Memory Inflation During Chronic Viral Infection is Maintained by Continuous Production of Short-Lived Functional T Cells (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 1.
Summary
During persistent murine cytomegalovirus (MCMV) infection the T cell response is maintained at extremely high levels for the life of the host. These cells closely resemble human CMV-specific cells which comprise a major component of the peripheral T cell compartment in most people. Despite a phenotype that suggests extensive antigen-driven differentiation, MCMV-specific T cells remain functional and respond vigorously to viral challenge. We hypothesized that a low rate of antigen-driven proliferation would account for the maintenance of this population. Instead, we found that most of these cells divide only sporadically in chronically infected hosts and have a short half-life in circulation. The overall population is supported, at least in part, by memory cells primed early in infection as well as recruitment of naïve T cells at late times. These data show that memory inflation is maintained by a continuous replacement of short-lived, functional cells during chronic MCMV infection.
Introduction
The development and maintenance of memory T cell responses during persistent or chronic infection is poorly understood. Most of our knowledge is derived from the LCMV-clone 13 model system, which involves very high levels of persistently replicating virus. In this environment, antigen specific T cells continuously divide and ultimately become non-functional, a phenomenon known as clonal exhaustion (Shin et al., 2007; Wherry et al., 2004; Wherry et al., 2003; Zajac et al., 1998). In contrast, cytomegalovirus (CMV) is a β-herpesvirus that undergoes systemic infection and establishes true latency but likely also maintains a low level of persistent infection. The T cell response to CMV in humans and mice is astonishingly large, comprising up to 10% of all CD8 T cells (Gillespie et al., 2000; Karrer et al., 2003; Lang et al., 2002; Munks et al., 2006a; Sylwester et al., 2005). Strikingly, most of these T cells are functional and the populations are maintained for the life of the host.
CMV-specific cells accumulate over time before stabilizing at a high level, a phenomenon known as memory inflation (Holtappels et al., 2000; Karrer et al., 2003; Karrer et al., 2004; Munks et al., 2006a; Sierro et al., 2005). Inflated CMV-specific T cells generally express low levels of the co-stimulatory molecules CD27 and CD28 and are also deficient in expression of the IL-7Rα and IL-15Rβ chains (Appay et al., 2002; Karrer et al., 2003; Munks et al., 2006a; Sierro et al., 2005; van Leeuwen et al., 2005; van Leeuwen et al., 2002; Weekes et al., 1999b). In addition, most cells express high levels of KLRG-1, an inhibitory molecule that has been linked to replicative senescence (i.e. failure to proliferate in response to antigen) (Ibegbu et al., 2005; Thimme et al., 2005). In total, this phenotype is indicative of extensive antigen driven differentiation.
Despite their phenotype, CMV-specific T cells can clearly be driven to divide and seem to respond to viral reactivation by expanding in vivo (Gamadia et al., 2004; Karrer et al., 2004; van Leeuwen et al., 2005; van Leeuwen et al., 2002; Waller et al., 2007). These cells can also kill targets ex vivo and provide protection in vivo (Cobbold et al., 2005; Holtappels et al., 2001; Holtappels et al., 2002; Karrer et al., 2004; Pahl-Seibert et al., 2005; Reddehase et al., 1988; Steffens et al., 1998; van Leeuwen et al., 2002). Perhaps most importantly, it is thought that the establishment and maintenance of these inflated T cell populations is critical to control the persistent viral infection (Avetisyan et al., 2006; Boeckh et al., 2003; Reusser et al., 1991; Simon et al., 2006; van Leeuwen et al., 2007). Thus, these cells are maintained at an astonishingly high level and remain functional despite expressing markers indicative of antigen-driven differentiation and possibly senescence.
These data have led to the general assumption that most CMV-specific T cells are long lived, and maintained by cell division resulting from infrequent, but repeated antigen stimulation (van Leeuwen et al., 2007). Surprisingly, our data suggest that the inflated T cell population primarily consists of functional, short-lived T cells that divide only a little and decay even in the presence of persistent infection. Naïve T cells can be recruited during chronic infection to replace the decaying short-lived pool, but maintenance of the total population is also largely dependent on the progeny of cells primed early in infection. Our data show that the inflated CMV-specific T cell population is extremely dynamic and suggest that CMV has found an ideal balance with the immune response that prevents T cell exhaustion.
Results
Inflationary CD8 T cells remain functional over time
MCMV infection drives two major types of T cell populations, stable memory and inflationary, which can be distinguished based on antigen specificity (Karrer et al., 2003; Munks et al., 2006a). T cells that will become stable memory decline rapidly after acute infection and then persist at low levels. In C57BL/6 mice (B6), the stable memory pool is typified by T cells specific for M45 and M57 [Figure 1A and (Munks et al., 2006a)]. In contrast, inflationary T cells increase in number after acute infection before stabilizing at a high level where they are maintained for the life of the host (Munks et al., 2006a; Munks et al., 2007). In B6 mice, populations specific for proteins encoded by m139, M38 and IE3 inflated dramatically over the 8 – 12 weeks following infection (Figure 1A). After the initial period of inflation, all three inflated populations became more stable and were maintained at high levels.
Figure 1.
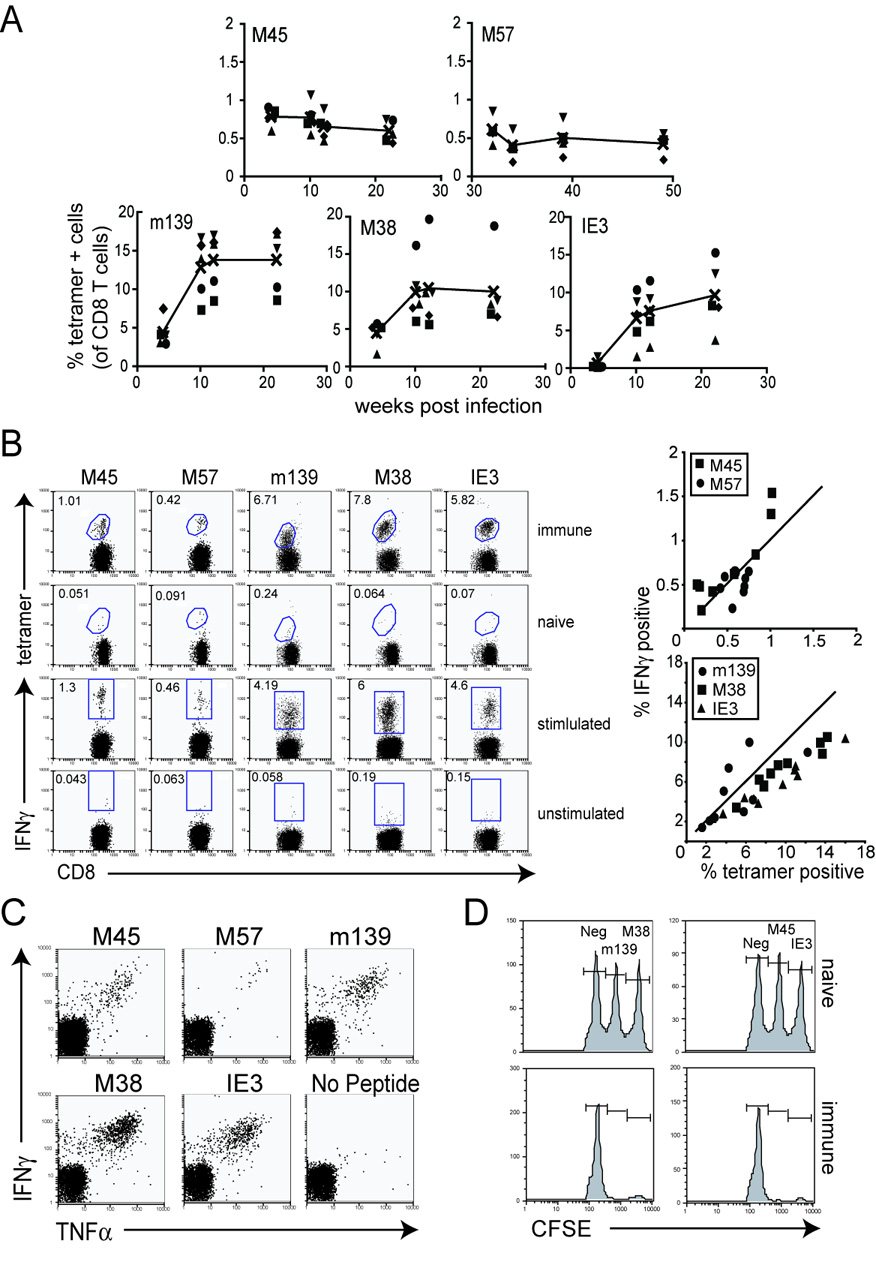
Inflationary T cells remain functional throughout chronic MCMV infection. A) T cell responses specific for m139, M38 and IE3 inflate over the first 3 months of infection. Shown is the percentage of MCMV-specific T cells with the indicated specificity over time as assessed by MHC-tetramer staining. Each symbol represents an individual mouse. The black line represents the average of all mice in this experiment. Note that the scale is different for M45 and M57-specific T cell populations. B) Similar percentages of antigen-specific T cells are found by both tetramer staining and intracellular cytokine analysis. Peripheral blood from individual mice infected for >3 months was divided and stained with the indicated tetramer or stimulated with the indicated peptide and assessed for IFNγ production. Except for the naïve, tetramer-staining control, the FACS plots shown in each column represent the same mouse. A summary of the data from multiple mice is shown in the graphs to the right. The solid line in each graph represents an ideal 1:1 correlation between tetramer staining and IFNγ production. C) Peripheral blood cells were stimulated as above and stained for IFNγ and TNFα production. D) MCMV-specific T cells can kill targets pulsed with peptides. Naïve CD45.1 congenic mice were stained with varying concentrations of CFSE, loaded with the indicated peptide and transferred i.v. into chronically infected C57BL/6 mice. FACS analysis of the surviving donor cells in the spleen of recipient mice was performed approximately 18 hours later.
