Systems Approaches to Preventing Transplanted Cell Death in Cardiac Repair (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 1.
Published in final edited form as: J Mol Cell Cardiol. 2008 Mar 19;45(4):567–581. doi: 10.1016/j.yjmcc.2008.03.009
Abstract
Stem cell transplantation may repair the injured heart, but tissue regeneration is limited by death of transplanted cells. Most cell death occurs in the first few days post-transplantation, likely from a combination of ischemia, anoikis and inflammation. Interventions known to enhance transplanted cell survival include heat shock, over-expressing anti-apoptotic proteins, free radical scavengers, anti-inflammatory therapy and co-delivery of extracellular matrix molecules. Combinatorial use of such interventions markedly enhances graft cell survival, but death still remains a significant problem. We review these challenges to cardiac cell transplantation and present an approach to systematically address them.
Most anti-death studies use histology to assess engraftment, which is time- and labor-intensive. To increase throughput, we developed two biochemical approaches to follow graft viability in the mouse heart. The first relies on LacZ enyzmatic activity to track genetically modified cells, and the second quantifies human genomic DNA content using repetitive Alu sequences. Both show linear relationships between input cell number and biochemical signal, but require correction for the time lag between cell death and loss of signal. Once optimized, they permit detection of as few as 1 graft cell in 40,000 host cells. Pro-survival effects measured biochemically at three days predict long-term histological engraftment benefits. These methods permitted identification of carbamylated erythropoietin (CEPO) as a pro-survival factor for human embryonic stem cell-derived cardiomyocyte grafts. CEPO’s effects were additive to heat shock, implying independent survival pathways. This system should permit combinatorial approaches to enhance graft viability in a fraction of the time required for conventional histology.
I. Review of the Literature
Introduction
Cell-based cardiac repair aims to correct the root cause of heart failure’s symptoms, namely the injured heart’s inability to adequately pump blood due to insufficient muscle mass. Many strategies being pursued to restore heart function involve repopulating the heart with exogenously delivered cells, either by direct injection [1, 2] or intravascular delivery. The first attempts to repopulate the scarred aftermath of myocardial infarction (MI) were performed over fifteen years ago [3–5]. Since that time, interest in cell transplantation for the prevention and treatment of heart failure has exploded, leading to multiple animal studies and clinical trials [2]. These studies showed that a panoply of cells injected into the myocardium both can change the nature of the scar and improve post-infarction cardiac function. Skeletal myoblasts [6–8], fetal or neonatal cardiomyocytes [9–11], fibroblasts [12], smooth muscle cells [13], hematopoietic stem cells [14, 15], mesenchymal stem cells [16], endothelial cells [17], resident cardiac progenitor cells [18, 19], and derivatives of human embryonic stem cells [20–24] have all been transplanted into animal hearts. While it is clear that functional benefit is conferred from numerous cell types, the mechanism of action is unclear. Electromechanical linkage has been disproven in some models [25], while mechanical buttressing [26] and the so-called paracrine effect [17, 27] may be responsible for improved myocardial function. No matter if synchronous graft contraction with host myocardium is the clinician’s goal or if other mechanisms of benefit are desired, efficient delivery of viable cells into the heart is a critical requirement for repopulating the MI, regardless of the cell source.
Several human clinical trials testing cell-based cardiac repair have been conducted, the progress of which has been reviewed elsewhere [2, 28]. Despite the promising preclinical benefits of cell therapy, success in these initial clinical trials has been modest, at best. For example, the MAGIC II trial of skeletal myoblast transplantation into chronic infarcts was terminated early by the study’s data safety monitoring board, because the treatment offered no benefit on ejection fraction compared to placebo [29]. Similarly, delivery of bone marrow cells to patients with acute MI has produced mixed results, with two negative trials [30, 31], one showing transient benefit [32] and one showing sustained benefit [33]. The reasons for this failure to translate preclinical results into benefits in humans are not known, but a good candidate is the vast difference in cell number required for human therapy compared to rodents.
In order to prevent or treat heart failure resulting from a myocardial infarct, the newly formed tissue needs to replace a substantial fraction of that lost to infarction. Indeed, studies in skeletal myoblast transplantation indicate that graft size is directly correlated with functional improvement [34, 35]. A few calculations are helpful in assessing the scope of regenerating a human myocardial infarct. Myocardium contains approximately 20 million cardiomyocytes per gram of tissue [36]. The average left ventricle is ~200g and therefore contains ~4 billion cardiomyocytes. In order to cause heart failure, an infarct needs to kill ~25% of the ventricle (for comparison, infarcting 40% of the ventricle results in acute cardiogenic shock [37]). Therefore, the myocyte deficit in infarction-induced heart failure is on the order of 1 billion cardiomyocytes.
When a proliferating population of cells is delivered to the heart, the final graft population can be estimated according to the equation:
Graftpopulation=(numbersuccessfullydelivered)(fractionsurviving)(1+prolif.fract.)n
where prolif. fract. is the fraction of those cells that proliferate after transplantation and n is the number of generations of proliferation.
When one injects cells directly into the heart, seeding efficiency is quite variable, ranging from 0–90%, with an average of 40–50% in the rat [38] and ~10% in the pig [39]. The survival fraction depends on the specific cell type, the number of cells injected (where the more one injects, the greater fraction dies off), and the status of the host tissue (with ischemic, inflamed tissues being less hospitable than normal myocardium [40]). The Yacoub group reported that 7% of skeletal myoblasts survive 3 days after grafting into the infarcted mouse heart [41], and Kloner’s group found 28% of a neonatal cardiomyocyte preparation (that also included non-myocytes) survived one week after grafting into normal rat hearts [38]. Similar results were found using different cell types. Using smooth muscle cells Yasuda et al reported viable graft cell counts of 15% at 1 week and 9% at 4 weeks after permanent LAD ligation in a rat infarction model [13, 42]. Similarly, Hayashi et al reported that 6% of unfractionated bone marrow cells survived at 3 days in infarcted rat [13, 42], and Freyman et al reported that ~5% of mesenchymal stem cells survived after transplanting into the infarcted porcine heart [43]. Clearly, there is considerable room for improvement, and solving the problem of cell death is central to successful remuscularization of the infarcted heart.
This review focuses on the causes of death in cell transplantation with a mind for developing interventions to increase graft survival. We also report a novel approach to test whether potential pro-survival candidates translate in vitro effects into an animal model. Our rapid system to evaluate graft size in vivo reduces the need to conduct lengthy histomorphometric analyses. This approach resulted in our identification of carbamylated erythropoietin as a new treatment strategy to increase human embryonic stem cell derived cardiomyocyte survival after injection into the infarcted heart.
Death in Cell Transplantation: Progression and Pathways
Understanding the timing of cell death after injection is important if it is to be appropriately targeted. We have shown previously that most cell death after transplantation occurs in the first week (Figure 1). Specifically, 32% of cardiomyocytes grafted into acutely necrotic myocardium were TUNEL-positive 24 hours after injection and 10% were positive at 4 days. A week after injection, only 1% of the graft consisted of dead cells [44]. Since cell attrition occurs principally in the first few days after engraftment, it seems reasonable that an early assessment of graft size could predict long term engraftment.

Three predominant pathways that lead to cell death during transplantation are illustrated in Figure 2. The first stress the cells encounter during the engraftment process is lack of matrix support, and this begins before transplantation. When cells that normally grow in attachment are kept in suspension, a pathway of cell death called anoikis is initiated [45]. Anoikis is a Greek word for homelessness and is defined in cell biology as programmed cell death induced by loss of matrix attachments [46]. Cardiomyocytes are normally surrounded by a basal lamina, to which they bind via integrins and other receptors. These receptors transduce survival signals, mediated in part through NF-kB pathways [47, 48] that if interrupted, lead to mitochondrial cytochrome c release and caspase-mediated cell death [45]. Typical cell preparations for injection involve enzymatically dispersed cells suspended in protein-free medium, stored for hours on ice. During this period, important adhesion-related survival signals could be absent and not reinitiated for many hours until the cells find themselves in the context of a recipient heart, and even then, the proper basal surface for the cells may not be present.
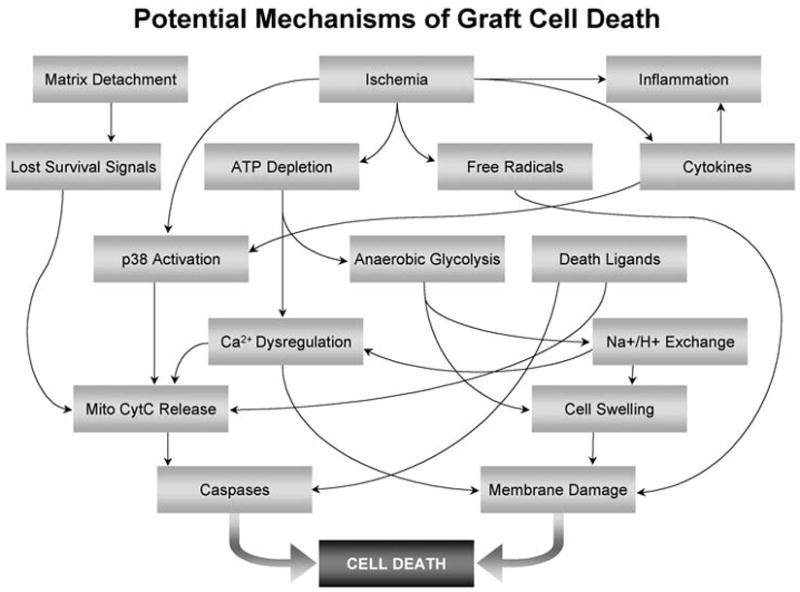
The next insult that grafted cells experience is ischemia. Injected cells tend to form various sized clumps that are forced into potential interstitial spaces between cardiomyocytes or connective tissue elements. Even in the context of well-vascularized normal heart tissue, these clumps are avascular, so diffusion is the only source of nutrient transport until angiogenesis provides a vasculature. The physical diffusion limit of ~150 microns means many cells deeper than 7 to 10 cell diameters toward the center of the graft become severely ischemic. This situation is exacerbated in the context of host tissue ischemia as occurs in MI, and in the reduced-perfusion environment of infarct scar. Electron microscopy has demonstrated that many cardiomyocytes show signs of irreversible ischemic injury, including formation of mitochondrial amorphous matrix densities, severe cytoplasmic and mitochondrial swelling, as well as signs of apoptotic death including nuclear fragmentation, all within a day of implantation [44]. Furthermore, survival of neonatal cardiomyocytes is directly proportional to tissue perfusion, with the greatest survival in the normal heart, intermediate survival in cardiac granulation tissue, and the lowest survival in acutely necrotic myocardium [49]. It seems likely that ischemic injury in transplanted cells follows the course characteristic of intact tissue, including ATP depletion, activation of anaerobic glycolysis with resulting acidosis, dysregulation of calcium and other ionic homeostasis, and swelling due to an osmotic load, culminating in membrane damage and cell death [50]. Ischemia also can lead to reactive oxygen species formation and subsequent mitochondrial permeability increases [51].
