The PPARγ agonist pioglitazone prevents the hyperglycemia caused by phosphoinositide-3-kinase pathway inhibition by PX-866 without affecting antitumor activity (original) (raw)
. Author manuscript; available in PMC: 2010 Jan 1.
Abstract
The PI-3-kinase/Akt signaling cascade is an important component of the insulin signaling in normal tissues leading to glucose uptake and homeostasis, and for cell survival signaling in cancer cells. Hyperglycemia is an on-target side effect of many inhibitors of PI-3-kinase/Akt signaling including the specific PI-3-kinase inhibitor PX-866. The PPARγ agonist pioglitazone, used to treat type 2 diabetes, prevents a decrease in glucose tolerance caused by acute administration of PX-866. Our studies have shown that pioglitazone does not inhibit the antitumor activity of PX-866 in A-549 non small cell lung cancer and HT-29 colon cancer xenografts. I_n vitro_ studies also showed that pioglitazone increases 2-[1-14C]deoxy-D-glucose uptake in L-6 muscle cells, and prevents inhibition of 2-deoxyglucose uptake by PX-866. Neither pioglitazone nor PX-866 had an effect on 2-deoxyglucose uptake in A-549 lung cancer cells. In vivo imaging studies using [18F]fluorodeoxyglucose (FDG) positron emission tomography showed that pioglitazone increases FDG accumulation by normal tissue but does not significantly alter FDG uptake by A549 xenografts. Thus, PPARγ agonists may be useful in overcoming the increase in blood glucose caused by inhibitors of PI-3-kinase signaling by preventing the inhibition of normal tissue insulin-mediated glucose uptake, without affecting antitumor activity.
Keywords: PX-866, PI-3-kinase, PPARγ, blood glucose, antitumor activity
INTRODUCTION
Phosphatidylinositol-3-kinase (PI-3-kinase)/ Akt (protein kinase B) signaling is an important cell survival signaling pathway that mediates the activity of many receptor and protein tyrosine kinases (1). A hallmark toxicity of inhibition of PI-3-kinase/Akt signaling is an increase in blood glucose due to inhibition of insulin signaling (2, 3) leading to insulin insensitivity and decreased glucose uptake by muscle and fat cells (4). Knockout of the p110α catalytic subunit of class I PI-3-kinase is embryonic lethal in mice due dysfunctional angiogenesis(5). However, mice with a heterozygous kinase-dead p110α knockin, have viable offspring that display a diabetic phenotype with defective insulin signaling and increased blood glucose, as do mice with a knockout of the regulatory p85 subunit of PI-3-kinase(6, 7). Mice with a knockout of the Akt2 isoform, a downstream target activated by PI-3-kinase, are viable and also display a diabetic phenotype (8). This role of Akt2 in preventing diabetes is conserved in humans, as shown in individuals with a mutationally inactivated Akt2 who exhibit severe insulin resistance manifested in diabetes (9). Together these studies establish that PI-3-kinase/ Akt signaling plays an important role in insulin signaling and is conserved throughout mammals.
In addition to this role in normal physiology, the PI-3-kinase/Akt signaling pathway has been shown to regulate several fundamental aspects of tumorgenesis including uncontrolled proliferation, angiogenesis, and resistance to apoptosis (10, 11). The PI-3-kinase/Akt pathway is frequently activated in human tumors by suppression or deletion of the PTEN tumor suppressor, or through mutational activation or amplification of PI-3-kinase (12, 13). Thus, inhibition of the PI-3-kinase / Akt signaling is an attractive target for agents to inhibit tumor growth, and a number of inhibitors of PI-3-K or Akt have, or will soon be introduced into clinical trial as antitumor agents (14). Disturbances in glucose metabolism have been observed in patients receiving some early examples of agents that inhibit PI-3-kinase/Akt signaling. UCN-01 which inhibits Akt signaling as well as cell cycle dependent kinases caused elevated glucose which had to be managed by insulin injection (15). More recently, clinical studies with rapamycin derivatives which inhibit mTOR a downstream target of Akt, have also shown hyperglycemia as an adverse event (16). Thus, disrupted glucose metabolism may become an important on-target dose limiting toxicity for agents that inhibit PI-3-kinase/Akt/mTOR signaling.