In contrast to the LCMV clone 13 model of chronic viral infection, MCMV-specific T cells remain functional during chronic infection. Most cells detected in the blood (Figure 1B) and spleen (not shown) by tetramer staining also secreted IFNγ, although some cells in the largest responses failed to secrete IFNγ. Though we have previously demonstrated that very few MCMV-specific T cells secrete IL-2 at these time points (Munks et al., 2006a), most cells that secreted IFNγ also produced TNFα (Figure 1C). In addition, peptide-pulsed targets were readily killed in vivo indicating that the T cells also retain their cytotoxic capacity (Figure 1D).
Phenotype of stable and inflationary T cells
Sierro et al. described a phenotypic dichotomy between inflationary and stable memory cells (Sierro et al., 2005). Consistent with that report, we found that the prototypical stable memory populations (M45 and M57-specific T cells) generally expressed a central memory-like phenotype (Figure 2). This is best illustrated by the expression of high levels of the cytokine receptors CD127 (IL-7Rα) and CD122 (IL-15Rβ) as well as high levels of the costimulatory molecule CD27. In addition, these cells did not express the NK cell inhibitory molecules NKG2A or KLRG-1 and a significant portion of the cells in the blood and spleen expressed high levels of CD62L (L-selectin).
Figure 2.
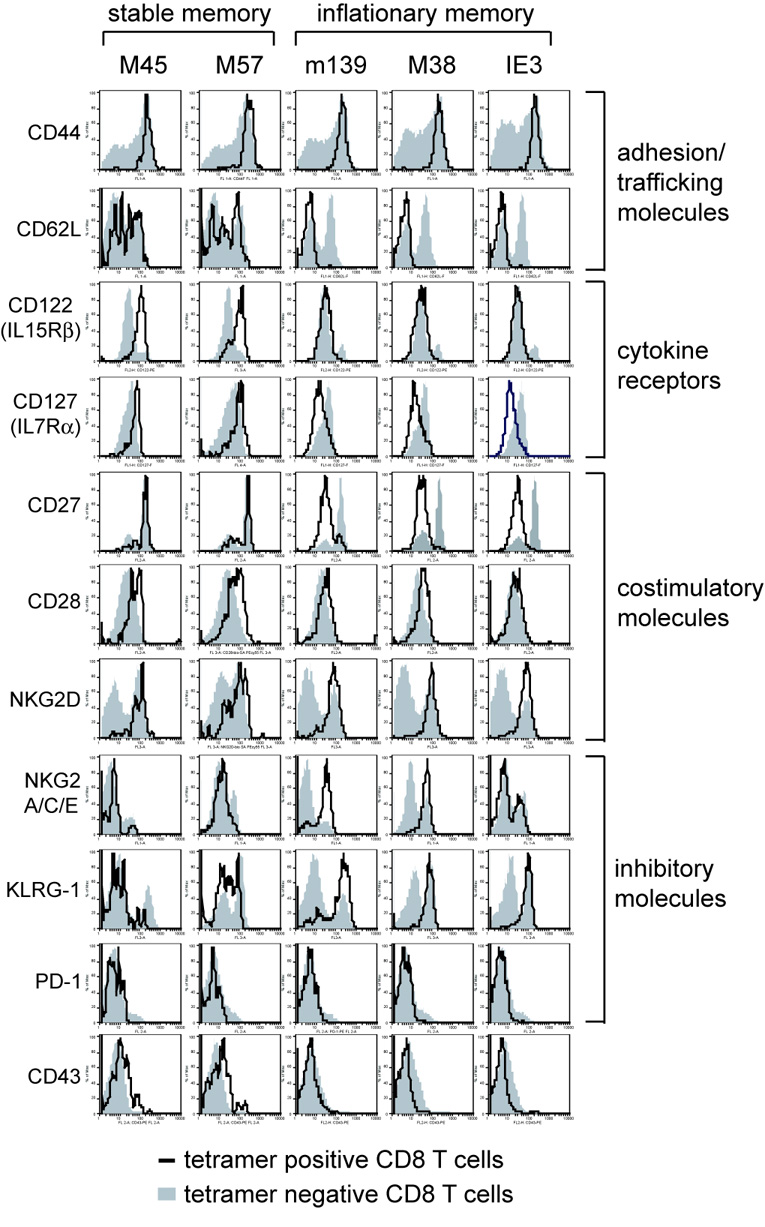
Phenotype of stable and inflationary CD8 T cells. Peripheral blood and/or splenocytes from mice infected for >3 months was stained with the indicated tetramer and antibodies specific for CD8 and the indicated cell surface molecule. The plots shown are gated on tetramer+ CD8 T cells (black line) or tetramer negative CD8 T cells from the same sample (shaded histogram). Data is compiled from multiple experiments and multiple time points during chronic infection.
In contrast, the prototypical inflating populations (m139 and M38-specific T cells) were almost uniformly differentiated. Almost all of these cells expressed low levels of CD62L, CD27, CD127 and CD122 and high levels of the NK cell inhibitory molecules NKG2A and KLRG-1. IE3-specific cells, which are almost undetectable during acute infection but inflate dramatically thereafter, were almost identical to the m139 and M38-specific populations with one exception: approximately 30–60% of IE3-specific cells were NKG2A-negative. It is unclear what this might mean since these NKG2A negative cells were otherwise indistinguishable from the NKG2A-positive cells. Similar results were seen with the NKG2A-specific antibody (clone 16a11) that does not cross-react with the NKG2C and E molecules (not shown). Interestingly, most of the inflationary T cells retained expression of CD28, although we sometimes observed CD28 downregulation (Figure 2 and Supplemental Figure 1A). HCMV-specific T cells and inflationary T cells in BALB/c mice are typically CD28-negative (Appay et al., 2002; Gamadia et al., 2001; Khan et al., 2002a; Sierro et al., 2005; Weekes et al., 1999a). The phenotype of m139, M38 and IE3-specific T cells is consistent with that seen after repeated antigen stimulation (Jabbari and Harty, 2006; Masopust et al., 2006). It is worth noting however, that a very small fraction of the m139, M38 and IE3-specific populations express a more central memory-like phenotype, similar to most M45 and M57-specific T cells (Supplemental Figure 1B).
Both stable and inflationary cells expressed high levels of the costimulatory molecule NKG2D and no cells expressed the inhibitory molecule PD-1 (Figure 2), in agreement with data from HCMV-specific T cells (Day et al., 2006; Trautmann et al., 2006). Finally, most cells were not stained with the CD43-specific antibody 1B11, although there was usually a fraction of M45 and M57-specific T cells that were stained. This antibody has recently been used to distinguish memory T cells with a reduced capacity to proliferate after challenge (Hikono et al., 2007).
T cells within the inflating population do not respond to homeostatic signals but can respond to a new viral infection
To address how inflationary T cell populations are maintained during chronic infection, we performed a series of adoptive transfer experiments. Responsiveness to homeostatic signals was investigated by transferring CFSE-labeled splenocytes from chronically infected mice (>3 months post infection) into SCID recipients and tracking tetramer positive cells (Figure 3A). As predicted from their low expression of CD122 and CD127, m139, M38 and IE3-specific T cells mostly failed to divide in the lymphopenic host while tetramer negative cells underwent extensive division in the same time period (Figure 3B).
Figure 3.
