expressed high levels of IFN-γ mRNA. These results corroborated our earlier results on the close association between regional promoter demethylation and high IFN-γ mRNA expression (Fig. 6), and are consistent with the well-described augmented IFN-γ expression status of CD44high T cells (49, 50). In contrast, more variability in IFN-γ gene methylation and expression was seen among and within the families derived from CD44low cells. Interestingly, this variability was less marked for the CpG sites at positions −205, −191, and −53, whose status was shown (Fig. 6 and see above) to correlate most closely with IFN-γ mRNA expression. As a result, the previously determined relationships between methylation pattern and mRNA expression (Fig. 6) were largely maintained in this panel. In addition, partial methylation at certain sites was perpetuated in some related siblings. This may reflect the maintenance of a balance between methylation and demethylation similar to a previously reported phenomenon for the adenosine phosphoribosyltransferase gene in a panel of teratocarcinoma stem cell lines (51).
Figure 8.


Heritability of IFN-γ promoter methylation patterns and IFN-γ mRNA expression levels in families of CD8+ T cells. CD8+ LN T cells from normal mice were separated by FACS® into CD44low and CD44high subpopulations, and seeded by automated deposition as single cells into stimulation cultures for 4 d as described in Materials and Methods. Parental CD8+ T cell clones were then subcloned by micromanipulation, with harvesting of progeny subclones after 2–3 d of further growth. Families of clones derived from CD44low cells are shown in A, whereas those derived from CD44high cells are shown in B. Each family has a separate letter code with the parent clone (0) and sibling subclones (1–6) numbered. The parent clone for family C did not survive micromanipulation. Methylation patterns and mRNA levels for the families of clones are shown as described in the legends to Figs. 4 and 5.
These data indicate that demethylation of multiple sites in the IFN-γ promoter is different in clones derived from the CD44high and CD44low subsets of mouse CD8+ T cells, and that these differences, in conjunction with quantitatively distinct levels of IFN-γ expression, can be passed on to the progeny of individual T cell clones.
Discussion
The above results demonstrate that cytokine gene methylation exhibits many of the features of a mechanism that can regulate two T cell cytokine genes differentially, with heterogeneity between individual T cell clones, and with heritability in differentiated T cells with the CD44high phenotype. Therefore, these findings are germane to our understanding of the diversification of cytokine profiles in primary T cells, the maintenance of established cytokine expression patterns in differentiated T cells, and the molecular mechanisms involved in cytokine gene regulation.
This investigation was initiated upon finding that there was remarkable similarity in the potential CpG methylation sites of the mouse IFN-γ and IL-3 promoters, and that many of these sites overlapped with binding sites for transcription factors known to be directly affected by CpG methylation (Fig. 1; references 13, 38). These sequence-based inferences were supported by experiments with the cytosine methylation inhibitor 5-azacytidine, in which 5-azacytidine increased and accelerated both IL-3 and IFN-γ expression in bulk cultures of CD8+ T cells (Fig. 2). This result confirms previously reported results for the effect of 5-azacytidine on IFN-γ expression in long-term mouse CD4+ T cell clones or primary human CD4+ T cells (21,22), and extends them to both IFN-γ and IL-3 in primary mouse CD8+ T cells.
However, 5-azacytidine is a toxic agent and it is difficult to distinguish its direct methylation-mediated effects from other indirect actions, particularly in proliferating polyclonal cell populations. It is also thought that it acts via division-dependent interference in maintenance methylation, which is unlikely to resemble normal demethylation mechanisms (8, 9, 52). Therefore, we undertook experiments to characterize IFN-γ and IL-3 gene methylation, demethylation, and mRNA expression events as they occurred during the in vitro development of cytokine-producing T cells. Our approach incorporated use of (a) primary mouse CD8+ T cells analyzed during the early stages of growth and cytokine expression; (b) solid-phase anti-CD3ε, anti-CD8, and anti–LFA-1 mAb to stimulate high frequencies of T cells from normal mice in a TCR- and accessory molecule–dependent manner; (c) accessory cell–free conditions to eliminate non–T cell sources of DNA and mRNA; (d) clonal cultures to disclose the degree of heterogeneity obscured in bulk culture studies, allow visual quantitation of cell proliferation, and permit analyses of heritability by subcloning; (e) bisulfite genomic DNA sequencing to map comprehensively the sites of cytosine methylation on both strands of the IFN-γ and IL-3 promoters in small numbers of cells; and (f) sensitive competitive PCR-based quantitation of mRNA levels in small numbers of cells. This approach was thus designed to reveal the full range of cellular and molecular potentialities of the primary developing CD8+ T cell clones under study.
