Pirt, a Phosphoinositide-Binding Protein, Functions as a Regulatory Subunit of TRPV1 (original) (raw)
. Author manuscript; available in PMC: 2009 May 2.
Abstract
SUMMARY
Transient receptor potential vanilloid 1 (TRPV1) is a molecular sensor of noxious heat and capsaicin. Its channel activity can be modulated by several mechanisms. Here we identify a membrane protein, Pirt, as a regulator of TRPV1. Pirt is expressed in most nociceptive neurons in the dorsal root ganglia (DRG) including TRPV1-positive cells. Pirt null mice show impaired responsiveness to noxious heat and capsaicin. Noxious heat- and capsaicin-sensitive currents in Pirt-deficient DRG neurons are significantly attenuated. Heterologous expression of Pirt strongly enhances TRPV1-mediated currents. Furthermore, the C terminus of Pirt binds to TRPV1 and several phosphoinositides, including phosphatidylinositol-4,5-bisphosphate (PIP2), and can potentiate TRPV1. The PIP2 binding is dependent on the cluster of basic residues in the Pirt C terminus and is crucial for Pirt regulation of TRPV1. Importantly, the enhancement of TRPV1 by PIP2 requires Pirt. Therefore, Pirt is a key component of the TRPV1 complex and positively regulates TRPV1 activity.
INTRODUCTION
In mammals, pain sensation or nociception is initiated by a subset of sensory neurons, known as nociceptive neurons, in the dorsal root ganglia (DRG) (Scott, 1992). The essential functions of nociceptive neurons are detecting noxious thermal, mechanical, and chemical stimuli in the environment, converting them to electrochemical signals, and sending signals to the spinal cord. Nociceptive neurons can also become sensitized after tissue injury or inflammation such that innocuous stimuli can activate them (Scott, 1992).
In recent years, the family of transient receptor potential (TRP) channels has attracted interest due to its importance in various sensory systems including thermosensation and nociception (Montell, 2005). TRPV1, a nonselective cation channel of six transmembrane domains, was identified by its responsiveness to noxious heat (>43°C) and capsaicin from chili peppers (Caterina et al., 1997; Tominaga et al., 1998). TRPV1 is expressed primarily in small-to-medium diameter nociceptive neurons. _TRPV1_−/− mice show a complete absence of behavioral responses to capsaicin and impaired responses to painful heat (Caterina et al., 2000). In addition, cultured sensory neurons from _TRPV1_−/− mice show a loss of capsaicin responses and thermal (42°C–52°C) sensitivity (Caterina et al., 2000; Davis et al., 2000). TRPV1 also plays a critical role in thermal inflammatory hyperalgesia since its activity can be potentiated by inflammatory mediators such as nerve growth factor and bradykinin (Chuang et al., 2001). These mediators act through various mechanisms, including phosphorylation of intracellular residues or increased membrane expression (Huang et al., 2006).
The activity of TRPV1 can also be modulated by phosphoinositides (PIPs). For example, TRPV1 can be negatively regulated by phosphatidylinositol-4,5-bisphosphate (PIP2)(Chuang et al., 2001). A putative PIP2-binding domain in the C terminus of TRPV1 has been identified, and mutations in this domain result in increased TRPV1 baseline currents and reduced thermal threshold and sensitivity to capsaicin (Prescott and Julius, 2003). However, recent studies have shown that PIP2 can have an activating effect on TRPV1 (Liu et al., 2005). When PIP2 (or other PIPs) is added heterologously to excised inside-out membrane patches, TRPV1-mediated current is enhanced (Stein et al., 2006; Lukacs et al., 2007). These data suggest that PIP2 plays a dual role in TRPV1 regulation. More recently, it was shown that TRPV1 can directly bind PIPs and that this binding disrupts the association of calmodulin (CaM) with TRPV1 (Kwon et al., 2007).
Despite the clear physiological importance of TRPV1, little is known about TRPV1-associated proteins and the subunit structure of TRPV1 complexes. Here we describe a membrane protein, called _p_hosphoinositide _i_nteracting _r_egulator of _T_RP (Pirt), which is expressed specifically in the peripheral nervous system (PNS), predominantly in nociceptive neurons. Pirt null (_Pirt_−/−) mice exhibit phenotypes that resemble those observed in _TRPV1_−/− mice, though they are less severe. _Pirt_-deficient DRG neurons have significantly lower noxious heat- and capsaicin-evoked currents than those observed in wild-type (WT) neurons. Consistently, coexpression of Pirt and TRPV1 in HEK293 cells significantly enhances TRPV1-mediated currents. Furthermore, the C terminus of Pirt binds TRPV1 and several PIPs including PIP2 and is sufficient to potentiate TRPV1 activity. Lastly, we found that Pirt and PIP2 require each other to enhance TRPV1. These data suggest that Pirt is a component of the TRPV1 complex and positively regulates the channel activity via PIP2.
RESULTS
Identification of Pirt: A Gene Strongly Expressed in DRG but Not in the CNS
A cDNA subtractive screen using neonatal WT and _Ngn1_−/− mouse DRG was performed to find genes specifically expressed in nociceptive neurons (Dong et al., 2001). From this screen, we isolated a previously uncharacterized gene encoding a 135 amino acid membrane protein named Pirt based on its function. The predicted Pirt protein sequence is highly conserved among vertebrates (Figure 1A). Using the TMHMM program we predicted that Pirt has two transmembrane domains. It is likely that Pirt is inserted into the membrane as a hairpin with both N and C termini in the cytoplasm (Figure 1E). Pirt protein does not contain any known functional domains with the exception of clusters of positively charged residues, characteristic of PIP-binding motifs (Wang et al., 2001), at its C terminus (Figure 1A). In situ hybridization revealed that Pirt is strongly expressed in most DRG (Figures 1B and 1C) and trigeminal neurons (data not shown). Weaker staining was also found in other parts of the PNS including sympathetic (Figure 1D) and enteric (data not shown) neurons. Strikingly, there was no Pirt expression in the spinal cord (Figure 1B). Pirt is first expressed in DRG neurons around embryonic day 11.5, and expression is maintained throughout adulthood. Consistently, Pirt protein is specifically detected in the PNS (Figure S1 available online).
Figure 1. Pirt Encodes a Two Transmembrane Domain Protein and Is Expressed Predominantly in DRG.
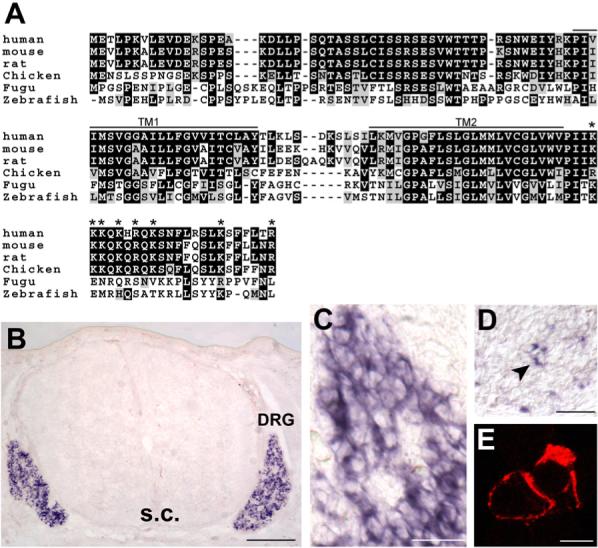
(A) Alignment of amino acid sequences of Pirt from various vertebrates. Residues shaded in black are identical in >50% of the predicted proteins; similar residues are highlighted in gray. The predicted transmembrane domains (TM1 and TM2) and positively charged residues in the C-terminal region (asterisk) are indicated. (B–D) In situ hybridization with cRNA probes detecting Pirt. (B) Cross-section through the trunk region of E14.5 mouse embryo. Strong Pirt signal was present in DRG but absent in the spinal cord (s.c.). (C) A higher magnification view of the DRG region. (D) Cross-section of sympathetic ganglia from E14.5 embryo. A small subset of neurons was stained (arrowhead). (E) Membrane localization of Pirt. Pirt cDNA was tagged with a myc epitope at the C terminus of the protein and expressed in PC12 cells. When the plasma membrane was permeabilized by detergent, membrane localization of the Pirt-myc fusion protein was observed by anti-myc antibody staining (two adjacent cells shown). Under nondetergent conditions no specific staining was found (data not shown). Similar results were obtained using an antibody against the N-terminus of Pirt (data not shown). Scale bars in (B), (C), (D), and (E) represent 100, 50, 40, and 10 μm, respectively.