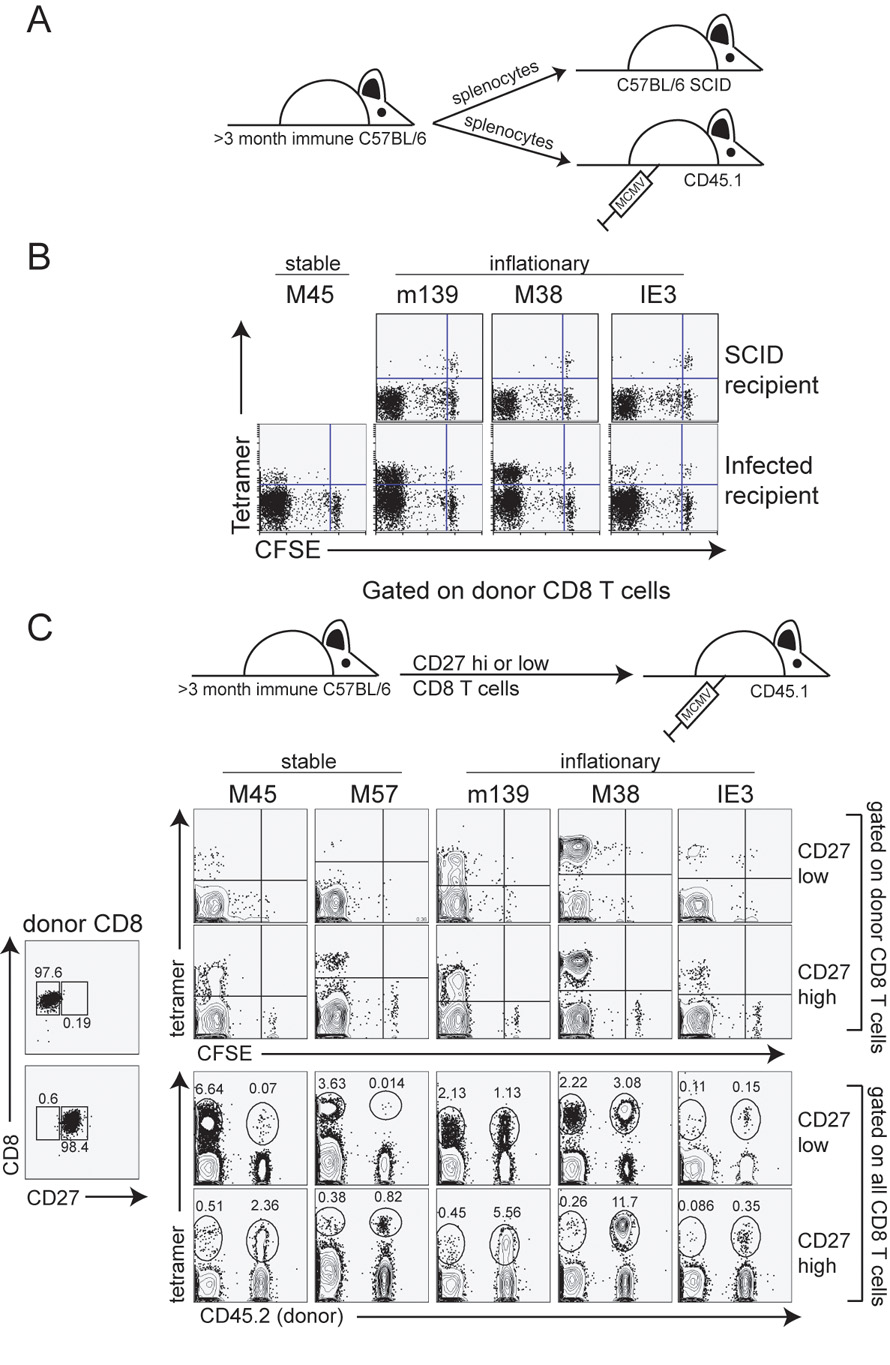
Inflationary T cells fail to divide in SCID recipients, but can respond to a new viral infection. A) Schematic of the adoptive transfer. Splenocytes from C57BL/6 mice infected for >3 months were transferred into C57BL/6-SCID mice or naïve CD45.1 congenic mice. Naïve, CD45.1 congenic recipients were infected with MCMV 2 days later. B) Peripheral blood from SCID recipients (d12 post transfer) or splenocytes from CD45.1 congenic recipients (d7 post infection) were stained with the indicated tetramers. The FACS plots have been gated on CD8+, donor T cells in all cases. C) Cells were FACS sorted for CD8+, CD27 high and low cells. Sorted cells were CFSE labeled and transferred into naïve, congenic mice. Recipients were infected one hour later and donor cells were analyzed in the peripheral blood on day 7 post transfer. Numbers represent the percent of all CD8 T cells that fall into the indicated gates.
To test whether these inflationary cells had lost the capacity to divide, we transferred CFSE labeled cells from chronically infected mice into naïve congenic mice and infected recipients 1 day later (Figure 3A). Strikingly, a large fraction of donor T cells specific for all antigens had proliferated extensively during the first week of an acute infection (CFSE negative, Figure 3B), although there were also cells that had failed to divide. To test whether all of the divided inflationary T cells were derived from the small proportion of the inflationary population that is less differentiated, we sorted the total CD8 pool into CD27 high (less differentiated) and CD27 low (more differentiated) populations, transferred them into naïve congenic mice, and infected the recipients (Figure 3C). Inflationary T cells from both donor populations divided in the recipient mice (CFSE negative, top panels). However, the CD27hi cells appeared to have a greater proliferative capacity. Since so few inflationary T cells are CD27hi (Figure 2), many more cells specific for inflationary epitopes were transferred in the CD27lo donor pool than the CD27hi donor pool. Nevertheless, donor cells from the CD27hi pool contributed >90% the entire inflationary response in the recipient mouse 7 days later, whereas donor cells from the CD27lo pool comprised only approximately half of the overall inflationary response (Figure 3C lower panel). Together, these data suggest that CD27lo inflationary cells retain some ability to divide in response to viral antigen, but are likely not as responsive as the few CD27hi inflationary T cells within each population.
Inflationary T cells undergo sporadic antigen-dependent division
Since inflationary T cells are capable of dividing in response to infection, we wanted to asses whether inflationary T cells undergo antigen-driven proliferation in chronically infected mice. CFSE-labeled splenocytes from chronically infected mice were transferred into infection matched (infected on the same day) or naïve CD45 congenic recipients (Figure 4A). Tetramer+ donor cells were tracked in the peripheral blood of recipients (e.g. Supplemental Figure 2). At these late time points post infection, there is no detectible replicating virus in the spleen of an infected mouse. Nevertheless, to control for the transfer of virus to the naïve recipients, splenocytes were simultaneously transferred into SCID mice which were monitored for survival for 3 months. All mice survived and no plaque forming virus was detected in any organ by plaque assay at the end of the experiment (not shown).
Figure 4.
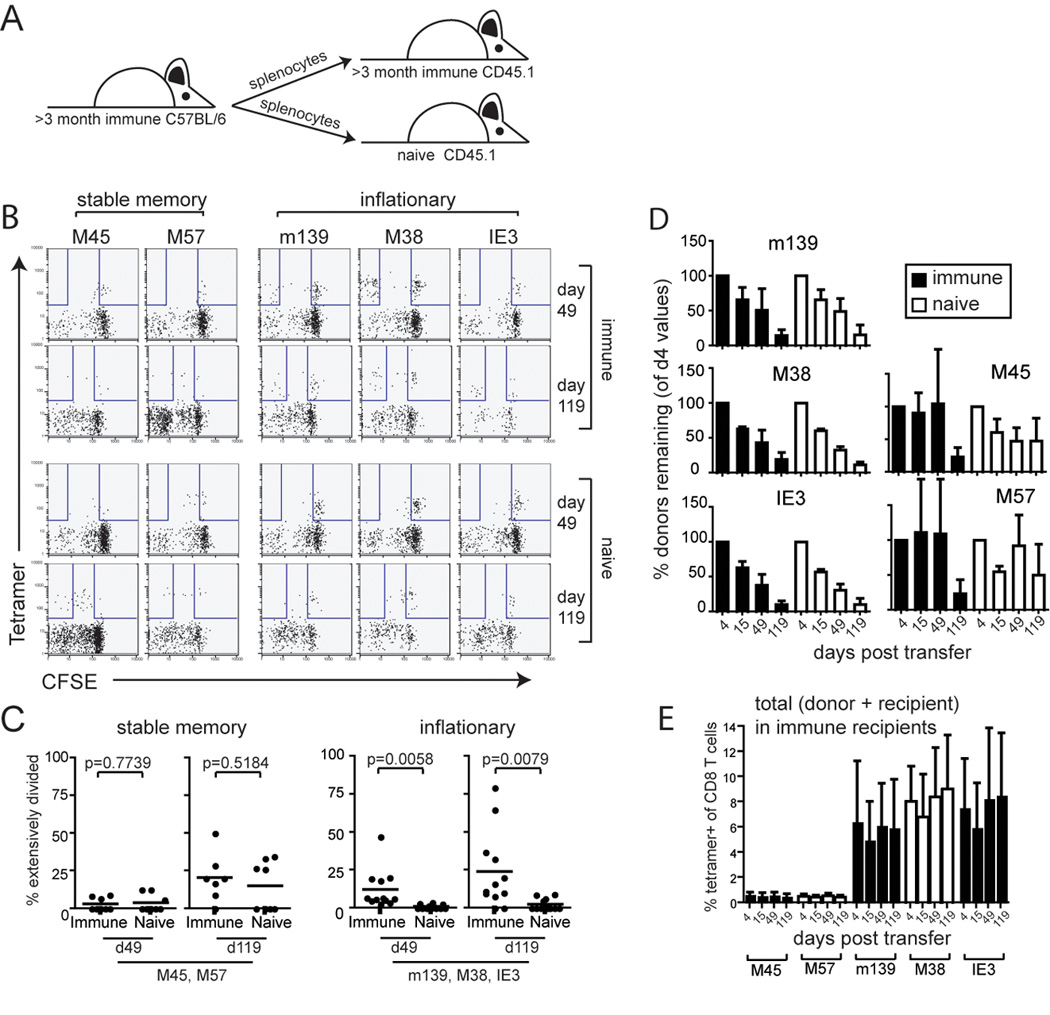
Inflationary T cells decline over time in both immune and naïve recipients. A) Splenocytes from C57BL/6 mice infected for >3 months were harvested and transferred into infection matched CD45.1 congenic recipients. Donor, tetramer-positive cells were tracked over time as a percentage of all CD8 cells in the peripheral blood of recipients. B) Extensively proliferated populations of inflationary T cells (CFSE negative based on recipient cells) can be detected in infected recipients, but fail to accumulate over time. Shown are FACS plots at two time points from the same individual mice gated on donor CD8 cells. C) Extensively proliferated populations within the inflated memory T cell pools are only evident in infected animals. Shown is the percentage of extensively divided tetramer positive T cells (CFSE negative as shown in 4B) specific for either stable (left panels) or inflationary (right panels) T cells. The Student's t test was used to assess significance. D) Inflationary T cells decline over time in both infected and naïve recipients. When measured as a percentage of all CD8 T cells, donor CD8 T cells engrafted ~1.5 to 2x better in naïve recipients compared to immune recipients. To correct for these differences in the "take" of the donor population, the percentage of the donor MCMV-specific T cells was normalized to 100% at the first time point. The error bars represent the standard deviation from the average. T cell half-life was estimated by the length of time it took for the population to be reduced by 50% in circulation, relative to all CD8 T cells. E) The total percentage of MCMV-specific T cells is stable after transfer. Shown is the total (donor + recipient) tetramer staining population from the same mice as in D. Error bars represent standard deviation.