Clonal heterogeneity, in the levels and combinations of cytokines produced, is a prominent feature of cytokine expression in primary T cells (2, 4, 53). Broad heterogeneity in IFN-γ and IL-3 gene CpG methylation patterns was seen in the panel of clones examined in this study (Figs. 4,6, and 7). Indeed, when the methylation patterns of all the sites in the two genes were considered together (e.g., Fig.4), nearly every clone in the panel possessed a unique pattern. These results showed that there was not a uniform demethylation pathway or program for each gene in all cells, or for two sequences with similar CpG arrays such as the IFN-γ and IL-3 promoters, even when both cytokines were expressed (e.g., Figs. 6 and 7; clones 1383, 1412, 1415, and 1418). Instead, differential demethylation of the two induced genes occurred in the same T cell clone. Nevertheless, the methylation status of individual CpG sites studied here was not entirely autonomous or independent since apparent linkages between the status of neighboring CpG sites in the IFN-γ and IL-3 promoters were seen (Figs. 6 and 7). This may, in part, reflect maintenance methylase activity since this enzyme, while restoring symmetrical methylation to hemi-methylated CpG sites, has also been found to initiate de novo methylation of neighboring symmetrically nonmethylated CpG cytosines (54, 55).
Regional or site-specific demethylation of the IFN-γ or IL-3 promoters, respectively, was associated with different levels of the mRNAs derived from these genes. In particular, there was a striking association between promoter-wide demethylation of the IFN-γ gene and expression of high levels of IFN-γ mRNA (Fig. 6). However, large differences in demethylation frequencies of CpG sites in IFN-γ mRNA-negative clones also occurred and revealed a subset of CpG sites, including the previously characterized −53 site, whose methylation was closely associated with the absence of detectable IFN-γ mRNA. Similarly, site-specific effects in the IL-3 promoter were pronounced with most of the demethylation activity focussed on the −164 CpG site, which was almost invariably demethylated in IL-3 mRNA-positive clones (Fig. 7). A small proportion of clones possessing a demethylated −164 CpG site lacked detectable IL-3 mRNA, suggesting that demethylation at this site may precede mRNA expression and/or that demethylation of this site alone may be either unnecessary or insufficient for IL-3 gene expression in some T cells.
Together, the above findings justify several levels of studies required to elucidate the role of DNA methylation in the regulation of cytokine gene expression in T cells. The dissection of methylation-mediated regulatory mechanisms is an intensely active field that is currently in a state of flux, with many long-standing views being reconsidered in light of unanticipated new information (10, 15, 52, 55). We believe that similar reconsideration arises from comprehensive bisulfite sequencing methylation analysis of clonal cultures of primary cells.
First, the above mapping in primary T cells of the site-specific CpG differences that correlate with specific levels of mRNA expression, and with known transcription factor binding sites, has identified a revised set of sites and transcription factors to be probed for direct methylation-dependence. One study has been conducted for the −53 CpG site in the human IFN-γ promoter where methylation was shown to affect directly the binding of the factors CREB, ATF-2, and the c-jun component of AP-1 (23). The results presented here indicate that similar studies are warranted for the −205 and −191 CpG sites in the mouse IFN-γ promoter and the −164 and −62 sites in the mouse IL-3 promoter. Moreover, the transcription factors to be assayed can be provisionally extended to include two members of the GATA family, YY1 (which can interact directly with CREB and ATF members; reference 56) and Sp1. The factors YY1 and Sp1 are reportedly unaffected by CpG methylation in some sequence contexts (13, 38), but, to our knowledge, the sensitivity of members of the GATA family has not yet been reported. Conversely, recent description of the unanticipated creation of an AP-1 factor–binding site by CpG methylation within a nonconsensus AP-1 sequence (57) underlines the importance of further analyses at this level.