The third potential initiator of injury is inflammation. Healing infarcts are highly inflamed environments; neutrophils predominate the innate cellular response in the first few days and macrophages populate the area thereafter [52]. These leukocytes produce oxygen-derived free radicals and inflammatory cytokines that can directly damage graft cells via cell membrane damage, but can also initiate signaling pathways that result in caspase activation. This effect is most severe in acutely injured heart, but could also have a considerable effect on cell grafting independent of infarction, since macrophages readily localize to grafted regions in healthy hearts.
Strategies to Increase Cell Survival
Each of the major pathways of cell death will need to be addressed if cell-based regenerative medicine is to become a successful therapy. Two general strategies can be employed. The first is targeting a specific molecular pathway, e.g. through expression of an anti-apoptotic protein or blocking a caspase. The second is inducing a broader spectrum cytoprotective state, e.g. through heat shock or induction of a hypoxia response. These broad spectrum approaches can be quite powerful in their ability to protect, but they are less informative regarding the pathogenesis of cell death. In addition to strong pro-survival effects, an ideal treatment should not induce permanent changes in the graft’s physiology nor induce undesired host responses such as fibrosis, excess inflammation or blocking angiogenesis. Chemical or biological inhibitors offer the advantage of transient application but could generate off-target side effects if delivered systemically. On the other hand, gene therapy can yield large increases of specific pro-survival molecules, but many approaches result in permanent over-expression, a feature that may be undesired in the long term. Each of these delivery options is a legitimate approach to increasing survival, at the levels of both pre-clinical and clinical evaluation.
A positive side to the problem of graft cell death is that there is a large body of literature describing cardioprotective interventions, both in vitro and in the context of ischemic injury to the heart. Similarly, there is a large database on anti-apoptotic interventions. These provide many possibilities for approaches to reduce graft cell death. Several interventions are already known to enhance survival of transplanted cells in the heart, and these are summarized in Table 1. We identified heat shock as a potent strategy to enhance survival of neonatal cardiomyocyte grafts [44], and heat shock treatment has subequently been shown to be effective for skeletal myoblast survival [53, 54] and hESC-derived cardiomyocytes [21, 22]. Heat shock is typically achieved by heating cells to 43°C for 30–60 minutes one day before transplantation, and this results in induction of the heat shock proteins (hsps) 60, 70 and 90 [21, 44]. Of these, hsp70 has implicated by several groups [55, 56] as limiting cell death by inhibiting pro-apoptotic signaling [57], acting as molecular chaperones for damaged proteins and during translation [58, 59], or opposing oxidative stress [60]. Interestingly, similar expression patterns for hsp70 were seen after thawing from frozen cell storage [54]. Heat shock is an attractive intervention because of its simplicity, low cost and potency.
Akt is a serine-threonine kinase in the PI 3-kinase signaling pathway that had been shown to by cytoprotective in vitro [61]. Akt is activated by hypoxia and a variety of other stimuli, including cytokines [61] and reduces death in part by targeting apoptotic molecules Bcl-2 and caspases. In addition, Akt can also increase intracellular glucose metabolism [61] and enhance energy production during hypoxia. The multifactorial effects of Akt make it a good pro-survival candidate. We initially demonstrated that viral Akt over-expression improves neonatal cardiomyocyte graft survival [44], and Dzau’s group subsequently showed that Akt enhances mesenchymal stem cell survival after transplantation [62]. Interestingly, Akt-transduction stimulates MSCs to produce paracrine factors such as secreted frizzled-related protein 2 that exert a beneficial effect on the infarcted heart post-engraftment [63]. IGF-1, an upstream activator of Akt, has been shown to enhance survival of transplanted neonatal cardiomyocytes or smooth muscle in the injured heart [64, 65], and we have used IGF-1 as part of our pro-survival cocktail [66], described below.
Bcl-2 is the prototypical member of a family of pro- or anti-apoptotic proteins with BH-3 domains. Bcl-2 inhibits cell death by blocking cytochrome C release from the mitochondria [67], that otherwise leads to caspase activation and cell death. Bcl-2 is up-regulated in surviving ventricular myocytes in the context of MI [68], and transgenic over-expression of Bcl-2 in the engrafted cells limits myocardial infarct size after ischemia/reperfusion [69]. Li and colleagues have demonstrated the pro-survival effects of transduced Bcl-2 in two cell types: smooth muscle cells [69] and mesenchymal stem cells [70]. In each case, engraftment was improved as measured by cell graft size and ventricular function.
A potentially large contributor to graft cell death in cell-based cardiac repair is anoikis. As a contractile cell type, cardiomyocytes are normally tightly attached to basement membrane structures composed of extracellular matrix including laminin, collagen IV, fibronectin and entactin. Most cell injection strategies to date have not accounted for this potential need for extracellular substrate. Memon et al found that myoblast cell sheets implanted with culture matrix intact onto the surface of the infarcted rat heart improve cardiac outcomes compared to direct injection of myoblasts [71]. The cell sheet technology also has the advantage of maintaining cell-cell contacts that promote survival. Two recent studies have established that successful engraftment of cardiomyocytes is enhanced by co-delivery of the cells with matrix. Kutschka et al demonstrated that grafts were larger and ventricular function was improved in infarcted rats when human embryonic stem cell-derived cardiomyocytes were delivered with collagen matrices, including GelFoam and the basement membrane preparation, Matrigel [72]. As will be discussed below, Laflamme et al found that Matrigel significantly enhanced survival of hESC-derived cardiomyocytes in infarcted hearts of athymic rats [21, 22].
Free radical species are formed as a natural component of oxygen respiration. Normally, cells employ a contingent of free-radical scavenging molecules to quench the labile species. In the context of injury, cells can be exposed to large concentrations of free radical species, especially from inflammatory leukocytes [73] and improper metabolite washout from ischemia. Co-injecting the free radical scavenger superoxide dismutase with skeletal myoblasts yielded a 2-fold increase in graft survival [53] three days after engraftment. This effect was concurrent with a reduced inflammatory response, suggesting that superoxide is an important factor in the initial graft death and in mediating recruitment of inflammatory cells.
The ischemic environment in the infarct is a persistent problem for cardiac cell transplantation. A number of strategies have been pursued to generate an enriched vascular supply that will enable better graft survival. The most challenging problem with this approach is the relatively long time required to form new blood vessels compared to the rapid death of transplanted cells. One approach to this challenge is a two-stage process, where pro-vascularization interventions are employed initially, followed by seeding of a myogenic population. In any case, vascularization is clearly required for long term graft survival, and several studies have found over-expression of vascular endothelial growth factor (VEGF) enhances grafted cell survival [74–76]. Interestingly, adenoviral over-expression of hypoxia inducible factor-1α (HIF-1α) [77] with grafted skeletal myoblasts increased graft cell number, blood vessel formation and cardiac function. Although direct pro-survival effects of HIF-1α expression in myoblasts may have occurred, another potential benefit is increased vascularization by angiogenic molecules induced by HIF-1α, such as VEGF.
It seems logical that most of the mechanisms driving death of transplanted cells in the heart would be generic responses of cells to transplantation rather than being specific to a single cell types or target organ. If this is true, findings from other cell transplantation systems should be informative. Grafting of skeletal myoblasts in models of muscular dystrophy indicate a significant role for inflammation and immunity in mediating graft cell death. In those studies, myoblast survival was enhanced by blocking leukocyte adhesion [78], depleting complement [79] or by depleting NK cells [80]. In our own experiments with hESC-derived cardiomyocyte transplantation in the heart, we tried broad spectrum immunosuppression with steroids, complement depletion with cobra venom factor and NK cell depletion using lytic antibodies, as well as blocking pro-inflammatory “danger signals” from uric acid using allopurinol and uricase, but unfortunately none of these interventions improved cell survival [66]. Approaches to boosting cell survival for grafting applications in different tissues and organ systems have also been evaluated. Overexpression of Bcl-2 has been shown to improve survival of endothelial [81, 82] and cardiac [83] cells implanted subcutaneously. Caspase inhibition provided to cultured cells, and delivered systemically after injection enhances survival of islet cells [84] and dopaminergic neurons [85, 86]. Similarly, treatment of islet cells in vitro (but not after injection) with simvistatin, an HMG-CoA reductase inhibitor, enhanced survival after transplantation [87].