We have previously reported an increase in both blood insulin and glucose in mice treated acutely with the irreversible, specific PI-3-kinase inhibitor PX-866 which has recently entered Phase I clinical trial (2). Additionally, it was found that the thiazolidinedone PPARγ agonist pioglitazone, approved for the treatment of type 2 diabetes, prevented the increase in blood glucose caused by PX-866 in animals through reversal of a decreased glucose tolerance (2). However, whether pioglitazone might also antagonize the antitumor effects of a PI-3-kinase inhibitor and the mechanism for the reversal of the PI-3-kinase mediated hyperglycemia was not established. We now show that pioglitazone does not inhibit the antitumor activity of PX-866. We also show that PI-3-kinase inhibition by PX-866 inhibits insulin-sensitive glucose uptake by normal tissue but not by tumor tissue, and the inhibition can be overcome by pioglitazone. This suggests a mechanism by which pioglitazone may prevent the increase in blood glucose associated with PI-3-kinase signaling inhibition without inhibiting antitumor activity.
MATERIALS AND METHODS
Cells
A-549 human non small cell (nsc) lung cancer, HT-29 human colon cancer and rat L-6 cells, a differentiating myoblast cell line, were obtained from the American Tissue Type Collection (Rockville, MD). The cells were grown in humidified 95% air, 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), or for the L-6 cells in Hams F12 medium with 10% FBS. L-6 cells were differentiated by transferring confluent cells to DMEM with 2% FBS for 4-6 days until the fusion of myoblasts into myotubes. All cell lines were tested to be mycoplasma free using a PCR ELISA kit (Roche Diagnostics Inc, Indianapolis) For intravenous (iv) administration to mice PX-866 (acetic acid (1_S_,4_E_,10_R_,11_R_,13_S_,14_R_)-[4-diallylaminomethylene-6-hydroxy-1-methoxymethyl-10,13-dimethyl-3,7,17-trioxo-1,3,4,7,10,11,12,13,14,15,16,17-dodecahydro-2-oxa-cyclopenta[a]phenanthren-11-yl ester) was dissolved at 10 mg/ml in 5% ethanol in 0.9% NaCl and for oral (po) administration by gavage, at 5 mg/ml in 5% ethanol in water. Pioglitazone hydrochloride and human recombinant insulin were obtained from Sigma Chemical (St. Louis, MO) Pioglitazone was administered to mice po by gavage suspended in 0.1% Tween 20 in water. Rabbit purified anti-phosphoSer473-Akt antibody, anti-Akt antibody and anti-glut4 were obtained from Cell Signaling Technology (Beverly, MA).
Glucose Measurements and Antitumor Studies
Approximately 107 A-549 nsc lung, or HT-29 colon cancer cells in log phase growth were injected subcutaneously (sc) in 0.1ml phosphate buffered saline (PBS) into the flanks of female severe combined immunodeficient (scid) mice. When the tumors reached 100 to 200 mm3 the mice were stratified into groups of 6 or 8 animals having approximately equal mean tumor volumes and drug administration begun. Mice were dosed daily with pioglitazone po at 17.5 or 20 mg/kg, and PX-866 was administered po every other day at 2.5 mg/kg. On days when pioglitazone was given in combination it was administered 4 hr before PX-866. Animals were weighed weekly and tumor diameters were measured twice weekly at right angles (dshort and dlong) with electronic calipers and converted to volume by the formula volume = (dshort)2 × (dlong). Animals were euthanized when the tumor volume reached 2,000 mm3. Blood glucose was measured using the FreeStyle Freedom (Therasense, Alameda, CA).
Cellular glucose uptake
107 differentiated L-6 myoblasts or A-549 nsc lung cancer cells at 80% confluence were serum starved overnight in Hams F12 medium or DMEM, respectively and incubated with or without 10μM pioglitazone for 12 hours, followed by a further 5 hr in serum-free Hanks’ buffer (pH 7.4). Fresh Hanks buffer containing 300 nM insulin, 10 μM pioglitazone or 100 nM PX-866 in various combinations was added to the cells. Cytochalasin B, 10 μM, was used as a positive control to inhibit glucose uptake. For glucose uptake measurements, 0.2 μCi 2-[1-14C]deoxy-D-glucose (Perkin Elmer, Waltham, Massachusetts) per sample was added to medium containing 1 mM nonradioactive 2-deoxyglucose. The cells were incubated for 20 min at room temperature with or without 300 nM human recombinant insulin, the medium aspirated and the cells rapidly washed with 4 × 1ml PBS at 4°C. The cells were solubilized with I ml 0.5 M NaOH and radioactivity measured by liquid scintillation counting.