Second, similar transcription factor–binding studies will also need to be conducted in the presence or absence of proteins such as MeCP2 and histone H1 (13, 14). These well-characterized ubiquitous proteins can bind methylated cytosines in any sequence context and indirectly inhibit transcription factor–binding in a competitive manner or via active repression (15). These effects are methylation density dependent (58, 59) and compatible with the clustered distribution of the key CpG sites in the IFN-γ and IL-3 promoters.
The third level of studies requires analyses of the effect of gene methylation on chromatin structure. Several recent reports show that a medium to high density of methylated CpG sites, in the presence of MeCP2 or histones, can promote the formation of a higher-order nucleosome-like structure that is inaccessible to DNase I and is transcriptionally silent (15, 60, 61). The regional patterns of methylation and promoter-wide demethylation that we have described here for the IFN-γ promoter are also compatible with the involvement of such a mechanism in the switch between undetectable and high-level IFN-γ mRNA expression.
The results of these three substantial levels of study will determine to what extent DNA methylation-mediated mechanisms are necessary and/or sufficient for regulation of expression of different cytokine genes in T cells. Moreover, the results we have presented here suggest that conclusions on the necessity or sufficiency of individual mechanisms may need to be qualified according to the level of gene expression and the phenotype of the T cell (see below). The potential regulatory diversity arising from this pretranscriptional/transcriptional level is likely to be great given the differences described here between two cytokine genes with similar CpG site densities and distributions, the broad range of promoter CpG methylation patterns in other cytokine genes expressed by T cells, and the possible contributions from methylation in distant enhancer elements (reference 62 and Fitzpatrick, D.R., unpublished data).
Equally important is investigation of the pathways that regulate methylation and demethylation processes. Two reports have linked activation of the Ras signaling pathway to DNA methylation or demethylation activities (63, 64), and a role for the transcription factor NF-κB in demethylation of the immunoglobulin κ gene in a B cell line has also been shown (65). Since the former pathway would tend to link cytokine gene demethylation to activation of the TCR, whereas the latter molecular mechanism may be more dependent on costimulatory signals (66), it will be pivotal to determine which signals are involved in the induction of cytokine expression in T cells. We have recently shown that activation of the extracellular signal-regulated kinase branch of the Ras-activated cascade is required for maximal TCR-mediated IFN-γ and IL-3 expression in primary mouse CD8+ T cells (67, 68). Although these bulk culture studies are not directly comparable to the clonal studies described here, it will be interesting to determine whether this signaling pathway exerts some of its downstream effects through cytokine gene demethylation.
The importance of the above multi-faceted investigations is emphasized by the experiments herein, which characterize the heritability of IFN-γ methylation patterns and mRNA expression levels in T cells at different stages of in vivo differentiation: previously activated (also referred to as memory/effector) CD44high cells and resting (or naive) CD44low cells. In clones derived from CD44high T cells, regional IFN-γ promoter demethylation and high-level expression of IFN-γ mRNA were inherited characteristics shared by all parent and progeny clones. In contrast, in clones derived from CD44low T cells, IFN-γ gene methylation and expression patterns were variable both between and within different families of clones. Therefore, over the two to six cell division time frame of these studies, the IFN-γ promoter CpG methylation pattern and the fidelity of its vertical transfer were closely associated with both the initial surface marker phenotype of the parental T cells and with the final IFN-γ expression phenotype of both the parent and progeny clones.
This result is germane to four sets of previously published work. First, it confirms and extends the report that the −53 CpG site in the human IFN-γ promoter may be preferentially demethylated in the recently activated CD45RAlow/CD45ROhigh subset of primary human CD4+T cells (22). Second, it corroborates our previous findings on similar kinetic patterns of GM-CSF and IL-3 expression in the progeny of some CD4+ T cells (27), and on the retention of IFN-γ mRNA expression by the progeny of some CD8+ T cells (29). Third, it compares favorably with the previously described flexibility of resting or “naive” CD44low T cells in contrast to the more restricted differentiative potential of previously activated or “memory/effector” CD44high T cells (4, 29, 50). Fourth, it supports earlier studies on the heritability of methylated CpG sites in long-term cell lines transfected with plasmid or viral DNA (69,70), and the variable fidelity of this process in some primary cells (51, 71, 72). Our data add to each of these aspects by demonstrating differential short-term heritability of multiple contiguous methylation sites in the IFN-γ gene, an endogenous induced gene, and linking this difference to the surface marker phenotypes of two subpopulations of primary CD8+ T lymphocytes.