These numerous reports of increased cell survival and improved ventricular function by various unrelated interventions suggest that death in the context of cell transplantation occurs by multiple pathways. Our recent experiments designed to remuscularize infarcted rat hearts with hESC-derived cardiomyocytes confirmed this point [66]. When these cells were delivererd to uninjured hearts, we observed human myocardial grafts in ~90% of attempts. In contrast, when these cells were delivered to infarcted rat hearts, we observed grafts in only 18% of cases, and these grafts often consisted of small clusters of a few cells. We screened multiple potential pro-survival interventions, but none of the interventions in singlicate enhanced survival. This experience led us to prepare a “pro-survival cocktail” that targeted multiple points in apoptotic and oncotic death pathways. The cocktail as currently formulated includes Matrigel to block anoikis, IGF-1 to activate Akt, pinacidil to open K-ATP channels and mimic preconditioning, cyclosporine to block mitochondrial cyclophilin D pathways, the broad spectrum caspase inhibitor ZVAD-fmk, and a cell-permeant TAT peptide from Bcl-XL to block apoptotic pathways. Cells also were heat shocked one day prior to engraftment to further “toughen” them for transplantation. When the pro-survival cocktail was used, we achieved engraftment in nearly 100% of animals, reaching up to 11% of the infarcted heart. Thus, a multifactorial approach yielded successful engraftment when simpler interventions failed.
Several lessons emerged from these experiments. The first is that, while the use of the pro-survival cocktail significantly enhanced human cardiomyocyte engraftment, most of the cardiomyocytes are still dying. The second is that, although studying graft size and cell death by histology in the infarcted rat heart is a gold standard assay, it makes for a very inefficient screening system. Transplantation in the rat requires 10 million cardiomyocytes per animal [22]. These cells require about 4 weeks to expand in the undifferentiated cells and an additional 2–3 weeks of differentiation [23]. Finally, using histological graft size as an endpoint requires an incubation period upwards of 4 weeks after implantation. Add another 2 weeks for tissue processing and morphometry per group to measure graft size, and it is 2–3 months before one knows if a given intervention is successful. Alternatively, a screening model with a much higher throughput could identify only the most promising interventions for inclusion in long term experiments testing for physiological benefit of cardiac engraftment. In the next section we describe development and validation of two such screening models.
II. Development of Rapid Assays For Graft Size
We sought to develop graft size screening assays with the following characteristics:
- The graft cell must be a muscle cell type.
- There must be a short duration from cell injection to graft measurement to promote rapid turnaround.
- The assay must be simple, reliable and have good sensitivity.
- A mouse model is preferred due to lower required cell number and cost.
- Positive hits need to be confirmed in longer term studies using histology as a gold standard.
We reasoned that, since most of the cell death occurred in the first few days post-transplantation, positive effects detected at 3 days would be likely to persist long term. We chose biochemical strategies rather than a histological assay, because of the potential for high precision and rapid turnaround [13, 38, 41, 88]. (We recognize the loss of structural information using biochemistry, but this can be obtained in follow up validation studies.) The biochemical assays were based on the presence of graft-specific labels in the context of an entire heart homogenate. In vivo marker quantity was calculated based on standard curves of known cell quantities in the range expected for grafts. Our goal of developing a high throughput mouse assay necessitated injection of cells immediately after coronary occlusion, because the mouse infarct wall thins too much to deliver cells in the granulation tissue or scar tissue phases of infarct healing. The acute infarct is likely the least hospitable environment for transplanted cells, but we reasoned that factors enhancing cell survival in the acute infarct likely will predict cell survival in different pre-clinical models of cell engraftment.
We used β-gal and Alu based assay systems to evaluate the effect on cell graft size by two interventions: heat shock and erythropoietin. The effect measured from heat shock could be compared to previously established effects seen in other cardiac cell transplantation studies. Erythropoietin (EPO) and its chemically modified derivative, carbamylated erythropoietin (CEPO) [89], were evaluated as a wholly new cell survival intervention. Heat shock, EPO and CEPO were used independently and in combination in comparison to untreated control injections. The assays were validated by histomorphometric analysis of long term human embryonic stem cell-derived cardiomyocyte grafts.
Methods
Animal Models
All animals were housed under specific pathogen-free conditions and used in accordance with NIH guidelines as overseen by the Institutional Animal Care and Use Committee at the University of Washington approved this study according to protocol 2225-04. The two graft measurement assays presented here follow the same timeline, test the same interventions and require similar validation experiments and controls. They use different cell types (C2C12 vs. hESC-derived cardiomyocytes), different host animals (C3H vs. Rag2/γc double knockout mice) and different assay systems (β-gal vs. Alu PCR). Treated and control cells were delivered into mice following thoracotomy and direct left anterior descending coronary artery occlusion [90]. In brief, Avertin-anesthetized mice were ventilated with room air supplemented with oxygen and underwent left-sided thoracotomy under aseptic conditions. The left anterior descending coronary artery was ligated permanently to induce infarction. Immediately after ligation, we injected 105 cells in a volume of 5 μl into each mouse heart, after which the chest wall was closed. Rehydrated animals were permitted to recover in a warming chamber. The survival rate for this procedure was 85%. Three days after grafting we collected the hearts, gently washed them with isotonic saline and snap froze the samples in liquid nitrogen. Hearts were then homogenized and diluted to produce samples to be assayed for a marker molecule (β-gal or Alu). Final cell number calculations accounted for the signal contributed by dead cells. Statistics for the graft size assay used two-tailed unequal variance Student’s T-tests.
Graft assay findings were validated against histomorphometry data from hESC derived cardiomyocytes engrafted into ischemia-reperfused Rag2/γc DKO mouse hearts. Histological measurements were performed by a blinded observer, and statistical comparison between groups was conducted with a Mann-Whitney rank-sum test. Significance was defined as a p value of less than 0.05.
β-Galactosidase as a Marker for Grafted Skeletal Myoblasts
Our high throughput platform quantifies grafted cells in a way that definitively discriminates graft material from the host. The first system employs a syngeneic transplant model of C2C12 immortalized skeletal myoblasts (satellite cells) grafted into C3H mice. Clonal populations of C2C12 cells stably transduced with a retrovirus encoding nuclear-localized β-gal [91] were expanded for use in this assay. β-gal labeled C2C12 cells have been previously delivered to the heart and measured by histology [92, 93], establishing β-gal as a high-fidelity marker for grafted cells.
Detailed description of the nLacZ C2C12 assay is presented in the online supplement to this report. Briefly, a robust, clonal population of β-gal expressing C2C12 skeletal myoblasts [94, 95] was expanded and passaged before confluency to prevent myotube formation. Cell injectates were prepared at 40,000 cells per μl and stored on ice until grafted. A standard curve for nLacZ C2C12 cells using a chemiluminescent β-gal assay was generated for a range of 5000 to 500,000 cells in the context of whole heart homogenates. The linear relationship between cells per well and luminescence is shown in Figure 3B.

After three days, hearts were extracted from euthanized mice, washed and snap-frozen in liquid nitrogen. Homogenized whole hearts were dissolved in 1 ml of cold lysis buffer, which was sonicated on ice for 10 seconds. From this solution, 5 μl of the final solution was assayed using a luminometer and compared to a standard curve. To control for residual β-gal signal contribution from dead cells, a cell injectate with 100,000 cells underwent three freeze-thaw cycles in liquid nitrogen before being injected into the injured living mouse heart. Hearts were collected at multiple points after dead cell injection and assayed as above forβ-gal content. Figure 3C shows that the half-life of β-gal from dead cells is 32 hours. From the relationships of cell number to luminometric signal and the half life of the signal, total living graft cell number is calculated as described below.
Human Alu Sequences to Track hESC-Derived Cardiomyocytes
We also developed an assay using human embryonic stem cell-derived cardiomyocytes grafted into severely immune compromised Rag2/γc double knockout mice. This model was developed to assay interventions in a more clinically applicable cell population. The human Alu sequence was first used in the forensics field to identify evidence of human signs or remains from crime scenes [96]. With over one million copies per diploid genome and comprising 10% of total genomic mass, the non-coding 280 base pair Alu insertions are the most abundant elements in the human genome. [97]. We identified an Alu primer set that had no homology in the mouse genome.
The Alu system used the same general outline as the β-gal study, but with hESCs grafted into Rag2/γc DKO mouse hearts. Detailed methods including primer information and hESC cardiomyocyte derivation is included in the online supplement. The female hESC line H7 [98] was cultured and differentiated into cardiomyocytes [23] as described previously. 100,000 cells were injected in 5 μl of serum free medium into acutely infarcted hearts, and animals were allowed to recover.