Generation of _Pirt_−/− Mice
To study Pirt function in vivo, we generated Pirt null (_Pirt_−/−) mice. In these mice, the entire open reading frame of Pirt was replaced by the axonal tracer farnesylated enhanced green fluorescent protein (EGFPf) (Figure 2A). Expression of the knockin EGFPf is under control of the endogenous Pirt promoter. The resulting _Pirt_−/− animals are viable, fertile, and indistinguishable from WT littermates in appearance. They show normal locomotion (Figure S4D) and overt behavior. Mating between heterozygous animals produced WT, heterozygous, and homozygous mutant offspring with expected Mendelian ratios. The deletion of Pirt protein in _Pirt_−/− mice was confirmed by western blot of DRG lysates using anti-Pirt antibody (Figure 2B). In contrast, TRPV1 protein levels of Pirt+/+ and _Pirt_−/− DRG neurons are similar (Figure 2B). WT and _Pirt_−/− mice also have the same proportions of CGRP-, IB4-, and NF200-immunoreactive neurons (i.e., different subtypes of sensory neurons) to the total number of DRG neurons (Figure 2C). Furthermore, the percentages of TRPV1+ neurons are comparable in WT and _Pirt_−/− mice (Figure 2C). The immunoreactivity and projection patterns of TRPV1+, CGRP+, and IB4+ primary afferents to the spinal cord are similar between the two groups (Figure S2). These results suggest that Pirt is not involved in the development of nociceptive neurons.
Figure 2. Generation of Pirt Null Mice.
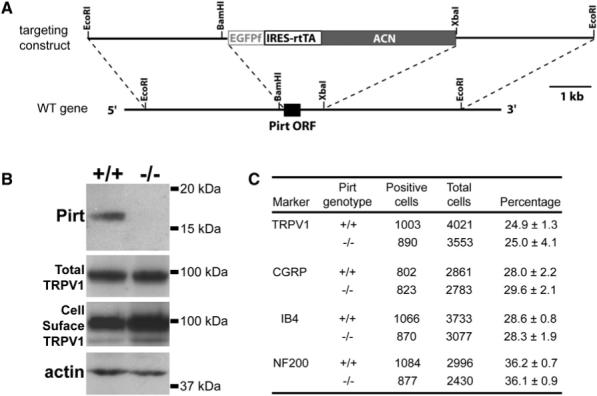
(A) Targeting strategy for disrupting the Pirt gene. The entire ORF of Pirt (black box) was replaced by farnesylated enhanced GFP (EGFPf) followed by IRES-reverse-tetracycline-transactivator (IRES-rtTA). ACN is a self-excising neomycin expression cassette. (B) Western blots of DRG lysates derived from Pirt+/+ and _Pirt_−/− mice using antibodies specifically against Pirt, TRPV1, and β-actin demonstrate that Pirt protein is specifically deleted in _Pirt_−/− mice. TRPV1 expression in cultured DRG neurons was measured by western blot after plasma membrane proteins were biotinylated and purified with streptavidin. The molecular weights are indicated on the right. (C) Immunofluorescence staining of DRG sections show normal proportions of TRPV1-, CGRP-, IB4-, and NF200-expressing neurons in _Pirt_−/− mice (n = 3). The total number of DRG neurons was determined by costaining with the pan-neuronal marker NeuN (Lo et al., 2002). Values are mean ± standard error.
Pirt Is Expressed by Most CGRP+ and IB4+ DRG Neurons
Since the expression of the knockin EGFPf is under control of the endogenous Pirt promoter, EGFPf should be expressed by Pirt+ neurons in Pirt-EGFPf knockin mice. Anti-GFP antibody staining revealed that GFP is widely expressed in the DRG isolated from these mice, labeling 83.9% of all neurons. This staining pattern reflects the results from in situ hybridization using Pirt riboprobe. We then assessed whether Pirt is expressed by peptidergic (CGRP+) and/or nonpeptidergic (IB4+) neurons, two major subtypes of unmyelinated nociceptive C-fibers (Caterina and Julius, 1999). Double immunofluorescence staining revealed that most if not all CGRP+ and IB4+ neurons express Pirt (Figures S3A and S3B). Myelinated neurons, visualized by anti-neurofilament 200 (NF200) antibody, partially overlap with Pirt+ neurons (Figure S3C). The _Pirt_-expressing myelinated neurons may include thinly myelinated nociceptive Aδ fibers and other myelinated non-nociceptive neurons. Importantly, subsets of Pirt+ neurons express TRPV1 (Figure S3D). Most _Pirt_-negative neurons are NF200+ and have large-diameter cell bodies (36.7 ± 1.8 mm). Furthermore the axons of Pirt-expressing neurons project to both laminae I and II of the dorsal spinal cord, the central projection sites for most nociceptive fibers (Figures S3E and S3F).
Pirt Null Mice Have Impaired Behavioral Responses to Noxious Heat and Capsaicin
Since Pirt is expressed in most C-fiber nociceptive neurons, we performed a battery of nociceptive behavioral experiments on _Pirt_−/− mice and WT littermates to assess the possible involvement of Pirt in nociception. To study the behavioral responses to noxious heat, we first performed the tail immersion test, where the latency time for the mice to withdraw their tails after tail immersion into heated water baths was recorded. _Pirt_−/− mice exhibited significantly prolonged tail-withdrawal latency at temperatures of 48°C, 50°C, and 52°C as compared to WT controls (Figure 3A). The tail-withdrawal latency at 46°C was not significantly different between the two groups (data not shown). We then performed the hot-plate assay (Figure 3B). Mice were placed on a hot surface and the latency time for shaking or licking of the hindpaw was measured. The _Pirt_−/− mice showed a significant increase in the latency time at temperatures of 50°C and 52.5°C. At 48°C (data not shown) and 55°C (Figure 3B), mutant mice still showed increased latency time compared to WT mice, but the difference was not significant. In the Hargreaves assay (radiant heat), a similar reduction in heat sensitivity was seen in _Pirt_-deficient animals (Figure S4A). Furthermore, thermal hyperalgesia (elevated pain sensitivity) under inflamma-tory conditions was reduced in the mutant mice (Figure S4). These results indicate that _Pirt_−/− mice have impaired behavioral responses to noxious heat.
Figure 3. _Pirt_−/− Mice Show Impaired Behavioral Responses to Noxious Heat and Capsaicin.

(A) Tail-withdrawal latency of _Pirt_−/− mice (black bars) in the tail immersion test was significantly increased as compared with Pirt+/+ mice (white bars) when water bath temperature was at 48°C (Pirt+/+: n = 15; _Pirt_−/−: n = 16), 50°C (+/+: n = 21; −/−: n = 21), and 52°C (+/+: n = 9; −/−: n = 10). (B) Paw-withdrawal latency of _Pirt_−/− mice on hot plate was significantly longer than in WT mice at 5C (+/+: n = 21; −/−: n = 22) and 52.5°C (+/+: n = 15; −/−: n = 17) but not at 55°C (+/+: n = 11; −/−: n = 14). (C) Behavioral responses to intraplantar injection of capsaicin (0.6 μg) were measured for Pirt+/+ (n = 18) and _Pirt_−/− (n = 18) mice for 10 min after injection. (D) Responsiveness to mechanical stimulation as measured by von Frey test was the same in WT (n = 8) and mutant (n = 9) mice. (E) _Pirt_−/− mice (n = 18) showed a similar number of writhes in response to intraperitoneal acetic acid injection compared with WT mice (n = 16). (F) Paw licking times during phase I (0−10 min) and phase II (10−60 min) of the nociceptive response to formalin injection (Tjolsen et al., 1992) were not significantly different between the two genotypes (n = 14/genotype). (G) Behavioral responses to intraplantar injection of mustard oil (6 μl of 2.0%) were measured for Pirt+/+ and _Pirt_−/− mice (n = 13/genotype) for 10 min after injection. *p < 0.05; **p < 0.01; ***p < 0.001 for Pirt+/+ versus _Pirt_−/−; two-tailed unpaired t test. All error bars represent standard error of the mean (SEM).