Thus, differences in cytokine gene methylation represent a molecular correlate of other well-characterized markers of T cell differentiation. Whether these correlations are preserved over longer periods and in vivo, for example during antigen stimulation followed by a return to quiescence (48), merits additional study. Whether DNA methylation can reflect, establish, and/or maintain the commitment or reversibility of T cell cytokine expression patterns, particularly under polarizing priming or restimulation conditions (73), is also worthy of further investigation.
Acknowledgments
We thank Grace Chojnowski for expert FACS® assistance; David Sester and Daphne Macaranas for contributions to the early stages of this work; Macky Edmundson for invaluable help with automated DNA sequence analysis; and Dr. Sue Clark for constructive comments on the manuscript.
This work was supported by the National Health and Medical Research Council of Australia, the Queensland Cancer Fund, the Clive and Vera Ramaciotti Foundations, and the Queensland Institute of Medical Research Trust.
Abbreviations used in this paper
AP
activator protein
ATF
activating transcription factor
CREB
cyclic AMP response element binding protein
QCPCR
quantitative competitive PCR
References
- 1.Bucy RP, Karr L, Huang G, Li J, Carter D, Honjo K, Lemons JA, Murphy KM, Weaver CT. Single cell analysis of cytokine gene coexpression during CD4+T-cell phenotype development. Proc Natl Acad Sci USA. 1995;92:7565–7569. doi: 10.1073/pnas.92.16.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelso A. Th1 and Th2 subsets: paradigms lost? . Immunol Today. 1995;16:372–379. doi: 10.1016/0167-5699(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 3.Kelso A, Groves P, Troutt AB, Francis K. Evidence for the stochastic acquisition of cytokine profile by CD4+ T cells activated in a T helper type 2-like response in vivo. . Eur J Immunol. 1995;25:1168–1175. doi: 10.1002/eji.1830250506. [DOI] [PubMed] [Google Scholar]
- 4.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–791. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 5.Ernst P, Smale ST. Combinatorial regulation of transcription I: general aspects of transcriptional control. Immunity. 1995;2:311–319. doi: 10.1016/1074-7613(95)90139-6. [DOI] [PubMed] [Google Scholar]
- 6.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy JEG, Kollmus H. Cytoplasmic mRNA-protein interactions in eukaryotic gene expression. Trends Biochem Sci. 1995;20:191–197. doi: 10.1016/s0968-0004(00)89006-4. [DOI] [PubMed] [Google Scholar]
- 8.Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev. 1991;55:451–458. doi: 10.1128/mr.55.3.451-458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird A. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 10.Siegfried Z, Cedar H. DNA methylation: a molecular lock. Curr Biol. 1997;7:R305–R307. doi: 10.1016/s0960-9822(06)00144-8. [DOI] [PubMed] [Google Scholar]
- 11.Keshet I, Lieman-Hurwitz J, Cedar H. DNA methylation affects the formation of active chromatin. Cell. 1986;44:535–543. doi: 10.1016/0092-8674(86)90263-1. [DOI] [PubMed] [Google Scholar]
- 12.Kass SU, Goddard JP, Adams RLP. Inactive chromatin spreads from a focus of methylation. Mol Cell Biol. 1993;13:7372–7379. doi: 10.1128/mcb.13.12.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 14.McArthur M, Thomas JO. A preference of histone H1 for methylated DNA. EMBO (Eur Mol Biol Organ) J. 1996;15:1705–1714. [PMC free article] [PubMed] [Google Scholar]
- 15.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 16.Hendrich BD, Willard HF. Epigenetic regulation of gene expression: the effect of altered chromatin structure from yeast to mammals. Hum Mol Genet. 1995;4:1765–1777. doi: 10.1093/hmg/4.suppl_1.1765. [DOI] [PubMed] [Google Scholar]
- 17.Ballas ZK. The use of 5-azacytidine to establish constitutive interleukin 2-producing clones of the EL4 thymoma. J Immunol. 1984;133:7–9. [PubMed] [Google Scholar]
