Regional CNS responses to IFN-γ determine lesion localization patterns during EAE pathogenesis (original) (raw)
Abstract
The localization of inflammatory foci within the cerebellum is correlated to severe clinical outcomes in multiple sclerosis (MS). Previous studies of experimental autoimmune encephalomyelitis (EAE), a model of MS, revealed distinct clinical outcomes correlated with the capacity of the animal to produce IFN-γ. Outcomes were linked to localization of inflammatory cells in either the spinal cord (wild type [WT]) or the cerebellum and brain stem (IFN-γ deficient). We demonstrate, using an adoptive transfer system, that the ability of the central nervous system (CNS) to sense pathogenic T cell–produced IFN-γ during EAE initiation determines the sites of CNS pathogenesis. Transfer of WT Th1 cells into IFN-γ receptor–deficient mice results in pathogenic invasion of the brain stem and cerebellum with attendant clinical symptoms, which are identical to the disease observed after transfer of IFN-γ–deficient T cells to WT hosts. Inflammation of the spinal cord associated with classical EAE is abrogated in both IFN-γ–deficient systems. Cotransfer of CNS antigen-specific WT Th1 cells with IFN-γ–deficient T cells is sufficient to restore spinal cord invasion and block cerebellar and brain stem invasion. These data demonstrate that interaction between IFN-γ and host CNS cells during the initiation of EAE can selectively promote or suppress neuroinflammation and pathogenesis.
The clinical course and prognosis of multiple sclerosis (MS) and other encephalitic diseases is highly variable between patients. In MS, both symptoms and prognosis are correlated with the inflammation of specific regions of the central nervous system (CNS). Clinical studies have found that disease affecting the cerebellum has a particularly poor prognosis and rapid progression (1–5). Previous work in the well studied mouse MS model experimental autoimmune encephalomyelitis (EAE) demonstrated that antigen and genetic background influence the distribution of lesions and clinical signs (6–8).
Recent work demonstrated that neuroinflammation is associated with a “conversation” or exchange of signals between the inflammatory cells and cells in the CNS (9). “Classical” or “typical” EAE is dominated by clinical signs associated with inflammatory lesions in the white matter of the spinal cord (10). Further, in classical EAE, myelin-specific Th1 cells can effectively penetrate the parenchyma of the spinal cord but not the cerebrum, brain stem, or cerebellum (11). Instead, in the cerebellum, there is perivascular monocyte and T cell accumulation with little attendant pathogenesis as indicated by either histological analysis or clinical symptoms (8).
However, experiments with a TCR transgenic animal found that the clinical symptoms of spontaneous disease changed from classical to atypical when the transgenic TCR was bred to the IFN-γ–null background (12). The site of pathology was shifted from the spinal cord to the cerebellum and brain stem, resulting in disturbances in balance and coordination, suggesting that characteristics of the initiating T cells other than antigen specificity play an important role in directing the site of pathogenesis (7).
Several previous models in which pathogenesis was redirected to the cerebellum and brainstem involved cells from an IFN-γ–null background (7, 12). These studies postulated that the absence of IFN-γ's negative effect on lymphocyte proliferation or IFN-γ's effects on antigen presentation or sensitivity to apoptosis may explain the importance of the IFN-γ in directing regional pathogenesis. In this paper, we have explored two competing explanations: first, IFN-γ production may directly influence the capacity of particular areas of the CNS to support inflammation and pathogenesis; or second, the effects of IFN-γ on the lineage commitment of cells produced by immunization with CFA may determine the capacity of cells to form lesions at specific anatomical locations. The latter hypothesis overlaps with the antigen presentation hypothesis but also includes the possibility that the newly described Th17 lineage may be involved, as IFN-γ has been demonstrated to be a negative factor in Th17 development (13–16). The possibility that increased levels of IL-17 production were involved was supported by a recent study that suggested a significant role for Th17 cells in determination of lesion localization (17).
To address these possibilities, we have produced myelin-specific CD4+ T cell lines from WT and IFN-γ–deficient mice and expanded these lines in vitro under either Th1 or Th17 polarizing conditions, before transferring these cells to examine the pathological and clinical characteristics of neurological disease induced in either WT or IFN-γ receptor (IFN-γR)–deficient mice. Our results indicate that it is the absence of IFN-γ rather than increased numbers of IL-17–producing cells that accounts for pathogenesis in the cerebellum and brainstem.
RESULTS
Increased numbers of IL-17–producing cells are not sufficient to drive atypical EAE
We generated cell lines specific for the MOG35-55 peptide in the context of I-Ab in both WT and IFN-γ–deficient mice to examine the mechanisms involved in the induction of atypical EAE previously observed in IFN-γ–deficient mice. We used the adoptive transfer system both to allow dissection of the components involved in IFN-γ signaling during EAE and to avoid use of the exogenous inflammatory mediators (mycobacteria and pertussis toxin) used to induce active EAE.
As previously reported, we found that transfer of IFN-γ–deficient Th1-polarized CNS antigen-specific activated CD4+ T cells resulted in an atypical clinical course of EAE (7, 12). The most common clinical symptoms noted were vertigo/dysequilibrium, as measured by a mouse's tendency to lean, turn, and/or roll to one side, and ataxia (Table I). Some mice also demonstrated symptoms of limb dysfunction associated with dystonia of the tail and limbs rather than the flaccid paralysis associated with classical EAE (Table I). Mice that received WT Th1 (IL12 treated) displayed classical EAE symptoms of ascending paralysis (Table I). The ascending paralysis was associated with monocytic inflammation of white matter tracts in the spinal cord, with negligible parenchymal infiltration of the cerebellum (Fig. 1 A).
Table I.
Pathogenic IFN-γ–deficient T cells induce atypical EAE
| Injected cells | WT-IL12–treatedcells | WT-IL23–treatedcells | IFN-γ KO-IL12–treated cells | IFN-γ KO-IL23–treatedcells |
|---|---|---|---|---|
| Incidence of disease | 5/5 | 5/5 | 14/15 | 15/15 |
| Incidence of limb dysfunction | 5/5 | 5/5 | 5/15 | 2/15 |
| Incidence of flaccid paralysis | 5/5 | 5/5 | 0/15 | 0/15 |
| Incidence of dystonia | 0/5 | 0/5 | 5/15 | 2/15 |
| Mean day of onset of clinical signs of limb dysfunction | 7.6 ± 0.6 | 20.8 ± 1.5 | 13.4 ± 4.2 | 9.0 ± 0 |
| Mean peak limb dysfunction clinical score | 2.5 ± 0 | 3.8 ± 0.5 | 1.5 ± 0.7 | 1.0 ± 0 |
| Incidence of non-classical EAE | 0/5 | 0/5 | 14/15 | 15/15 |
| Mean day of onset of clinical signs of non-classical EAE | NA | NA | 14.4 ± 4.2 | 13.1 ± 3.4 |
| Vertigo | 0/5 | 0/5 | 11/14 | 12/15 |
| Ataxia | 0/5 | 0/5 | 9/14 | 9/15 |
Figure 1.