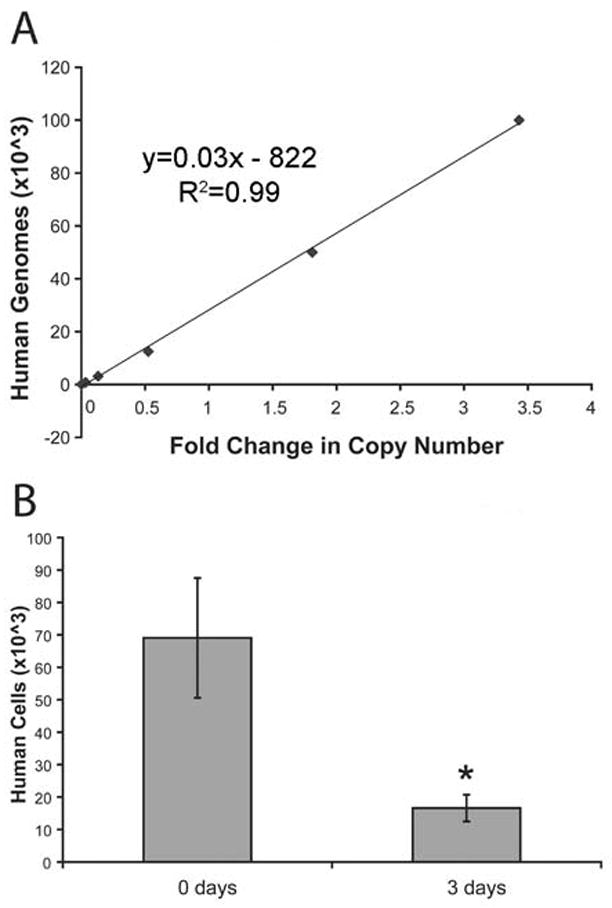
Sample collection for these animals was similar to the β-gal based assay. Three days after cell injections into the infarcted myocardium of Rag2/γc mouse hearts, the hearts were removed, snap frozen in liquid nitrogen and homogenized using a dismembranator (Braun, Melsungen, Germany). The samples were resuspended in 200 uL RNase/DNase-free water supplemented with proteinase K and Chelex beads. After incubation the samples were spun, and a 2 μl sample of the DNA-containing supernatant was carefully removed and used in subsequent SYBR green quantitative PCR using Alu-specific primers. Data were compared to a calibration curve of known human DNA quantity (Figure 5A) to identify the total number of initial transcript copies in the sample. As with the β-gal assay, some human DNA from dead cells persists at three days, so the rate of DNA loss from human cells subjected to freeze-thaw was similarly determined, yielding a half-life of 23 hours for the Alu sequences (Figure 5B).
Long Term Engraftment of hESC-derived cardiomyocytes
Previous reports in rats suggest that long-term engraftment of hESC-derived cardiomyocytes is more successful in hearts that have undergone ischemia-reperfusion as compared to permanent artery occlusion [22]. This led us to use an ischemia- reperfusion (IR) model of cardiac injury for our long term engraftment studies. This procedure followed established surgical techniques [90, 99, 100], but myocardial infarction was induced by ligating the left anterior descending coronary artery (LAD) with an 8-0 suture tied around a small piece of silicone tubing placed between the coronary artery and the suture to compress the LAD. After 45 minutes, the suture and tubing were removed to initiate reperfusion. Cell injections of 106 treated or control hESC-derived cardiomyocytes were delivered directly into the IR left ventricular free, and animals were allowed to recover for two weeks.
Erythropoietin Studies
C2C12 erythropoietin receptor and the common cytokine β-chain receptor(CβR) expression were evaluated by RT-PCR. Akt phosphorylation was assessed in response to treatments of CEPO, EPO and heat shock, alone and in combination, as detailed in supplemental information. CEPO and EPO treatment include a 30-minute incubation at 100 ng/ml of cells in culture [89] before preparation for injection followed by each recipient animal receiving doses of 10 ng of drug per gram of animal weight at days 0 and 1.
EPO and CEPO treatment were initially tested in groups of at least 6 animals using the β-gal protocol. Graft size was determined at three days as well as effects of CEPO and EPO on hematocrit. After demonstrating equivalent effects on cell survival (Figure 4A), but no change in hematocrit (Figure 7) in the CEPO group, subsequent grafting experiments only used CEPO treatment in the experimental groups. A second set of experiments evaluated whether an effect from heat shock could be independently detected and if CEPO’s pro-survival effect was additive to that of heat shock. This experiment consisted of four groups: no treatment, heat shock only, CEPO only and CEPO with heat shock treatment. Each group contained at least 7 animals.

The experimental groups for the Alu-based hESC derived cardiomyocyte grafting study included untreated controls, CEPO treatment alone and CEPO with heat shock. Fewer groups were used because of the decreased availability of hESC-derived cardiomyocytes compared to the C2C12 cell line. The hESC study used sample sizes of 7 or more animals each. Because of the greater time and cell number required for the long-term engraftment study we studied a more limited number of interventions. All groups were heat shocked, and the experimental group received CEPO treatment. Sample sizes were at least 8 animals for this study.
Histology
Five-micron sections were stained with hematoxylin and eosin, or subjected to immunohistochemistry as previously described [8, 49, 99, 101]. Hematoxylin was used as the nuclear counterstain in immunohistochemical studies. Immunostaining of tissue sections and of chamber slides for cardiomyocyte purity was performed with antibodies directed against the β-myosin heavy chain (β-MHC) isoform (clone A4.951; both from American Type Culture Collection). Human cells were identified using in situ hybridization for a human-specific pan-centromeric repeat sequence [102], labeled with digoxigenin through nick-translation (Cytocell: Cambridge, UK). The pan-centromeric probe is hereafter referred to as PCP. All evaluated sections were either double-labeled with this human-specific marker or an adjacent section was so-labeled. When performing the double-labeling, immunostaining for the cell type of interest preceded in situ hybridization [101, 103].
Human ESC-derived cardiac implants have a pronounced immunoreactivity for β-MHC. Healthy mouse host ventricular myocardium predominantly expresses α-myosin heavy chain. Although infarction induces some β-MHC in surviving host cardiomyocytes, this is of much weaker intensity compared to the grafted cells. Furthermore, the graft cells’ distinct morphology enables accurate discrimination from graft and host. Even so, adjacent sections with PCP/β-MHC double-labeled sections were used to confirm each suspected graft. Graft size was measured on each engrafted heart identified from the by β-MHC staining. Grafts were small enough that total cell number could be counted for each section. Two 5 μm sections 50 μm apart were stained for β-MHC, and cell numbers were summed for analysis.
Results
Assay Validation
The number of β-gal expressing C2C12 cells in vitro exhibits a strong linear relationship (R2 = 0.99) to the luminometric readings expected with graft sizes between 103 and 105 cells (Figure 3A). Each cell generated an average of 36 lumens. An ex vivo calibration curve using myoblasts injected into isolated, non-beating hearts generated a linear curve in the expected graft size range, as shown in Figure 3B. Compared to the in vitro curve, each cell produced 22 lumens; the difference could be attributed to light dispersion or absorption by the heart homogenate. To estimate the contribution that dead cells make to this assay, we injected cells killed by freeze-thaw into infarcts. Figure 3C shows that β-gal from dead cells exhibits a monoexponential decay with a half-life of approximately 32 hours. These data indicate that nLacZ C2C12 cells can be measured biochemically after injection into whole heart tissue, yielding the expected graft sizes, and that once cells die, the amount of β-gal measurable in the tissue declines following a standard pattern of decay. In reality, graft cell death is not immediate or synchronous (Figure 1), so this calculation slightly under-corrects for contributions from dead cells. Given that we are seeking to identify interventions that give >2-fold differences in graft size, residual signal from dead cells should not be a significant problem.
Basic principles of linearity and half-life may be combined to generate an equation relating measured signal to graft size:
Cellsatto=Injectate∗RetentionTotalGraftSignal=SignalLivingCells+SignalDeadCellsSignalDeadCells=(Cellsatto−GraftSize)∗(1/2)(implantduration/halflife)
The relevant relationships for half-life, initial injection size and implant duration can be combined to calculate graft size. The total graft signal may be derived from the standard curve in Figure 3B, which is performed for each experiment. This equation is combined with the governing equation in Figure 3C to generate a final graft size determination. (Note that the specific numeric values vary depending on results of the standard curves.)
TotalGraftSignal=16.9∗(Lumens)−5135GraftSize=16.3_∗(Lumens)−13904_
A similar set of data was derived for the QPCR Alu-based analysis. Ex vivo mixing data again demonstrated an excellent linear correlation between input cell number and the biochemical readout (Figure 5A). The threshold for detection was one human cell in 4×104 rodent cells. Assuming a monoexponential decay similar to the C2C12 cells, we measured residual DNA at 3 days from human cells killed by freeze-thaw and calculated a half-life of 23 hours (Figure 5B). These values were combined with the same set of equations above with the exception of graft signal being determined by qPCR fold change. The subsequent equations correlating fold change to surviving graft size are:
TotalGraftSignal=0.029∗(FoldChange)−822GraftSize=0.00402∗(FoldChange)−14200
These equations represent the derivation of graft size for the data presented in Figures 4 and 6. Each subsequent experiment requires calibration to a new curve using the stored extract from the ex vivo injection study to account for run-to-run variability.

Cell Signaling
EPO and CEPO exert their cytoprotective effects through a heterodimeric receptor, comprised of one chain of the EPOR and a second CβR chain. This is in contrast to EPO’s effect on hematocrit, which is mediated through homodimerization of the EPOR. Activation of heterodimeric receptor is reported to result in Akt phosphorylation in other cell types [104]. We performed RT-PCR for both EPOR and CβR in C2C12 cells and found both transcripts were readily detected (Figure 3F). Weaker signals for the two receptors were seen in infarcted mouse hearts (data not shown). To explore signaling responses following cytoprotective treatments, we studied Akt phosphorylation after treatment with EPO, CEPO, heat shock, or CEPO plus heat shock. These experiments revealed that Akt in CEPO- and EPO- treated C2C12 cells undergoes phosphorylation within 30 minutes after treatment (Figure 3G). Conversely, heat shock alone shows no Akt induction. CEPO and EPO show different effects on erythropoiesis. Figure 3E shows that EPO treatment increased hematocrit 3.9 points (p = 0.012) in 3 days, while CEPO and saline treatment both showed no change from day 0. This is consistent with EPO acting through the EPOR homodimer (which regulates red cell production) and CEPO acting through the EPOR/CβR heterodimer.