- 18.Kochanek S, Radbruch A, Tesch H, Renz D, Doerfler W. DNA methylation profiles in the human genes for tumor necrosis factors α and β in subpopulations of leukocytes and in leukemias. Proc Natl Acad Sci USA. 1991;88:5759–5763. doi: 10.1073/pnas.88.13.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takei S, Fernandez D, Redford A, Toyoda H. Methylation status of 5′-regulatory region of tumor necrosis factor-α gene correlates with differentiation stages in monocytes. Biochem Biophys Res Commun. 1996;220:606–612. doi: 10.1006/bbrc.1996.0450. [DOI] [PubMed] [Google Scholar]
- 20.Fukunaga R, Matsuyama M, Okamura H, Nagata K, Nagata S, Sokawa Y. Undermethylation of the interferon-γ gene in human T cell lines and normal T lymphocytes. Nucleic Acids Res. 1986;14:4421–4436. doi: 10.1093/nar/14.11.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard J, Penix L, Wilson CB, Melvin AJ, McGurn M, et al. Differentiation of T helper phenotypes by analysis of the methylation state of the IFN-γ gene. J Immunol. 1994;153:3603–3610. [PubMed] [Google Scholar]
- 22.Melvin AJ, McGurn ME, Bort SJ, Gibson C, Lewis DB. Hypomethylation of the interferon-γ gene correlates with its expression by primary T-lineage cells. Eur J Immunol. 1995;25:426–430. doi: 10.1002/eji.1830250218. [DOI] [PubMed] [Google Scholar]
- 23.Penix LA, Sweetser MT, Weaver WM, Hoeffler JP, Kerppola TK, Wilson CB. The proximal regulatory element of the IFN-γ promoter mediates selective expression in T cells. J Biol Chem. 1996;271:31964–31972. doi: 10.1074/jbc.271.50.31964. [DOI] [PubMed] [Google Scholar]
- 24.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDonald LE, Kay GF. Methylation analysis using bisulfite genomic sequencing: application to small numbers of intact cells. Biotechniques. 1997;22:272–274. doi: 10.2144/97222bm16. [DOI] [PubMed] [Google Scholar]
- 27.Fitzpatrick DR, Kelso A. Dissociated expression of granulocyte-macrophage CSF and IL-3 in short-term T cell clones from normal mice. J Immunol. 1995;155:5140–5150. [PubMed] [Google Scholar]
- 28.Maraskovsky E, Troutt AB, Kelso A. Co-engagement of CD3 with LFA-1 or ICAM-1 adhesion molecules enhances the frequency of activation of single murine CD4+ and CD8+T cells and induces synthesis of IL-3 and IFN-γ but not IL-4 or IL-6. Int Immunol. 1992;4:475–485. doi: 10.1093/intimm/4.4.475. [DOI] [PubMed] [Google Scholar]
- 29.Kelso A, Groves P. A single peripheral CD8+T cell can give rise to progeny expressing type 1 and/or type 2 cytokines and can retain its multipotentiality through many cell divisions. Proc Natl Acad Sci USA. 1997;94:8070–8075. doi: 10.1073/pnas.94.15.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciulla TA, Sklar RM, Hauser SL. A simple method for DNA purification from peripheral blood. Anal Biochem. 1988;174:485–488. doi: 10.1016/0003-2697(88)90047-4. [DOI] [PubMed] [Google Scholar]
- 31.Brady G, Iscove NN. Construction of cDNA libraries from single cells. Methods Enzymol. 1993;225:611–623. doi: 10.1016/0076-6879(93)25039-5. [DOI] [PubMed] [Google Scholar]
- 32.Smith KGC, Nossal GJV, Tarlinton DM. FAS is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Proc Natl Acad Sci USA. 1995;92:11628–11632. doi: 10.1073/pnas.92.25.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Don RH, Cox PT, Wainwright BT, Baker K, Mattick JS. “Touchdown” PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991;19:4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Troutt AB, Kelso A. Enumeration of lymphokine mRNA-containing cells in vivo in a murine graft-versus-host reaction using the PCR. Proc Natl Acad Sci USA. 1992;89:5276–5280. doi: 10.1073/pnas.89.12.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrick T, Qian L, Wilkinson MF. TCR-α mRNA accumulation does not dictate cell surface TCR/ CD3 expression. Mol Immunol. 1992;29:531–536. doi: 10.1016/0161-5890(92)90011-l. [DOI] [PubMed] [Google Scholar]