In addition to thermosensation, we evaluated chemically and mechanically evoked pain responses in _Pirt_−/− mice. Intraplantar injection of capsaicin was performed on WT and _Pirt_−/− mice. The time spent by the mice licking and biting of the hindpaw was recorded. A significant reduction in the response time was observed in _Pirt_−/− mice (15.7 ± 2.3 s) as compared to WT mice (24.2 ± 2.5 s) (Figure 3C). When sensitivity to acid was tested on the WT and _Pirt_−/− mice, the mutant mice showed a slight, but statistically insignificant, increase in sensitivity (Figure 3E). We also injected mustard oil into the mouse hindpaw and recorded time spent licking the affected paw, which is a behavior mediated by TRPA1 (Bautista et al., 2006; Kwan et al., 2006). WT and _Pirt_−/− mice showed similar responses in this test (Figure 3G). Licking behavior induced by intraplantar injection of formalin into the hindpaw was also similar between _Pirt_−/− and WT mice (Figure 3F). Furthermore, the two groups exhibited similar mechanical responses under normal or inflammatory conditions (Figures 3D and S4C). These results show that Pirt is required for capsaicin-induced pain behavior but not for other chemically or mechanically evoked responses.
TRPV1-Mediated Currents Are Attenuated in DRG Neurons Lacking Pirt
The behavioral phenotypes of _Pirt_−/− mice resemble those observed in _TRPV1_−/− mice (Caterina et al., 2000). We hypothesized that the altered mouse behavior could be attributed to defects in the ability of the TRPV1 channel to conduct current in the absence of Pirt. We tested this hypothesis by performing whole-cell patch clamp recordings on dissociated DRG neurons isolated from WT and _Pirt_−/− mice. Since TRPV1 is expressed in small- and medium-diameter nociceptive neurons (Caterina et al., 2000), we recorded DRG neurons with cell body diameters ranging from 18 μm to 25 μm (means at 21.4 μm for both genotypes). Inward currents generated by noxious heat were found in 53.0% of DRG neurons (n = 76) from Pirt+/+ mice. A similar number of DRG cells (52.8%; n = 70) from _Pirt_−/− mice also responded to noxious heat, consistent with the fact that percentages of TRPV1-expressing cells are similar in WT and _Pirt_−/− mice (Figure 2C). Although the percentages of DRG neurons responding to noxious heat in these two genotypes are similar, the current sizes of responsive neurons from _Pirt_−/− mice are significantly smaller than those of WT mice (Figures 4A and 4B). The average currents in WT and _Pirt_−/− neurons were 3.0 ± 0.3 nA and 1.9 ± 0.2 nA, respectively (p = 0.008). When the average current size was divided by cell capacitance to normalize the current to cell size, the current over capacitance values were 94.8 ± 8.7 pA/pF and 56.6 ± 4.0 pA/pF for the WT and _Pirt_−/− neurons, respectively (p = 0.0002). The heat activation threshold was not significantly different between the two groups (WT: 43.2°C ± 0.8°C; _Pirt_−/−: 44.7°C ± 0.7°C). These data show that the absence of Pirt reduces the ability of TRPV1 to conduct current in response to noxious temperatures.
Figure 4. Heat- and Capsaicin-Evoked Currents Were Reduced in Pirt-Deficient DRG Neurons.
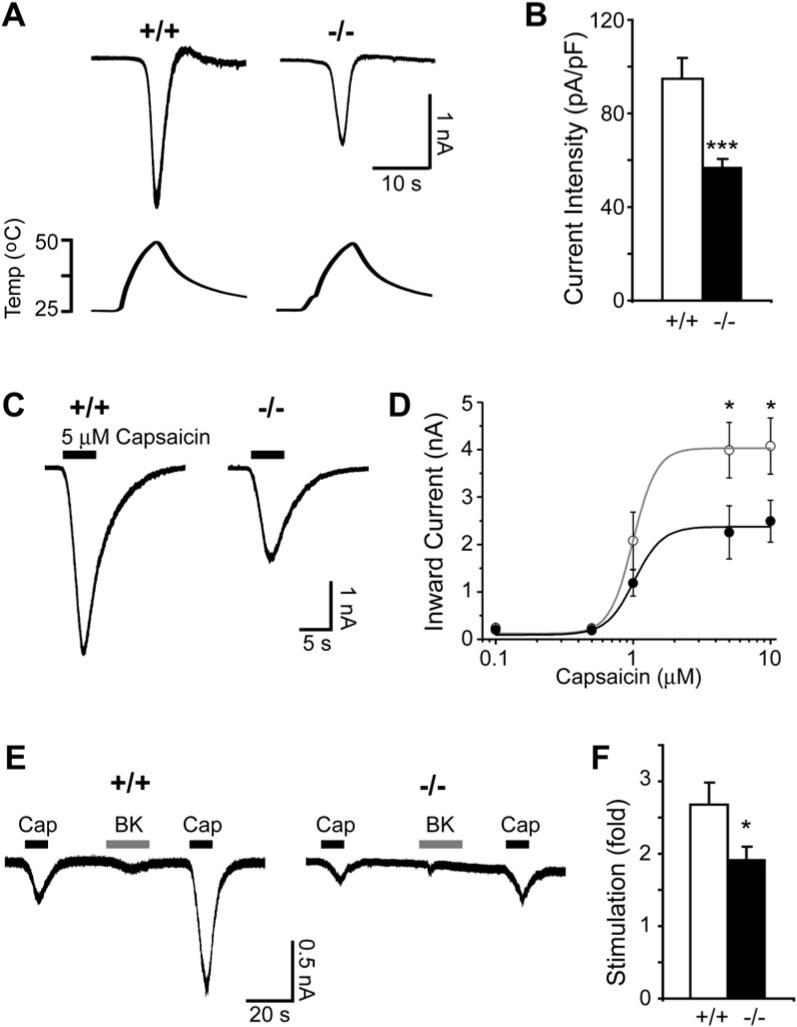
(A) Responses of DRG neurons from Pirt+/+ (+/+) and _Pirt_−/− (−/−) mice to heat. Current trace and temperature ramp (25°C –50°C) are shown on the top and bottom, respectively. (B) Current intensities of DRG neuron response to heat. Current intensities of WT (open bar, n = 39) and _Pirt_−/− (black bar, n = 33) neurons were significantly different (***p < 0.005). (C) Inward current response to application of 5 μM capsaicin (black line) in DRG neurons from Pirt+/+ (+/+) and _Pirt_−/− (−/−) mice. (D) The concentration-response relationship expressed as inward current versus concentration of capsaicin in WT (open circles) and _Pirt_−/− (closed circles) neurons. Data points were fitted to the Hill equation. When 5 μM and 10 μM capsaicin were applied to DRG culture neurons of WT (n = 26 in 5 μM, n = 25 in 10 μM) and _Pirt_−/− (n = 26 at 5 μM, n = 20 at 10 μM) mice, respectively, the current sizes (*p < 0.05) and current intensities (data not shown) were significantly different. (E) Representative current traces from Pirt+/+ (n = 15) and _Pirt_−/− (n = 16) neurons stimulated with capsaicin (0.1 μM, black line) followed by bradykinin (1 μM, gray line) and capsaicin (0.1 μM) again. (F) Potentiation by bradykinin was calculated by dividing post-treatment capsaicin-evoked currents by pre-treatment values. Wild-type neurons showed stronger potentiation by bradykinin than _Pirt_−/− neurons (p = 0.041). All error bars represent SEM.