Western blotting for plasma membrane glucose transporters
Cells were prepared and treated with agents as described for the glucose uptake measurements before being incubated with 0.5 mg/ml Sulfo-NHS-Biotin (Pierce, Rockford, IL) in PBS for 30 min at 4°C to label cell surface proteins. The cells were washed with PBS containing 100 mM glycine at 4°C to quench unreacted biotin before being harvested in 200 μl lysis buffer containing 50mM Tris pH7.5, 1% Triton X-100, 0.5% NP-40, 2mM EDTA, 150mM NaCl, 20 μM aprotinin, 20 μM leupeptin, 0.2 mM sodium fluoride, 0.2 mM sodium PPi, and 0.2 mM sodium vanadate. Plasma membrane bound proteins were captured by 4 hr room temperature incubation with 2 units of streptavidin bound to agarose beads (Pierce, Rockford, IL). The streptavidin beads were then washed 4 times with lysis buffer by centrifugation. Streptavidin bound proteins were solubilized by boiling in NuPage LDS sample buffer (Invitrogen Carlsbad, CA) with 0.05M DTT and separated by SDS—PAGE before being transferred to a polyvinylidene fluoride membrane, preincubated with a blocking buffer of 137 mM NaCl, 2.7 mM KCl, 897 mM CaCl2, 491 mM MgCl2, 3.4 mM Na2HPO4, 593 mM KH2PO4, and 5% bovine serum albumin, and incubated overnightwith anti-Glut1, (Chemicon, Temecula, CA) or anti-Glut4 (Cell Signaling, Danvers,MA) polyclonal antibody. Detection used donkey anti-rabbit IgG peroxidase-coupled secondaryantibody and the Renaissance chemiluminescence system on KodakX-Omat Blue ML films (Eastman Kodak, New Haven, CT).
In vivo glucose uptake
In vivo glucose uptake was measured by 2-[18F]-deoxyglucose positron emission tomography (FDG PET) imaging. Scid mice with 300 mm3 subcutaneous A-549 nsc lung cancer xenografts in the left back shoulder were dosed 7 days with 9mg/kg pioglitazone. The mice were fasted overnight and injected with 100 uCi FDG by tail vein 45 minutes prior to imaging. A whole animal field of view was obtained over 10 minutes on a micro PET R4 (Concorde Microsystems, Knoxville, TN) under 2% isoflurane inhalation anesthesia. Maximum tissue (tumor, hind leg muscle) was measured in triplicate with standardized (3 pixel diameter for tumor, 7 pixels for muscle) 2D circular regions-of-interest on contiguous transverse image projections with ASIProVM software (Concorde Microsystems, Knoxville, TN). Tissue tracer uptake was expressed as a ratio relative to baseline brain FDG uptake, measured in triplicate with 7 pixel 2D ROIs on contiguous transverse projections.
RESULTS
PX-866 decreases glucose tolerance in mice
We have previously reported that a single dose of the PI-3-kinase inhibitor PX-866 administered acutely to mice causes a decrease in glucose tolerance [14]. We now show that chronic administration of PX-866 to mice over 30 days, to model the chronic administration of PX-866 usd clinically, causes an increase in blood glucose from 50 to 220 mg/dl associated with a decrease in glucose tolerance, measured 24 hours after the last dose in fasted mice. This decrease in glucose tolerance was reversible with a partial recovery 5 days after the last treatment with PX-866 and a complete recovery with a glucose tolerance similar to the control mice, by 12 days (Figure 1A). The decrease in glucose tolerance was accompanied by an increase in serum insulin in both non-fasted and fasted mice, the non-fasted animals having a slower increase in serum insulin than the fasted animals (Figure 1B). The results suggests that the increase in serum glucose by PI-3-kinase inhibition is due to insulin insensitivity as seen in Type 2 diabetes, and as would be expected from the dependency of insulin activity on PI-3-kinase signaling (17).
Figure 1. Effect of PX-866 on glucose tolerance and plasma insulin.