Very little division was seen in any population at early times. At late times post transfer, most of the M45 and M57-specific T cells had undergone some division, but only a few cells had fully diluted their CFSE. The extent of division was the same in naïve and infected recipients and is consistent with homeostatic turnover (Figure 4B and C).
The inflationary T cells (m139, M38 and IE3-specific) failed to divide significantly in naïve recipients at any time point. In contrast, there was clear evidence of antigen-driven proliferation in immune recipients as evidenced by extensive CFSE dilution in a fraction of the inflationary donor cells (Figure 4B and C). Yet division was infrequent and sporadic. Even 4 months post transfer, the majority of the transferred inflationary T cells had not divided (Figure 4B and C) and division within one epitope-specific inflationary population did not correlate with division in the other inflationary populations in the same mouse (Supplemental Figure 3A).
The inflationary T cell populations are exchanged regularly
Despite the sporadic antigen-driven proliferation in infected recipients, the donor inflationary T cells (m139, M38 and IE3-specific) disappeared from the blood of both immune and naïve recipients with an approximate half-life of 45–60 days (Figure 4D). A similar half-life was estimated regardless of whether we counted cells based on IFNγ production or tetramer staining (not shown). The fact that inflationary T cells parked in both immune and naïve recipients decayed at a similar rate suggests that this decay occurred in an antigen independent manner and that the sporadic antigen-driven proliferation seen in Figure 4B had little impact on the size of the total inflationary population. This is supported by the data in Figure 4B, which shows that the divided T cells did not accumulate. The FACS plots for each epitope shown in Figure 4B represent the same recipient at different time points, and it can be seen that there was not a noticeable shift of cells from the undivided to the divided fraction over time. Interestingly, we occasionally saw some mouse to mouse variation in the rate of decline of an individual inflationary population, particularly in immune recipients (Supplemental Figure 3B). However, in only one experiment was an accumulation of any transferred inflationary T cells observed (Supplemental Figure 3C). This example of inflation in the M38-specific population correlated with a large antigen-specific population that had undergone division (CFSE negative). But this was only seen once out of all 3 inflationary T cell responses followed in 15 infected recipients from 4 independent experiments. It is important to note that the overall inflationary populations (donor + recipient) were maintained in all immune mice (Figure 4E), which suggests that the circulating cells are being replaced as they decay.
In contrast to the inflationary donor cells, donor stable memory populations (M45 and M57-specific), largely persisted in naïve recipients, which is consistent with homeostatic cell division (Figure 4D, right side). Interestingly, at very late times, there appeared to be some loss of donor M45 and M57-specific cells in immune recipients.
Similar experiments were set up in parallel to control for antigen-independent effects of viral infection (i.e. cytokine environment, etc.) using a recombinant MCMV that expresses Ovalbumin (MCMV-Ova). The SIINFEKL-specific T cells after MCMV-Ova infection act like inflationary T cells and are extensively differentiated (Supplemental Figure 4A–B). Splenocytes from mice chronically infected with MCMV-Ova were transferred into mice that were naïve, or were infected with wild-type MCMV or MCMV-Ova. As above, extensive division of donor cells was only seen in the presence of antigen (MCMV-Ova infected recipients), but antigen-specific T cells decayed after transfer in all mice (Supplemental Figure 4C–E).
Together these data show that the inflationary T cell populations decay from circulation during chronic MCMV infection. Though we found evidence of antigen-dependent T cell division within the inflated populations, this appeared to be due to sporadic events that may have reduced the rate of decay, but only involved a small number of cells and did not sustain the populations.
Recruitment of naïve cells at late time contributes to the inflated CD8 T cell populations
If most cells in the circulating inflationary population decay, how is the overall population maintained? It has been recently shown that naïve T cells can be recruited during chronic polyomavirus and LCMV clone 13 infections (Kemball et al., 2005; Vezys et al., 2006). To test whether recruitment of naïve T cells occurs during chronic MCMV infection, chronically infected mice were treated with busulfan, a DNA-alkylating agent used to condition recipients for bone marrow transplant. One day after busulfan treatment, mice were given bone marrow from naïve, congenic donors (Figure 5A). In this way, we could track donor-derived lymphocytes that developed in the presence of chronic infection. Importantly, busulfan is thought to have minimal impact on the established memory T cells, allowing us to analyze recruitment of new cells without dramatically disrupting the overall T cell compartment (Kemball et al., 2005; Vezys et al., 2006).
Figure 5.