- 36.Jarnicki A, Fitzpatrick DR, Robinson BWS, Bielefeldt-Ohmann H. Altered CD3 chain and cytokine gene expression in tumor infiltrating T lymphocytes (TIL) during the development of mesothelioma. Cancer Lett. 1996;103:1–9. doi: 10.1016/0304-3835(96)04178-x. [DOI] [PubMed] [Google Scholar]
- 37.Hockett RD, Janowski KM, Bucy RP. Simultaneous quantitation of multiple cytokine mRNAs by RT-PCR utilizing plate based EIA technology. J Immunol Methods. 1995;187:273–285. doi: 10.1016/0022-1759(95)00195-5. [DOI] [PubMed] [Google Scholar]
- 38.Gaston K, Fried M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res. 1995;23:901–909. doi: 10.1093/nar/23.6.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrar WL, Ruscetti FW, Young HA. 5-azacytidine treatment of a murine cytotoxic T cell line alters interferon-γ gene induction by interleukin-2. J Immunol. 1985;135:1551–1554. [PubMed] [Google Scholar]
- 40.Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat Genet. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- 41.Paul CL, Clark SJ. Cytosine methylation: quantitation by automated sequencing and GENESCAN™ analysis. Biotechniques. 1996;21:126–133. doi: 10.2144/96211rr04. [DOI] [PubMed] [Google Scholar]
- 42.Kelso A. Frequency analysis of lymphokine-secreting CD4+ and CD8+T cells activated in a graft-versus-host reaction. J Immunol. 1990;145:2167–2176. [PubMed] [Google Scholar]
- 43.Kelso A, Troutt AB. Survival of the myeloid progenitor cell line FDC-P1 is prolonged by interferon-γ or interleukin-4. Growth Factors. 1992;6:233–242. doi: 10.3109/08977199209026930. [DOI] [PubMed] [Google Scholar]
- 44.Fong TA, Mosmann TR. Alloreactive murine CD8+T cell clones secrete the Th1 pattern of cytokines. J Immunol. 1990;144:1744–1752. [PubMed] [Google Scholar]
- 45.Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271–279. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 46.Akbar A, Salmon M, Janossy G. The synergy between naive and memory T cells during activation. Immunol Today. 1991;12:184–188. doi: 10.1016/0167-5699(91)90050-4. [DOI] [PubMed] [Google Scholar]
- 47.Sprent J, Tough DF. Lymphocyte life-span and memory. Science. 1994;265:1395–1400. doi: 10.1126/science.8073282. [DOI] [PubMed] [Google Scholar]
- 48.Pihlgren M, Dubois PM, Tomkowiak M, Sjogren T, Marvel J. Resting memory CD8+T cells are hyperreactive to antigenic challenge in vitro. J Exp Med. 1996;184:2141–2151. doi: 10.1084/jem.184.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehlers S, Smith KA. Differentiation of T cell lymphokine gene expression: the in vitro acquisition of T cell memory. J Exp Med. 1991;173:25–36. doi: 10.1084/jem.173.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swain S, Croft M, Dubey C, Haynes L, Rogers P, Zhang X, Bradley LM. From naive to memory T cells. Immunol Rev. 1996;150:143–167. doi: 10.1111/j.1600-065x.1996.tb00700.x. [DOI] [PubMed] [Google Scholar]
- 51.Turker MS, Swisshelm K, Smith AC, Martin GM. A partial methylation profile for a CpG site is stably maintained in mammalian tissues and cultured cell lines. J Biol Chem. 1989;264:11632–11636. [PubMed] [Google Scholar]
- 52.Weiss A, Keshet I, Razin A, Cedar H. DNA demethylation in vitro: involvement of RNA. Cell. 1996;86:709–718. doi: 10.1016/s0092-8674(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 53.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2, and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 54.Christman JK, Sheikhnejad G, Marasco CJ, Sufrin JR. 5-methyl-2′-deoxycytidine in single-stranded DNA can act in cis to signal de novo DNA methylation. Proc Natl Acad Sci USA. 1995;92:7347–7351. doi: 10.1073/pnas.92.16.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tollefsbol TO, Hutchinson CA. Control of methylation spreading in synthetic DNA sequences by the murine DNA methyltransferase. J Mol Biol. 1997;269:494–504. doi: 10.1006/jmbi.1997.1064. [DOI] [PubMed] [Google Scholar]