A. Female scid mice were administered (Δ) vehicle alone po QOD; or (●) PX-866 2.5 mg/kg po, for 30 days (15 doses) and glucose tolerance measurements made 24 hours, 5 days, or 12 days after the last dose. Values are the mean of 6 mice and bars SE. B, Female scid mice were fasted for16 hr and administered (▴) vehicle alone, or (●) PX-866 10 mg/kg iv; or not fasted and administered (Δ) vehicle alone, or (○) PX-866 10 mg/kg iv, and plasma insulin measured at the times shown. Values are the means of 4 mice and bars are S.E..
Pioglitazone restores normal glucose uptake in mice without affecting anti-tumor activity in response to PX-866 treatment
The effect of pioglitazone on the antitumor activity of PX-866 was studied in A-549 nsc lung cancer and HT-29 colon cancer xenografts. (Figures 2A and 2B). We observed no antitumor activity due to pioglitazone alone and a comparable response between PX-866 treated mice and mice treated with PX-866 and pioglitazone. Mice were allowed unrestricted access to food while treated with vehicle, PX-866, pioglitazone, or a combination of PX-866 and pioglitazone for 30 days (Figure 2B). Twenty four hr after the last treatment blood glucose was measured in each group. The group treated with PX-866 alone had a blood glucose level significantly higher than control (214 mg/dl compared to 105 mg/dl), while both the pioglitazone and combination groups had a glucose levels similar to control (Figure 2C).
Figure 2. Effect of pioglitazone and PX-866 in mice bearing A-549 non small cell lung or HT-29 colon cancer xenografts.

A. Female scid mice were injected sc with 107 A-549 human non small cell lung cancer cells. When the tumors were 100 mm3 mice were administered (○) vehicle alone po QOD × 10; (∎) pioglitazone 20 mg/kg po QD × 30 beginning 7 days before the start of treatment with other drugs; (Δ) PX-866 2.5 mg/kg po QOD × 10; and (▴) pioglitazone 20 mg/kg po QD × 30 and PX-866 2.5 mg/kg po QOD × 10. B. Female scid mice were injected sc with 107 HT-29 human colon cancer cells. On day 11 when the tumors were 180 mm3 mice were administered (○) vehicle alone po QOD × 10; (∎) pioglitazone 17.5 mg/kg po QD × 20; (Δ) PX-866 2.5 mg/kg po QOD × 10; (▴) PX-866 2.5 mg/kg po QOD × 10 and pioglitazone 17.5 mg/kg po QD × 20. The pioglitazone was given 4 hr before PX-866. Values are the means of 8 mice and bars are SE. * = p < 0.05 and ** p < 0.01 compared to vehicle treated mice. C. Mice were treated with vehicle alone po QOD × 15; pioglitazone 10 mg/kg po QD × 30, PX-866 2.5 mg/kg po QOD × 15; or pioglitazone 10 mg/kg po QD × 30 and PX-866 2.5 mg/kg po QOD × 15 and blood glucose levels measured. Values are the mean of 9 mice per group and bars are SE. * = p < 0.05 compared to control value.
2-Deoxyglucose uptake in cells
The effect of PX-866 and pioglitazone on the uptake of the non-metabolizable 2-[1-14C]deoxy-D-glucose was studied in L-6 muscle cells and in A-549 nsc lung cancer cells. Preliminary studies showed that 2-deoxyglucose uptake was linear in both L-6 and A-549 cells for at least 2 hr (data not shown). With L-6 muscle cells in serum free media insulin stimulated 2-deoxyglucose uptake, while PX-866 decreased the uptake (Figure 3A, upper panel). Pioglitazone alone did not significantly alter 2-deoxyglucose uptake but blocked the decrease in 2-deoxyglucose uptake caused by PX-866 in the L-6 cells. In contrast, 2-deoxyglucose uptake by A-549 tumor cells was insensitive to insulin, PX-866, pioglitazone or the combination of PX-866 and pioglitazone (Figure 3A, lower panel). Western blotting following labeling of cell surface proteins with non-cell permeant biotin showed the presence of the glucose transporter Glut-4 at the plasma membrane in differentiated L-6 myoblasts exposed to insulin, but not with A-549 non small cell lung cancer cells (Figure 3B). PX-866 inhibited the expression of Glut-4 in the L-6 myoblasts which was reversed by pioglitazone treatment. Glut-1 was expressed at the plasma membrane of both L-6 myoblasts and A-549 nsc lung cancer cells. In L-6 myoblast Glut-1 was decreased by PX-866, an effect that was not reversed by pioglitazone, In A-549 nsc lung cancer cells Glut-1 levels were not affected by either PX-866 or pioglitazone. Glut-4 could not be detected in A-549 nsc lung cancer cells. PX-866 inhibited cellular PI-3-kinase signaling measured by phosphor-Ser473-Akt in both L6-myoblasts and A-549 nsc lung cancer cells (Figure 3B).