We also examined capsaicin-evoked currents in Pirt+/+ and _Pirt_−/− neurons. Again, the proportion of DRG neurons responding to capsaicin was the same in WT (56.0%; n = 294) and mutant (57.0%; n = 268) neurons. We applied different doses of capsaicin to dissociated neurons to determine TRPV1 sensitivity. Although _Pirt_−/− neurons exhibited less capsaicin-activated current than WT neurons at every concentration tested, statistically significant reductions were only found at higher capsaicin concentrations (5 and 10 μM; Figures 4C and 4D). The half-maximal effective doses (EC50) for activating capsaicin-sensitive currents were similar between WT (0.99 ± 0.03 μM) and _Pirt_−/− (1.02 ± 0.07 μM) neurons, suggesting that Pirt affects the efficacy but not potency of capsaicin-induced current. These results are not due to a Pirt-dependent reduction in total expression of TRPV1 and TRPV1 membrane trafficking in DRG neurons (Figure 2B). These electrophysiological experiments demonstrate that Pirt is required for normal noxious heat- and capsaicin-evoked currents.
TRPV1 can be potentiated by bradykinin via both protein kinase C (PKC) and phospholipase C (PLC) dependent pathways (Hardie, 2007). _Pirt_−/− neurons exhibited a reduction in bradykinin-mediated TRPV1 potentiation (Figures 4E and 4F), suggesting that Pirt plays a role in this process (see Discussion).
We next addressed whether deletion of Pirt affects the function of other ion channels in nociceptive neurons. We found that _Pirt_−/− neurons respond normally to neurotransmitter γ-amino-butyric acid (GABA) (Figures S5A and S5B). Voltage-sensitive inward currents are similar between WT and _Pirt_−/− neurons (Figures S5C–S5E). Consistent with our behavioral data, _Pirt_−/− neurons responded normally to mustard oil (TRPA1 agonist) as assessed by calcium imaging (Figure S5H). These results suggest that although TRPV1-mediated currents are greatly reduced in _Pirt_−/− neurons, other ion channels function normally.
Pirt Enhances TRPV1 Activity
The _Pirt_−/− behavioral and cellular loss-of-function phenotypes strongly suggest that Pirt positively regulates TRPV1 channel activity. To further test this hypothesis, we examined whether Pirt itself has the ability to enhance TRPV1 activity in a heterologous system. We transfected Pirt cDNA into a HEK293 cell line that stably expresses TRPV1 (Guler et al., 2002) and measured currents in response to noxious temperatures in TRPV1- or TRPV1/Pirt-expressing cells. We used the TRPV1 stably expressing cell line instead of transient transfection of TRPV1 in order to keep the level of TRPV1 more consistent. When the temperature was raised from 25°C to 45°C, cells expressing both TRPV1 and Pirt exhibited significantly larger inward currents than cells expressing TRPV1 alone (3.2 ± 0.4 nA versus 1.6 ± 0.2 nA; p = 0.0004) (Figure 5A). We also examined whether coexpression of Pirt in these cells can increase TRPV1-mediated current activated by capsaicin. Consistent with the DRG neuron experiment, addition of Pirt significantly enhanced capsaicin-activating current only at higher concentrations (5 and 10 μM; Figure 5C). Again, the EC50 values of capsaicin for these two groups of HEK293 cells were not significantly different (0.78 ± 0.03 and 0.67 ± 0.04 μM for Pirt/TRPV1 and TRPV1 cells, respectively). The TRPV1 antagonist capsazepine completely inhibited capsaicin activation in HEK293 cells expressing both TRPV1 and Pirt, suggesting that the increased current is not due to activation of another channel (Figure S6). Furthermore, current-voltage relationships for capsaicin-evoked currents in HEK293 cells coexpressing TRPV1 and Pirt showed a relationship characteristic of TRPV1 (Figures S6B–S6D). The total protein levels and cell-surface expression of TRPV1 in HEK293 cells were not altered by coexpression of Pirt (Figure 5D). Together, these results indicate that Pirt alone is sufficient to enhance TRPV1-mediated currents in the absence of any other DRG-specific proteins.
Figure 5. TRPV1-Mediated Currents Are Enhanced by Expression of Pirt.
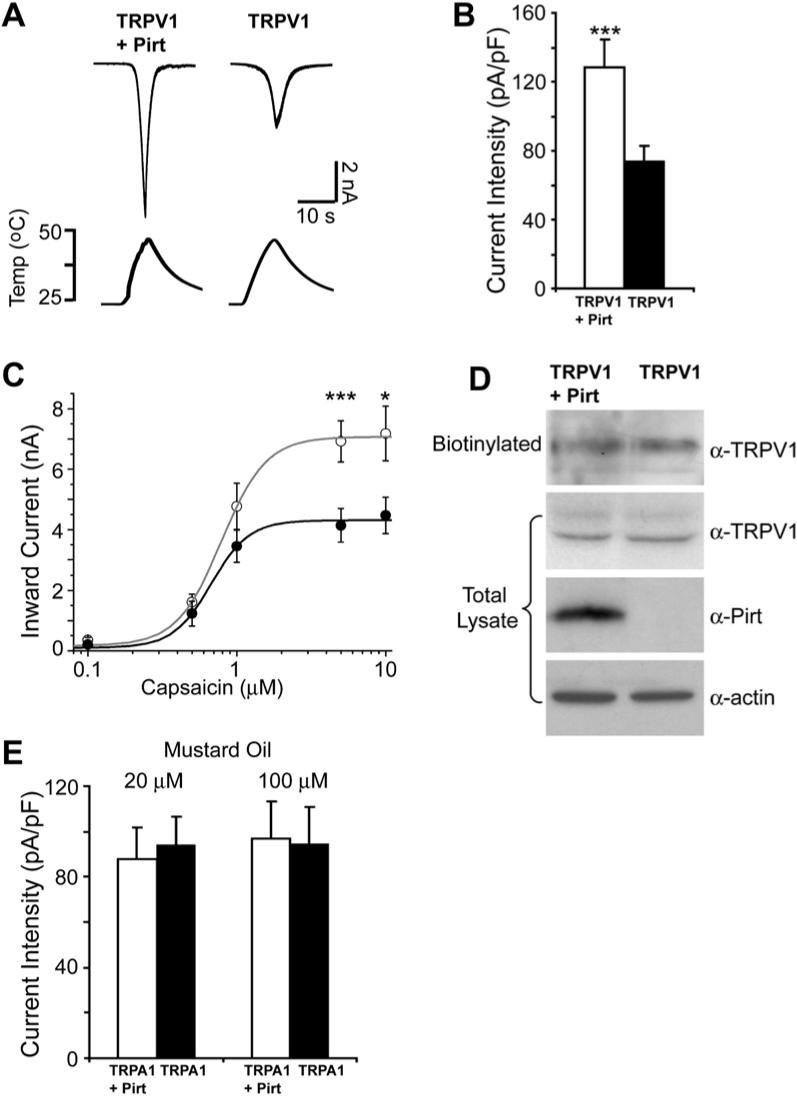
(A) Heat-evoked current traces in TRPV1 stably expressing HEK293 cells. Top shows current traces in TRPV1 cells cotransfected with Pirt and GFP (TRPV1 + Pirt) and TRPV1 cells transfected with GFP alone (TRPV1); bottom shows the ramp of heated buffer application. (B) Heat-evoked current intensities of TRPV1 cells expressing Pirt (open bar, n = 30) or TRPV1 alone (black bar, n = 30). Pirt significantly increases TRPV1 current in response to heat stimulation (***p < 0.005). (C) Capsaicin dose-response curves of TRPV1 stably expressing cells cotransfected with Pirt and GFP (open circles) and TRPV1 cells transfected with GFP alone (closed circles). When 5 μM and 10 μM capsaicin were applied to TRPV1/Pirt cells (n = 26 at 5 μM, n = 16 at 10 μM) and TRPV1 cells (n = 26 at 5 μM, n = 15 at 10 μM), respectively, the current sizes and current intensities (data not shown) indicated significantly different responses. Holding potential = −60 mV. (D) The expression level of TRPV1 in the TRPV1 stable HEK293 cells was measured by western blot after plasma membrane proteins were biotinylated and purified with streptavidin. β-actin was used as a loading control. (E) Mustard oil-evoked current intensities in TRPA1 stably expressing HEK293 cells. Twenty and one hundred micromolars of mustard oil were used to stimulate the cells. The current intensities of TRPA1 cells expressing Pirt (open bar, n = 12) and TRPA1 alone (black bar, n = 12) were almost identical at both concentrations. All error bars represent SEM.