- 56.Zhou Q, Gedrich RW, Engel DA. Transcriptional repression of the c-fos gene by YY1 is mediated by a direct interaction with ATF/CREB. J Virol. 1995;69:4323–4330. doi: 10.1128/jvi.69.7.4323-4330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tulchinsky EM, Georgiev GP, Lukanidin EM. Novel AP-1 binding site created by DNA-methylation. Oncogene. 1996;12:1737–1745. [PubMed] [Google Scholar]
- 58.Boyes J, Bird A. Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. EMBO (Eur Mol Biol Organ) J. 1992;11:327–333. doi: 10.1002/j.1460-2075.1992.tb05055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsieh C-L. Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994;14:5487–5494. doi: 10.1128/mcb.14.8.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davey C, Pennings S, Allan J. CpG methylation remodels chromatin structure in vitro. J Mol Biol. 1997;267:276–288. doi: 10.1006/jmbi.1997.0899. [DOI] [PubMed] [Google Scholar]
- 61.Kass SU, Landsberger N, Wolffe AP. DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol. 1997;7:157–165. doi: 10.1016/s0960-9822(97)70086-1. [DOI] [PubMed] [Google Scholar]
- 62.Osborne CS, Vadas MA, Cockerill PN. Transcriptional regulation of mouse granulocyte-macrophage colony-stimulating factor/IL-3 locus. J Immunol. 1995;155:226–235. [PubMed] [Google Scholar]
- 63.MacLeod AR, Rouleau J, Szyf M. Regulation of DNA methylation by the Ras signaling pathway. J Biol Chem. 1995;270:11327–11337. doi: 10.1074/jbc.270.19.11327. [DOI] [PubMed] [Google Scholar]
- 64.Szyf M, Theberge J, Bozovic V. Ras induces a general DNA demethylation activity in mouse embryonal P19 cells. J Biol Chem. 1995;270:12690–12696. doi: 10.1074/jbc.270.21.12690. [DOI] [PubMed] [Google Scholar]
- 65.Kirillov A, Kistler B, Mostoslavsky R, Cedar H, Wirth T, Bergman Y. A role for NF-κB in B-cell-specific demethylation of the Igκ locus. Nat Genet. 1996;13:435–441. doi: 10.1038/ng0895-435. [DOI] [PubMed] [Google Scholar]
- 66.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 67.Egerton M, Fitzpatrick DR, Catling AD, Kelso A. Differential activation of T cell cytokine production by the extracellular-signal regulated kinase (ERK) signalling pathway. Eur J Immunol. 1996;26:2279–2285. doi: 10.1002/eji.1830261002. [DOI] [PubMed] [Google Scholar]
- 68.Egerton M, Fitzpatrick DR, Kelso A. Activation of the extracellular signal-regulated kinase (ERK) pathway is differentially required for T cell antigen receptor-stimulated production of six cytokines in normal T lymphocytes. Int Immunol. 1998;10:223–229. doi: 10.1093/intimm/10.2.223. [DOI] [PubMed] [Google Scholar]
- 69.Wigler M, Levy D, Perucho M. The somatic replication of DNA methylation. Cell. 1981;24:33–40. doi: 10.1016/0092-8674(81)90498-0. [DOI] [PubMed] [Google Scholar]
- 70.Stein R, Gruenbaum Y, Pollack Y, Razin A, Cedar H. Clonal inheritance of the pattern of DNA methylation in mouse cells. Proc Natl Acad Sci USA. 1982;79:61–65. doi: 10.1073/pnas.79.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldstein S, Shmookler RJ, Reis Methylation patterns in the gene for the alpha subunit of chorionic gonadotropin are inherited with variable fidelity in clonal lineages of human fibroblasts. Nucleic Acids Res. 1985;13:7055–7064. doi: 10.1093/nar/13.19.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yen PH, Mohandas T, Shapiro LJ. Stability of DNA methylation of the human hypoxanthine phosphoribosyltransferase gene. Somat Cell Mol Genet. 1986;12:153–161. doi: 10.1007/BF01560662. [DOI] [PubMed] [Google Scholar]
- 73.Trinchieri G, Peritt D, Gerosa F. Acute induction and priming for cytokine production in lymphocytes. Cytokine Growth Factor Rev. 1996;7:123–132. doi: 10.1016/1359-6101(96)00018-4. [DOI] [PubMed] [Google Scholar]