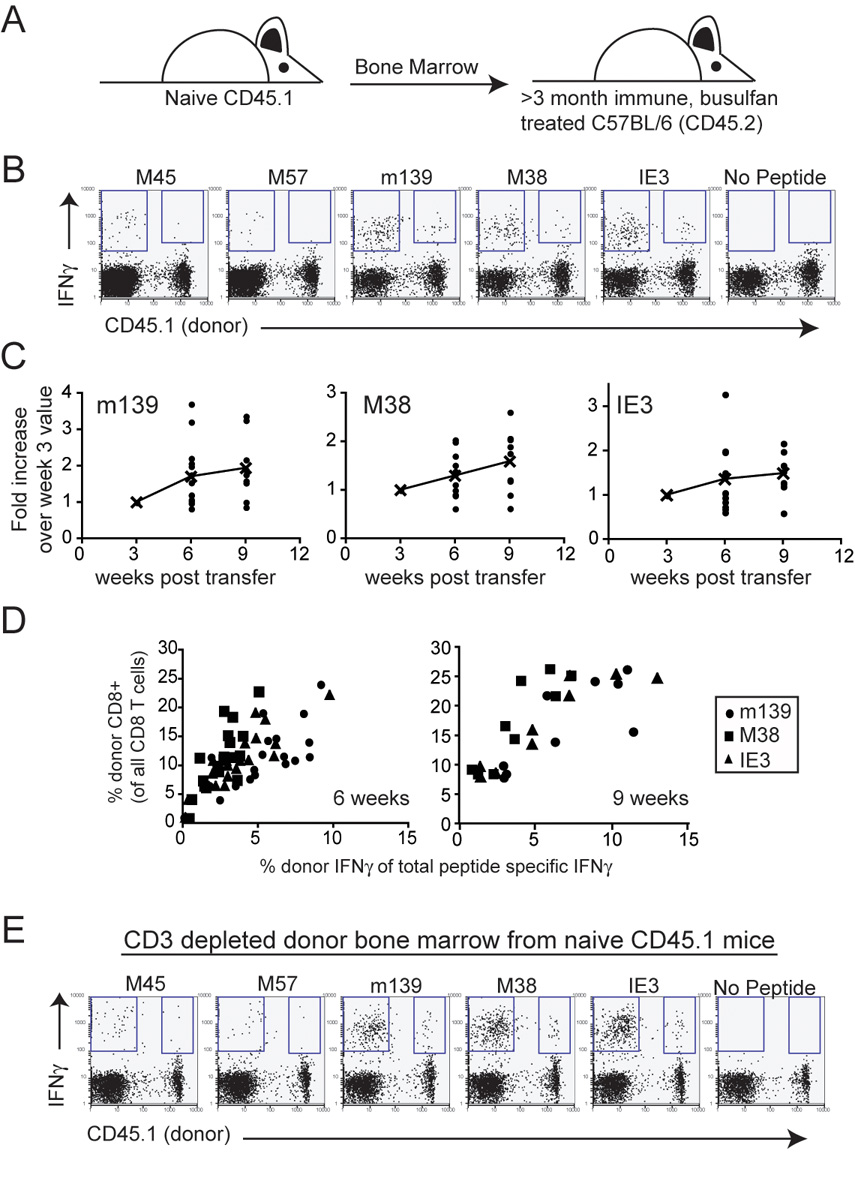
Recruitment of naïve T cells occurs during chronic infection. A) C57BL/6 mice that were infected for >3 months were treated with busulfan and injected with bone marrow from naïve CD45.1 congenic donors. Donor, MCMV-specific T cells in the peripheral blood were tracked by intracellular cytokine staining over time. B) Donor-derived MCMV-specific T cells can be found in all recipients. Shown are representative FACS plots gated on all CD8 T cells from mice that received naïve bone marrow 9 weeks earlier. C) Donor MCMV-specific cells represent an increasing percentage of the total peptide-specific response over time. The graphs show donor-derived cells that produced IFNγ in response to the indicated peptides as a percentage of the total peptide-specific IFNγ response. The data was normalized to the amount of donor-derived IFNγ found at the first time point. The lines represent the average of the donor responses over time. Combined data from three independent experiments is shown. D) The percentage of peptide-specific response that is donor derived correlates with the donor CD8 T cell engraftment. Shown is the same data as in C, but graphed with respect to the percent of all CD8 T cells that are donor-derived in the peripheral blood. E) Naïve T cells that develop in the infected host can be recruited into the inflationary pools. The experiment was carried out as in B, but with CD3-depleted donor bone marrow.
The data shown in Figure 5 clearly demonstrate that naïve T cells are readily recruited into the inflationary T cell pool. As early as 3 weeks after bone marrow transfer, a significant population of donor-derived CD8 T cells could be detected and these increased as a percentage of the total MCMV-specific pool over time (Figure 5B and C). Importantly, the overall amount of recruitment seemed to be dependent on the degree of chimerism achieved in a given mouse (Figure 5D) and the overall T cell responses were not affected by the busulfan treatment (Supplemental Figure 5A). In contrast to the inflationary populations, few if any M45 and M57-specific T cells were derived from the donor, as expected since these populations don't inflate (Figure 5B). Recruitment of cells from the donor marrow into the inflationary pool was also seen when mature T cells were depleted from the donor bone marrow prior to transplantation (Figure 5E and Supplemental 5B). Thus, T cells that develop in the presence of chronic MCMV infection can respond to the ongoing infection and join the inflationary populations.
Cells primed early in infection also contribute to the maintenance of the inflationary populations
If recruitment of naïve T cells were solely responsible for maintaining the inflationary populations, then eventually all inflationary T cells should be replaced from the naïve T cell pool. Thus in mice that developed a high degree of chimerism (most new CD8 T cells were donor-derived), most MCMV-specific T cells eventually should become donor-derived as well. However, we found that even though the majority of all thymocytes were donor-derived in some mice, the percentage of donor MCMV-specific T cells remained low 10 – 11 months post transplant (Figures 6A and B). Although animals with a higher percentage of donor thymocytes had more donor-derived MCMV-specific cells (not shown), the two populations did not equalize, even after more than 5 half-lives of the circulating, differentiated T cells had elapsed. This result suggests that naïve T cells are not essential to maintain the inflationary populations. In addition, memory inflation was not abrogated when mice were infected after thymectomy, confirming these results (Andrea Loewendorf, Ramon Arens and Chris Benedict, personal communication). These results were initially puzzling, but an experiment originally performed for a different reason suggested an explanation. We transferred splenocytes from 7 day infected mice into infection matched, congenic recipients. This time point represents the peak of T cell expansion after acute infection. Donor cells were measured in the peripheral blood of recipients 2 weeks and 6–7 months post transfer (Figures 6C, D and E). As expected, donor M45-specific T cells persisted in most recipients. Strikingly however, many donor inflationary T cells also persisted or inflated in these recipients, even after what should have been 3 – 4 half lives based on our previous findings (compare Figure 6E to Figure 4D). This was most evident for m139-specific T cells, which underwent dramatic inflation or were maintained in most mice (Figure 6E). Persistence of M38-specific donor T cells was less obvious, but was also seen in several animals. Strikingly, donor-derived IE3-specific T cells, which are almost undetectable at day 7 post infection when the transfer was performed, were found to have inflated in several mice. Thus, at day 7 post infection, we were able to transfer MCMV-specific cells whose progeny could replenish the inflationary pool, whereas such cells were not transferred with splenocytes from chronically infected mice. Together, these data show that the circulating, short-lived inflationary T cell populations can be replaced by the progeny of cells primed during acute infection as well as naïve T cells primed at late time points.
Figure 6.
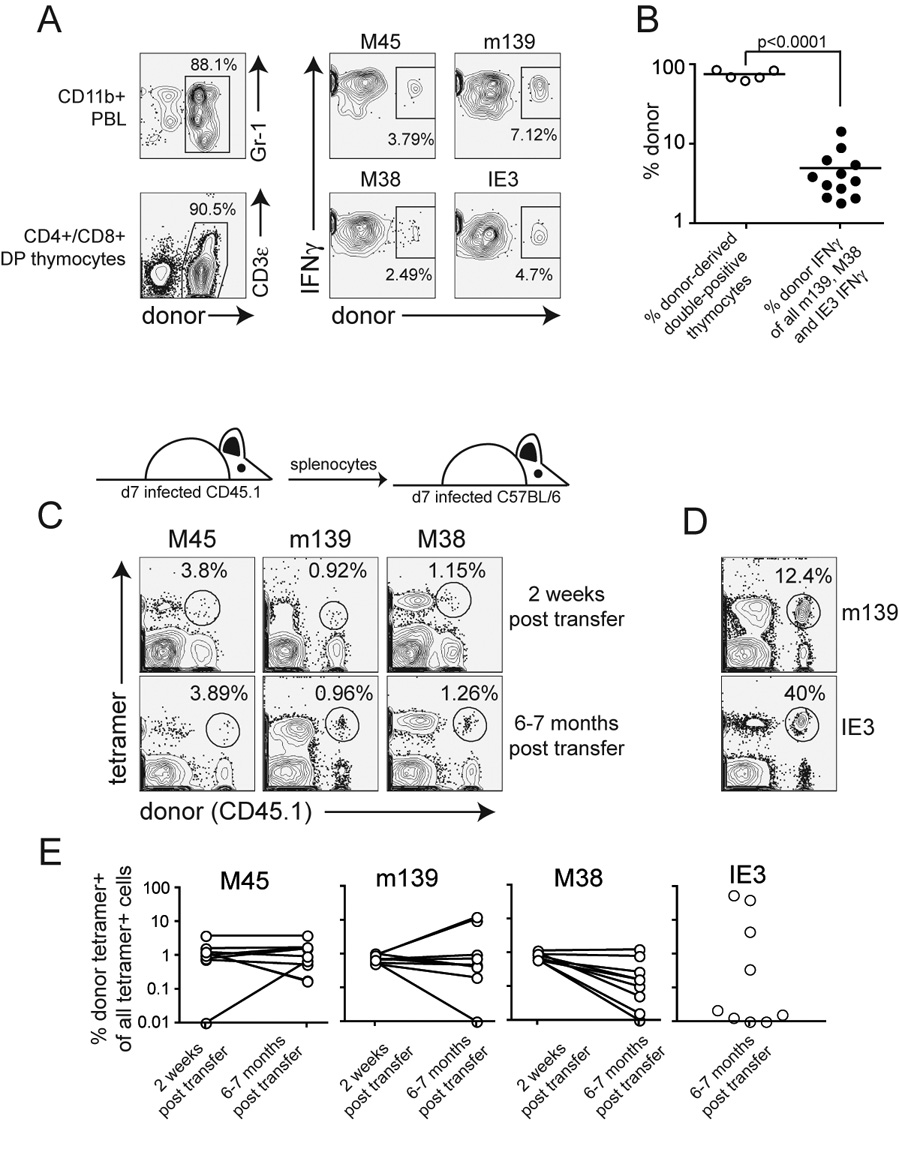
Naive T cells do not account for the replacement of all decaying circulating differentiated T cells. A) One mouse in which most of the newly produced T cells were donor derived, but most MCMV-specific T cells were recipient derived 10 months post transplant. B) MCMV-specific chimerism is shown for several mice with a high level of chimerism within double-positive thymocytes C) Donor MCMV-specific cells transferred at the peak of T cell expansion can persist or inflate. Sample FACS plots are from the same mouse at each time point. The numbers indicate the percent of all tetramer+ cells that are donor derived. D) Two examples of dramatic inflation of the donor MCMV-specific T cell populations 7 months after transfer. E) Combined data (as in C and D) from multiple mice and two independent experiments. Shown is the donor tetramer+ population as a percent of all tetramer+ T cells to adjust for inflation of the m139 and M38 populations over time. Lines connect individual mice at either 2 weeks post transfer (3 weeks post infection) or 6–7 months post transfer. IE3-specific T cells were not measured at 2 weeks post transfer.