Effects of EPO, CEPO and Heat Shock on Skeletal Muscle Graft Size
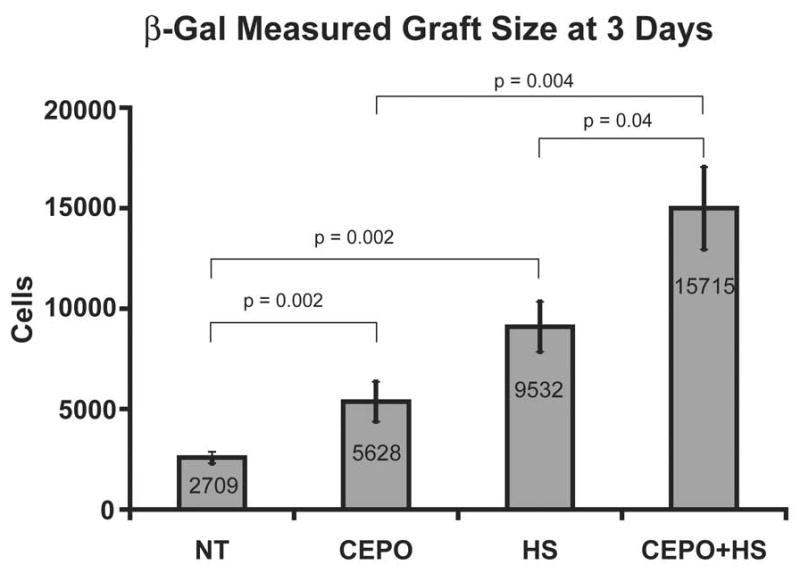
Pilot studies revealed that EPO and CEPO had comparable protective effects on myoblast graft size, each enhancing LacZ signal by 2.2-fold over vehicle control (p<0.05 for each; Figure 3D). This initial pilot study showed 25% cell retention at Day 0 (Figure 3C); subsequent studies routinely achieved between 50% and 75% retention. Because CEPO had no confounding effect on hematocrit, we studied it further for combinatorial treatments. As reviewed above, heat shock has been reported to increase survival of skeletal myoblasts [54, 93], neonatal rat cardiomyocytes [40] and hESC-derived cardiomyocytes [105] following transplantation into the injured heart. In accordance with these data, heat shock resulted in a 5.1-fold increase in myoblast graft size determined by β-gal luminometry at 3 days (Figure 4). Importantly, if cells are treated with both CEPO and heat shock, graft size increases 8.8-fold (Figure 4; p<0.05 vs. heat shock-only or CEPO-only), implying the interventions work through separate mechanisms.
It is noteworthy that the time elapsed between treating the cells, performing the experiments and compiling the data into the graphs shown in Figure 4 was significantly less than two weeks. The speed by which CEPO was first verified to be a pro-survival factor suggests that other candidates to reduce death could be tested with equal efficiency.
Alu-Based measurement of hESC-Derived Cardiomyocyte Engraftment
The human Alu sequence based assay provided similar findings for hESC-derived cardiomyocyte grafts as for C2C12 cells. Figure 6 shows the graft size measurements from the Alu-based rapid graft size assay. CEPO administration to grafted hESC-derived cardiomyocytes increases graft size by a factor of two over untreated controls (p<0.05). Similar to the β-gal assay, CEPO in combination with heat shock were additive to yield a 3.5-fold graft size increase. The gains in survival were not as large for human cardiomyocytes as with the C2C12 cells, perhaps reflecting the greater susceptibility of the cardiomyocytes to injury. Because of the reduced availability of hESC-derived cardiomyocytes, fewer combinatorial treatment strategies were evaluated.
Effects of CEPO on Long Term Engraftment of hESC Derived Cardiomyocytes
We found that successful long term engraftment of hESC-derived cardiomyocytes in the Rag2/γc DKO mouse requires injecting 106 hESCs, with a cardiomyocyte purity >50%, into hearts injured by 45 minutes of ischemia followed by reperfusion. Delivering fewer cells, cells of lower purity or the use of permanent coronary ligation was associated with minimal cardiomyocyte engraftment. In contrast, when these criteria were followed, hearts studied at 2 weeks showed engraftment in more than half of the animals. Figure 7 shows representative images from a heart containing a human cardiac cell graft two weeks after injury and transplantation. Note that the grafted cells are significantly smaller than the host cardiomyocytes and stain intensely for β-MHC, in contrast to the much lighter β-MHC expression by peri-infarct host cardiomyocytes. This host expression can be accounted for by the common occurrence of β-MHC upregulation in the context of injury. At two weeks, every cell that hybridized with our human pan-centromeric probe also stained for β-MHC, suggesting all remaining human cells were cardiomyocytes. Grafts were found in 4 of 9 animals from the heat shock treated group and 5 of 8 mice from the group receiving heat shock and CEPO treatment. If all animals are considered (including animals without grafts that were assigned a value of 0), there is a 2.3-fold increase in graft size in heat shock + CEPO treatment compared to hearts receiving cells treated with heat shock only (p < 0.05 by the Mann-Whitney rank-sum test). This compares favorably to the increases seen in the short term biochemical assays.
Discussion
We have designed two high-throughput assays to measure cell graft size in the infarcted mouse heart three days after injection. One assay uses a reliable β-gal expressing skeletal myoblast cell line in a syngeneic host, while the other employs a xenograft model measuring Alu sequences unique to human embryonic stem cell-derived cardiomyocytes injected into a severely immunocompromised host. The β-gal assay is fast, reliable, inexpensive and produces statistically significant results with small animal group sample sizes. The Alu assay is extremely sensitive, has an excellent internal control, measures engraftment of a clinically relevant cell type (hESC-derived cardiomyocytes), and has been validated with histological measurements in a long-term engraftment study. Three different models indicate that heat shock and CEPO treatments each increase cell survival. Results from the 3-day assay correlate well with the 2-week assay, especially when the same cell type is used. Engraftment of heat shocked cells into infarcts induced by ischemia-reperfusion showed a 2.4-fold increase in graft size at two weeks when the only difference between groups was CEPO treatment. This matches the 2-fold improvement seen in the rapid Alu-based assay using human cells.
The extent to which engraftment occurs in hESC-derived cardiomyocytes is less than that of C2C12 skeletal myoblasts. Several conditions could explain this. First, C2C12 cells are particularly ischemia-resistant; large grafts have been produced in mouse hearts infarcted by permanent coronary artery occlusion [106] without any pro-survival interventions. Additionally, hESC-derived cardiomyocyte viability by Trypan blue exclusion after long periods on ice is lower than C2C12s (60% after 5 hours versus 90% for C2C12s), and plating efficiency of the human cardiomyocytes at the end of a grafting experiment is correspondingly lower. This increased fragility for hESCs could be due to several factors, most notably the more intensive cell purification process that hESCs are put through. C2C12s are also an immortalized cell line and better adapted to the conditions of tissue culture. A second reason why C2C12 grafts are larger after these treatments is the potential for proliferation. C2C12s could establish grafts and rapidly resume proliferation in vivo after engraftment. The C2C12 doubling time in vitro is about 12 hours, which may carry over into an in vivo environment. It is known that hESCs divide after injection into the heart, but much more slowly; in 4 weeks, graft size increases 7-fold [21], for a doubling time of about 10 days.
The time from cell injection to data read-out with the rapid techniques is between one and two weeks. This short turn-around period contrasts with the two months it typically takes to assess graft size by standard histological methods. Rapid quantification is a valuable tool for constructing a panel of cell survival boosting techniques from the potential interventions listed in the first portion of this review. It is our hope that this approach will enable refinement of the current pro-survival cocktail that facilitates engraftment of human cardiomyocytes [22]. Such interventions must yield several-fold increases in survival and graft size, and the immense repopulation required to restore heart tissue depends on a large initial population of viable cells. Survival is not the only aspect of cardiac repair to which these assays could be applied. Other areas in need of improvement include cell delivery systems and strategies to enhance cell proliferation. Both problems could be studied using a higher throughput screening system.
Limitations of the Rapid Assay Systems
The rapid assay we describe is a destructive procedure that prevents histological data collection. The assay’s short period between engraftment and data collection removes any opportunity for physiological assessments. Like any screening method, these have risks of false-positive outcomes. For example, an intervention might delay cell death without influencing final graft size, or alternatively, it might prevent clearance of the LacZ or human Alu sequences from dead cells without true protection. For this reason, it is important that initial positive results be confirmed in longer term assays.
In our study of human cardiomyocyte engraftment, we observed a significant difference in engraftment at 3 days vs. the two week time point. At 3 days, the Alu-based assay measured a graft size of almost 12,000 human cells from grafts of only 105 cells delivered to the cohort receiving heat shock and CEPO. Conversely, CEPO treatment with heat shock generated a long term graft measuring about 10 cells per section, despite an input of 10-fold more cells. A direct comparison requires consideration of sampling associated with tissue sectioning: The section on the slide is 5 μm thick, sampled from a slice of tissue 2 mm thick. This means that there are potentially 400 sections per heart slice. If 10 cells per section are distributed in a thin patch through the entire slice, it is conceivable that 10 cells per section could represent a graft of around 4,000 cells total. This suggests that there is ongoing cell death between 3 days and 2 weeks in this system, although we emphasize that the protective effects of CEPO and heat shock could still be detected.