Figure 3. Effect of PX-866 and pioglitazone on glucose uptake in normal muscle and tumor cells.

A, upper panel, L-6 muscle cells; and lower panel, A-549 non small cell lung cancer cells were incubated with 2-[ 1-14C]deoxy-D-glucose in serum free medium and cellular accumulation of radioactivity measured after 1 hr in cells treated with insulin, 300 nM, PX-866 100 nM, pioglitazone 10 μM, a combination of PX-866 and pioglitazone, or cytochalasin 10 μM as a positive control to inhibit 2-deoxglucose uptake. Values are the mean of 4 experiments an bars are SE. * = p < 0.05 compared to control value. B, Western blot showing the expression at the cell membrane of glucose transporters Glut-1 and Glut-4 in L-6 myoblasts and A-549 cells. Cell membrane proteins were labeled by biotin and collected on streptavidin beads. It was not possible to obtain labeled cell membranes proteins in the absence of insulin because of cell lysis by the reactive biotin. The effects of PX-866 and pioglitazone treatment are shown. Also shown is total cellular phospho-Ser473-AKT and total AKT as a loading control.
Fluoro-deoxyglucose accumulation in vivo
The effect of PX-866 and pioglitazone on the accumulation of [18F]-2-deoxyglucose measured by PET imaging was studied in normal tissue and A-549 nsc lung cancer xenografts in mice (Figure 4A). The accumulation of [18F]-2-deoxyglucose (FDG) in the tissues was normalized to FDG accumulation in the brain, a high glucose uptake tissue that shows minimal insulin sensitivity (18). PX-866 treatment showed a trend towards decreased FGD uptake in normal tissue, but the effects were not significant (Figure 4B). Pioglitazone treatment significantly increased FDG accumulation by the normal tissue, an effect that was reversed by PX-866 to a level that was not significantly different to the non-treated control value (Figure 4B). In A-549 nsc lung cancer xenografts pioglitazone had no effect on FDG uptake, while PX-866 decreased FDG uptake which was not reversed by pioglitazone (Figure 4C).
Figure 4. Effect of PX-866 and pioglitazone on FDG-PET imaging in normal tissue and tumor xenografts in mice.

A, Female scid mice with 300 mm3 sc A-4549 nsc lung cancer xenografts were administered vehicle alone (control), pioglitazone 20 mg/kg po QD × 5 starting 5 days before the study; PX-866 9 mg/kg iv, or the combination of pioglitazone treatment and PX-866. Four hr later 18F-deoxyglucose (FDG) PET imaging was performed as described in the Methods. Dorsal views are shown with the head uppermost. B, The signal from muscle was normalized to the signal from the brain in each animal, and expressed as a percent of the mean control value. Values are the mean of 4 animals and bars are SE. * = p < 0.05 of PX-866 and pioglitazone treated tumor to pioglitazone treated tumor C, The signal from tumor was normalized to the signal from the brain in each animal, and expressed as a percent of the mean control value. Values are the mean of 4 animals and bars are SE. * = p < 0.05 of PX-866 and pioglitazone treated animal to the pioglitazone alone treated animal
DISCUSSION
A hallmark on target side effect of PI-3-kinase/Akt inhibitors is an increase in blood glucose due to inhibition of insulin signaling (2, 3). The toxicity is similar to Type 2 diabetes and could potentially limit the use of such agents, particularly for long term use. We have investigated ways that might be used clinically to limit this toxicity without inhibiting the antitumor activity of the agents. To inhibit PI-3-kinae we used PX-866 a selective inhibitor of the Type 1 PI-3-kinase family that has recently entered Phase I clinical trial. PX-866 has been shown to down regulate tumor phospho-Akt and to exhibit antitumor activity in a number of human tumor xenograft models when administered either intravenously or orally (19). Previous studies have shown that PX-866 at the doses used in this study inhibits PI-3-kinase measured by phospho- Akt levels in tumor and in normal tissue (20).