Discussion
Our work demonstrates that the inflationary MCMV-specific T cell populations are highly dynamic. Most inflationary T cells in the spleen and blood appear to be functional but are short-lived, disappearing from circulation with a half-life of approximately 45 to 60 days. As they decay, these cells are constantly being replaced by an influx of differentiated cells that derive from at least two sources: cells that were primed early in infection but are not transferred at late times, and naïve T cells. In our model (Figure 7), cells primed early in infection will consistently produce progeny that can replace the decaying short-lived T cells in circulation. This may be the primary way that inflationary populations are maintained, resulting in the persistence of some clones over time even as individual cells within those clones decay. However, naïve T cells can also be primed during chronic infection and may effectively alter the clonality of the population over time. Although naïve T cells are not essential to maintain inflationary populations, the impact of naïve T cell recruitment is unclear and may depend on the time post infection and the age of the animal. Regardless, it is clear that the populations are highly dynamic and individual analyses of circulating cells reveal only snapshots of a complex process that also includes a small amount of antigen driven cell division. It is possible that the disappearance of the short-lived populations from the blood indicates a migration into tissues over this time. Indeed, we have seen large numbers of inflationary T cells in the lungs (not shown). However if this was the cause of the loss of donor cells from the circulation, we would have to conclude that these cells rarely re-enter the blood after extravasation and likely die in the tissues. Significantly, most HCMV-specific T cells also bear a highly differentiated phenotype, similar to that described here for the majority of inflationary T cells (Gamadia et al., 2001; Kuijpers et al., 2003; van Leeuwen et al., 2005; van Leeuwen et al., 2004; Weekes et al., 1999a; Weekes et al., 1999b) and some, but not all HCMV-specific T cell clones, have been shown to persist over time (Day et al., 2007; Weekes et al., 1999a), agreeing with our results.
Figure 7.
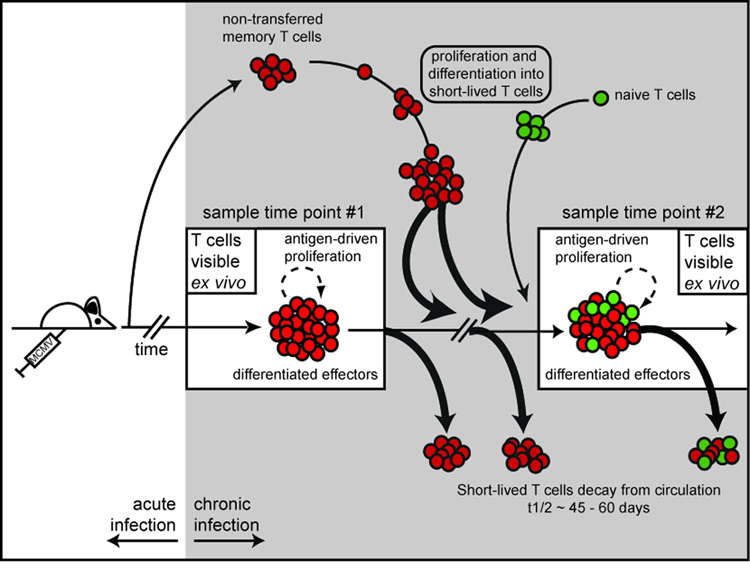
Model of the dynamics of the inflationary MCMV-specific T cell populations. Ex vivo measurement of MCMV specific T cells at a given time point represents a snap-shot of a highly dynamic population. These T cells undergo a small amount of antigen-driven cell division, but most cells disappear from the blood with a half-life of ~45 to 60 days. The decaying cells are replaced primarily by the progeny of cells that were primed early in infection. We would expect these clones to persist as the individual decaying cells are replaced by the progeny of identical clones that were primed early in infection. However the population is also supported by the recruitment of naïve T cells at late times. As new cells are recruited, we predict that the clonal composition of each inflationary population will change over time. Though the inflationary populations may appear similar at any given time point, our evidence shows that these populations are constantly in flux.
We can imagine three possible explanations for our failure to transfer memory from chronically infected mice. First, it is possible that the memory cells are not present in the spleen at late times. They may reside selectively in some other compartment, perhaps the bone marrow or lymph nodes, and continuously produce the differentiated cells that we see in circulation at late times. Second, it is possible that the memory cells cannot find a niche to survive in after transfer. If all of the available niches are occupied in chronically, but not acutely infected mice, we would expect donor memory cells to die shortly after transfer. Finally, it is possible that the memory cells are not extracted from the spleen before transfer, perhaps remaining associated with the splenic stroma when the spleens were crushed through a filter. Experiments are ongoing to discriminate between these possibilities, and should enhance our understanding of the nature of T cell memory.
The phenotype of the circulating inflationary MCMV-specific T cells resembles cells that have been repeatedly stimulated with antigen and not allowed to rest (Jabbari and Harty, 2006; Masopust et al., 2006). However, we saw no reversion of these cells to a more "central memory" phenotype after transfer into naïve animals suggesting that the inflationary T cells may be terminally differentiated (not shown). Yet, these inflationary cells clearly retained the capacity to divide in response to viral antigen (Figure 3) even though they largely failed to do so during chronic infection (Figure 4). This lack of proliferation may reflect the fact that differentiated inflationary T cells express low levels of costimulatory molecules and high levels of inhibitory molecules (Figure 2), a combination that should increase the activation threshold for these cells. Recent data indicating that expression of 4-1BBL by APCs is essential to drive the proliferation of HCMV-specific T cells supports the idea that CMV-specific T cells have "special" stimuation requirements (Waller et al., 2007).
It is interesting to speculate that MCMV, which is a natural murine pathogen, may have evolved to allow cells to be replaced as they become extensively differentiated, rather than overstimulating an existing population. This could allow T cell immunity to be maintained without exhaustion so that the virus and its host can both persist. It is likely however, that many factors influence this equilibrium. First, the inflammatory environment of the host clearly increases viral reactivation from latency (Cook et al., 2006; Haagmans et al., 1994a; Haagmans et al., 1994b; Prosch et al., 1995; Simon et al., 2005; Stein et al., 1993). Therefore, any ongoing infections are likely to drive more viral replication. In addition, there are likely to be age-associated effects on the host-virus relationship. As thymic output decreases, the host may be more dependent on the memory and short-lived T cell populations. Indeed, CMV infection is associated with dramatic T cell clonal expansions in aged people (Khan et al., 2002b; Ouyang et al., 2003).
In contrast to the inflationary populations, cells specific for M45 and M57 appear to be more similar to classical central memory T cells and we were clearly able to transfer self-maintaining populations into naïve mice. While the cause for this dichotomy between stable and inflationary cells is not clear, we speculate that it reflects different patterns of viral gene expression during acute infection and latency. Interestingly, upon viral reactivation, patterns of gene expression vary dramatically in different tissues (Streblow et al., 2007).
Overall, our data describe a dynamic host-virus balance that may shed light on the behavior of T cells in the face of chronic antigen. Our data suggests that antigen persistence alone is insufficient to induce T cell dysfunction and that the context of antigen presentation (that which determines whether previously primed T cells are driven to proliferate) is likely to play a major role in determining T cell fate during chronic infections.
Experimental Procedures
Mice
C57BL/6 mice were purchased from The Jackson Laboratory. B6.SJL-Ptprca Pepcb/BoyJ (B6.SJL - CD45.1 congenic) and B6.CB17-Prkdcscid/SzJ (B6.SCID) were also initially purchased from The Jackson Laboratory and then bred in house. Mice were between the ages of 6 and 16 weeks and all studies were approved by the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee at OHSU.
Virus Strains and Infections
Unless otherwise indicated, mice were infected i.p. with 1×106 pfu of MCMV strain MW97.01, which is derived from a bacterial artificial chromosome of the Smith strain (Wagner et al., 1999). In some experiments, recombinant MCMV expressing ovalbumin (Ova) was used. This virus was produced as described elsewhere (Lloyd et al., 2003). In all cases, virus stocks were grown on mouse embryo fibroblasts which were sonicated or dounced to release the viral particles.