Carbamylated Erythropoietin as a Treatment for Cell Graft Survival
In C2C12 cells we observed expression of the two receptor domains required to transduce cytoprotection by EPO and CEPO, namely the EPO receptor and the common β-chain receptor. We observed that CEPO initiates Akt phosphorylation, which is likely a key component in protection. It should be noted that Chanséaume et al recently published a report indicating that systemic EPO administration did not increase skeletal myoblast engraftment or physiological function in rats [107]. This study differs from ours in several ways. Chanséaume et al used cells injected into two week old infarcts in Lewis rats, and the cells used for injection were primary isolated skeletal myoblasts. They studied EPO, whereas our studies emphasized CEPO (but did detect protection by EPO in short term myoblast grafts). Finally, in their study, only the animals were given EPO; cells were not pre-treated. Of these differences, the last could be the most important factor. We pretreated cells 30 minutes before preparation for injection and injected them anywhere from 1 hour later to 6 hours later into a mouse pre-dosed with CEPO. In our system, Akt phosphorylation occurs even before the cells are removed from the plate, initiating the pro-survival pathway, presumably through Bcl-XL upregulation [89, 108]. Failure to give the transplanted cells a head start in survival signaling could be responsible for that study’s negative finding.
Summary
We developed two rapid screening systems for detection of pro-survival factors for cells transplanted into the heart. Using these systems, a candidate factor from the literature (carbamylated erythropoietin) was screened for a short term effect and evaluated for effects on long-term engraftment using standard histological techniques. Two independent rapid assays indicated that CEPO improves cardiac graft size, and that the effect from CEPO is additive to another common pro-survival technique, heat shock. Protection at 3 days predicted long term benefits, although there appears to be ongoing death with hESC derivatives in the mouse infarct. CEPO treatment may be a useful addition to cell engraftment studies and should be tested in larger animal models. The rapid graft size assays described here should be useful to identify new candidate pro-survival factors.
Supplementary Material
01
Acknowledgments
The authors thank Ms. Veronica Muskheli for assistance with histology and immunostaining and Ms. Sarah Dupras for assistance with animal handling and surgery. This work was supported in part by NIH grants R01 HL084642, P01 HL03174, and R24 HL64387 and a research grant from Geron Corp.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Murry CE, Field LJ, Menasche P. Cell-based cardiac repair: reflections at the 10-year point. Circulation. 2005 Nov 15;112(20):3174–83. doi: 10.1161/CIRCULATIONAHA.105.546218. [DOI] [PubMed] [Google Scholar]
- 2.Laflamme MA, Zibinden S, Epstein SE, Murry CE. Cell-Based Therapy for Myocardial Ischemia and Infarction: Pathophysiological Mecanisms. Annu Rev Pathol Mech Dis 2007. 2007;2:307–39. doi: 10.1146/annurev.pathol.2.010506.092038. [DOI] [PubMed] [Google Scholar]
- 3.Marelli D, Desrosiers C, el-Alfy M, Kao RL, Chiu RC. Cell transplantation for myocardial repair: an experimental approach. Cell Transplant. 1992;1(6):383–90. doi: 10.1177/096368979200100602. [DOI] [PubMed] [Google Scholar]
- 4.Chiu RC, Zibaitis A, Kao RL. Cellular cardiomyoplasty: myocardial regeneration with satellite cell implantation. Ann Thorac Surg. 1995 Jul;60(1):12–8. [PubMed] [Google Scholar]
- 5.Koh GY, Klug MG, Soonpaa MH, Field LJ. Differentiation and long-term survival of C2C12 myoblast grafts in heart. J Clin Invest. 1993 Sep;92(3):1548–54. doi: 10.1172/JCI116734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leobon B, Garcin I, Menasche P, Vilquin JT, Audinat E, Charpak S. Myoblasts transplanted into rat infarcted myocardium are functionally isolated from their host. Proc Natl Acad Sci U S A. 2003 Jun 24;100(13):7808–11. doi: 10.1073/pnas.1232447100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain M, DerSimonian H, Brenner DA, Ngoy S, Teller P, Edge AS, et al. Cell therapy attenuates deleterious ventricular remodeling and improves cardiac performance after myocardial infarction. Circulation. 2001 Apr 10;103(14):1920–7. doi: 10.1161/01.cir.103.14.1920. [DOI] [PubMed] [Google Scholar]
- 8.Murry CE, Kay MA, Bartosek T, Hauschka SD, Schwartz SM. Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with MyoD. J Clin Invest. 1996 Nov 15;98(10):2209–17. doi: 10.1172/JCI119030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scorsin M, Hagege A, Vilquin JT, Fiszman M, Marotte F, Samuel JL, et al. Comparison of the effects of fetal cardiomyocyte and skeletal myoblast transplantation on postinfarction left ventricular function. J Thorac Cardiovasc Surg. 2000 Jun;119(6):1169–75. doi: 10.1067/mtc.2000.104865. [DOI] [PubMed] [Google Scholar]
- 10.Leor J, Patterson M, Quinones MJ, Kedes LH, Kloner RA. Transplantation of fetal myocardial tissue into the infarcted myocardium of rat. A potential method for repair of infarcted myocardium? Circulation. 1996 Nov 1;94(9 Suppl):II332–6. [PubMed] [Google Scholar]
- 11.Li RK, Jia ZQ, Weisel RD, Mickle DA, Zhang J, Mohabeer MK, et al. Cardiomyocyte transplantation improves heart function. Ann Thorac Surg. 1996 Sep;62(3):654–60. doi: 10.1016/s0003-4975(96)00389-x. discussion 60–1. [DOI] [PubMed] [Google Scholar]
- 12.Hutcheson KA, Atkins BZ, Hueman MT, Hopkins MB, Glower DD, Taylor DA. Comparison of benefits on myocardial performance of cellular cardiomyoplasty with skeletal myoblasts and fibroblasts. Cell Transplant. 2000 May-Jun;9(3):359–68. doi: 10.1177/096368970000900307. [DOI] [PubMed] [Google Scholar]
- 13.Yasuda T, Weisel RD, Kiani C, Mickle DA, Maganti M, Li RK. Quantitative analysis of survival of transplanted smooth muscle cells with real-time polymerase chain reaction. J Thorac Cardiovasc Surg. 2005 Apr;129(4):904–11. doi: 10.1016/j.jtcvs.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 14.Penn MS, Francis GS, Ellis SG, Young JB, McCarthy PM, Topol EJ. Autologous cell transplantation for the treatment of damaged myocardium. Prog Cardiovasc Dis. 2002 Jul-Aug;45(1):21–32. doi: 10.1053/pcad.2002.123466. [DOI] [PubMed] [Google Scholar]
- 15.Nygren JM, Jovinge S, Breitbach M, Sawen P, Roll W, Hescheler J, et al. Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med. 2004 May;10(5):494–501. doi: 10.1038/nm1040. [DOI] [PubMed] [Google Scholar]
- 16.Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation. 2002 Jan 1;105(1):93–8. doi: 10.1161/hc0102.101442. [DOI] [PubMed] [Google Scholar]
- 17.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001 Apr;7(4):430–6. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 18.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A. 2003 Oct 14;100(21):12313–8. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003 Sep 19;114(6):763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 20.Klug MG, Soonpaa MH, Koh GY, Field LJ. Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J Clin Invest. 1996 Jul 1;98(1):216–24. doi: 10.1172/JCI118769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laflamme MA, Gold J, Xu C, Hassanipour M, Rosler E, Police S, et al. Formation of human myocardium in the rat heart from human embryonic stem cells. Am J Pathol. 2005 Sep;167(3):663–71. doi: 10.1016/S0002-9440(10)62041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007 Aug 26; doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 23.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002 Sep 20;91(6):501–8. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 24.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001 Aug;108(3):407–14. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinecke H, MacDonald GH, Hauschka SD, Murry CE. Electromechanical coupling between skeletal and cardiac muscle. Implications for infarct repair. J Cell Biol. 2000 May 1;149(3):731–40. doi: 10.1083/jcb.149.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Etzion S, Battler A, Barbash IM, Cagnano E, Zarin P, Granot Y, et al. Influence of embryonic cardiomyocyte transplantation on the progression of heart failure in a rat model of extensive myocardial infarction. J Mol Cell Cardiol. 2001 Jul;33(7):1321–30. doi: 10.1006/jmcc.2000.1391. [DOI] [PubMed] [Google Scholar]
- 27.Reffelmann T, Dow JS, Dai W, Hale SL, Simkhovich BZ, Kloner RA. Transplantation of neonatal cardiomyocytes after permanent coronary artery occlusion increases regional blood flow of infarcted myocardium. J Mol Cell Cardiol. 2003 Jun;35(6):607–13. doi: 10.1016/s0022-2828(03)00081-6. [DOI] [PubMed] [Google Scholar]
- 28.Laflamme MA, Murry CE. Regenerating the heart. Nat Biotechnol. 2005 Jul;23(7):845–56. doi: 10.1038/nbt1117. [DOI] [PubMed] [Google Scholar]
- 29.Menasche P. MAGIC II Report. American Heart Association. 2006 2006; Scientific Sessions [Google Scholar]
- 30.Lunde K, Solheim S, Aakhus S, Arnesen H, Abdelnoor M, Egeland T, et al. Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. N Engl J Med. 2006 Sep 21;355(12):1199–209. doi: 10.1056/NEJMoa055706. [DOI] [PubMed] [Google Scholar]
- 31.Janssens S, Dubois C, Bogaert J, Theunissen K, Deroose C, Desmet W, et al. Autologous bone marrow-derived stem-cell transfer in patients with ST-segment elevation myocardial infarction: double-blind, randomised controlled trial. Lancet. 2006 Jan 14;367(9505):113–21. doi: 10.1016/S0140-6736(05)67861-0. [DOI] [PubMed] [Google Scholar]
- 32.Meyer GP, Wollert KC, Lotz J, Steffens J, Lippolt P, Fichtner S, et al. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months' follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006 Mar 14;113(10):1287–94. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- 33.Assmus B, Honold J, Schachinger V, Britten MB, Fischer-Rasokat U, Lehmann R, et al. Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med. 2006 Sep 21;355(12):1222–32. doi: 10.1056/NEJMoa051779. [DOI] [PubMed] [Google Scholar]
- 34.Tambara K, Sakakibara Y, Sakaguchi G, Lu F, Premaratne GU, Lin X, et al. Transplanted skeletal myoblasts can fully replace the infarcted myocardium when they survive in the host in large numbers. Circulation. 2003 Sep 9;108(Suppl 1):II259–63. doi: 10.1161/01.cir.0000087430.17543.b8. [DOI] [PubMed] [Google Scholar]
- 35.Pouzet B, Vilquin JT, Hagege AA, Scorsin M, Messas E, Fiszman M, et al. Factors affecting functional outcome after autologous skeletal myoblast transplantation. AnnThoracSurg. 2001 3/2001;71(3):844–50. doi: 10.1016/s0003-4975(00)01785-9. [DOI] [PubMed] [Google Scholar]
- 36.Olivetti G, Capasso JM, Sonnenblick EH, Anversa P. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. CircRes 1990. 1990;67(1):23–34. doi: 10.1161/01.res.67.1.23. [DOI] [PubMed] [Google Scholar]
- 37.Caulfield JB, Leinbach R, Gold H. The relationship of myocardial infarct size and prognosis. Circulation. 1976 Mar;53(3 Suppl):I141–4. [PubMed] [Google Scholar]
- 38.Muller-Ehmsen J, Whittaker P, Kloner RA, Dow JS, Sakoda T, Long TI, et al. Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol. 2002 Feb;34(2):107–16. doi: 10.1006/jmcc.2001.1491. [DOI] [PubMed] [Google Scholar]
- 39.Hudson W, Collins MC, deFreitas D, Sun YS, Muller-Borer B, Kypson AP. Beating and arrested intramyocardial injections are associated with significant mechanical loss: implications for cardiac cell transplantation. J Surg Res. 2007 Oct;142(2):263–7. doi: 10.1016/j.jss.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 40.Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE. Cardiomyocyte grafting for cardiac repair: graft cell death and anti- death strategies. JMolCell Cardiol. 2001 5/2001;33(5):907–21. doi: 10.1006/jmcc.2001.1367. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki K, Murtuza B, Beauchamp JR, Brand NJ, Barton PJ, Varela-Carver A, et al. Role of interleukin-1beta in acute inflammation and graft death after cell transplantation to the heart. Circulation. 2004 Sep 14;110(11 Suppl 1):II219–24. doi: 10.1161/01.CIR.0000138388.55416.06. [DOI] [PubMed] [Google Scholar]
- 42.Hayashi M, Li TS, Ito H, Mikamo A, Hamano K. Comparison of intramyocardial and intravenous routes of delivering bone marrow cells for the treatment of ischemic heart disease: an experimental study. Cell Transplant. 2004;13(6):639–47. doi: 10.3727/000000004783983558. [DOI] [PubMed] [Google Scholar]
- 43.Freyman T, Polin G, Osman H, Crary J, Lu M, Cheng L, et al. A quantitative, randomized study evaluating three methods of mesenchymal stem cell delivery following myocardial infarction. Eur Heart J. 2006 May;27(9):1114–22. doi: 10.1093/eurheartj/ehi818. [DOI] [PubMed] [Google Scholar]
- 44.Zhang M, Methot D, Poppa V, Fujio Y, Walsh K, Murry CE. Cardiomyocyte grafting for cardiac repair: graft cell death and anti-death strategies. J Mol Cell Cardiol. 2001 May;33(5):907–21. doi: 10.1006/jmcc.2001.1367. [DOI] [PubMed] [Google Scholar]
- 45.Zvibel I, Smets F, Soriano H. Anoikis: roadblock to cell transplantation? Cell Transplant. 2002;11(7):621–30. doi: 10.3727/000000002783985404. [DOI] [PubMed] [Google Scholar]
- 46.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005 Sep;24(3):425–39. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 47.Zhan M, Zhao H, Han ZC. Signalling mechanisms of anoikis. Histol Histopathol. 2004 Jul;19(3):973–83. doi: 10.14670/HH-19.973. [DOI] [PubMed] [Google Scholar]
- 48.Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J Cell Biol. 1998 May 18;141(4):1083–93. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reinecke H, Zhang M, Bartosek T, Murry CE. Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation. 1999 Jul 13;100(2):193–202. doi: 10.1161/01.cir.100.2.193. [DOI] [PubMed] [Google Scholar]
- 50.Jennings RB, Reimer KA, Steenbergen C. Myocardial ischemia revisited. The osmolar load, membrane damage, and reperfusion. JMolCell Cardiol. 1986 8/1986;18(8):769–80. doi: 10.1016/s0022-2828(86)80952-x. [DOI] [PubMed] [Google Scholar]
- 51.Mitch WE, Goldberg AL. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med. 1996 Dec 19;335(25):1897–905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 52.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007 Nov 26;204(12):3037–47. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki K, Murtuza B, Beauchamp JR, Smolenski RT, Varela-Carver A, Fukushima S, et al. Dynamics and mediators of acute graft attrition after myoblast transplantation to the heart. Faseb J. 2004 Jul;18(10):1153–5. doi: 10.1096/fj.03-1308fje. [DOI] [PubMed] [Google Scholar]
- 54.Maurel A, Azarnoush K, Sabbah L, Vignier N, Le Lorc’h M, Mandet C, et al. Can cold or heat shock improve skeletal myoblast engraftment in infarcted myocardium? Transplantation. 2005 Sep 15;80(5):660–5. doi: 10.1097/01.tp.0000172178.35488.31. [DOI] [PubMed] [Google Scholar]
- 55.Jayakumar J, Suzuki K, Sammut IA, Smolenski RT, Khan M, Latif N, et al. Heat shock protein 70 gene transfection protects mitochondrial and ventricular function against ischemia-reperfusion injury. Circulation. 2001 Sep 18;104(12 Suppl 1):I303–7. doi: 10.1161/hc37t1.094932. [DOI] [PubMed] [Google Scholar]
- 56.Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995 Apr;95(4):1446–56. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997 Sep;17(9):5317–27. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998 Jul 27;83(2):117–32. doi: 10.1161/01.res.83.2.117. [DOI] [PubMed] [Google Scholar]
- 59.Mosser DD, Caron AW, Bourget L, Meriin AB, Sherman MY, Morimoto RI, et al. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000 Oct;20(19):7146–59. doi: 10.1128/mcb.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Su CY, Chong KY, Owen OE, Dillmann WH, Chang C, Lai CC. Constitutive and inducible hsp70s are involved in oxidative resistance evoked by heat shock or ethanol. J Mol Cell Cardiol. 1998 Mar;30(3):587–98. doi: 10.1006/jmcc.1997.0622. [DOI] [PubMed] [Google Scholar]
- 61.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999 Nov 15;13(22):2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 62.Mangi AA, Noiseux N, Kong D, He H, Rezvani M, Ingwall JS, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat Med. 2003 Sep;9(9):1195–201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- 63.Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, et al. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci U S A. 2007 Jan 30;104(5):1643–8. doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis ME, Hsieh PC, Takahashi T, Song Q, Zhang S, Kamm RD, et al. Local myocardial insulin-like growth factor 1 (IGF-1) delivery with biotinylated peptide nanofibers improves cell therapy for myocardial infarction. Proc Natl Acad Sci U S A. 2006 May 23;103(21):8155–60. doi: 10.1073/pnas.0602877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu TB, Fedak PW, Weisel RD, Yasuda T, Kiani G, Mickle DA, et al. Enhanced IGF-1 expression improves smooth muscle cell engraftment after cell transplantation. Am J Physiol Heart Circ Physiol. 2004 Dec;287(6):H2840–9. doi: 10.1152/ajpheart.00439.2004. [DOI] [PubMed] [Google Scholar]
- 66.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotech. 2007 doi: 10.1038/nbt1327. Advance Online Publication August 26, 2007. [DOI] [PubMed] [Google Scholar]
- 67.Reed JC. Bcl-2 family proteins. Oncogene. 1998 Dec 24;17(25):3225–36. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 68.Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T, Fujiwara H. Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation. 1996 Oct 1;94(7):1506–12. doi: 10.1161/01.cir.94.7.1506. [DOI] [PubMed] [Google Scholar]
- 69.Nakamura Y, Yasuda T, Weisel RD, Li RK. Enhanced cell transplantation: preventing apoptosis increases cell survival and ventricular function. Am J Physiol Heart Circ Physiol. 2006 Aug;291(2):H939–47. doi: 10.1152/ajpheart.00155.2006. [DOI] [PubMed] [Google Scholar]
- 70.Li W, Ma N, Ong LL, Nesselmann C, Klopsch C, Ladilov Y, et al. Bcl-2 engineered MSCs inhibited apoptosis and improved heart function. Stem Cells. 2007 Aug;25(8):2118–27. doi: 10.1634/stemcells.2006-0771. [DOI] [PubMed] [Google Scholar]