Insulin signaling is relayed predominantly through PI-3-kinase p110α, although other PI-3-kinase class 1 isoforms have been implicated in specific tissues (4, 21). We have found that prolonged administration of PX-866 to mice decreases glucose tolerance and at the same time plasma insulin levels increase suggesting a diminished sensitivity to insulin. This is similar to Type 2 diabetes, and also similar to the phenotype of mice deficient in the Akt2 isoform that show marked hyperglycemia, hyperinsulinemia and an impaired ability of insulin to lower blood glucose(8). We found that the thiazolidinedione drug pioglitazone, used to treat Type 2 diabetes, reverses the inhibitory effects of both acute and chronic PX-866 administration on glucose tolerance. However, another drug used to treat Type 2 diabetes, metformin, did not reverse the decrease in glucose tolerance by PX-866 (2).
Thiazolidenediones are ligands for the peroxisome proliferator-activated receptor-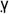 (PPA
(PPA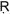 )γ transcription factor present at high levels in adipose tissue, and they sensitize the body to the effects of insulin (22). An important question remaining to be answered was whether pioglitazone also inhibits the antitumor activity associated with PI-3-kinase inhibition by PX-866. PPAR
)γ transcription factor present at high levels in adipose tissue, and they sensitize the body to the effects of insulin (22). An important question remaining to be answered was whether pioglitazone also inhibits the antitumor activity associated with PI-3-kinase inhibition by PX-866. PPAR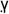 activation has been reported have tumor suppressing effects of its own through experiments using genetic manipulation of the target (23). PPAR
activation has been reported have tumor suppressing effects of its own through experiments using genetic manipulation of the target (23). PPAR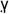 activation by thiazolidinediones, including pioglitazone, has been reported to inhibit the growth of some tumors including A-549 nsc lung cancer xenograft in scid mice and to augment the effects of cytotoxic agents (24, 25). However, this activity could not be confirmed in clinical trials using this class of drugs which have been largely disappointing (26, 27). In our study we found that pioglitazone by itself did not affect the growth of A-549 nsc lung cancer or of HT-29 colon cancer xenografts and did not block the antitumor effects of PX-866, but did restore insulin signaling in treated mice.
activation by thiazolidinediones, including pioglitazone, has been reported to inhibit the growth of some tumors including A-549 nsc lung cancer xenograft in scid mice and to augment the effects of cytotoxic agents (24, 25). However, this activity could not be confirmed in clinical trials using this class of drugs which have been largely disappointing (26, 27). In our study we found that pioglitazone by itself did not affect the growth of A-549 nsc lung cancer or of HT-29 colon cancer xenografts and did not block the antitumor effects of PX-866, but did restore insulin signaling in treated mice.
Studies in cells using the non metabolizable glucose analogue 2-deoxyglucose showed that PX-866 significantly inhibited glucose uptake in L-6 rat differentiated myotubes and that the inhibition was reversed by pioglitazone. There was no effect of either PX-866 or pioglitazone on glucose uptake by A-549 nsc lung cancer cells. Glut-4 is the primary glucose transporter in muscle and fat where it translocates to the cell membrane from intracellular stores in response to insulin (28, 29). The expression of cell membrane Glut-1 is not insulin responsive and is seen in both normal and transformed tissues (30). We found that the expression of Glut-4 at the cell membrane of differentiated L-6 myoblasts was decreased by PX-866, and the effect was reversed by pioglitazone. Glut-4 could not be detected at the cell membrane of A-549 nsc lung cancer cells. Glut-1 was present at the cell membrane of both L-6 myoblasts and A-549 cells and while its expression was decreased by PX-866 in L-6 myoblasts, this was not reversed by pioglitazone, and there was no regulation by PX-866 in A-549 cells. The results are consistent with the inhibition of glucose uptake in L-6 myoblasts being due to the inhibition of Glut-4 translocation to the cell membrane by PX-866.
2-[18F]-deoxyglucose (FDG) PET imaging studies in mice with A-549 nsc lung cancer xenografts suggested a mechanism through which pioglitazone blocks the increase in blood glucose caused by PX-866. A-549 xenografts showed a decrease in FDG uptake when treated with PX-866 which was not reversed by treatment with pioglitazone. This is contrary to what was seen in cell culture, a phenomena that may result from the reported differential effects of PX-866 in 2-D and 3-D cultures (31), or the well established relationship between PI-3-kinase signaling and angiogenesis (32). Pioglitazone increased the uptake of glucose by normal tissue, presumably muscle and adipose tissue which are known to take up glucose in response to insulin (33). PX-866 showed a small inhibition of glucose uptake by normal tissue by itself, and reversed the increase by pioglitazone to values that were the same as in untreated animals.