Besides TRPV1, we also tested whether Pirt can modulate TRPA1-mediated current. We transfected Pirt cDNA into TRPA1 stably expressing HEK293 cells (Hill and Schaefer, 2007) and measured mustard oil-evoked currents. Unlike the TRPV1 experiments, expression of Pirt has no effect on TRPA1-mediated current (Figure 5E).
Pirt Binds to TRPV1
Pirt positively regulates TRPV1 activity, raising the possibility that TRPV1 and Pirt may form a complex. To address this possibility, we tested whether Pirt can coimmunoprecipitate TRPV1 when both proteins are expressed in HEK293 cells. When cell lysates of HEK293 cells transfected with TRPV1 and Pirt-myc were immunoprecipitated with anti-myc antibody, TRPV1 was specifically precipitated in the coexpressing cells (Figure 6A). We also examined binding of Pirt-myc to other TRPs. Interestingly, Pirt could bind TRPM8 but not TRPA1 (Figure 6A).
Figure 6. Pirt Binds to TRPV1 and PIPs.
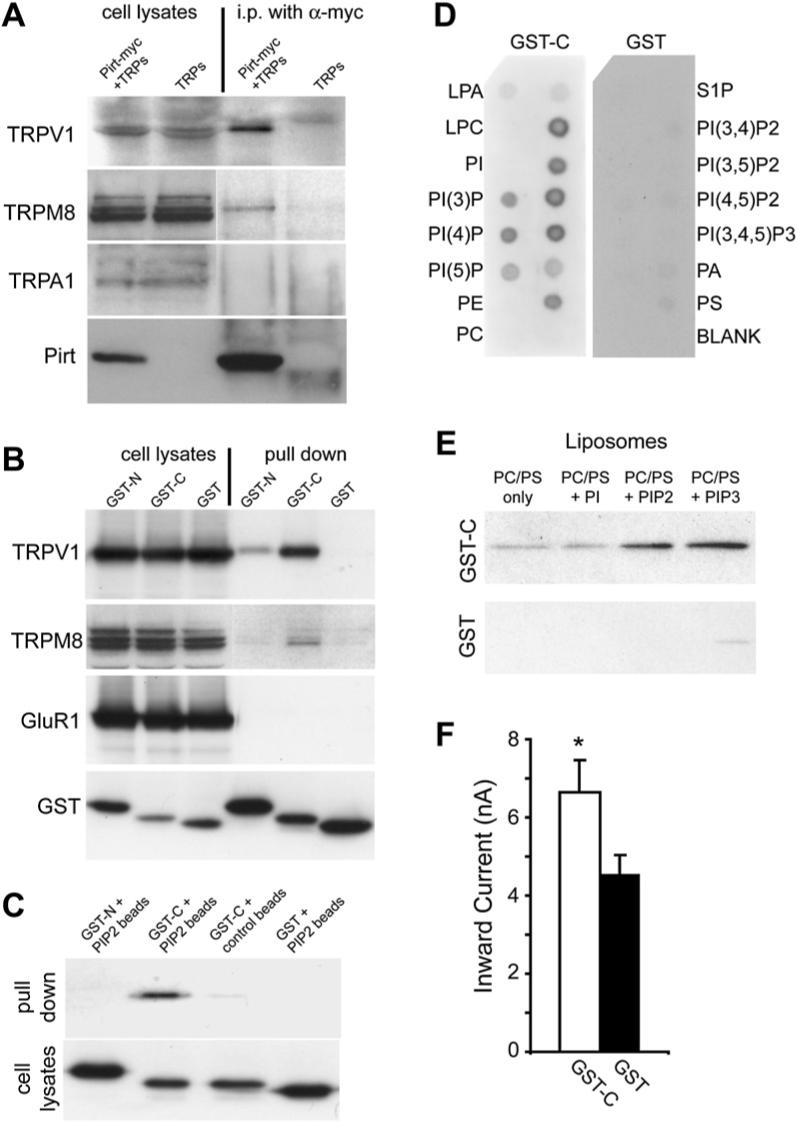
(A) Coimmunoprecipitation of TRP channels and Pirt in HEK293 cells. HEK293 cells transfected with different TRPs and Pirt-myc were immunoprecipitated with anti-myc antibody. Immunoprecipitates were detected by antibodies against corresponding TRPs and Pirt. The immunoblots of TRPs and Pirt in cell lysates indicate their expression levels before immunoprecipitation. Pirt forms a complex with TRPV1 and TRPM8 but not TRPA1. (B) GST pull-down assay of HEK293 cell lysates coexpressing TRPV1 with GST-N, GST-C, or GST alone and visualized by western blot analysis with anti-TRPV1 antibody. GST pull down of HEK293 cell lysates coexpressing either TRPM8 or HA-tagged GluR1 with GST-N, GST-C, and GST alone and visualized by western blot analysis with anti-TRPM8 or anti-HA antibodies, respectively. (C) GST-C binds PIP2-conjugated agarose beads. PIP2-conjugated agarose beads were used to pull down GST-N, GST-C, and GST from HEK293 cell lysates expressing these proteins. Control beads without PIP2 were also used to pull down GST-C. (D) Protein-lipid overlays. Cell lysates from GST-C fusion protein comprised of the Pirt C terminus (left panel) or GST only (right panel) transfected HEK293 cells were used to probe PIP strips. For each strip, lipids were deposited in two rows and only labeled outside. The proteins were detected by ECL after probing with anti-GST antibodies. Strong binding of GST-C to PIPs, PIP2, and PIP3 was observed but no binding was found with control lipids such as phosphatidylinositol (PI) and phosphatidylethanolamine (PE). (E) Liposomal pulldown assay. Liposomes composed of phosphatidylcholine (PC), phosphatidylserine (PS), and different PIPs were prepared as indicated in Experimental Procedures. PI-, PIP2-, or PIP3-containing or control (composed of PC and PS only) liposomes were incubated with the same amount (0.1 μg) of purified GST-C or GST and pelleted by centrifugation. Associated proteins were visualized by western blot with anti-GST antibody. (F) Heat-evoked current sizes of TRPV1 cells expressing GST-C (open bar, n = 29) or GST alone (black bar, n = 33). The C terminus of Pirt can significantly increase TRPV1 current response to heat stimulation (*p < 0.05). All error bars represent SEM.
To determine the TRPV1-binding site in the intracellular regions of Pirt, we made GST fusion constructs, which include sequences encoding the N- (first 53 amino acids of Pirt; GST-N) or C-terminal (last 26 amino acids of Pirt; GST-C) region of Pirt. The constructs were individually transfected with TRPV1 into HEK293 cells and the GST pull-down assay was performed to study the protein-protein interaction. The C terminus of Pirt binds strongly to TRPV1, whereas the N terminus binds weakly and GST alone does not bind (Figure 6B). Again, the C terminus binds TRPM8 (Figure 6B). However, Pirt fails to bind GluR1, a subunit of the AMPA-responsive class of glutamate receptors (Figure 6B). These results support the idea that Pirt interacts with specific TRPs.
Pirt Binds to Phosphoinositides
Since the C terminus of Pirt contains a cluster of positively charged residues (Figure 1A), a characteristic of PIP-binding domains, we examined the interaction between these two molecules using PIP2-conjugated agarose beads in protein pull-down experiments. GST-C bound to the PIP2 beads, (Figure 6C), while GST-N or GST alone did not interact with PIP2 beads. We further tested whether Pirt binds other PIPs using PIP strips, nitrocellulose membranes containing various immobilized PIPs.
We found that the GST-C, but not GST alone, bound most PIPs, whereas no or weak interaction was detected with control phospholipids or blank areas (Figure 6D). To study the binding of Pirt and PIPs in a more physiological context (i.e., a bilayer), we performed a liposomal binding assay on a series of liposomal preparations containing different PIPs. The purified GST-C showed strong association with PIP2- and PIP3-containing liposomes but weak binding to phosphatidylinositide (PI)-containing liposome or control liposome composed of phosphatidylserine (PS) and phosphatidylcholine (PC) only (Figure 6E). Similar results were obtained using cytosolic fractions of HEK293 cells expressing GST-C (data not shown). GST alone did not show strong binding to these liposomes. The data from these different approaches clearly show that Pirt is a PIP-binding protein and it binds various PIPs, including PIP2, but not PI and other negative controls.