- 71.Memon IA, Sawa Y, Fukushima N, Matsumiya G, Miyagawa S, Taketani S, et al. Repair of impaired myocardium by means of implantation of engineered autologous myoblast sheets. J Thorac Cardiovasc Surg. 2005 Nov;130(5):1333–41. doi: 10.1016/j.jtcvs.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 72.Kutschka I, Chen IY, Kofidis T, Arai T, von Degenfeld G, Sheikh AY, et al. Collagen matrices enhance survival of transplanted cardiomyoblasts and contribute to functional improvement of ischemic rat hearts. Circulation. 2006 Jul 4;114(1 Suppl):I167–73. doi: 10.1161/CIRCULATIONAHA.105.001297. [DOI] [PubMed] [Google Scholar]
- 73.Kumar V, editor. Robbins’ Pathologic Basis of Disease. 7. Philadelphia: Elsevier Saunders; 2005. [Google Scholar]
- 74.Retuerto MA, Schalch P, Patejunas G, Carbray J, Liu N, Esser K, et al. Angiogenic pretreatment improves the efficacy of cellular cardiomyoplasty performed with fetal cardiomyocyte implantation. J Thorac Cardiovasc Surg. 2004 Apr;127(4):1041–9. doi: 10.1016/j.jtcvs.2003.09.049. discussion 9–51. [DOI] [PubMed] [Google Scholar]
- 75.Wang Y, Haider HK, Ahmad N, Xu M, Ge R, Ashraf M. Combining pharmacological mobilization with intramyocardial delivery of bone marrow cells over-expressing VEGF is more effective for cardiac repair. J Mol Cell Cardiol. 2006 May;40(5):736–45. doi: 10.1016/j.yjmcc.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 76.Matsumoto R, Omura T, Yoshiyama M, Hayashi T, Inamoto S, Koh KR, et al. Vascular endothelial growth factor-expressing mesenchymal stem cell transplantation for the treatment of acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2005 Jun;25(6):1168–73. doi: 10.1161/01.ATV.0000165696.25680.ce. [DOI] [PubMed] [Google Scholar]
- 77.Azarnoush K, Maurel A, Sebbah L, Carrion C, Bissery A, Mandet C, et al. Enhancement of the functional benefits of skeletal myoblast transplantation by means of coadministration of hypoxia-inducible factor 1alpha. J Thorac Cardiovasc Surg. 2005 Jul;130(1):173–9. doi: 10.1016/j.jtcvs.2004.11.044. [DOI] [PubMed] [Google Scholar]
- 78.Guerette B, Skuk D, Celestin F, Huard C, Tardif F, Asselin I, et al. Prevention by anti-LFA-1 of acute myoblast death following transplantation. J Immunol. 1997 Sep 1;159(5):2522–31. [PubMed] [Google Scholar]
- 79.Hodgetts SI, Grounds MD. Complement and myoblast transfer therapy: donor myoblast survival is enhanced following depletion of host complement C3 using cobra venom factor, but not in the absence of C5. Immunol Cell Biol. 2001 Jun;79(3):231–9. doi: 10.1046/j.1440-1711.2001.01006.x. [DOI] [PubMed] [Google Scholar]
- 80.Hodgetts SI, Spencer MJ, Grounds MD. A role for natural killer cells in the rapid death of cultured donor myoblasts after transplantation. Transplantation. 2003 Mar 27;75(6):863–71. doi: 10.1097/01.TP.0000053754.33317.4B. [DOI] [PubMed] [Google Scholar]
- 81.Karl E, Warner K, Zeitlin B, Kaneko T, Wurtzel L, Jin T, et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res. 2005 Jun 15;65(12):5063–9. doi: 10.1158/0008-5472.CAN-05-0140. [DOI] [PubMed] [Google Scholar]
- 82.Nor JE, Christensen J, Liu J, Peters M, Mooney DJ, Strieter RM, et al. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001 Mar 1;61(5):2183–8. [PubMed] [Google Scholar]
- 83.Kutschka I, Kofidis T, Chen IY, von Degenfeld G, Zwierzchoniewska M, Hoyt G, et al. Adenoviral human BCL-2 transgene expression attenuates early donor cell death after cardiomyoblast transplantation into ischemic rat hearts. Circulation. 2006 Jul 4;114(1 Suppl):I174–80. doi: 10.1161/CIRCULATIONAHA.105.001370. [DOI] [PubMed] [Google Scholar]
- 84.Nakano M, Matsumoto I, Sawada T, Ansite J, Oberbroeckling J, Zhang HJ, et al. Caspase-3 inhibitor prevents apoptosis of human islets immediately after isolation and improves islet graft function. Pancreas. 2004 Aug;29(2):104–9. doi: 10.1097/00006676-200408000-00004. [DOI] [PubMed] [Google Scholar]
- 85.Schierle GS, Hansson O, Leist M, Nicotera P, Widner H, Brundin P. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat Med. 1999 Jan;5(1):97–100. doi: 10.1038/4785. [DOI] [PubMed] [Google Scholar]
- 86.Cicchetti F, Costantini L, Belizaire R, Burton W, Isacson O, Fodor W. Combined inhibition of apoptosis and complement improves neural graft survival of embryonic rat and porcine mesencephalon in the rat brain. Exp Neurol. 2002 Oct;177(2):376–84. doi: 10.1006/exnr.2002.8007. [DOI] [PubMed] [Google Scholar]
- 87.Contreras JL, Smyth CA, Bilbao G, Young CJ, Thompson JA, Eckhoff DE. Simvastatin induces activation of the serine-threonine protein kinase AKT and increases survival of isolated human pancreatic islets. Transplantation. 2002 Oct 27;74(8):1063–9. doi: 10.1097/00007890-200210270-00001. [DOI] [PubMed] [Google Scholar]
- 88.Blaveri K, Heslop L, Yu DS, Rosenblatt JD, Gross JG, Partridge TA, et al. Patterns of repair of dystrophic mouse muscle: studies on isolated fibers. Dev Dyn. 1999 Nov;216(3):244–56. doi: 10.1002/(SICI)1097-0177(199911)216:3<244::AID-DVDY3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 89.Fiordaliso F, Chimenti S, Staszewsky L, Bai A, Carlo E, Cuccovillo I, et al. A nonerythropoietic derivative of erythropoietin protects the myocardium from ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005 Feb 8;102(6):2046–51. doi: 10.1073/pnas.0409329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Virag JI, Murry CE. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol. 2003 Dec;163(6):2433–40. doi: 10.1016/S0002-9440(10)63598-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994 Jun 15;8(12):1434–47. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- 92.Robinson SW, Cho PW, Levitsky HI, Olson JL, Hruban RH, Acker MA, et al. Arterial delivery of genetically labelled skeletal myoblasts to the murine heart: long-term survival and phenotypic modification of implanted myoblasts. Cell Transplant. 1996 Jan-Feb;5(1):77–91. doi: 10.1177/096368979600500113. [DOI] [PubMed] [Google Scholar]
- 93.Suzuki K, Smolenski RT, Jayakumar J, Murtuza B, Brand NJ, Yacoub MH. Heat shock treatment enhances graft cell survival in skeletal myoblast transplantation to the heart. Circulation. 2000 Nov 7;102(19 Suppl 3):III216–21. doi: 10.1161/01.cir.102.suppl_3.iii-216. [DOI] [PubMed] [Google Scholar]
- 94.Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977 Dec 22–29;270(5639):725–7. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 95.Yaffe D, Saxel O. A myogenic cell line with altered serum requirements for differentiation. Differentiation. 1977;7(3):159–66. doi: 10.1111/j.1432-0436.1977.tb01507.x. [DOI] [PubMed] [Google Scholar]
- 96.Nicklas JA, Buel E. Development of an Alu-based, real-time PCR method for quantitation of human DNA in forensic samples. J Forensic Sci. 2003 Sep;48(5):936–44. [PubMed] [Google Scholar]
- 97.Roy-Engel AM, Carroll ML, Vogel E, Garber RK, Nguyen SV, Salem AH, et al. Alu insertion polymorphisms for the study of human genomic diversity. Genetics. 2001 Sep;159(1):279–90. doi: 10.1093/genetics/159.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998 Nov 6;282(5391):1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 99.Reinecke H, Poppa V, Murry CE. Skeletal muscle stem cells do not transdifferentiate into cardiomyocytes after cardiac grafting. J Mol Cell Cardiol. 2002 Feb;34(2):241–9. doi: 10.1006/jmcc.2001.1507. [DOI] [PubMed] [Google Scholar]
- 100.Robey TE, Murry CE. Absence of Regeneration in the MRL Mouse Heart Following Infarction or Cryoinjury. Cardiovasc Pathol. 2007 November-December;16(6) doi: 10.1016/j.carpath.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murry CE, Wiseman RW, Schwartz SM, Hauschka SD. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest. 1996 Dec 1;98(11):2512–23. doi: 10.1172/JCI119070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weier HU, Lucas JN, Poggensee M, Segraves R, Pinkel D, Gray JW. Two-color hybridization with high complexity chromosome-specific probes and a degenerate alpha satellite probe DNA allows unambiguous discrimination between symmetrical and asymmetrical translocations. Chromosoma. 1991 Jul;100(6):371–6. doi: 10.1007/BF00337515. [DOI] [PubMed] [Google Scholar]
- 103.Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002 Apr 5;90(6):634–40. doi: 10.1161/01.res.0000014822.62629.eb. [DOI] [PubMed] [Google Scholar]
- 104.Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood. 2006 Feb 1;107(3):907–15. doi: 10.1182/blood-2005-06-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007 Sep;25(9):1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 106.Stevens KR, Rolle MW, Minami E, Ueno S, Nourse MB, Virag JI, et al. Chemical dimerization of fibroblast growth factor receptor-1 induces myoblast proliferation, increases intracardiac graft size, and reduces ventricular dilation in infarcted hearts. Hum Gene Ther. 2007 May;18(5):401–12. doi: 10.1089/hum.2006.161. [DOI] [PubMed] [Google Scholar]
- 107.Chanseaume S, Azarnoush K, Maurel A, Bellamy V, Peyrard S, Bruneval P, et al. Can erythropoietin improve skeletal myoblast engraftment in infarcted myocardium? Interact Cardiovasc Thorac Surg. 2007 Jun;6(3):293–7. doi: 10.1510/icvts.2006.144014. [DOI] [PubMed] [Google Scholar]
- 108.Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci U S A. 2004 Oct 12;101(41):14907–12. doi: 10.1073/pnas.0406491101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01