Tetramer Staining, Antibodies, Intracellular Cytokine Stimulation Assay and FACS analysis
Tetramers were synthesized by the NIH tetramer core facility (http://www.niaid.nih.gov/reposit/tetramer/overview.html). Tetramer staining was performed on 50µl of whole blood or 1×106 splenocytes. Red blood cells were lysed with BD lysis buffer after staining (Becton Dickenson). Various fluorescently conjugated antibodies were used [CD8α(53-6.7), CD27 (LG.7F9), CD28 (37.51), CD43 (1b11), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD62L (MEL-14), CD122 (TM-b1), CD127 (A7R34), KLRG-1 (2F1), NKG2A/C/E (20d5), NKG2D (C7), PD-1 (RMP1-30)]. For measurement of intracellular IFNγ, red blood cells were lysed with 3 ml of lysis buffer (150mM NH4Cl, 10mM NaHCO3) and the remaining cells were stimulated and stained as described previously (Munks et al., 2006a; Munks et al., 2006b). In all cases, samples were acquired on a LSR II or FACSCalibur (both from BD) and analyzed with FlowJo software (Tree Star).
Adoptive Transfers and Bone Marrow Chimeras
For in vivo CTL assays, CD45.1 congenic splenocytes were stained with 2µM, 0.5µM or 0.1µM CFSE, loaded with 2µM of the indicated peptide and i.v. transferred into C57BL/6 recipients that were infected for >3 months or naïve controls. FACS analysis of the surviving donor cells (CD45.1 positive) in the spleen of recipient mice was performed approximately 18 hours later.
For adoptive transfer of mature T cells, splenocytes from congenic mice were labeled with 1 µM CFSE, suspended in PBS and injected i.v. into recipient mice. Typically, recipients were given 2 – 5 × 107 unfractionated splenocytes per injection. In one experiment (Figure 3C), CD8 T cells were purified by negative selection (EasySep, Stem Cell Technologies) and then sorted for CD8+, CD27 hi and low cells using a FACSVantage cell sorter (Becton Dickenson). Sorted cells were i.v. transferred into naïve, congenic mice and recipients were infected 1 hour later.
To measure recruitment of naïve T cells, we adapted a protocol recently described (Kemball et al., 2005; Vezys et al., 2006). Briefly, chronically infected mice were injected i.p. with 600 µg of busulfan dissolved in 50% DMSO. The following day, 2×107 whole or CD3-depleted bone marrow cells from naïve CD45 congenic mice were injected i.v. into treated mice. CD3 depletion was carried out using 1.5µg of PE conjugated CD3ε per 1×108 bone marrow cells and the PE selection kit (EasySep, Stem Cell Technologies) following the manufacturer's recommended protocol.
Supplementary Material
01
Acknowledgments
This work was supported by an American Foundation for Aging Research grant and RO1 A I47206 awarded to A.B.H. as well as a post-doctoral training grant from the American Heart Association (# 0725786Z) awarded to C.M.S. The authors declare that they have no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- Avetisyan G, Larsson K, Aschan J, Nilsson C, Hassan M, Ljungman P. Impact on the cytomegalovirus (CMV) viral load by CMV-specific T-cell immunity in recipients of allogeneic stem cell transplantation. Bone Marrow Transplant. 2006;38:687–692. doi: 10.1038/sj.bmt.1705507. [DOI] [PubMed] [Google Scholar]
- Boeckh M, Leisenring W, Riddell SR, Bowden RA, Huang ML, Myerson D, Stevens-Ayers T, Flowers ME, Cunningham T, Corey L. Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood. 2003;101:407–414. doi: 10.1182/blood-2002-03-0993. [DOI] [PubMed] [Google Scholar]
- Cobbold M, Khan N, Pourgheysari B, Tauro S, McDonald D, Osman H, Assenmacher M, Billingham L, Steward C, Crawley C, et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J Exp Med. 2005;202:379–386. doi: 10.1084/jem.20040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook CH, Trgovcich J, Zimmerman PD, Zhang Y, Sedmak DD. Lipopolysaccharide, tumor necrosis factor alpha, or interleukin-1 beta triggers reactivation of latent cytomegalovirus in immunocompetent mice. J Virol. 2006;80:9151–9158. doi: 10.1128/JVI.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Day EK, Carmichael AJ, ten Berge IJ, Waller EC, Sissons JG, Wills MR. Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol. 2007;179:3203–3213. doi: 10.4049/jimmunol.179.5.3203. [DOI] [PubMed] [Google Scholar]
- Gamadia LE, Rentenaar RJ, Baars PA, Remmerswaal EB, Surachno S, Weel JF, Toebes M, Schumacher TN, ten Berge IJ, van Lier RA. Differentiation of cytomegalovirus-specific CD8(+) T cells in healthy and immunosuppressed virus carriers. Blood. 2001;98:754–761. doi: 10.1182/blood.v98.3.754. [DOI] [PubMed] [Google Scholar]
- Gamadia LE, van Leeuwen EM, Remmerswaal EB, Yong SL, Surachno S, Wertheim-van Dillen PM, Ten Berge IJ, Van Lier RA. The size and phenotype of virus-specific T cell populations is determined by repetitive antigenic stimulation and environmental cytokines. J Immunol. 2004;172:6107–6114. doi: 10.4049/jimmunol.172.10.6107. [DOI] [PubMed] [Google Scholar]
- Gillespie GM, Wills MR, Appay V, O'Callaghan C, Murphy M, Smith N, Sissons P, Rowland-Jones S, Bell JI, Moss PA. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol. 2000;74:8140–8150. doi: 10.1128/jvi.74.17.8140-8150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans BL, Stals FS, van der Meide PH, Bruggeman CA, Horzinek MC, Schijns VE. Tumor necrosis factor alpha promotes replication and pathogenicity of rat cytomegalovirus. Journal of Virology. 1994a;68:2297–2304. doi: 10.1128/jvi.68.4.2297-2304.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans BL, van der Meide PH, Stals FS, van den Eertwegh AJ, Claassen E, Bruggeman CA, Horzinek MC, Schijns VE. Suppression of rat cytomegalovirus replication by antibodies against gamma interferon. Journal of Virology. 1994b;68:2305–2312. doi: 10.1128/jvi.68.4.2305-2312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med. 2007;204:1625–1636. doi: 10.1084/jem.20070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtappels R, Pahl-Seibert MF, Thomas D, Reddehase MJ. Enrichment of immediate-early 1 (m123/pp89) peptide-specific CD8 T cells in a pulmonary CD62L(lo) memory-effector cell pool during latent murine cytomegalovirus infection of the lungs. Journal of Virology. 2000;74:11495–11503. doi: 10.1128/jvi.74.24.11495-11503.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtappels R, Podlech J, Grzimek NK, Thomas D, Pahl-Seibert MF, Reddehase MJ. Experimental preemptive immunotherapy of murine cytomegalovirus disease with CD8 T-cell lines specific for ppM83 and pM84, the two homologs of human cytomegalovirus tegument protein ppUL83 (pp65) J Virol. 2001;75:6584–6600. doi: 10.1128/JVI.75.14.6584-6600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtappels R, Thomas D, Podlech J, Reddehase MJ. Two antigenic peptides from genes m123 and m164 of murine cytomegalovirus quantitatively dominate CD8 T-cell memory in the H-2d haplotype. J Virol. 2002;76:151–164. doi: 10.1128/JVI.76.1.151-164.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibegbu CC, Xu YX, Harris W, Maggio D, Miller JD, Kourtis AP. Expression of killer cell lectin-like receptor G1 on antigen-specific human CD8+ T lymphocytes during active, latent, and resolved infection and its relation with CD57. J Immunol. 2005;174:6088–6094. doi: 10.4049/jimmunol.174.10.6088. [DOI] [PubMed] [Google Scholar]
- Jabbari A, Harty JT. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J Exp Med. 2006;203:919–932. doi: 10.1084/jem.20052237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170:2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- Karrer U, Wagner M, Sierro S, Oxenius A, Hengel H, Dumrese T, Freigang S, Koszinowski UH, Phillips RE, Klenerman P. Expansion of protective CD8+ T-cell responses driven by recombinant cytomegaloviruses. Journal of Virology. 2004;78:2255–2264. doi: 10.1128/JVI.78.5.2255-2264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Lee ED, Vezys V, Pearson TC, Larsen CP, Lukacher AE. Late priming and variability of epitope-specific CD8+ T cell responses during a persistent virus infection. J Immunol. 2005;174:7950–7960. doi: 10.4049/jimmunol.174.12.7950. [DOI] [PubMed] [Google Scholar]