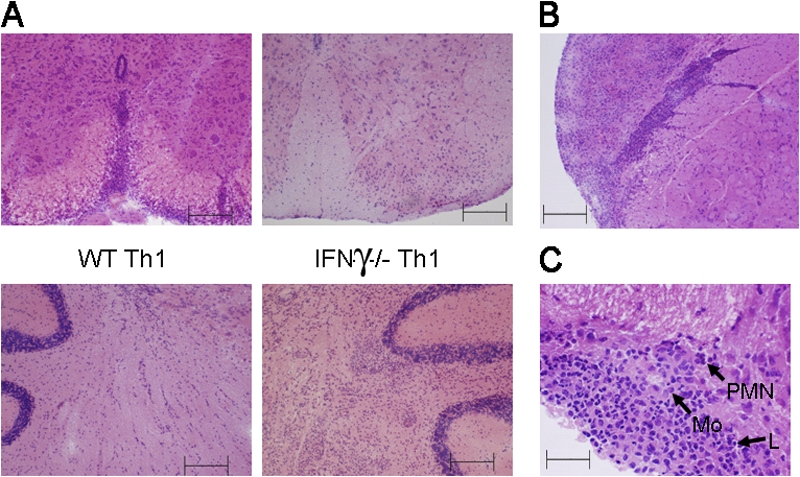
Adoptive transfer of Th1-polarized T cell lines generated in IFN-γ–deficient mice results in atypical CNS inflammation. MOG35-55-specific T cell lines were generated in either C57BL/6 (WT Th1) or IFN-γ–deficient (IFN-γ−/− Th1) mice. T cells were i.v. injected into WT mice at 5 × 106 cells/mouse. (A) 17 d after injection, spinal cord (top) and cerebellum (bottom) were collected and stained with hematoxylin and eosin (H&E) to reveal inflammation. (B) Brain stem sections from mice that received IFN-γ−/− Th1 were stained with H&E to reveal inflammation. (C) A higher magnification of brain stem sections stained with H&E was used to examine infiltrating cell composition. Arrows mark polymorphonuclear cells (PMN), monocytes (Mo), and lymphocytes (L). All slides shown are representative of sections taken from 12 mice per group over six separate experiments. Bars: (A and B) 200 μm; (C) 50 μm.
The induction of atypical EAE symptoms was associated with severe, often laterally asymmetrical, inflammation of the cerebellum and brain stem with facial and vestibulocochlear nerve involvement (Fig. 1, A and B). The inflammatory infiltrate consisted of mostly mononuclear cells, containing lymphocytes, monocytes, and macrophages, with a small number of accompanying granulocytic cells (Fig. 1 C and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20080155/DC1). In contrast, very little inflammation was found in the spinal cords of mice that received IFN-γ–deficient myelin/oligodendrocyte glycoprotein (MOG)–specific T cells (Fig. 1 A).
Previous work had suggested that the adjustment in regional specificity of encephalitogenic T cells may directly relate to changes in T cell lineage associated with the absence of IFN-γ during the initial activation of naive myelin-specific T cells (7). Recent studies have outlined the complicated relationship between IFN-γ and IL-17 lineage commitment (13–16). Specifically, previous studies demonstrated a marked increase in IL-17–producing cells differentiated under IFN-γ–deficient conditions. We hypothesized that an increase in IL-17–producing cells associated with IFN-γ deficiency was responsible for the change in cell localization during EAE. To address this hypothesis we produced MOG35-55 peptide-specific T cell lines from both WT and IFN-γ–deficient mice and cultured the lines under conditions previously reported to polarize cells to either Th1 or Th17 phenotypes.
It is clear that immunization with CFA produces a population of cells skewed toward the Th1 phenotype (Fig. 2, WT cells expanded in IL-2 alone). However, Th17 conditions (IL-23, IL-6, and TGF-β) increase the fraction of cells expressing IL-17, especially IFN-γ IL-17 double producers. As expected, MOG-specific T cell lines produced from IFN-γ–deficient mice contained greater numbers of IL-17–producing cells than Th1 lines produced from WT mice (Fig. 2). However, IFN-γ–deficient lines cultured under Th1 polarizing conditions contained numbers of IL-17–producing T cells far lower than the numbers of IL-17–producing cells present in WT cell lines developed under Th17 polarizing conditions (Fig. 2). An increase in IL-17–producing cells to levels greater than that associated with IFN-γ deficiency was not sufficient to induce changes in encephalitogenic cell localization equivalent to those associated with IFN-γ deficiency (Table I). Instead, WT T cells polarized to produce IL-17–induced clinical symptoms identical to those produced by WT cells cultured under Th1 polarizing conditions, albeit with delayed onset (Table I). Further, in vitro polarization played no role in determining the clinical outcome of transferred IFN-γ–deficient cell lines, with equivalent atypical clinical manifestations observed in both IFN-γ–deficient cell lines regardless of previous conditioning (Table I).
Figure 2.

T cell lines treated with IL-23 produce high levels of IL-17 MOG-specific T cell lines were generated in either C57BL/6 (WT) or IFN-γ–deficient (IFN-γ−/−) mice. T cell lines were produced in the presence either of IL-12 or IL-23+TGF-β+IL-6+IL-1 as described in Materials and methods. Cells were tested for cytokine production after activation with PMA and Ionomycin. Data shown were previously gated by forward and side scatter to include lymphocytes. The data shown are representative of lines developed in four separate experiments.
Cotransferred Th1 cells prevent cerebellar disease in a dose-dependent manner
As the clinical outcomes of classical and atypical disease appeared to be mutually exclusive, we wished to examine the relative dominance of the trafficking patterns associated with both WT Th1 cells and IFN-γ–deficient cells. To do so, we mixed MOG35-55-specific encephalitogenic cell lines from IFN-γ–deficient and WT mice at varying ratios and examined the mixed cell populations for their capacity to induce atypical and/or classical EAE clinical symptoms. Cotransfer of WT and IFN-γ–deficient MOG-specific Th1-polarized cells at a 1:1 ratio resulted in induction of classical EAE in the majority of mice treated (Table II). Histological examination revealed no difference in cell localization between mice with classical EAE that received either 1:1 mixed transfer populations or WT Th1 cells alone (Fig. 3). Cotransfer of WT non-CNS antigen-specific (anti-OVA) Th1-polarized cells and IFN-γ–deficient MOG-specific Th1-polarized cells at a 1:1 ratio resulted in atypical disease, demonstrating that antigen-specific signaling of transferred Th1 cells was required to determine the site of lesion localization (Table II). Thus, IFN-γ production in a large fraction of CNS antigen-specific transferred cells is sufficient to localize inflammatory infiltration to the spinal cord and prevent inflammation of the cerebellum.
Table II.