Taken together, the results suggest that PX-866 inhibits insulin signaling, thus, blocking the ability of muscle and presumably other normal tissues, to take up glucose, resulting in hyperglycemia. Pioglitazone, a thiazolinedione, increases sensitivity to insulin thorough modulation of the PPARγ response or other mechanisms (22). Pioglitazone is able to prevent the decreased glucose uptake by muscle and other normal tissue by PX-866, thus restoring normal blood glucose levels. In contrast to normal tissue, PX-866 decreased glucose uptake by tumor and neither the decreased glucose uptake or the antitumor activity of PX-866 was blocked by pioglitazone. Glut-4 is the primary glucose transporter in muscle and fat where it translocates to the cell membrane from intracellular stores in response to insulin (28, 29). The expression of cell membrane Glut-1 is not insulin responsive and is seen in both normal and transformed tissues (30). Early studies showed that cells transformed with an active Ras or Src oncogene showed pronounced upregulation of the Glut 1 high affinity glucose tranporter and that tumors showed dramatically increased glucose uptake, famously noted by Warburg (34, 35). Our current study serves to confirm that cancer cells rely on high affinity glucose transporters such as Glut-1 to maximize glucose uptake while insulin responsive tissues rely on Glut 4. Pioglitazone restores insulin induced Glut 4 translocation in normal tissues, without directly effecting cancer cells which rely on more efficient, but less regulated mechanisms for glucose uptake. It is unlikely that the antitumor activity of PX-866 relies solely on the decrease in glucose uptake by tumors, but it may contribute to the inhibition of the many other well known effects of tumor PI-3-kinase on tumor cell survival and proliferation,or serve as a marker for these processes (1,2,3)
In summary, we have shown that thiazolidinedone PPARγ agonist pioglitazone can prevent the increase in blood glucose caused by the PI-3-kinase inhibitor PX-866. We have also shown that pioglitazone does not inhibit the antitumor activity of PX-866. Neither PX-866 nor pioglitazone in combination with PX-866 showed altered glucose accumulation by tumor cells in vitro. In vivo a modest inhibition in the FDG glucose was seen, which was not reversed by the addition of pioglitazone. However, PX-866 inhibited insulin-sensitive glucose uptake by normal tissue, and the inhibition was overcome by pioglitazone both in vitro and in vivo, suggesting a mechanism by which pioglitazone is able to prevent the increase in blood glucose associated with PI-3-kinase signaling inhibition without altering antitumor activity in vivo.
Acknowledgments
Supported by NIH grants CA52995, CA17094,CA95060 and CA610159.
REFRENCES
- 1.Cantley LC. Science. 5573. Vol. 296. New York, NY: May 31, 2002. The phosphoinositide 3-kinase pathway; pp. 1655–7. [DOI] [PubMed] [Google Scholar]
- 2.Ihle NT, Paine-Murrieta G, Berggren MI, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Molecular cancer therapeutics. 2005 Sep;4(9):1349–57. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondapaka SB, Zarnowski M, Yver DR, Sausville EA, Cushman SW. 7-hydroxystaurosporine (UCN-01) inhibition of Akt Thr308 but not Ser473 phosphorylation: a basis for decreased insulin-stimulated glucose transport. Clin Cancer Res. 2004 Nov 1;10(21):7192–8. doi: 10.1158/1078-0432.CCR-04-0772. [DOI] [PubMed] [Google Scholar]
- 4.Knight ZA, Gonzalez B, Feldman ME, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006 May 19;125(4):733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graupera M, Guillermet-Guibert J, Foukas LC, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008 May 29;453(7195):662–6. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 6.Foukas LC, Claret M, Pearce W, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006 May 18;441(7091):366–70. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 7.Luo J, Sobkiw CL, Hirshman MF, et al. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell metabolism. 2006 May 3;(5):355–66. doi: 10.1016/j.cmet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. The Journal of clinical investigation. 2003 Jul;112(2):197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.George S, Rochford JJ, Wolfrum C, et al. Science. 5675. Vol. 304. New York, NY: May 28, 2004. A family with severe insulin resistance and diabetes due to a mutation in AKT2; pp. 1325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cellular signalling. 