- Khan N, Cobbold M, Keenan R, Moss PA. Comparative analysis of CD8+ T cell responses against human cytomegalovirus proteins pp65 and immediate early 1 shows similarities in precursor frequency, oligoclonality, and phenotype. J Infect Dis. 2002a;185:1025–1034. doi: 10.1086/339963. [DOI] [PubMed] [Google Scholar]
- Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002b;169:1984–1992. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- Kuijpers TW, Vossen MT, Gent MR, Davin JC, Roos MT, Wertheim-van Dillen PM, Weel JF, Baars PA, van Lier RA. Frequencies of circulating cytolytic, CD45RA+CD27-, CD8+ T lymphocytes depend on infection with CMV. J Immunol. 2003;170:4342–4348. doi: 10.4049/jimmunol.170.8.4342. [DOI] [PubMed] [Google Scholar]
- Lang KS, Moris A, Gouttefangeas C, Walter S, Teichgraber V, Miller M, Wernet D, Hamprecht K, Rammensee HG, Stevanovic S. High frequency of human cytomegalovirus (HCMV)-specific CD8+ T cells detected in a healthy CMV-seropositive donor. Cell Mol Life Sci. 2002;59:1076–1080. doi: 10.1007/s00018-002-8488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd ML, Shellam GR, Papadimitriou JM, Lawson MA. Immunocontraception is induced in BALB/c mice inoculated with murine cytomegalovirus expressing mouse zona pellucida 3. Biol Reprod. 2003;68:2024–2032. doi: 10.1095/biolreprod.102.012880. [DOI] [PubMed] [Google Scholar]
- Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006a;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- Munks MW, Gold MC, Zajac AL, Doom CM, Morello CS, Spector DH, Hill AB. Genome-wide analysis reveals a highly diverse CD8 T cell response to murine cytomegalovirus. J Immunol. 176:3760–3766. doi: 10.4049/jimmunol.176.6.3760. [DOI] [PubMed] [Google Scholar]
- Munks MW, Pinto AK, Doom CM, Hill AB. Viral interference with antigen presentation does not alter acute or chronic CD8 T cell immunodominance in murine cytomegalovirus infection. J Immunol. 2007;178:7235–7241. doi: 10.4049/jimmunol.178.11.7235. [DOI] [PubMed] [Google Scholar]
- Ouyang Q, Wagner WM, Voehringer D, Wikby A, Klatt T, Walter S, Muller CA, Pircher H, Pawelec G. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1) Exp Gerontol. 2003;38:911–920. doi: 10.1016/s0531-5565(03)00134-7. [DOI] [PubMed] [Google Scholar]
- Pahl-Seibert MF, Juelch M, Podlech J, Thomas D, Deegen P, Reddehase MJ, Holtappels R. Highly protective in vivo function of cytomegalovirus IE1 epitope-specific memory CD8 T cells purified by T-cell receptor-based cell sorting. J Virol. 2005;79:5400–5413. doi: 10.1128/JVI.79.9.5400-5413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosch S, Staak K, Stein J, Liebenthal C, Stamminger T, Volk HD, Kruger DH. Stimulation of the human cytomegalovirus IE enhancer/promoter in HL-60 cells by TNFalpha is mediated via induction of NF-kappaB. Virology. 1995;208:197–206. doi: 10.1006/viro.1995.1143. [DOI] [PubMed] [Google Scholar]
- Reddehase MJ, Jonjic S, Weiland F, Mutter W, Koszinowski UH. Adoptive immunotherapy of murine cytomegalovirus adrenalitis in the immunocompromised host: CD4-helper-independent antiviral function of CD8-positive memory T lymphocytes derived from latently infected donors. J Virol. 1988;62:1061–1065. doi: 10.1128/jvi.62.3.1061-1065.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reusser P, Riddell SR, Meyers JD, Greenberg PD. Cytotoxic T-lymphocyte response to cytomegalovirus after human allogeneic bone marrow transplantation: pattern of recovery and correlation with cytomegalovirus infection and disease. Blood. 1991;78:1373–1380. [PubMed] [Google Scholar]
- Shin H, Blackburn SD, Blattman JN, Wherry EJ. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med. 2007;204:941–949. doi: 10.1084/jem.20061937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro S, Rothkopf R, Klenerman P. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. European Journal of Immunology. 2005;35:1113–1123. doi: 10.1002/eji.200425534. [DOI] [PubMed] [Google Scholar]
- Simon CO, Holtappels R, M. TH, V. B, T. D, A. O-KS, B. K, A. R, D. S, Podlech J, et al. CD8 T Cells Control Cytomegalovirus Latency by Epitope- Specific Sensing of Transcriptional Reactivation. Journal of Virology. 2006 doi: 10.1128/JVI.01248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CO, Seckert CK, Dreis D, Reddehase MJ, Grzimek NK. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. Journal of Virology. 2005;79:326–340. doi: 10.1128/JVI.79.1.326-340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens HP, Kurz S, Holtappels R, Reddehase MJ. Preemptive CD8 T-cell immunotherapy of acute cytomegalovirus infection prevents lethal disease, limits the burden of latent viral genomes, and reduces the risk of virus recurrence. J Virol. 1998;72:1797–1804. doi: 10.1128/jvi.72.3.1797-1804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein J, Volk HD, Liebenthal C, Kruger DH, Prosch S. Tumour necrosis factor alpha stimulates the activity of the human cytomegalovirus major immediate early enhancer/promoter in immature monocytic cells. Journal of General Virology. 1993;74:2333–2338. doi: 10.1099/0022-1317-74-11-2333. [DOI] [PubMed] [Google Scholar]
- Streblow DN, van Cleef KW, Kreklywich CN, Meyer C, Smith P, Defilippis V, Grey F, Fruh K, Searles R, Bruggeman C, et al. Rat cytomegalovirus gene expression in cardiac allograft recipients is tissue specific and does not parallel the profiles detected in vitro. J Virol. 2007;81:3816–3826. doi: 10.1128/JVI.02425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimme R, Appay V, Koschella M, Panther E, Roth E, Hislop AD, Rickinson AB, Rowland-Jones SL, Blum HE, Pircher H. Increased expression of the NK cell receptor KLRG1 by virus-specific CD8 T cells during persistent antigen stimulation. J Virol. 2005;79:12112–12116. doi: 10.1128/JVI.79.18.12112-12116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- van Leeuwen EM, de Bree GJ, Remmerswaal EB, Yong SL, Tesselaar K, ten Berge IJ, van Lier RA. IL-7 receptor alpha chain expression distinguishes functional subsets of virus-specific human CD8+ T cells. Blood. 2005;106:2091–2098. doi: 10.1182/blood-2005-02-0449. [DOI] [PubMed] [Google Scholar]
- van Leeuwen EM, Gamadia LE, Baars PA, Remmerswaal EB, ten Berge IJ, van Lier RA. Proliferation requirements of cytomegalovirus-specific, effector-type human CD8+ T cells. J Immunol. 2002;169:5838–5843. doi: 10.4049/jimmunol.169.10.5838. [DOI] [PubMed] [Google Scholar]
- van Leeuwen EM, Remmerswaal EB, Vossen MT, Rowshani AT, Wertheim-van Dillen PM, van Lier RA, ten Berge IJ. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J Immunol. 2004;173:1834–1841. doi: 10.4049/jimmunol.173.3.1834. [DOI] [PubMed] [Google Scholar]
- van Leeuwen EM, ten Berge IJ, van Lier RA. Induction and maintenance of CD8+ T cells specific for persistent viruses. Adv Exp Med Biol. 2007;590:121–137. doi: 10.1007/978-0-387-34814-8_9. [DOI] [PubMed] [Google Scholar]
- Vezys V, Masopust D, Kemball CC, Barber DL, O'Mara LA, Larsen CP, Pearson TC, Ahmed R, Lukacher AE. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection. J Exp Med. 2006;203:2263–2269. doi: 10.1084/jem.20060995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Jonjic S, Koszinowski UH, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus econstitution. Journal of Virology. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller EC, McKinney N, Hicks R, Carmichael AJ, Sissons JG, Wills MR. Differential costimulation through CD137 (4 1BB) restores proliferation of human virus-specific "effector memory" (CD28 CD45RAHI) CD8+ T cells. Blood. 2007;110:4360–4366. doi: 10.1182/blood-2007-07-104604. [DOI] [PubMed] [Google Scholar]
- Weekes MP, Carmichael AJ, Wills MR, Mynard K, Sissons JG. Human CD28-CD8+ T cells contain greatly expanded functional virus-specific memory CTL clones. J Immunol. 1999a;162:7569–7577. [PubMed] [Google Scholar]
- Weekes MP, Wills MR, Mynard K, Hicks R, Sissons JG, Carmichael AJ. Large clonal expansions of human virus-specific memory cytotoxic T lymphocytes within the CD57+ CD28- CD8+ T-cell population. Immunology. 1999b;98:443–449. doi: 10.1046/j.1365-2567.1999.00901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci U S A. 2004;101:16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01