IFN-γ production in a fraction of pathogenic T cells prevents development of atypical EAE
| Transferred cells | Incidence of nonclassical disease | Incidence of classical EAE | Incidence of both diseases |
|---|---|---|---|
| WT Th1 | 0/25 | 21/25 | 0/25 |
| IFNγ−/− Th1 | 22/25 | 0/25 | 0/25 |
| WT Th1 + IFNγ−/− Th1 (1:1) | 2/25 | 24/25 | 2/25 |
| OVA Th1 + IFNγ−/− Th1 (1:1) | 17/17 | 0/17 | 0/17 |
| WT Th1 + IFNγ−/− Th1 (0.1:1) | 11/15 | 1/15 | 0/15 |
| WT Th1 + IFNγ−/− Th1 (0.2:1) | 19/30 | 16/30 | 6/30 |
Figure 3.

IFN-γ production in a fraction of pathogenic T cells protects the cerebellum from inflammation. C57BL6/J mice received either 5 × 106 MOG35-55-specific Th1 cells generated in C57BL6/J (WT Th1; A and B), 5 × 106 MOG35-55-specific Th1 cells generated in IFN-γ–deficient (IFN-γ KO) mice (C and D), 5 × 106 WT and 5 × 106 IFN-γ KO (E and F), or 106 WT and 5 × 106 IFN-γ KO (G–J) as a single i.v. injection. 17 d after injection and after development of classical EAE (A, B, and E–H), nonclassical EAE (C and D), or both classical and nonclassical disease (I and J), spinal cord and cerebellum were collected and stained with H&E to reveal inflammation. Representative slides from five independent experiments are shown. Bars, 200 μm.
We further explored the threshold of cotransferred Th1 pathogenic cells necessary to determine lesion localization by administering populations of WT Th1 and IFN-γ–deficient MOG-specific T cells mixed at lower ratios. Reduction of the ratio of Th1/IFN-γ–deficient cells to 1:10 largely ablated the capacity of Th1 cells to prevent inflammation of the cerebellum (Table II). Interestingly, injection of Th1/IFN-γ–deficient cells at a 1:5 ratio gave mixed clinical results, with some mice demonstrating classical EAE symptoms, some mice displaying atypical symptoms, and a fraction of mice that exhibited both sets of clinical symptoms (Table II). Immunohistochemical analysis of the cerebellum and spinal cord revealed severe inflammation in the spinal cord of mice with classical symptoms and lesions within the cerebellum of mice that exhibited atypical disease, with infiltration in both tissues of mice that demonstrated both classical and atypical symptoms (Fig. 3). Perivascular noninfiltrating lesions were also observed in the cerebellum of mice that received a 1:5 ratio of Th1/IFN-γ–deficient pathogenic cells but did not demonstrate symptoms of atypical disease. This suggested that the reduced lesion area was insufficient to drive atypical clinical disease. Thus, although reduced numbers of IFN-γ–producing invading cells were sufficient to generate inflammation within the spinal cord, the numbers of Th1 cells present were insufficient to completely prevent development of lesions within the cerebellum. These data demonstrate that the capacity of IFN-γ–producing cells to induce inflammation within the spinal cord and prevent inflammation within the cerebellum are separable functions, with different thresholds of Th1 numbers necessary for operation.
IFN-γ signaling determines CNS lesion localization
As lineage commitment appeared to play less of a role in determining lesion localization than the cells' capacity to produce IFN-γ, we hypothesized that IFN-γ signaling itself was responsible for the differences in clinical course observed, independent of the effects IFN-γ has on T cell differentiation and function. To address this hypothesis, we examined the capacity of WT MOG-specific encephalitogenic T cells to induce classical and/or atypical clinical symptoms in an IFN-γR–deficient host. These experiments allowed us to determine if the observed differences in cell trafficking were caused by changes in T cell selection and/or activation or by disruption of cytokine signaling during the invasion process itself.
After transfer of WT MOG-specific pathogenic T cells, IFN-γ receptor (IFN-γR)–deficient mice developed an atypical disease identical to that observed with transfer of IFN-γ–deficient effector T cells into WT hosts (Table III). In addition to the appearance of identical clinical symptoms, transfer of WT encephalitogenic T cells resulted in histopathology very similar to that seen after transfer of IFN-γ–deficient cells into WT hosts (Fig. 4). Thus, the IFN-γ–IFN-γR interactions involved in determining regions of inflammatory pathology within the CNS occur long after T cell lineage commitment. These data indicate that the determination of regional inflammatory sites most likely occurs during the invasion process itself and involves input from the site of tissue inflammation.
Table III.
Host expression of IFN-γR determines clinical outcome after adoptive transfer of CNS pathogenic T cells
| Mouse genotype | C57BL6/J | IFN-γR KO |
|---|---|---|
| Incidence of clinical signs | 18/20 | 19/19 |
| Incidence of clinical signs of limb dysfunction | 18/20 | 14/19 |
| Mean day of onset of clinical limb dysfunction | 7 ± 1 | 10 ± 2 |
| Incidence of flaccid paralysis | 18/20 | 0/19 |
| Incidence of dystonia | 0/20 | 14/19 |
| Mean peak limb dysfunction clinical score | 2.6 ± 1 | 1.2 ± 0.6 |
| Incidence of clinical signs of nonclassical EAE | 0/20 | 19/19 |
| Mean day of onset of nonclassical signs of EAE | NA | 10 ± 1 |
| Vertigo/dysequilibrium | NA | 19/19 |
| Ataxia | NA | 19/19 |
Figure 4.

Deficiency of either IFN-γ or IFN-γR results in inflammation of the cerebellum after adoptive transfer of CNS pathogenic T cells. MOG35-55-specific T cell lines were generated in either C57BL/6 (WT Th1) or IFN-γ–deficient (IFN-γ KO Th1) mice in the presence of IL-12. T cells were i.v. injected into either C57BL/6 (WT) or IFN-γR–deficient (IFN-γR KO) mice at 5 × 106 cells/mouse. 17 d after injection, spinal cord and cerebellum were collected and stained with H&E to reveal inflammation. Perivascular lesions and invasive parenchymal lesions are indicated with an arrow and the letters P and I, respectively. Representative slides from three independent experiments are shown. Bars, 200 μm.
The induction of atypical disease in IFN-γR–deficient mice allowed us to examine the roles of the radiosensitive cells of the hematopoetic system and the radioresistant cells of the CNS in induction of typical and atypical EAE disease using BM chimeras. Examination of the chimeric mice revealed severe symptoms of both typical and atypical EAE in mice lacking expression of IFN-γR in either radiosensitive cells of the hematopoetic system or radioresistant cells of the CNS (Table S1, available at http://www.jem.org/cgi/content/full/jem.20080155/DC1).
We then used real-time PCR to examine the cerebellum and spinal cord of mice with either classical or atypical EAE for tissue expression of a variety of molecules previously implicated in CNS autoimmunity (Fig. 5). Global differences in cerebellar expression of the IFN-γ–inducible chemokines CXCL9 and CXCL10 were observed when tissue from typical and atypical EAE were compared (Fig. 5). In addition there were significant reductions in the production of IFN-γ in mice that received IFN-γ–deficient cells and reduction in the production of TNF-α in IFN-γR–deficient mice (Fig. 5).