2002 May;14(5):381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proceedings of the National Academy of Sciences of the United States of America. 1998 Dec 22;95(26):15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005 Dec;4(12):988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 14.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008 Jan;1784(1):159–85. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Kortmansky NS S, O’Reilly E, Shah M, Abou-Alfa GK, Winkelmann J, Yi S, Gonen M, Kelsen DP, Schwartz GK. Management of hyperglycemia in patients with metastatic pancreatic cancer receiving UCN-01 and fluorouracil Journal of Clinical Oncology; 2004 ASCO Annual Meeting Proceedings (Post-Meeting Edition); Jul 15, 2004. p. 2140. [Google Scholar]
- 16.Bellmunt J, Szczylik C, Feingold J, Strahs A, Berkenblit A. Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann Oncol. 2008 Apr 2; doi: 10.1093/annonc/mdn066. [DOI] [PubMed] [Google Scholar]
- 17.Musi N, Goodyear LJ. Insulin resistance and improvements in signal transduction. Endocrine. 2006 Feb;29(1):73–80. doi: 10.1385/ENDO:29:1:73. [DOI] [PubMed] [Google Scholar]
- 18.Bingham EM, Hopkins D, Smith D, et al. The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes. 2002 Dec;51(12):3384–90. doi: 10.2337/diabetes.51.12.3384. [DOI] [PubMed] [Google Scholar]
- 19.Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Molecular cancer therapeutics. 2004 Jul;3(7):763–72. [PubMed] [Google Scholar]
- 20.Williams R, Baker AF, Ihle NT, Winkler AR, Kirkpatrick L, Powis G. The skin and hair as surrogate tissues for measuring the target effect of inhibitors of phosphoinositide-3-kinase signaling. Cancer chemotherapy and pharmacology. 2006 Oct;58(4):444–50. doi: 10.1007/s00280-006-0190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaussade C, Rewcastle GW, Kendall JD, et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J. 2007 Jun 15;404(3):449–58. doi: 10.1042/BJ20070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng AY, Fantus IG. Oral antihyperglycemic therapy for type 2 diabetes mellitus. CMAJ. 2005 Jan 18;172(2):213–26. doi: 10.1503/cmaj.1031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004 Jan;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 24.Keshamouni VG, Reddy RC, Arenberg DA, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene. 2004 Jan 8;23(1):100–8. doi: 10.1038/sj.onc.1206885. [DOI] [PubMed] [Google Scholar]
- 25.Girnun GD, Naseri E, Vafai SB, et al. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell. 2007 May;11(5):395–406. doi: 10.1016/j.ccr.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003 Jun;79(3):391–7. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- 27.Kulke MH, Demetri GD, Sharpless NE, et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J. 2002 Sep-Oct;8(5):395–9. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Hashiramoto M, James DE. Snareing GLUT4 at the plasma membrane in muscle and fat. Adv Exp Med Biol. 1998;441:47–61. doi: 10.1007/978-1-4899-1928-1_5. [DOI] [PubMed] [Google Scholar]
- 29.Ishiki M, Klip A. Minireview: recent developments in the regulation of glucose transporter-4 traffic: new signals, locations, and partners. Endocrinology. 2005 Dec;146(12):5071–8. doi: 10.1210/en.2005-0850. [DOI] [PubMed] [Google Scholar]
- 30.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005 Mar;202(3):654–62. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 31.Howes AL, Chiang GG, Lang ES, et al. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Molecular cancer therapeutics. 2007 Sep;6(9):2505–14. doi: 10.1158/1535-7163.MCT-06-0698. [DOI] [PubMed] [Google Scholar]
- 32.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008 Feb;8(1):19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 33.Elmendorf JS, Damrau-Abney A, Smith TR, David TS, Turinsky J. Insulin-stimulated phosphatidylinositol 3-kinase activity and 2-deoxy-D-glucose uptake in rat skeletal muscles. Biochem Biophys Res Commun. 1995 Mar 28;208(3):1147–53. doi: 10.1006/bbrc.1995.1453. [DOI] [PubMed] [Google Scholar]
- 34.Murakami T, Nishiyama T, Shirotani T, et al. Identification of two enhancer elements in the gene encoding the type 1 glucose transporter from the mouse which are responsive to serum, growth factor, and oncogenes. J Biol Chem. 1992 May 5;267(13):9300–6. [PubMed] [Google Scholar]
- 35.Warburg O. On the origin of cancer cells. Science. 1956 Feb 24;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]