Since the regulation of TRPV1 by PIP2 is well documented in the literature, we focused our subsequent analysis on PI(4,5)P2. To determine whether Pirt binding to PIP2 is dependent on the cluster of basic residues in the C terminus, we generated two mutant forms of GST-Pirt C-terminal fusion protein. In one mutated protein (GST-C-3Q, Figure 7A), three lysines were changed to three glutamines, whereas the second mutant (GST-C-6A, Figure 7A) had five lysines and one arginine mutated to six alanines. As predicted, both mutated C termini failed to bind PIP2 beads, suggesting that these positively charged residues are necessary for PIP2 binding (Figure 7B). Surprisingly, both mutated fusion proteins retained the ability to bind TRPV1 (Figure 7C).
Figure 7. Pirt and PIP2 Are Dependent on Each Other to Enhance TRPV1 Activity.
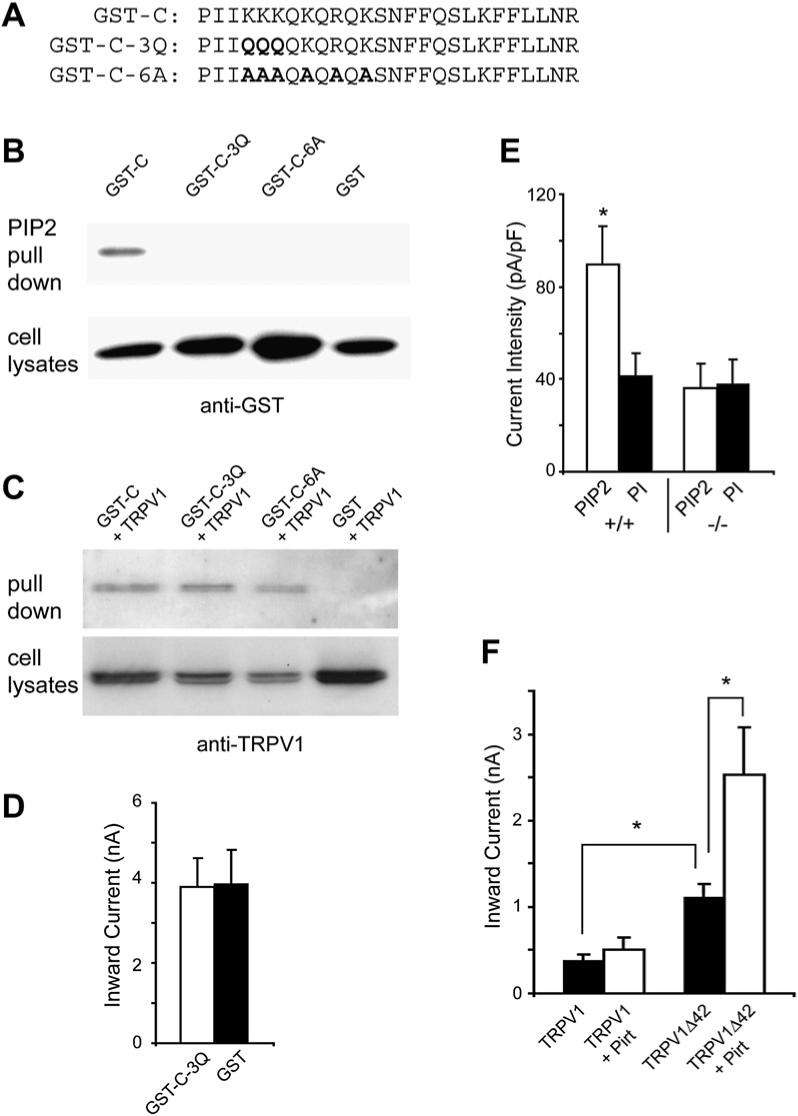
(A) Sequences of WT and mutant forms of the Pirt C terminus, which were fused with GST. Bold letters indicate the mutated basic residues. (B) PIP2-conjugated agarose beads failed to pull down GST-C-3Q and GST-C-6A from HEK293 cell lysates expressing these proteins. The pull-down products were immunoblotted by antibody against GST. (C) Binding of TRPV1 with GST-C-3Q or GST-C-6A in HEK293 cells. HEK293 cells transfected with TRPV1 and either GST-C, GST-C-3Q, or GST-C-6A were pulled down with glutathione-agarose beads. Immunoprecipitates were immunoblotted by anti-TRPV1 antibody. The mutant C termini still formed a complex with TRPV1. (D) Heat-evoked current sizes of TRPV1 stably expressing cells expressing GST-C-3Q (open bar, n = 18) or GST alone (black bar, n = 18). The mutant C terminus has no effect on TRPV1 activity. (E) Current densities of DRG neurons from Pirt+/+ (+/+) and _Pirt_−/− (−/−) mice in response to 5 μM capsaicin. Whole-cell recordings were performed with either water-soluble PIP2-diC8 (10 μM; Echelon Biosciences; open bars) or PI-diC8 (10 μM; black bar) added to the pipette solution. In WT neurons, PIP2 (n = 27) significantly enhanced TRPV1-mediated current as compared to PI (n = 25; p = 0.017). In _Pirt_−/− neurons, PIP2 (n = 25) did not show any effect on TRPV1 current as compared to PI (n = 19). (F) Currents of HEK293 cells transfected with WT TRPV1 or TRPV1Δ42 in the presence or absence of Pirt in response to 0.1 μM capsaicin. Cells expressing TRPV1Δ42 alone (n = 9) showed significantly stronger current than those expressing WT channels (n = 11). Coexpressing Pirt can significantly potentiate TRPV1Δ42- (n = 10) but not WT TRPV1- (n = 12) mediated currents (*p < 0.05). All error bars represent SEM.
Pirt and PIP2 Require Each Other to Enhance TRPV1
Since the C terminus of Pirt can bind both TRPV1 and PIP2, we tested whether the C terminus alone is sufficient to modulate TRPV1 activity. Strikingly, when GST-C was expressed in TRPV1 stably expressing HEK293 cells, it significantly enhanced heat-evoked current as compared to control cells expressing GST alone (Figure 6F). However, when the mutated C terminus of GST-C-3Q was expressed in TRPV1 stably expressing cells, it showed heat-evoked current identical to cells expressing GST alone (Figure 7D; compare to Figure 6F). Together these data suggest that the basic residues in the last 26 amino acids of Pirt are crucial for PIPs’ binding and Pirt function.
Since several studies have indicated that PIP2 enhances TRPV1 activity, it is important to determine if the positive effects of PIP2 on TRPV1 are dependent on Pirt (Liu et al., 2005; Stein et al., 2006; Lukacs et al., 2007). We added 10 μM PIP2-diC8 to the patch pipette solution and recorded whole-cell capsaicin-evoked currents in WT and _Pirt_−/− DRG neurons. In parallel, we used 10 μM PI-diC8 in the pipette solution as a negative control. Recent studies have shown that PI does not have a strong effect on TRPV1 activity (<10% of that of PIP2; Lukacs et al., 2007) and does not interfere with the association between TRPV1 and CaM (Kwon et al., 2007). Furthermore, our data showed that Pirt does not bind to PI (Figure 6D). In WT neurons, PIP2 strongly enhanced capsaicin-induced currents, as compared to PI controls. Importantly, PIP2 had no effect on capsaicin–evoked currents in _Pirt_−/− neurons (Figure 7E). Thus, the potentiation of TRPV1 activity by PIP2 requires Pirt.