Figure 5.

CNS expression of EAE-associated inflammatory proteins changes in the absence of an IFN-γ signal MOG35-55-specific T cell lines were generated in either C57BL/6 or IFN-γ–deficient mice and polarized to a Th1 phenotype. T cells were injected IV into either C57BL/6 or IFN-γR–deficient mice at 1 × 107 cells/mouse. Between 24 and 48 h after disease onset, mRNA was extracted from spinal cord and cerebellum and examined for inflammatory expression patterns using real time PCR as described in Materials and methods. Moderate Th1 tissue was extracted from animals that exhibited classical clinical scores of 1–2 in the 24–48 h after onset. Severe Th1 tissue was extracted from mice that exhibited classical clinical scores of 2.5–3.5 during the same period. The cells used to induce the severe EAE were injected in parallel into IFN-γR–deficient mice (IFN-γR null). The tissue in the IFN-γ–null group was extracted from WT mice that received IFN-γ–deficient pathogenic T cells. The data are presented as mean ± SEM of 4–5 mice/group from two independent experiments.
The severe Th1 and IFN-γR–null were injected in parallel with the same Th1 pathogenic T cells. There was similar induction of IFN-γ in the cerebellum of both groups, indicating similar levels of antigenic stimulation of the transferred lymphocytes (Fig. 5). In turn, the difference in spinal cord IFN-γ expression between the two groups supports the hypothesis of a strongly proinflammatory role for IFN-γ in increasing inflammatory cell infiltration of the spinal cord in WT mice.
Increased numbers of adoptively transferred IL-17–producing cells are not required to drive atypical EAE
A recent study suggested that changes in the ratio of IL-17 and IFN-γ–producing pathogenic T cells determined CNS lesion localization (17). To determine if changes in pathogenic T cell cytokine profiles coincided with development of atypical EAE in IFN-γR KO mice, we examined the cytokine profile of leukocytes recovered from the CNS of WT mice or IFN-γR–deficient mice that received WT pathogenic cells (Fig. 6). Interestingly, a high number of the recently described (18) host CD4−CD8− IL-17–producing T cells were found in the cerebellum of mice with atypical EAE, suggesting that these cells may play a role in the pathogenesis of atypical EAE (Fig. 6). However, examination of adoptively transferred WT cells revealed no change in IL-17 production capacity regardless of the host's IFN-γR expression level and clinical manifestation (Fig. 6). Thus, the changes in lesion localization associated with IFN-γR deficiency are not a result of changes within the pathogenic T cells but instead reflect the inability of the host to respond to IFN-γ produced by the invasive T cell.
Figure 6.

Atypical inflammation in IFN-γR–deficient mice does not require increased Il-17 production in adoptively transferred cells. MOG35-55-specific T cell lines were generated in CD90.1+ C57BL/6 mice and retroorbitally injected into either C57BL/6 (WT) or IFN-γR–deficient (γR−/−) mice at 5 × 106 cells/mouse. After disease onset, T cells were recovered from either the cerebellum and brainstem or the spinal cord. T cells were restimulated with PMA and Ionomycin and tested for their capacity to produce IFN-γ and IL-17. Perivascular lesions and invasive parenchymal lesions are indicated with an arrow and the letters P and I, respectively. Data shown is representative of three separate experiments that used tissue pooled from two mice.
DISCUSSION
In this study, we used an adoptive transfer system to examine the capacity of IFN-γ–IFN-γR interactions to determine the sites of inflammation within the CNS. IFN-γ–expressing WT Th1 cells were found to initiate similar lesions in IFN-γR–null hosts as IFN-γ–null cells in WT hosts. These data suggested that IFN-γ produced by the effector T cell population was selectively antiinflammatory in the cerebellum and brainstem. In the same mice, IFN-γ–IFN-γR interactions were necessary for inflammatory infiltration of the spinal cord. Together, these data demonstrate discrete regional responses to IFN-γ during neuroinflammation and suggest that these responses can shape the clinical manifestation of CNS autoimmunity.
The experiments reported here reveal that the previously reported effects on EAE clinical outcome associated with IFN-γ deficiency are linked to two discrete functions of IFN-γ in EAE induction. First, recognition of pathogenic T cell–produced IFN-γ is necessary to prevent inflammation of the cerebellum and brain stem. Adoptively transferred pathogenic Th1 cells appear to possess the necessary components for invasion of the cerebellum and brain stem as, in the absence of a host response to IFN-γ, these cells are fully capable of infiltrating and initiating cerebellar disease. However, under ordinary circumstances the IFN-γ produced by the encephalitogenic cells is sufficient to prevent entry of the same cells into the cerebellum and brainstem. As such, IFN-γ–mediated protection of the cerebellum and brain stem represents an active process, acting under ordinary circumstances to prevent cerebellar inflammation. Second, recognition of encephalitogenic T cell–produced IFN-γ is required for the induction of inflammation within the spinal cord. That is, in the absence of an IFN-γ signal, adoptively transferred cells are not capable of initiating spinal cord inflammation. Interestingly, some mice showed symptoms of paralysis in the absence of measurable spinal cord infiltration. Unlike the ascending paralysis observed in classical EAE, the limb dysfunction that occurred in mice with the atypical clinical course was not necessarily caudal to rostral, as mice could demonstrate hind- and/or forelimb weakness and/or dystonia in the absence of tail involvement. These findings suggest that the observed dystonia is induced by either inflammatory lesions within the spinal cord too small to be detected using standard histological techniques or, more likely, is induced by lesions within the cerebellum and/or brain stem. Together, these data reveal that encephalitogenic effector T cell cytokine production and concomitant host recognition of the cytokine plays a vital role in localizing the lesions associated with classical EAE.
Previous reports suggested that differences in cell localization followed from recognition of distinct antigens (6, 19). Alternatively, it has been suggested that differences in cellular localization reflected anatomical differences in the capacity to support T cell invasion through breakdown of the blood–brain barrier (10, 20, 21). Several groups previously demonstrated the induction of atypical axial-rotary disease in a variety of EAE models, and particularly within IFN-γ–deficient mice (6, 7, 22–24). Reports suggested that the nature of the disease in IFN-γ–deficient systems reflected biased selection of the T cell repertoire, either through direct manipulation of the T cells themselves or by influencing the range of antigens presented to T cells during initiation of the immune response (7). However, another group used a TCR transgenic mouse incapable of mediating T cell receptor recombination to demonstrate that T cells with identical specificities were capable of inducing either classical or atypical EAE depending on the conditions under which the cells were generated (12). Although clearly demonstrating the capacity of a single TCR to initiate either atypical disease or classical EAE, the study did not determine if the difference in pathogenesis observed was a result of specific programs induced in the T cells themselves or if pathogenesis resulted from differential cytokine production and thus target tissue susceptibility.
In this study we demonstrate, using adoptive transfer of WT Th1 cells into IFN-γR–deficient mice, that localization of T cells to specific regions of the brain relies on IFN-γ–IFN-γR interactions during the invasion process. A single T cell preparation is able to induce atypical and/or classical disease depending on whether cells within the host are capable of recognizing IFN-γ. Thus, regional differences in T cell invasion represent regional differences in cytokine responsiveness rather than any intrinsic differences in the individual initiating T cells.
The mixing experiments demonstrate that the capacity for IFN-γ expression in only 50% of initiating T cells is sufficient to drive both protection of the cerebellum and infiltration of the spinal cord. Further, protective IFN-γ production required antigenic stimulation of invading Th1 cells. (Table II) This suggests that both protection effects and infiltration are initiated by host cell recognition of an IFN-γ signal produced by a portion of CNS antigen-specific invading cells. Invading cells are not necessarily discriminated on an individual basis, rather the overall group makeup determines tissue receptiveness to invasion.
This finding is particularly intriguing in light of a recent study that suggested that IL-17/IFN-γ–producing tissue invasive cell ratios determine the localization of T cell invasion and lesion formation (17). In contrast to the results demonstrated in the current study, Stromnes et al. (17) found that IL-23–treated MOG35-55 cells were capable of inducing atypical EAE in WT mice. This capacity was linked to the change in the ratio of IL-17/IFN-γ T cell ratios as found in the CNS. Given the large number of IFN-γ–producing cells still found in our IL-23–treated pathogenic T cells, it is perhaps not surprising that we found no capacity for IL-23–treated cells to induce atypical EAE.
The data presented here clearly demonstrate that interruption of IFN-γ/IFN-γR signaling resulted in redistribution of lesions from the classical pattern of spinal cord to the cerebellum and brain stem. However, it remained possible that the disruption of IFN-γ/IFN-γR signaling lay upstream of a change in cytokine production capacity that resulted in the skewed IL-17/IFN-γ ratios reported by Stromnes et al. (17) Examination of cytokine production in WT pathogenic T cells after transfer into IFN-γR revealed almost no change in IL-17/IFN-γ ratio, demonstrating that in our system an increased IL-17/IFN-γ ratio is not required to induce atypical disease. As such, although the demonstration that decreasing the fraction of IFN-γ–producing cells increases atypical disease is in accord with the recent observations of Stromnes et al. (17), the induction of atypical disease in IFN-γR–deficient mice given WT cells indicates a very different mechanism active in our system. Although their results suggested that IL-17 was essential to the induction of atypical disease, the data shown by Stromnes et al. (17) could also support the finding that dilution of IFN-γ–producing pathogenic T cells with pathogenic cells incapable of producing IFN-γ results in atypical disease, as reported in this paper.
Interestingly, Stromnes et al. (17) found that administration of IL-17RA–Fc protein preferentially blocked atypical disease induction. Combined with the cerebellar recruitment of host IL-17–producing cells seen in atypical disease, these data suggest that IL-17 plays a vital role in atypical EAE. However, these data do not demonstrate that IL-17 is responsible for lesion localization to the cerebellum and brainstem but only that the atypical disease process requires IL-17 at some time during the pathogenesis process. Indeed, it is perhaps more surprising that administration of the IL-17RA–Fc protein had so little effect on classical EAE incidence and symptoms in this model, a finding that contrasts with studies that reported a profound effect of IL-17 blockade on classical EAE induction (25, 26), than that IL-17 is required to drive atypical EAE. Although it remains possible that IL-17 production is required for lesion localization to the brain after immunization with MOG97–114, it is equally possible that the necessary role for IL-17 in pathogenesis within the brain occurs after site selection.
The differences in cell trafficking presented in the cell mixing experiments illustrate the discrete nature of the pro- and antiinflammatory (protective) aspects of IFN-γR signaling. Although initial experiments left open the possibility that spinal cord invasion and cerebellar invasion were mutually exclusive, the histological and clinical data from the cell mixing experiments and BM chimeras clearly demonstrate the capacity for these two inflammatory pathways to occur simultaneously. Thus, the IFN-γ–meditated protection of the cerebellum and brain stem and inflammation of the spinal cord are separable events.
The data presented here support previous studies that demonstrated differential responses to IFN-γ in discrete portions of the brain (27, 28). These studies clearly demonstrated a differential capacity for IFN-γ responses within various sections of the brain after exogenous cytokine administration. In contrast, the data presented here delineate differential responses to physiological levels of IFN-γ production. This difference in methodology may explain the apparent discrepancies between IFN-γ–induced cell trafficking into the brainstem in the earlier study (27) and the IFN-γ–induced protection from cell trafficking observed in the brainstem in this study.
The idea that regional CNS responses to cytokines are vital to the development of neuroinflammation also fits well with previous MS studies that demonstrated anti-myelin T cell responses in clinically silent individuals (29–33). The lack of clinical manifestation in individuals that are clearly capable of a vigorous antimyelin immune response could be explained by CNS responses that are either insufficient to allow inflammation or that are actively suppressive of inflammation. Under this model, development of MS would rely on both induction of CNS specific autoimmune responses and the capacity of the CNS to respond to initial immune invasion.
Interestingly, initial examination of IFN-γR1 within the cerebellum and spinal cord of both naive mice and mice with either typical or atypical EAE revealed little to no difference in expression between the two tissues. These data suggest that the capacity to bind IFN-γ is equivalent between the two tissues and that differential responses to IFN-γ within different regions reflect downstream components of the IFN-γ signaling pathway (unpublished data).
The expression of both CXCL9 and CXCL10 were completely ablated in the CNS of mice that did not receive an IFN-γ signal from invading T cells. These data demonstrate that in our model system, alternative inflammatory agents such as TNF-α are not sufficient to drive expression of CXCL9 and CXCL10. The complete absence of these chemokines in the cerebellum of mice with atypical disease supports recent suggestions that these chemokines restrain T cells within the perivasculature of the cerebellum preventing development of atypical disease (34).
The data presented here demonstrate the sufficiency and necessity for IFN-γ signaling during the invasion process to determine cellular localization within various anatomical components of the CNS. The atypical EAE model may give useful information for cerebellar MS but it may also tell us about other inflammatory diseases. The atypical EAE findings described here may be a particularly good model of the human disease, Bickerstaff's brainstem encephalitis (BBE) (35). This is usually a monophasic inflammatory disease of the brainstem and cerebellum. Patients have decreased consciousness, corticospinal tract abnormalities, multiple cranial nerve dysfunctions, and ataxia. Its less severe form appears to be Miller Fisher syndrome (MFS), which has much less brainstem involvement and is defined by ophthalmoplegia, ataxia, and areflexia. The differences between BBE and MFS, although controversial, may be a matter of the intensity of the immune response, given that both affect the same target areas of the brain (36, 37). MFS in turn overlaps with Guillain Barre syndrome (GBS). It is of interest that BBE, MFS, and a subset of GBS can have seemingly similar immune responses (all can have similar anti-GQ1B ganglioside antibodies) (38–41), yet have different inflammatory targeting (42). Differences in IFN-γ production in these diseases could account for these differences if they follow the same paradigm as described in our atypical EAE model. As would be predicted by our disease paradigm, patients with MFS have lower IFN-γ production than patients with GBS (43). Also, cases have been described that started as one of these diseases that then changed or relapsed as a different disease (44, 45). Fluctuations in IFN-γ production might be expected to produce exactly this scenario.
The data presented here demonstrate the sufficiency and necessity for IFN-γ signaling during the invasion process to determine regional localization within various anatomical sites of the CNS. An understanding of the exact mechanisms involved in cellular localization to the CNS and PNS may allow development of better methods for determining prognosis and modifying pathogenesis in many neurological diseases such as MS, BBE, MFS, and GBS.
MATERIALS AND METHODS
Mice.
C57BL/6J, congenic CD45.1 mice, IFN-γR KO, and IFN-γ–deficient B6.129S7-ifngtm1Ts/J mice were purchased from Jackson ImmunoResearch Laboratories. All experiments were approved by the Washington University Animal Care Committee and all mice were housed in an Association for Assesment and Accreditation of Laboratory Animal Care–approved facility.
T cell lines.
To generate MOG-specific cell lines, C57BL/6J or IFN-γ–deficient B6.129S7-ifngtm1Ts/J mice were immunized s.c. with 50 μg MOG35-55 peptide (Sigma-Aldrich) emulsified in IFA and supplemented with 500 μg/ml Mycobacterium tuberculosis. CD4+ cells were isolated from the spleen as previously described (46). Cells were then stimulated at a concentration of 106 cells/ml in the presence of 5 × 106 cells/ml of irradiated C57BL/6 splenocytes, 10 μg/ml MOG35-55 peptide, 10 U/ml IL-12, and 10 U/ml IL-2, in 10% FCS containing RPMI complete media to polarize cells to a Th1 phenotype or in the presence of 5 × 106 cells/ml of irradiated C57BL/6 splenocytes, 10 μg/ml MOG35-55 peptide, 1 μg/ml of human TGF-β1, 10 μg/ml of mouse IL-6, 10 μg/ml of recombinant mouse IL-23, 1 mg/ml of neutralizing antibody to IFN-γ, and 10 U/ml IL-2, in 10% FCS containing RPMI complete media to polarize cells to a Th17 phenotype. Cells were isolated 7 d later by Histopaque 1.077 (Sigma-Aldrich) and then restimulated in the absence of IL-12 under the other conditions outlined earlier in the section. Cells underwent two rounds of stimulation before being stored at −70°C in 10% DMSO containing FCS. Before usage, cells were thawed and restimulated for 7 d with MOG peptide and IL-2. Before injection, cells were restimulated for 4 d with MOG peptide and IL-2, separated by Histopaque 1.077, and then washed and resuspended in HBSS. To induce EAE, 5 × 106 cells in 300 μl HBSS was transferred by retroorbital i.v. injection to 6–8-wk-old mice. Ovalbumin-specific (OVA) T cell lines were produced using the same protocol with 900 μg of whole ovalbumin protein substituted for MOG35-55 peptide.
Clinical scoring.
Mice were examined daily for symptoms. Symptoms of limb dysfunction were scored based on areas affected (1, partial or total inability to move tail; 2, hind limb weakness/disrupted righting reflex; 3, complete inability to move hind limb; 4, complete inability to move both hind limbs; 5, moribund/dead) and type of dysfunction (flaccid or dystonic). Incidence of atypical EAE was assigned if limb dysfunction associated with dystonia occurred or if symptoms of either ataxia or vertigo/dysequilibrium were recorded on any given day. Incidence of classical EAE was determined based on appearance of limb dysfunction associated with flaccid paralysis. Symptoms of ataxia were determined separately for the head, each individual limb quarter, and the tail. Symptoms of vertigo/dysequilibrium were scored based on side affected and severity.
Histological examination.
Mice were perfused with 30 ml of ice-cold saline. CNS tissues were extracted through mechanical removal of upper skull and vertebrae. Desired tissues were immediately removed from the whole CNS and placed in embedding media before being frozen on dry ice. Tissues were kept at −80°C until being sectioned in 10-μm slices and stained with H&E.
Intracellular cytokine staining.
Intracellular staining was performed, with some modification, as previously described (47). In brief, 106 cells were restimulated on plates precoated with anti-CD3 (10 μg/ml PBS; eBioscience) or by incubation with 50 ng/ml PMA and 1 μM ionomycin (Sigma-Aldrich), with concurrent blockade of cytokine secretion by treatment with 1 μg/ml brefeldin A (Sigma-Aldrich) for 2–4 h at 37°C. Cells were stained with anti-CD4 on ice for 15 min with continual agitation. Cells were then washed three times with PBS supplemented with 0.5% BSA and 0.1% sodium azide (SB), fixed with 5% buffered formalin phosphate (5% paraformaldehyde in PBS) for 20 min at room temperature, washed with PBS, and permeabilized with PBS supplemented with 0.5% BSA, 0.1% sodium azide, and 0.1% saponin (PB) for 10 min at room temperature. Cells were then washed once with PB, and stained with 5 μg/ml of anti–IL-17A-PE or APC and/or anti–IFN-γ-FITC or APC or isotype controls (eBioscience) in PB for 5 min at room temperature with continual agitation. After washing twice with PB and twice with SB, cells were immediately collected using a FACSCalibur (BD Biosciences) and were analyzed using CellQuest software (BD Biosciences).
Quantitative PCR.
All experiments were performed as previously described (48). Previously described primer sets were used, including IFN-γ, CXCL10, and GAPDH (48); CXCL2 (11); CXCL9 (49); IL-12p35 and IL-23p19 (50); and IL-17 (51). Individual copy numbers were normalized to copies of GAPDH.
Statistics.
All statistics were performed using Prism 4 software (GraphPad Software, Inc.).
Online supplemental material.
Fig. S1 shows the immune cell constituents of CNS tissue from typical and atypical disease. Table S1 shows that deficiency of IFN-γR in either radioresistant or BM-derived cells results in a loss of cerebellar protection. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080155/DC1.
Supplementary Material
[Supplemental Material Index]
Acknowledgments
This work was supported in part by a National Multiple Sclerosis Society Fellowship (FG 1757-A-1), by research grants (RG3314 and RG 3732) from the National Multiple Sclerosis Society, and by a National Institutes of Health training grant (5 T32 NS07205-22).
The authors have no conflicting financial interests.
Abbreviations used: BBE, Bickerstaff's brainstem encephalitis; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; GBS, Guillain Barre syndrome; IFN-γR, IFN-γ receptor; MFS, Miller Fisher syndrome; MOG, myelin/oligodendrocyte glycoprotein; MS, multiple sclerosis.
References
- 1.Citterio, A., G. Azan, R. Bergamaschi, A. Erbetta, and V. Cosi. 1989. Multiple sclerosis: disability and mortality in a cohort of clinically diagnosed patients. Neuroepidemiology. 8:249–253. [DOI] [PubMed] [Google Scholar]
- 2.Langdon, D.W., and A.J. Thompson. 1999. Multiple sclerosis: a preliminary study of selected variables affecting rehabilitation outcome. Mult. Scler. 5:94–100. [DOI] [PubMed] [Google Scholar]
- 3.Naismith, R.T., K. Trinkaus, and A.H. Cross. 2006. Phenotype and prognosis in African-Americans with multiple sclerosis: a retrospective chart review. Mult. Scler. 12:775–781. [DOI] [PubMed] [Google Scholar]
- 4.Phadke, J.G. 1987. Survival pattern and cause of death in patients with multiple sclerosis: results from an epidemiological survey in north east Scotland. J. Neurol. Neurosurg. Psychiatry. 50:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riise, T., M. Gronning, O. Fernandez, K. Lauer, R. Midgard, J.M. Minderhoud, H. Nyland, G. Palffy, S. Poser, and J.A. Aarli. 1992. Early prognostic factors for disability in multiple sclerosis, a European multicenter study. Acta Neurol. Scand. 85:212–218. [DOI] [PubMed] [Google Scholar]
- 6.Muller, D.M., M.P. Pender, and J.M. Greer. 2000. A neuropathological analysis of experimental autoimmune encephalomyelitis with predominant brain stem and cerebellar involvement and differences between active and passive induction. Acta Neuropathol. 100:174–182. [DOI] [PubMed] [Google Scholar]
- 7.Abromson-Leeman, S., R. Bronson, Y. Luo, M. Berman, R. Leeman, J. Leeman, and M. Dorf. 2004. T-cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am. J. Pathol. 165:1519–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobel, R.A. 2000. Genetic and epigenetic influence on EAE phenotypes induced with different encephalitogenic peptides. J. Neuroimmunol. 108:45–52. [DOI] [PubMed] [Google Scholar]
- 9.Gimenez, M.A., J. Sim, A.S. Archambault, R.S. Klein, and J.H. Russell. 2006. A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am. J. Pathol. 168:1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cross, A.H., T. O'Mara, and C.S. Raine. 1993. Chronologic localization of myelin-reactive cells in the lesions of relapsing EAE: implications for the study of multiple sclerosis. Neurology. 43:1028–1033. [DOI] [PubMed] [Google Scholar]
- 11.Archambault, A.S., J. Sim, E.E. McCandless, R.S. Klein, and J.H. Russell. 2006. Region-specific regulation of inflammation and pathogenesis in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 181:122–132. [DOI] [PubMed] [Google Scholar]
- 12.Wensky, A.K., G.C. Furtado, M.C. Marcondes, S. Chen, D. Manfra, S.A. Lira, D. Zagzag, and J.J. Lafaille. 2005. IFN-gamma determines distinct clinical outcomes in autoimmune encephalomyelitis. J. Immunol. 174:1416–1423. [DOI] [PubMed] [Google Scholar]
- 13.Iwakura, Y., and H. Ishigame. 2006. The IL-23/IL-17 axis in inflammation. J. Clin. Invest. 116:1218–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver, C.T., L.E. Harrington, P.R. Mangan, M. Gavrieli, and K.M. Murphy. 2006. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 24:677–688. [DOI] [PubMed] [Google Scholar]
- 15.Harrington, L.E., R.D. Hatton, P.R. Mangan, H. Turner, T.L. Murphy, K.M. Murphy, and C.T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 16.Mangan, P.R., L.E. Harrington, D.B. O'Quinn, W.S. Helms, D.C. Bullard, C.O. Elson, R.D. Hatton, S.M. Wahl, T.R. Schoeb, and C.T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 441:231–234. [DOI] [PubMed] [Google Scholar]
- 17.Stromnes, I.M., L.M. Cerretti, D. Liggitt, R.A. Harris, and J.M. Goverman. 2008. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat. Med. 14:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lees, J.R., Y. Iwakura, and J.H. Russell. 2008. Host T cells are the main producers of IL-17 within the central nervous system during initiation of experimental autoimmune encephalomyelitis induced by adoptive transfer of Th1 cell lines. J. Immunol. 180:8066–8072. [DOI] [PubMed] [Google Scholar]
- 19.Berger, T., S. Weerth, K. Kojima, C. Linington, H. Wekerle, and H. Lassmann. 1997. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab. Invest. 76:355–364. [PubMed] [Google Scholar]
- 20.Juhler, M., D.I. Barry, H. Offner, G. Konat, L. Klinken, and O.B. Paulson. 1984. Blood-brain and blood-spinal cord barrier permeability during the course of experimental allergic encephalomyelitis in the rat. Brain Res. 302:347–355. [DOI] [PubMed] [Google Scholar]
- 21.Namer, I.J., J. Steibel, P. Poulet, J.P. Armspach, M. Mohr, Y. Mauss, and J. Chambron. 1993. Blood-brain barrier breakdown in MBP-specific T cell induced experimental allergic encephalomyelitis. A quantitative in vivo MRI study. Brain. 116:147–159. [DOI] [PubMed] [Google Scholar]
- 22.Kerlero de Rosbo, N., I. Mendel, and A. Ben-Nun. 1995. Chronic relapsing experimental autoimmune encephalomyelitis with a delayed onset and an atypical clinical course, induced in PL/J mice by myelin oligodendrocyte glycoprotein (MOG)-derived peptide: preliminary analysis of MOG T cell epitopes. Eur. J. Immunol. 25:985–993. [DOI] [PubMed] [Google Scholar]
- 23.Gordon, F.L., K.B. Nguyen, C.A. White, and M.P. Pender. 2001. Rapid entry and downregulation of T cells in the central nervous system during the reinduction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 112:15–27. [DOI] [PubMed] [Google Scholar]
- 24.Greer, J.M., R.A. Sobel, A. Sette, S. Southwood, M.B. Lees, and V.K. Kuchroo. 1996. Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J. Immunol. 156:371–379. [PubMed] [Google Scholar]
- 25.Uyttenhove, C., and J. Van Snick. 2006. Development of an anti-IL-17A auto-vaccine that prevents experimental auto-immune encephalomyelitis. Eur. J. Immunol. 36:2868–2874. [DOI] [PubMed] [Google Scholar]
- 26.Hofstetter, H.H., S.M. Ibrahim, D. Koczan, N. Kruse, A. Weishaupt, K.V. Toyka, and R. Gold. 2005. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell. Immunol. 237:123–130. [DOI] [PubMed] [Google Scholar]
- 27.Phillips, L.M., and L.A. Lampson. 1999. Site-specific control of T cell traffic in the brain: T cell entry to brainstem vs. hippocampus after local injection of IFN-gamma. J. Neuroimmunol. 96:218–227. [DOI] [PubMed] [Google Scholar]
- 28.Phillips, L.M., P.J. Simon, and L.A. Lampson. 1999. Site-specific immune regulation in the brain: differential modulation of major histocompatibility complex (MHC) proteins in brainstem vs. hippocampus. J. Comp. Neurol. 405:322–333. [DOI] [PubMed] [Google Scholar]
- 29.Kitze, B., M. Pette, E. Rohrbach, D. Stadt, L. Kappos, and H. Wekerle. 1988. Myelin-specific T lymphocytes in multiple sclerosis patients and healthy individuals. J. Neuroimmunol. 20:237. [DOI] [PubMed] [Google Scholar]
- 30.Pette, M., K. Fujita, B. Kitze, J.N. Whitaker, E. Albert, L. Kappos, and H. Wekerle. 1990. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology. 40:1770–1776. [DOI] [PubMed] [Google Scholar]
- 31.Zhang, J.W., C.H. Chou, G. Hashim, R. Medaer, and J.C. Raus. 1990. Preferential peptide specificity and HLA restriction of myelin basic protein-specific T cell clones derived from MS patients. Cell. Immunol. 129:189–198. [DOI] [PubMed] [Google Scholar]
- 32.Pelfrey, C.M., L.R. Tranquill, A.B. Vogt, and H.F. McFarland. 1996. T cell response to two immunodominant proteolipid protein (PLP) peptides in multiple sclerosis patients and healthy controls. Mult. Scler. 1:270–278. [DOI] [PubMed] [Google Scholar]
- 33.Trotter, J.L., C.M. Pelfrey, A.L. Trotter, J.A. Selvidge, K.C. Gushleff, T. Mohanakumar, and H.F. McFarland. 1998. T cell recognition of myelin proteolipid protein and myelin proteolipid protein peptides in the peripheral blood of multiple sclerosis and control subjects. J. Neuroimmunol. 84:172–178. [DOI] [PubMed] [Google Scholar]
- 34.Muller, M., S.L. Carter, M.J. Hofer, P. Manders, D.R. Getts, M.T. Getts, A. Dreykluft, B. Lu, C. Gerard, N.J. King, and I.L. Campbell. 2007. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J. Immunol. 179:2774–2786. [DOI] [PubMed] [Google Scholar]
- 35.Willison, H.J. 2002. Anti-glycolipid antibodies in the diagnosis of autoimmune neuropathies. Rev. Neurol. (Paris). 158:S16–S20. [PubMed] [Google Scholar]
- 36.Ogawara, K., S. Kuwabara, and N. Yuki. 2002. Fisher syndrome or Bickerstaff brainstem encephalitis? Anti-GQ1b IgG antibody syndrome involving both the peripheral and central nervous systems. Muscle Nerve. 26:845–849. [DOI] [PubMed] [Google Scholar]
- 37.Tezer, F.I., G. Gurer, H. Karatas, G. Nurlu, and O. Saribas. 2002. Involvement of the central nervous system in Miller Fisher syndrome: a case report. Clin. Neurol. Neurosurg. 104:377–379. [DOI] [PubMed] [Google Scholar]
- 38.Paparounas, K. 2004. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders: pathophysiologic role and clinical relevance. Arch. Neurol. 61:1013–1016. [DOI] [PubMed] [Google Scholar]
- 39.Nagashima, T., M. Koga, M. Odaka, K. Hirata, and N. Yuki. 2004. Clinical correlates of serum anti-GT1a IgG antibodies. J. Neurol. Sci. 219:139–145. [DOI] [PubMed] [Google Scholar]
- 40.Ilyas, A.A., S.D. Cook, F.A. Mithen, T. Taki, T. Kasama, S. Handa, H. Hamasaki, B.S. Singhal, S.C. Li, and Y.T. Li. 1998. Antibodies to GT1a ganglioside in patients with Guillain-Barre syndrome. J. Neuroimmunol. 82:160–167. [DOI] [PubMed] [Google Scholar]
- 41.Alaedini, A., C. Briani, I. Wirguin, G. Siciliano, C. D'Avino, and N. Latov. 2002. Detection of anti-ganglioside antibodies in Guillain-Barre syndrome and its variants by the agglutination assay. J. Neurol. Sci. 196:41–44. [DOI] [PubMed] [Google Scholar]
- 42.Lo, Y.L. 2007. Clinical and immunological spectrum of the Miller Fisher syndrome. Muscle Nerve. 36:615–627. [DOI] [PubMed] [Google Scholar]
- 43.Hohnoki, K., A. Inoue, and C.S. Koh. 1998. Elevated serum levels of IFN-gamma, IL-4 and TNF-alpha/unelevated serum levels of IL-10 in patients with demyelinating diseases during the acute stage. J. Neuroimmunol. 87:27–32. [DOI] [PubMed] [Google Scholar]
- 44.Arai, M., M. Odaka, N. Yuki, and K. Hirata. 2002. A patient with overlapping Bickerstaff's brainstem encephalitis, Miller Fisher syndrome and Guillain-Barre syndrome during the clinical course. Eur. J. Neurol. 9:115–116. [DOI] [PubMed] [Google Scholar]
- 45.Mochizuki, A., K. Ota, M. Iijima, T. Yamauchi, and M. Iwata. 1996. (A case of relapsing Guillain-Barre syndrome following Miller Fisher syndrome). Rinsho Shinkeigaku. 36:675–679. [PubMed] [Google Scholar]
- 46.Archambault, A.S., J. Sim, M.A. Gimenez, and J.H. Russell. 2005. Defining antigen-dependent stages of T cell migration from the blood to the central nervous system parenchyma. Eur. J. Immunol. 35:1076–1085. [DOI] [PubMed] [Google Scholar]
- 47.Yu, J.J., C.S. Tripp, and J.H. Russell. 2003. Regulation and phenotype of an innate Th1 cell: role of cytokines and the p38 kinase pathway. J. Immunol. 171:6112–6118. [DOI] [PubMed] [Google Scholar]
- 48.Klein, R.S., E. Lin, B. Zhang, A.D. Luster, J. Tollett, M.A. Samuel, M. Engle, and M.S. Diamond. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belperio, J.A., M.P. Keane, M.D. Burdick, J.P. Lynch III, D.A. Zisman, Y.Y. Xue, K. Li, A. Ardehali, D.J. Ross, and R.M. Strieter. 2003. Role of CXCL9/CXCR3 chemokine biology during pathogenesis of acute lung allograft rejection. J. Immunol. 171:4844–4852. [DOI] [PubMed] [Google Scholar]
- 50.Wakabayashi, H., N. Takakura, K. Yamauchi, and Y. Tamura. 2006. Modulation of immunity-related gene expression in small intestines of mice by oral administration of lactoferrin. Clin. Vaccine Immunol. 13:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferretti, S., O. Bonneau, G.R. Dubois, C.E. Jones, and A. Trifilieff. 2003. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170:2106–2112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Material Index]