PIP2 has also been shown to have an inhibitory effect on TRPV1, particularly at a low stimulus strength (Chuang et al., 2001; Lukacs et al., 2007). A site (42 amino acids) within the C terminus of TRPV1 was found to be required for PIP2-mediated inhibition (Prescott and Julius, 2003). A mutant form of TRPV1 without this site exhibited stronger current than WT TRPV1 and failed to be potentiated by PLC-coupled receptor activation as observed for the WT channel. These data suggest that this is the only site required for the PIP2 inhibitory effect. To determine whether Pirt plays a role in PIP2 inhibitory effect, we tested the ability of Pirt to modulate mutant TRPV1 (TRPV1D42)-mediated current. When 0.1 μM capsaicin was applied, significantly stronger currents were found in HEK293 cells expressing TRPV1Δ42 than those found in cells expressing WT TRPV1, confirming the previous finding. Strikingly, coexpression of Pirt further enhances TRPV1Δ42-mediated current (Figure 7F). Again, at this low level of capsaicin, Pirt has no significant effect on WT TRPV1-mediated current. Furthermore, Pirt can still bind to TRPV1Δ42 in vitro, suggesting that another region of TRPV1 is required for binding Pirt (Figure S7). Together, these data indicate that Pirt is unlikely to be involved in the PIP2-mediated inhibitory effect on TRPV1.
DISCUSSION
Phosphoinositides such as PIP2 are emerging as important modulators of TRP channel activity. Here, we demonstrate that Pirt, a membrane protein, binds both PIP2 and TRPV1 and positively regulates TRPV1 via PIP2. Pirt is highly conserved among vertebrates, and no closely related invertebrate homologs were found by a genome-wide sequence search, suggesting that vertebrates have acquired additional regulatory subunits for TRP channels. The expression of Pirt is restricted to the PNS (predominantly DRG and trigeminal ganglia) and is not found in the central nervous system (CNS). Therefore, the modulation of TRPV1 activity by Pirt likely occurs in these peripheral neurons but not in higher-order neurons within the CNS. Since Pirt is also expressed in TRPV1-negative neurons in DRG, it may be involved in regulating other TRP channel activity.
_Pirt_−/− mice show noxious heat and capsaicin behavioral phenotypes that resemble, but are less severe than, those observed in _TRPV1_−/− mice. These data support the hypothesis that TRPV1 works as a primary sensor of noxious heat and capsaicin, while Pirt regulates TRPV1 activity. Pirt itself does not exhibit any channel activity upon noxious heat or capsaicin treatment (data not shown). Whole-cell recordings from DRG neurons and TRPV1 stably expressing HEK293 cells suggest that Pirt enhances TRPV1-mediated currents. This enhancement is not due to altered cell surface level expression of TRPV1 by Pirt based on our surface biotinylation data. Single-channel recording experiments should give us more detailed information on how Pirt affects conductance, open time, or open probability of TRPV1.
Consistent with a role for Pirt in potentiating TRPV1 sensation, we found that Pirt physically interacts with TRPV1. Therefore, it is likely that Pirt functions as a regulatory subunit of the TRPV1 complex. More importantly, the C terminus of Pirt alone can bind PIPs and TRPV1 and enhance heat-evoked current through TRPV1. The basic residues in the C terminus of Pirt are required for PIP2 but not TRPV1 binding, suggesting that the 26 amino acids of the C terminus contain separate binding domains for PIP2 and TRPV1. In addition, the mutant form of the Pirt C terminus failed to modulate TRPV1 function, indicating that PIP2 binding is essential for Pirt regulation of TRPV1 and that the binding to TRPV1 alone is not sufficient to enhance the channel's activity. The C terminus of Pirt can bind to PIPs other than PIP2 in vitro, suggesting that nonspecific electrostatic interactions contribute strongly to the binding between the basic residues and the negatively charged inositides. Such interactions with PIPs are well characterized for several proteins, such as MARCKS, which contain clusters of positively charged residues (Wang et al., 2001). The broad PIP binding of Pirt is consistent with other findings that TRPV1 can be modulated by various PIPs besides PIP2 (Kwon et al., 2007; Lukacs et al., 2007). Therefore, it is probable that other PIPs may also participate in Pirt's regulation of TRPV1.
It has been shown that PIP2 has an inhibitory effect on heat- and capsaicin-evoked TRPV1 current (Chuang et al., 2001; Prescott and Julius, 2003). However, other studies have found that PIP2 has an activating effect on TRPV1. For example, direct application of PIP2 to the inner leaflet of TRPV1-expressing membranes led to channel activation, while sequestering PIP2 with polylysine inhibited channel function (Stein et al., 2006). A more recent study may reconcile the discrepancy (Lukacs et al., 2007). These authors found that PIP2 has a dual effect on TRPV1: inhibitory at low capsaicin concentrations and activating at high capsaicin concentrations. Our data support previous findings that PIP2 can have both activating and inhibitory effects on TRPV1 and highlight the role of Pirt in mediating the activating effect of PIP2.
Our PIP2 application experiments suggest that PIP2 has an activating effect on TRPV1 at high capsaicin concentrations (5 μM). Importantly, Pirt is required for this positive effect. Other groups have observed PIP2 activating effects on TRPV1 activity in heterologous systems where Pirt is unlikely to be present (Stein et al., 2006; Lukacs et al., 2007). In these cases, the experiments were performed using inside-out recording with direct application of PIP2 to the inner leaflets of membranes. Therefore, it is likely that the very high levels of PIP2 applied in their experimental systems can bypass the requirement for Pirt. Furthermore, in our capsaicin dose-response experiments, the activating effect of Pirt on TRPV1-mediated currents was only prominent when high concentrations of capsaicin (5 and 10 μM) were applied. This effect is also PIP2 dependent (Figure 7D). Therefore, both Pirt and PIPs are required for optimal TRPV1 function.
At low concentrations of capsaicin, PIP2 plays an inhibitory role on TRPV1. It has been proposed that the inhibitory effect maybe indirect and that other PIP2-binding protein(s) may be involved (Stein et al., 2006; Lukacs et al., 2007). Our data show that Pirt is not involved in this modulation since Pirt can further enhance the activity of TRPV1Δ42. Therefore, Pirt plays an essential role only in the activating effect of PIP2 but not in its inhibitory effect. These results also indicate that the inhibitory effect of PIP2 at low capsaicin concentration masks the activating effect of Pirt on TRPV1. However, removal of the inhibitory effect by bradykinin-activated PLC or deleting the last 42 amino acids of TRPV1 unmask the activating effect of Pirt. We have shown that PIP2 modulates TRPV1 in two ways: inhibition by interacting with the last 42 amino acids of TRPV1 and activation via association with Pirt at a different region of TRPV1. Furthermore, hydro-lysis of PIP2 by PLC may differ in these situations.
Our behavioral and cellular studies show that Pirt does not play a role in TRPA1 function, suggesting that Pirt does have specificity toward certain TRP channels. Interestingly, Pirt also fails to bind TRPA1. It will be interesting to test if the failure of Pirt to regulate TRPA1 is due to its inability to bind this channel. However, we cannot rule out the possibility that Pirt affects the activities of other TRPs, especially as PIP2 has been implicated in the regulation of many TRP channels (Hardie, 2007; Voets and Nilius, 2007). For example, TRPM8, the cold and menthol sensor (Colburn et al., 2007; Dhaka et al., 2007), requires PIP2 for activation (Rohacs et al., 2005). Interestingly, Pirt can also bind TRPM8. Therefore, Pirt may regulate the activities of other TRP channels via PIP2. Because TRP channels play critical roles in nociception, uncovering the interactions between Pirt, TRPs, and PIPs should provide a better understanding of pain transduction.
EXPERIMENTAL PROCEDURES
Molecular Biology
The arms of the Pirt targeting constructs were subcloned from a 129/SvJ genomic DNA lambda phage clone (Invitrogen) and ligated with an EGFPf-IRES-rtTA-CAN cassette. Pirt+/− mice were generated using the targeting construct by a transgenic mouse facility at the University of Cincinnati. Details of the generation of Pirt knockout mice are available in the Supplemental Data. The DNA fragments corresponding to the N- (1−53) and C- (110−135) terminal regions of Pirt were generated by PCR amplification of Pirt cDNA and cloned into pCMV-GST (Tsai and Reed, 1997) in frame with glutathione S-transferase (GST). The mutant forms of the Pirt C terminus were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene).
In Situ Hybridization
Nonisotopic in situ hybridization on frozen sections from WT E14.5 embryos was performed as previously described using cRNA probes (Dong et al., 2001).
Anti-Pirt Serum
Rabbit polyclonal antibody was raised against a synthetic peptide, EVLPKALEVDERSPESKDL, corresponding to the N terminus of mouse Pirt by Protein-tech Group, Inc.
Cell Culture
DRG neurons were collected from all spinal levels from 3- to 4-week-old mice and then were dissociated and plated on coated glass coverslips. Cells were plated for 24 hr before use. TRPV1 and TRPA1 stably expressing HEK cells were obtained from Drs. M. Caterina (Johns Hopkins University) and N. Tigue (GlaxoSmithKline), respectively. For transient transfection of HEK293 cells, Lipofectamine (Invitrogen) was used. Details of Neuronal and HEK cell culture conditions and transfection are available (see Supplemental Data).
Coimmunoprecipitation
HEK293 cells were transfected with a Pirt-myc fusion construct along with a TRPV1, TRPM8, or TRPA1 plasmid. After 24 hr, cell lysates were collected and supernatant was extracted. Myc antibody was added to the supernatant and incubated overnight. Gammabind Plus Sepharose beads (GE Healthcare Life Sciences) were added to the lysates, and the mixtures were incubated for an additional 3 hr and the proteins were eluted using 2× SDS-PAGE sample buffer (see Supplemental Data).
GST Pull Down
The GST-N, GST-C fusion, and GST constructs were separately transfected with various ion channel genes into HEK293 cells. After 24 hr, cell lysates were incubated using glutathione-agarose beads. Specifically bound proteins were eluted from the beads using 2× protein sample buffer. Details are available (see Supplemental Data).
Biotinylation of Cell-Surface Proteins
TRPV1 stably expressing HEK293 cells were transfected with Pirt-containing plasmid. After 24 hr, the HEK cells or cultured DRG neurons were then incubated with 1.0 mg/ml sulfo-NHS-SS-Biotin (Pierce) in PBS for 30 min at 4°C. After quenching, cells were lysed in 500 μl of RIPA buffer. Supernatant from cell lysate was added to BSA-Blocked ultralink-neutravidin beads (Pierce) and incubated for 2 hr. The proteins were eluted from the beads using 2× SDS-PAGE sample buffer and western blot was performed using the anti-TRPV1 antibody (from Dr. M. Caterina). Details are available (see Supplemental Data).
PIP2 Bead Binding and PIP Strips
HEK293 cells were singly transfected with GST-N, GST-C, or GST. The cells were lysed and spun down at 10,000 × g for 10 min. The supernatants were added to the PIP beads or PIP strips (Echelon Biosciences). Details are available (see Supplemental Data).
Liposomal Binding Assay
Liposomes were prepared by mixing PC, PS, and PIPs at the proportions of 49.5%, 49.5%, and 1.0%, respectively, drying the mixture under vacuum, and resuspending to a final concentration of 1 mg/ml of total phospholipid in a buffer composed of HEPES (50 mM), NaCl (100 mM), and EDTA (0.5 mM). After sonicating for 30 min, liposomes were collected by centrifugation at 16,000 × g for 10 min and resuspended in binding buffer. Fifty microliters of liposome suspension was mixed with 0.1 μg of purified protein and incubated at room temperature for 15 min. Liposomes were pelleted at 16,000 × g for 10 min and bound fraction was analyzed by western blot (see Supplemental Data).
Behavioral Assays
Behavioral tests were performed by individuals blind to genotype. The mice used in the tests were backcrossed to C57Bl/6 mice for at least five generations and were 2- to 3 month-old (20−30 g) males. All experiments were performed under protocol approved by the Animal Care and Use Committee of Johns Hopkins University School of Medicine. Details for all the behavior assays are available in the Supplemental Data.
Electrophysiology
Whole-cell voltage-clamp recordings were performed. For neurons, only small neurons were selected for recording. For HEK293 cells, transfected cells were identified by GFP fluorescence. Only one neuron or one HEK cell was tested per coverslip.
Capsaicin, mustard oil, and GABA were used as agonists to TRPV1, TRPA1, and GABA receptors, respectively. When examining heat-evoked current responses, a preheated solution was used to increase bath temperature; the heating was an in-line solution heater and used the TC-324B temperature controller (Warner Instruments). Statistical comparisons were made using un-paired Student's t test and differences were considered significant at p < 0.05. Details are available in the Supplemental Data.
Supplementary Material
supplemental d
ACKNOWLEDGMENTS
We thank Drs. M. Caterina for TRPV1 stable HEK293 cells, TRPV1, and TRPM8 cDNAs; P. Worley for GluR1-HA cDNA and pCMV-GST construct; N. Tigue for hTRPA1 stable HEK293 cells. We also thank Drs. M. Caterina, M. Chung, and A. Kolodkin for helpful comments on the manuscript. The cloning of Pirt and initial in situ hybridization using Pirt cRNA were carried out in the laboratory of Dr. D. Anderson (California Institute of Technology). The liposomal pulldown experiment was performed in the laboratory of Dr. S.H. Snyder (Johns Hopkins University). X.D. is supported by an NINDS grant (NS054791).
Footnotes
ACCESSION NUMBERS
The GenBank Accession Number for Pirt is EU447173.
REFERENCES
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. Sense and specificity: a molecular identity for nociceptors. Curr. Opin. Neurobiol. 1999;9:525–530. doi: 10.1016/S0959-4388(99)00009-4. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Lubin ML, Stone DJ, Jr., Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N. Attenuated cold sensitivity in TRPM8 null mice. Neuron. 2007;54:379–386. doi: 10.1016/j.neuron.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A. TRPM8 is required for cold sensation in mice. Neuron. 2007;54:371–378. doi: 10.1016/j.neuron.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M. Heat-evoked activation of the ion channel, TRPV4. J. Neurosci. 2002;22:6408–6414. doi: 10.1523/JNEUROSCI.22-15-06408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie RC. TRP channels and lipids: from Drosophila to mammalian physiology. J. Physiol. 2007;578:9–24. doi: 10.1113/jphysiol.2006.118372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K, Schaefer M. TRPA1 is differentially modulated by the amphipathic molecules trinitrophenol and chlorpromazine. J. Biol. Chem. 2007;282:7145–7153. doi: 10.1074/jbc.M609600200. [DOI] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Modulation of temperature-sensitive TRP channels. Semin. Cell Dev. Biol. 2006;17:638–645. doi: 10.1016/j.semcdb.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinosi-tide- and calmodulin-mediated regulation of TRPC6. Mol. Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zhang C, Qin F. Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005;25:4835–4843. doi: 10.1523/JNEUROSCI.1296-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo L, Dormand E, Greenwood A, Anderson DJ. Comparison of the generic neuronal differentiation and neuron subtype specification functions of mammalian achaete-scute and atonal homologs in cultured neural progenitor cells. Development. 2002;129:1553–1567. doi: 10.1242/dev.129.7.1553. [DOI] [PubMed] [Google Scholar]
- Lukacs V, Thyagarajan B, Varnai P, Balla A, Balla T, Rohacs T. Dual regulation of TRPV1 by phosphoinositides. J. Neurosci. 2007;27:7070–7080. doi: 10.1523/JNEUROSCI.1866-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C. The TRP superfamily of cation channels. Sci. STKE. 2005;2005:272. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- Rohacs T, Lopes CM, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005;8:626–634. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- Scott SA. Sensory neurons: Diversity, development and plasticity. Oxford University Press; Oxford: 1992. [Google Scholar]
- Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J. Gen. Physiol. 2006;128:509–522. doi: 10.1085/jgp.200609576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg A, Rosen TA, Gilbert H, Skinner K, Raumann B, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Tsai RYL, Reed RR. Using a eukaryotic GST fusion vector for proteins difficult to express in E. coli. Biotechniques. 1997;23:794–798. doi: 10.2144/97235bm06. [DOI] [PubMed] [Google Scholar]
- Voets T, Nilius B. Modulation of TRPs by PIPs. J. Physiol. 2007;582:939–944. doi: 10.1113/jphysiol.2007.132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Arbuzova A, Hangyas-Mihalyne G, McLaughlin S. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2001;276:5012–5019. doi: 10.1074/jbc.M008355200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental d