The Structural Immunology of Antibody Protection against West Nile Virus (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 1.
Summary
Recent investigations of the interaction between the West Nile virus (WNV) envelope protein (E) and monoclonal antibodies (mAbs) have elucidated fundamental insights into the molecular mechanisms of neutralization. Structural studies have defined an epitope on the lateral ridge of domain III (DIII-lr) of the WNV E protein that is recognized by antibodies with the strongest neutralizing activity in vitro and in vivo. Antibodies that bind this epitope are highly potent because they efficiently block at a post-entry step of viral infection with relatively low virion occupancy requirements. In this review, we will discuss the structural, molecular, and immunologic basis for antibody-mediated protection against WNV, and its implications for novel therapeutic or vaccine strategies.
Keywords: infectious diseases, antibodies, emerging infectious disease, antigens/peptides/epitopes, complement
Introduction
West Nile virus (WNV) is an 11-kilobase positive sense, single-stranded neurotropic RNA virus that has emerged globally as a significant cause of viral encephalitis. WNV is maintained in an enzootic cycle between mosquitoes and birds (reviewed in 1) but can also infect and cause disease in humans, horses, and other vertebrate animals. WNV causes a range of illness in humans from mild fever to acute flaccid paralysis and lethal encephalitis. Severe neuroinvasive disease is more frequent in elderly or immunocompromised individuals (2). Nucleotide sequencing separates WNV strains into two lineages (3). Lineage I viruses are emerging globally, and subsets of strains are associated with severe human and avian disease (4–6). In contrast, lineage II viruses isolated from central and southern Africa and parts of Asia have not been associated with severe human disease (7, 8). Historically, outbreaks of WNV disease occurred in the Middle East, Europe, and Africa. In 1999, WNV was introduced into North America (9), and over the last eight years, it has spread throughout the continental United States, as well as parts of Canada, Mexico, the Caribbean, and Central and South America (10, 11).
Because of the increased range of WNV, the number of human cases has continued to rise. During an epidemic, the seroconversion rate within the affected human population is estimated at ~3% (9), and the incidence of severe disease is ~7/100,000 (12). Overall, only a small percentage of humans (1/150) develop severe neurological disease upon WNV infection, which can include cognitive dysfunction, ocular manifestations, meningitis, encephalitis, and flaccid paralysis (reviewed in 2, 13). In the United States between 1999 and 2007, ~27,400 cases were diagnosed and associated with greater than 1,000 deaths (http://www.cdc.gov/ncidod/dvbid/westnile/index.htm). However, the spectrum of disease may be much larger. In 2003 alone, based on screening of blood-bank samples, there was an estimated 730,000 undiagnosed infections (14). No vaccines or specific therapies for WNV infection are currently approved for human use.
WNV is a member of the Flaviviridae family and is related closely to other human pathogens such as the dengue virus (DENV), tick borne encephalitis virus (TBEV), Japanese encephalitis virus (JEV), and yellow fever virus (YFV). Extensive work in small animal models has defined critical protective functions for the immune system including antibody, CD4+ and CD8+ T cells, CXCL10, CCR5, complement components, interferons (IFNs), and other innate immune modulators (reviewed in 15). The WNV genome is translated as a single polyprotein and subsequently cleaved by viral and cellular proteases. Three structural [capsid (C), pre-membrane/membrane (prM/M) and envelope (E)] and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins are encoded (16). A majority of the flavivirus-specific protective antibody response is directed against the E protein, although antibodies against other structural and non-structural proteins are detected. WNV-specific antibodies are required for viral clearance (17) and have therapeutic effects in vivo (18–27). Based on studies detailed below, protection by antibodies is a function of several parameters including epitope location and accessibility, the fractional occupancy or number of times the antibody can bind the virion at a given concentration, mechanism of inhibition, strength of binding, and effector function. A greater understanding of the dynamics of antibody protection against WNV may lead to the development of antibody-based therapeutics and novel immunization strategies that elicit potently inhibitory antibodies in vivo.
Pathogenesis and immune response
The pathogenesis of WNV and the immune responses that prevent central nervous system (CNS) dissemination have been characterized through studies in small animal models. Following peripheral inoculation, WNV is believed to initially replicate in skin dendritic cells (DCs) (28, 29). These cells then migrate to the draining lymph nodes (29, 30), where viral amplification occurs leading to viremia and spread to the visceral organs (e.g. kidney and spleen). DCs and macrophages in the lymph node likely initiate the innate immune response against WNV after recognition by Toll-like receptors (TLRs), RIG-I, and MDA5, and signaling through IFN-dependent and -independent mechanisms (31–35). Dissemination of WNV to the CNS occurs shortly before clearance of infectious virus from peripheral tissues (17). Infectious WNV is detected within the CNS at multiple sites including the cerebral cortex, hippocampus, basal ganglia, cerebellum, brain stem, and spinal cord (17, 36, 37). In most animals, CNS infection occurs primarily in neurons and is associated with their degeneration and loss of cell architecture and apoptosis (26, 38, 39). However, the mechanisms of WNV CNS seeding remain poorly understood. Earlier entry of WNV in the CNS has been observed in mice exhibiting increased levels of viremia, suggesting that hematogenous spread contributes to CNS seeding (40, 41). Yet, earlier viral invasion of the CNS has also been seen in complement-deficient mice that have normal levels of viremia (42, 43). These studies suggest that additional soluble inflammatory factors modulate blood-brain barrier (BBB) permeability and WNV CNS seeding. Indeed, recent evidence has suggested tumor necrosis factor-α (TNF-α) and macrophage inhibitory factor (MIF)-mediated changes in BBB permeability may enhance entry of WNV into the brain (44, 45). Retrograde neuronal transport and axonal spread also contributes to dissemination, especially in the spinal cord (26, 46) Clearly, additional studies are necessary to define the precise mechanism(s) for dissemination of WNV to the CNS.
Experiments in small animals suggest that both innate and adaptive immune responses orchestrate control of WNV dissemination and disease. Type I IFN (α/β) and its downstream effector molecules PKR and RNAse L are critical components of the innate immune response to WNV infection (40, 47–49). Pretreatment of cells with type I IFN in vitro prevents WNV infection (40, 50–53), and analogously, a deficiency of type I IFN signaling in vivo results in increased viral replication, expanded tropism, and uniform lethality (40, 50, 54). Similarly, type II IFN (IFN-γ) produced by γδ T cells also limits peripheral viral replication and early WNV dissemination into the CNS (55, 56). Additional innate immune responses including TLR3 and 2′5′ oligoadenylate synthetase also regulate WNV infection in vivo (44, 57, 58). Development of antiviral adaptive immunity is also necessary for protection from disease, as passive transfer of immune antibody protects wildtype and B-cell-deficient mice from lethal WNV challenge (see discussion below). However, antibody alone did not eradicate infection in recombination-activating gene 1 (RAG1)-deficient hosts, which lack both B and T cells (17, 18), indicating T cells likely have a critical role limiting severe WNV disease. Indeed, mice lacking CD8+ T cells exhibited increased viral burden and lethality following peripheral WNV infection (59, 60). Cytolytic T-cell responses are required for clearance of WNV infection, as persistence within the CNS was observed in mice that lacked either classical class I major histocompatibility complex (MHC), perforin, or functional Fas ligand molecules (61–63).
WNV structural biology
The E glycoprotein is the major surface protein on the WNV virion and is the principal antigen that elicits neutralizing antibodies. Through x-ray crystallography, cryo-electron microscopy, and other techniques, the atomic architecture and structural rearrangements that occur during the virus life cycle have begun to be defined (64). Based on crystallographic structures, the E proteins of DENV, TBEV, and WNV share common structural features (65–68). The ectodomain of E protein forms three structural domains (I, II, and III) (Fig 1A). Domain I (DI) is the central domain and consists of an eight-stranded β-barrel. Domain II (DII) is formed from two extended loops that project from DI and contains a highly conserved loop, amino acid residues 98–110, that has been implicated in the acid-catalyzed type II fusion event that has been observed for TBEV and DENV (69–71). DIII, located on the other side of DI, adopts a seven-stranded immunoglobulin (Ig)-like fold and has been implicated in receptor binding (72, 73). The integrin αvβ3 has been suggested as a potential WNV receptor (74, 75), and the C-type lectin DC-SIGNR can serve as an efficient attachment receptor for WNV (76, 77). Short, flexible linker regions connect the three domains and allow the conformational changes necessary for virus maturation and fusion (78). The C-terminus of the E protein consists of two α-helices, termed the stem region, and two anti-parallel coil-coil helices that span the lipid membrane (79). The cytoplasmic domain of the transmembrane region does not extend far beyond the lipid bilayer.
Fig. 1. E protein and mature WNV virion structure.
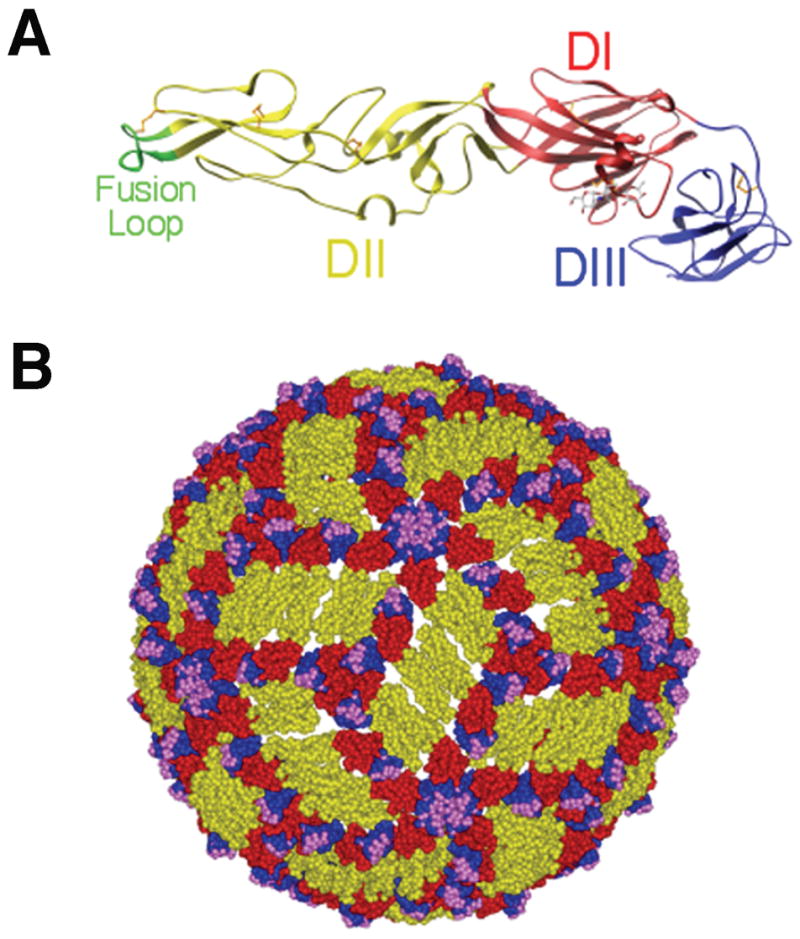
(A). Ribbon diagram of the WNV E protein crystal structure. Domains are labeled and the fusion loop is shown in green. (B). Pseudoatomic model of the mature WNV virion based on cryo-electron microscopy studies. E protein domains I, II, and III are indicated in red, yellow and blue, respectively. Residues critical for binding of E16, a DIII-lr mAb, are shown in magenta. Adapted from (21, 68, 117).
The structure of the mature WNV virion has been defined by cryo-electron microscopy and pseudo-atomic modeling (80). WNV particles are ~500 Å in diameter and have a smooth surface with no apparent spikes or large projections, as seen with other enveloped viruses. The 180 E monomers lay relatively flat along the virion surface as sets of three anti-parallel homodimers. This mature flavivirus virion has quasi-icosahedral symmetry, such that three E monomers are found in the asymmetric unit resulting in three distinct chemical environments that are available for antibody or receptor binding (Fig. 1B). When the E protein is in its homodimeric form on the mature virus particle, the fusion loop is shielded in a pocket at the DI-DIII interface of the adjacent E protein.
In immature virus particles, the E protein assumes a distinct conformation. There are 60 trimeric spikes, each consisting of 3 prM/E heterodimers with T = 1 icosahedral symmetry (81). In this position, prM may prevent low-pH induced conformational changes that would inactivate virus particle during egress through mildly acidic compartments of the secretory pathway (82). During the secretory process in the trans-Golgi network, prM undergoes cleavage by a furin-like protease that promotes viral maturation (83). This required cleavage step promotes a rearrangement of E protein on the surface of the virion from a heterodimer (prM-E) into an antiparallel homodimer (E-E) and the formation of a mature virus particle (reviewed in 64). Cleavage results in the formation of a small virion-associated M peptide and the release of the N-terminal ‘pr’ portion of the protein. In contrast to the spikes present on the immature precursor, the relatively smooth mature WNV virions are composed of 90 anti-parallel dimers arranged with T = 3 pseudo-icosahedral symmetry (Fig. 1B).
After attachment and endocytosis in a target cell, a reduction of pH in the early endosomes prompts a third structural transition that has been demonstrated for TBEV and DENV. This involves the dissociation of E protein homodimers present on the mature virion (a reversible step) and the formation of E protein trimers (an irreversible step). Structural studies indicate that DIII of the E protein moves ~36 Å towards the DII fusion loop in the transition from mature homodimer to post-fusion homotrimer (70, 71). This movement exposes the fusion peptide, which inserts into the target cell endosomal membrane. Subsequently, viral and cellular membranes are brought into close apposition as the E protein folds back upon itself with the stem anchor region fitting into grooves on the exterior of the trimer. These rearrangements represent a functionally analogous process to the well-characterized fusion process of class I fusion glycoproteins (reviewed in 84).
Humoral immunity against WNV infection
The majority of neutralizing antibodies against flaviviruses recognize the structural E protein, although a subset bind to the prM/M (85–88). Interestingly, antibodies to the NS1 protein, which is absent from the virion, also are protective against WNV in vivo (27, 89) (see below for discussion). Antibody responses to the intracellular proteins NS3 and NS5 have also been observed during WNV infection (90), although their functional significance remains uncertain.
At least 12 epitopes on the E protein of flaviviruses have been defined by antibody mapping techniques and are associated with distinct functions including cell attachment, dimerization, trimerization, and acid-catalyzed fusion (91–93). Virus type-specific epitopes elicit antibodies with the strongest neutralizing activity (92, 94), and animal protection studies with antibodies correlate with neutralizing activity in vitro (20, 41, 92, 95). Many of the most potent neutralizing antibodies against WNV recognize the upper lateral surface of DIII that protrudes off the surface of the virion (20, 96, 97). While humans can produce antibodies of this specificity in response to natural infection (98), recent studies indicate that the human humoral immune response to WNV infection is narrower than anticipated, with antibody specificity primarily focused on determinants around the fusion loop at the tip of DII. B-cell repertoire analysis of three WNV-infected humans revealed that only 8% of WNV-specific B-cell clones produced antibodies specific to DIII, whereas almost half produced antibody that bound determinants in DII, particularly the fusion loop (23). Functional studies of the polyclonal response of WNV-infected horses and humans indicate that the neutralization activity of sera is not dependent upon antibodies directed against the DIII-lateral ridge (lr) epitope (98, 99).
Priming of protective antiviral antibody responses
The priming of early effective neutralizing antiviral antibody responses is crucial for control of severe WNV infection. In C57BL/6 mice, the development of WNV-specific neutralizing IgM was consistently observed beginning on day 4 after subcutaneous infection (41, 42). Mice lacking secreted IgM (sIgM−/−) were highly susceptible to lethal WNV infection and exhibited sustained viremia, earlier viral entry into the CNS, and greater CNS viral accumulation (41). Transfer of serum from wildtype to sIgM−/− mice on day 4 post-infection significantly protected mice from lethal WNV infection. This observation suggested that amplification of early IgM-dependent neutralizing antibody was critical for the control of WNV-induced disease. Indeed, the level of WNV-specific IgM in serum on day 4 after infection predicts disease outcome in mice. Accordingly, immune deficiencies that impair antibody priming also predispose to WNV susceptibility. Mice lacking the C3 or C4 components of complement or complement receptors 1 and 2 exhibited blunted antiviral antibody priming and enhanced susceptibility to lethal WNV infection (42, 43). Additionally, the absence of CD4+ T cells, class II MHC expression, or CD40 signaling decreased neutralizing antiviral antibody responses and survival rates after WNV infection (100, 101).
Epitope localization of neutralizing antibodies
The specific binding epitopes of neutralizing antibodies have been examined for several members of the Flaviviridae family, including WNV. Epitope mapping has been evaluated using several different methods including nuclear magnetic resonance (NMR), X-ray crystallography, isolation of neutralization escape mutants, binding of antibodies to linear peptide binding, site-directed mutagenesis, and forward genetic screens with display of E proteins or domains on yeast or peptides on phage (reviewed in 102). Collectively, these studies suggest that virus type-specific antibodies that neutralize infection most efficiently bind to an epitope on the DIII-lr that is highly variable at the sequence level but structurally conserved among flaviviruses (Fig. 2A). One of the most potent neutralizing mAbs for WNV that was generated in our laboratory, E16, was crystallized in complex with DIII (103). E16 bound to four discontinuous regions of the DIII protein, encompassing residues 302–309, 330–333, 365–368, and 389–391 (Fig. 2A, B). Four residues centrally located in the antibody-antigen interface were initially identified by yeast surface display mutagenesis (S306, K307, T330, and T332), and each participates in an elaborated hydrogen-bonding network (20, 103). Amino acid substituted forms of recombinant DIII and neutralization escape studies by other groups also identified the same composite epitope as important for binding of antibodies with strong type-specific neutralizing activity (96, 97, 104).
Fig. 2. Strongly neutralizing DIII-lr specific antibodies define a single consensus epitope that is divergent in other flaviviruses.

(A). Sequence of the four segments of WNV DIII contacted by E16 aligned with the analogous residues of other flaviviruses. The DIII contact residues as determined by epitope mapping are highlighted in magenta, while those identified only structurally are highlighted in blue. Deletions are indicated with a hash symbol. (B). Structure of the WNV dominant neutralizing epitope as defined by the E16-DIII complex. Adapted from (68).
A major epitope that is recognized by anti-WNV neutralizing antibodies localizes to the fusion loop, located at the tip of DII (DII-fl). Unlike the DIII-lr epitope, this epitope is highly conserved among flaviviruses and elicits cross-reactive antibodies (21, 105–107) (Fig. 3). The level of neutralization observed with this class of antibodies is more variable among flaviviruses and appears to be somewhat virus-specific. Monoclonal antibodies (mAbs) that map to this epitope only weakly neutralize TBEV yet strongly inhibit DENV infectivity (107). Compared with DIII-lr mAbs, the DII-fl mAbs are less potent at neutralizing WNV in vitro and in vivo, and in some cases do not completely neutralize even at concentrations of mAb sufficient to saturate all available sites on the WNV virion (21). In studies with TBEV, these cross-reactive DII-fl antibodies bound weakly to native virions yet strongly to detergent-solubilized virions, implying that at least some of this epitope was obscured in the mature virus. However, some cross-reactive DII-fl antibodies have stronger neutralizing activity (22) and recognize additional amino acid residues in other domains (108). DII-fl antibodies that recognize residues in multiple domains may span across the E homodimer in the mature virion and possibly interfere with the dimer to trimer transition during viral fusion.
Fig. 3. . Epitopes of several different anti-WNV neutralizing mAbs as determined by yeast surface display screening of E protein mutants.

The backbone colors red, yellow, blue, and green indicate domains I, II, III, and the fusion loop respectively. Mutations that resulted in ≥ 50% reduction of mAb binding were mapped (shown in magenta and circled) onto the WNV E protein crystal structure. Epitopes are labeled using the nomenclature defined in Oliphant et al. (21).
Beyond the DIII-lr and DII-fl epitopes, neutralizing antibodies also recognize other regions on the E protein. We recently identified six additional epitopes in domains I and II of WNV E protein (the central interface, dimer interface, and lateral ridge of DII, the hinge region between DI and DII, the lateral ridge of DI, and the linker region between DI and DIII) and additional epitopes in DIII that bind mAbs with variable neutralizing activity (21) (Fig. 3). Analogously, additional epitopes in DI and DII also have been described for YFV and DENV neutralizing mAbs (109–111). Interestingly, one recently described anti-DENV-4 mAb that potently neutralizes infection localizes to amino acids 174 and 176 on a solvent-exposed loop DI and appears to inhibit at a post-attachment stage in pathogenesis (111).
Antibody occupancy and affinity determine antibody neutralization potency
Neutralization of WNV by antibodies is a “multiple” hit phenomena requiring engagement by more than a single antibody (112–116). Neutralization occurs when the number of antibodies bound to an individual virion exceeds a required threshold. Recent studies indicate for WNV that two factors primarily determine whether an antibody at a given concentration exceeds the stoichiometric requirements for neutralization: antibody affinity and the accessibility of epitopes on the surface of the virus (Fig. 4).
Fig. 4. Relationship between epitope accessibility and the occupancy requirements for neutralization.
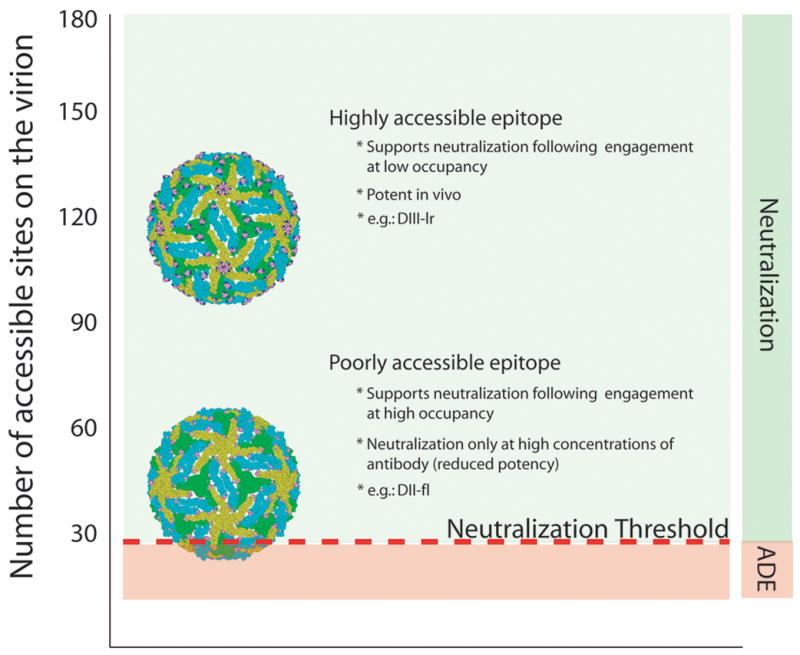
The accessibility of epitopes recognized by two different mAbs on the mature WNV virion is illustrated using molecular modeling: residues that comprise each determinant are illustrated as solid spheres. E proteins are colored according to their proximity to the 2-, 3-, or 5-fold symmetry axes (blue, green, and yellow, respectively). The number of accessible binding sites for each antibody is indicated on the left, whereas the ‘threshold’ for neutralization is indicated as a red line (modeled in this instance as 30 mAbs based on studies using the mAb E16). To exceed the threshold requirements for neutralization, only a fraction of highly accessible determinants must be simultaneously occupied by antibody (a low occupancy requirement). For cryptic epitopes (fewer accessible sites), a significantly greater percentage of accessible epitopes must be bound to achieve the same number of antibodies docked on the average virion (a high occupancy requirement). Adapted from (112, 139).
The strength of binding between antibody and viral antigen determines the fraction of epitopes on the virus particle occupied by antibody at any given concentration (defined as epitope occupancy) and is a primary determinant of neutralization potency of flaviviruses (112, 116). Thus, differences in neutralization potency between antibodies that bind similar epitopes can be accounted for by differences in the strength of antibody-antigen interactions. Integrating data from measurements of the avidity of antibody-virion interactions and the concentration of antibody required to inactivate 50% of the virus allows an estimate of antibody occupancy when the virus is neutralized. Data from both Fab docking models and cryo-electron microscopy studies indicate that the potent neutralizing mAb E16 engages a maximum of 120 of 180 sites at full occupancy as steric conflicts at the inner five-fold symmetry axis restrict complete occupancy (103, 117). Recent studies have evaluated directly how many antibodies for a given DIII epitope must be bound to a virion to achieve neutralization (112). mAbs that recognize the DIII-lr epitope block infection at concentrations that result in a low occupancy of the available sites on the virion. Neutralization of 50% of the virions by the most potent DIII-lr mAbs occurs when as few as 30 of 180 sites are occupied.
In contrast, more weakly neutralizing mAbs that bind a distinct epitope on DIII recognize fewer sites on the virion and require almost complete occupancy to inhibit WNV infection. High-affinity antibodies that recognize less exposed epitopes exhibit rather limited neutralization potency and inhibit infection only at very high concentrations relative to their affinity for viral antigens. Indeed, for some high affinity anti-DIII antibodies against WNV, even complete occupancy on the virion is not sufficient to exceed the threshold for neutralization (112). Preliminary experiments indicate that neutralization by antibodies that recognize DII-fl epitopes also requires engagement of these poorly accessible determinants with a high fractional occupancy.
Mechanisms of antibody neutralization
Beyond epitope occupancy and strength of antibody binding, the potency of neutralization may be independently modulated by the specific mechanism of neutralization. For enveloped flaviviruses including WNV, antibody-mediated virus neutralization can occur at several steps in the viral lifecycle, including attachment to receptors on the cell surface, internalization, or fusion within the endosomes (115). Antibodies that coat the virion surface could neutralize by directly by blocking receptor engagement or indirectly by inhibiting one of the conformational changes required for virus uncoating and nucleocapsid penetration into the cytoplasm.
There is still controversy as to how the potently inhibitory DIII-lr specific mAbs function. Indirect evidence suggest DIII plays in important role in virus attachment: (i) DIII protrudes the farthest from the surface of the virion; (ii) many of the mutations that impact tropism or virulence map to DIII (118, 119); and (iii) soluble forms of DIII can block WNV and TBEV infection (72, 73). Thus, blockade of the binding step is an attractive model for the neutralizing mechanism of DIII-specific mAbs. In contrast, 3H5-1, a DENV-2 DIII-lr mAb has been reported to neutralize virus by blocking entry and fusion (120). Our studies for WNV indicate that DIII-lr mAbs inhibit binding of virus to cells, albeit weakly compared to mAbs that localize to other regions on E (103). Because strongly neutralizing DIII-lr mAbs inhibited infection after virus was allowed to attach to cells, we suggested they must act predominantly at a post-attachment step. Studies now confirm that the most potently neutralizing DIII-lr mAbs (e.g. E16) against WNV block the pH-dependent fusion step: E16 efficiently enters cells in complex with the virus and prevents low pH inactivation of WNV virions in solution or low pH catalyzed fusion at the plasma membrane or with liposomes (B. Thompson, J. Smit, M. Diamond, and D. Fremont, unpublished results). Electron microscopy studies also have shown that target cells internalize WNV complexed with neutralizing polyclonal antibodies, suggesting a post-attachment mechanism of neutralization (121). Studies are underway to determine the structural mechanism of fusion inhibition, for example whether DIII-lr antibodies restrict the pH-dependent E protein trimer formation. Nonetheless, it remains possible that other particularly potent inhibitory DIII-lr mAbs could block at multiple steps in the viral lifecycle, including cellular attachment or trafficking.
The mechanism of antibody-mediated neutralization may be especially relevant for WNV because of the identification of several putative attachment and entry receptors on mammalian cells. Antibodies that neutralize by attachment blockade may show cell-specific effects: as different cell surface receptors engage distinct epitopes on the E protein, mAbs could block infection in one cell type but not another. Such a pattern was observed with DII-specific mAbs against WNV (21, 103).
Antibody-dependent enhancement of WNV infection
The mechanism of neutralization may also affect antibody-dependent enhancement (ADE) of infection. ADE, which has been observed for multiple viruses in vitro including WNV, occurs when virus-antibody complexes enter cells through Fc-γ receptor-mediated entry pathways (122, 123) and replicate to higher levels. This phenomenon has been hypothesized to contribute to the pathogenesis of dengue hemorrhagic fever (DHF) during secondary dengue infection (124), although it has not been implicated in WNV disease. The most direct link between ADE and the clinical outcome of DENV infection comes from investigations of the unusually large number of DHF cases following primary infection in infants during the first year of life (125). At birth, DENV-specific passively acquired antibodies are present at a relatively high concentration and exhibit neutralizing activity in vitro. However, as the infant ages, maternally acquired antibody wanes to levels that no longer neutralize virus and allows for enhancement of infection in vitro. The waning antibody titers of infants to levels that support ADE in vitro parallels the risk of DHF following primary DENV infection during the first year of life. Our recent studies suggest that the mechanism of neutralization and epitope specificity of antibodies impacts the extent of ADE observed in vitro. In cells expressing activating Fc-γ receptors, DII-fl mAbs that block attachment promote ADE over a wide range of concentrations whereas DIII-lr mAbs that block fusion promote ADE only at sub-neutralizing concentrations (103, 112).
Although the phenomenon of ADE has been established in vitro using several viral systems, it has been difficult to recapitulate in small or large animal models. Recent studies suggest a reason for this as the complement opsonin C1q inhibits ADE by WNV and DENV in vitro and in vivo (126, 127). IgG subclasses that bind C1q avidly (mouse IgG2a or IgG2b) induced minimal ADE of WNV infection in the presence of C1q, whereas subclasses that bind C1q weakly (mouse IgG1) strongly enhanced infection (Fig. 5). Thus, ADE can occur in vivo but does so under a very restricted set of conditions that is modulated in part by C1q binding to individual IgG subclasses (126). In C1q-sufficient mice, ADE was observed at relatively low levels and only with mAbs of the mouse IgG1 subclass, which poorly bind C1q. In contrast, in C1q−/− mice, robust ADE was observed with DII- or DIII-specific IgG2a mAbs; as these mAbs are predicted to bind C1q avidly, it makes sense that ADE was minimized in C1q+/+ wildtype mice. Although further studies are required, the observation that IgG2a but not IgG1 or IgG2b promote ADE in C1q−/− mice is most consistent with an interaction with the Fc-γRI (CD64), which primarily binds monomeric IgG2a with high affinity (128).
Fig. 5. C1q modulates mAb enhancement of WNV infection.

(A). Serial dilutions of E16 (mouse IgG2b) were mixed with PBS, 5% fresh or heat-inactivated mouse serum, incubated with WNV RVP, and added to FcγRIIa+ K562 cells. Forty-eight hours later, cells were analyzed by flow cytometry for GFP expression. The data are expressed as the fold enhancement of infection compared to no antibody. (B). Experiments were performed as in panel (A) except that fresh C1q−/− or C3−/− serum or purified C1q was mixed with the mouse E16 mAb. (C, D). Experiments were performed as in panel (B) except the epitope matched DIII-specific (C) E24 (mouse IgG2a) or (D) E34 (mouse IgG1) mAbs were used. Adapted from (126).
Although our experiments demonstrated ADE of WNV infection in vivo in C1q−/− mice, we did not observe a significant change in disease phenotype or survival. The linkage between ADE and severe disease as postulated for DENV infection may not occur for WNV, because one or more additional steps of viral pathogenesis are absent in the lifecycle of WNV. Indeed, an enhanced risk of severe WNV disease during secondary infection or vaccine challenge has never been described. Instead, ADE and severe disease may be more significant for other flaviviruses, such as DENV. As our recent experiments establish that C1q also modulates ADE by DENV infection, we plan to use C1q−/− mice and IgG2a mAbs along with specific adapted DENV strains (129) to evaluate the link between ADE and pathogenesis.
In vivo efficacy of neutralizing anti-WNV antibodies
While antibodies that recognize particular epitopes have neutralizing activity in vitro, a relevant test of their potency is protective activity in vivo in a passive transfer model. Neutralizing mAbs against flaviviruses are effective as pre-exposure prophylaxis in mice, with some mAbs having post-exposure therapeutic effects (reviewed in 92). Pooled γ globulin from immune and non-immune donors was therapeutic in mice infected with WNV, even when administered five days after infection (18, 19). More recent studies from our group and others have demonstrated that mouse and human mAbs against WNV protect in vivo in rodents (20–23). In vitro neutralizing activity correlated with therapeutic activity, as mAbs that bound the DIII-lr epitope had the greatest effect in vivo. In contrast, mAbs that bound to the DII-fl or other neutralizing epitopes in DI and DII protected only 18–60% of mice when given two days after infection compared to DIII-lr mAbs, which protected 80–100% of mice (21). Administration of human single-chain variable region antibody fragments converted to Fc fusion proteins (scFv-Fcs) that map to regions outside of DIII were also therapeutic in vivo (22).
If mAbs are to be an effective therapy for WNV encephalitis, they should function after the onset of symptoms and ideally, after infection in the CNS. In support of possible antibody therapy against WNV, case reports have suggested clinical improvement in patients treated with immune γ-globulin (130, 131). When mouse or humanized E16 was given as a single dose five or six days after infection, 90% of mice or hamsters were protected (20, 25, 132). In these rodent models, WNV enters and replicates in the CNS by four days after infection. Correspondingly, E16 administered on day 5 reduced or completely cleared WNV burden in the brain on day 9 after infection. Acute flaccid paralysis in hamsters also was blocked by treatment with E16 several days after infection (26). Thus, neutralizing antibody therapeutics show promise, as they directly inhibit transneuronal spread of WNV infection and prevent the development of paralysis in vivo.
Effector functions and protection by neutralizing antibodies
Antibodies may also inhibit WNV infection by activating Fc-dependent effector functions including complement activation and Fc-γ receptor targeting. Opsonization of enveloped RNA viruses with classical pathway complement components C1q, C4b, and C3b can inhibit receptor attachment and promote the formation of C5b-C9 membrane attack components that induce virolysis (133–135). Studies indicate that complement augments antibody-mediated neutralization of WNV in vitro (43). In the presence of WNV-specific mAbs, complement promotes lysis of BHK or mouse MC57GL cells that express surface E proteins (43). These data suggest antibodies that avidly fix complement may augment neutralizing activity in vivo against WNV.
More recent preliminary experiments show that C1q is necessary and sufficient to increase the potency of anti-WNV antibodies (E. Mehlhop, S. Nelson, M. Diamond, and T. Pierson, manuscript in preparation). In the presence of purified C1q, a ~25-fold reduction in the neutralizing titer of E16 was observed, indicating that C1q increased the efficacy of this mAb. This effect was entirely dependent on C1q, as C1q−/− mouse serum did not augment E16 neutralizing activity whereas C3−/− and C4−/− mouse serum did. Thus, C1q is necessary and sufficient for the complement-dependent increase in neutralization of WNV by specific IgG subclasses. Subsequent experiments in vivo with isotype switch variants of humanized E16 confirmed the beneficial effect of C1q binding in vivo. E16 isotypes that strongly bind C1q (hIgG1 and hIgG3) show greater protection against WNV, and the pre-exposure prophylactic potency of the IgG3 but not IgG2 variant of E16 was reduced in C1q−/− mice. Thus, in contrast to that observed for HIV (136), opsonization of antibodies by complement is functionally important in antibody protection against WNV. C1q may augment the neutralization potency of anti-WNV antibodies directly by modulating the occupancy requirements for neutralization: increasing antibody avidity or increasing the steric effects of bound antibody may more efficiently block virus attachment or fusion. This activity appears plausible, as C1q is a large multimeric protein (137).
Interaction of the antibody Fc region with Fc-γ receptors contributes to protection against WNV infection in vivo. Mice lacking activating Fc-γ receptors required significantly higher doses of a neutralizing anti-E mAb to maintain equivalent levels of protection against lethal WNV infection (20). Currently, the specific mechanism of protection (e.g. enhanced phagocytosis and destruction of viral particles or antibody-dependent cell-mediated cytotoxicity of infected cells) or the specific Fc-γ receptors that mediate this effect remain uncharacterized.
Protective activity of anti-NS1 antibodies
The flavivirus non-structural NS1 protein is a highly conserved secreted and cell surface-associated glycoprotein that does not package with the virion. Nonetheless, immunization with NS1 elicits a protective immune response against YFV, DENV, and TBEV through poorly defined mechanisms. To study how anti-NS1 antibodies could protect against infection we recently generated a large panel of NS1-specific mAbs (27). Prophylaxis of mice with several different anti-NS1 mAbs strongly protected against lethal WNV infection (75 to 97% survival, respectively) compared to saline-treated controls (17% survival). In contrast, other anti-NS1 mAbs of the same isotype provided no significant protection. Two of the anti-NS1 mAbs also demonstrated marked efficacy as post-exposure therapy, reaching 80% protection when co-administered as a single dose four days after infection. Virologic analysis showed these anti-NS1 mAbs limited viremia and viral entry into the CNS.
Because NS1 is absent from the virion, we speculated that protective antibodies would be inhibitory because of specific effector functions. To identify the specific mechanism, we re-evaluated the protective activity of anti-NS1 mAbs using mice with deficiencies of C1q or specific Fc-γ receptors. In C1q−/− mice, which cannot activate complement by the antibody-dependent classical pathway, virtually all of the protective activity of anti-NS1 mAbs was retained (27, 89). In contrast, in Fc-γ receptor I, III, and IV−/− mice, which lack the common signaling γ-chain and are impaired in antibody-dependent effector responses, the beneficial effect of anti-NS1 mAbs was lost. Protective effects were retained in Fc-γ receptor III−/− or NK cell-depleted mice, suggesting that NK cells did not contribute to anti-NS1 mAb-mediated protection against WNV.
We hypothesized that anti-NS1 mAbs might target infected cells that display high levels of cell surface NS1 for phagocytosis by tissue macrophages that express high levels of Fc-γ receptor I (138). Opsonization with anti-NS1 mAbs of the IgG2a subclass promoted efficient internalization of WNV-infected cells by wildtype and Fc-γ receptor III−/− but not Fc-γ receptor I, III, and IV−/− peritoneal macrophages. In contrast, internalization of WNV-infected cells by wildtype or deficient macrophages was not observed after addition of anti-NS1 mAbs that failed to recognize surface NS1 or were of the IgG1 subclass. Thus, our experiments suggest protective anti-NS1 mAbs of a given IgG subclass that bind to cell surface-associated NS1 facilitate phagocytosis and clearance of WNV-infected cells through Fc-γ receptors I and/or IV (Fig. 6).
Fig. 6. Mechanism of protection against WNV by anti-NS1 antibodies.

(A). Schematic diagram of NS1 fragments. (Top) A secondary structure prediction model, putative disulfide bonding patterns, and N-linked glycosylated sites are depicted. The secondary structure model is a consensus prediction based on NS1 from several different flaviviruses. (Bottom) Diagram of expression constructs used for yeast surface display and bacterial expression of NS1 fragments. NS1(125–352)B and NS1(158–352)Y indicate E. coli and yeast expressed FR-II-III, respectively. Adapted from (27). (B). Flow cytometry histograms showing immunoreactivity of NS1 on the surface of yeast, within permeabilized Raji-WNV cells, and on the surface of Raji-WNV-cells with individual anti-NS1 mAbs. Representative histograms are shown for four NS1 mAbs (8NS1, 15NS1, 10NS1, and 17NS1). The E1 mAb against WNV E protein was used as a negative control. Green and red arrows indicate the absence or presence of binding of mAbs to cell surface-associated NS1, respectively. The fragment localization of NS1 antibodies is indicated to the right. (C). Model of antibody and NS1-dependent clearance of WNV-infected cells by Fc-γ receptor I and IV-expressing phagocytes. WNV-infected cells express cell-associated forms of NS1 on their surface, which can be bound by subsets (fragment II and III-specific) antibodies. NS1-specific antibodies of a given IgG subclass (e.g. mouse IgG2a) are recognized by activating Fc-γ receptors (mouse Fc-γR I or IV) resulting in phagocytosis and clearance of infected cells.
Conclusions
Significant advances have been made in our understanding of the molecular and structural basis of antibody-mediated neutralization of WNV. Based on work by several groups, including our own, a composite picture has emerged as to the location of epitopes that are recognized by the most strongly neutralizing antibodies. Although preliminary experiments suggest the most potently inhibitory mAbs against WNV that map to the DIII-lr epitope block the pH-dependent fusion step, these results need to be confirmed with a larger panel of antibodies, including those that recognize related flaviviruses. The ongoing identification of bona fide attachment and entry receptors for flaviviruses will undoubtedly impact our understanding of antibody neutralization. Cell-specific differences in receptor usage will affect the mechanism and potency of antibody inhibition.
Despite much progress, many questions remain unanswered: (i) what is the mechanism of inhibition for the DI and DII-specific mAb neutralizing mAbs that localize outside of the fusion loop; (ii) what role do anti-prM and anti-M antibodies have in neutralization and protection; (iii) which E protein epitopes are immunodominant following natural WNV infection in humans and other animals; (iv) why are antibodies against poorly exposed epitopes still protective in vitro and in vivo; and (v) what is the structural basis of antibody blockade of WNV fusion by the DIII-lr antibodies.
An improved understanding of the mechanisms of antibody-mediated neutralization and protection has significant implications for the generation of novel antibody-based therapeutics, epitope-targeted vaccines, or chemical inhibitors of WNV infection. WNV and other related viruses may be well suited to ‘reverse vaccinology’, the identification and targeting of specific structural protein epitopes that elicit protective antibodies. In this strategy, epitopes that are poorly protective are eliminated or masked in favor of epitopes that elicit strongly protective antibodies. This could be achieved through selective epitope mutation or deletion, epitope masking with N-linked carbohydrates, subunit (i.e. DIII alone) vaccines, or through generation of novel variants that display desired epitopes. Vaccines that elicit potently neutralizing antibodies against the DIII-lr epitope that block fusion could be safer and more effective against a range of flaviviral infections.
Acknowledgments
We thank members of our laboratory for helpful discussions. Work in our laboratories is supported by the Pediatric Dengue Vaccine Initiative, the Burroughs Wellcome Fund, and the NIAID of the NIH (grants U01 AI061373, U54 AI057160, R01 AI073755, and NIH intramural funds).
References
- 1.Hayes EB, Komar N, Nasci RS, Montgomery SP, O’Leary DR, Campbell GL. Epidemiology and transmission dynamics of West Nile virus disease. Emerg Infect Dis. 2005;11:1167–1173. doi: 10.3201/eid1108.050289a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sejvar JJ, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511–515. doi: 10.1001/jama.290.4.511. [DOI] [PubMed] [Google Scholar]
- 3.Lanciotti RS, et al. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology. 2002;298:96–105. doi: 10.1006/viro.2002.1449. [DOI] [PubMed] [Google Scholar]
- 4.Beasley DW, Li L, Suderman MT, Barrett AD. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology. 2002;296:17–23. doi: 10.1006/viro.2002.1372. [DOI] [PubMed] [Google Scholar]
- 5.Beasley DW, et al. Genome sequence and attenuating mutations in West Nile virus isolate from Mexico. Emerg Infect Dis. 2004;10:2221–2224. doi: 10.3201/eid1012.040647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brault AC, et al. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat Genet. 2007;39:1162–1166. doi: 10.1038/ng2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berthet FX, Zeller HG, Drouet MT, Rauzier J, Digoutte JP, Deubel V. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J Gen Virol. 1997;78:2293–2297. doi: 10.1099/0022-1317-78-9-2293. [DOI] [PubMed] [Google Scholar]
- 8.Jupp PG. The ecology of West Nile virus in South Africa and the occurrence of outbreaks in humans. Ann NY Acad Sci. 2001;951:143–152. doi: 10.1111/j.1749-6632.2001.tb02692.x. [DOI] [PubMed] [Google Scholar]
- 9.Petersen LR, Marfin AA, Gubler DJ. West Nile virus. JAMA. 2003;290:524–528. doi: 10.1001/jama.290.4.524. [DOI] [PubMed] [Google Scholar]
- 10.Deardorff E, et al. Introductions of West Nile virus strains to Mexico. Emerg Infect Dis. 2006;12:314–318. doi: 10.3201/eid1202.050871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komar N, Clark GG. West Nile virus activity in Latin America and the Caribbean. Rev Panam Salud Publica. 2006;19:112–117. doi: 10.1590/s1020-49892006000200006. [DOI] [PubMed] [Google Scholar]
- 12.Huhn GD, et al. The Emergence of West Nile Virus During a Large Outbreak in Illinois in 2002. Am J Trop Med Hyg. 2005;72:768–776. [PubMed] [Google Scholar]
- 13.Davis LE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 14.Busch MP, et al. Screening the blood supply for West Nile virus RNA by nucleic acid amplification testing. N Engl J Med. 2005;353:460–467. doi: 10.1056/NEJMoa044029. [DOI] [PubMed] [Google Scholar]
- 15.Samuel MA, Diamond MS. Pathogenesis of West Nile virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80:9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinton MA. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol. 2002;56:371–402. doi: 10.1146/annurev.micro.56.012302.160654. [DOI] [PubMed] [Google Scholar]
- 17.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003;77:2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engle M, Diamond MS. Antibody prophylaxis and therapy against West Nile Virus infection in wild type and immunodeficient mice. J Virol. 2003;77:12941–12949. doi: 10.1128/JVI.77.24.12941-12949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Nathan D, Lustig S, Tam G, Robinzon S, Segal S, Rager-Zisman B. Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating west nile virus infection in mice. J Infect Dis. 2003;188:5–12. doi: 10.1086/376870. [DOI] [PubMed] [Google Scholar]
- 20.Oliphant T, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliphant T, et al. Determinants of West Nile virus envelope protein domains I and II antibody recognition and neutralization. J Virol. 2006;80:12149–12159. doi: 10.1128/JVI.01732-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gould LH, et al. Protective and therapeutic capacity of human single chain Fv-Fc fusion proteins against West Nile virus. J Virol. 2005;79:14606–14613. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Throsby M, et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile virus. J Virol. 2006;80:6982–6992. doi: 10.1128/JVI.00551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesh RB, Arroyo J, Travassos Da Rosa AP, Guzman H, Xiao SY, Monath TP. Efficacy of killed virus vaccine, live attenuated chimeric virus vaccine, and passive immunization for prevention of West Nile virus encephalitis in hamster model. Emerg Infect Dis. 2002;8:1392–1397. doi: 10.3201/eid0812.020229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrey JD, et al. Humanized monoclonal antibody against West Nile virus E protein administered after neuronal infection protects against lethal encephalitis in hamsters. J Infect Dis. 2006;194:1300–1308. doi: 10.1086/508293. [DOI] [PubMed] [Google Scholar]
- 26.Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci USA. 2007;104:17140–17145. doi: 10.1073/pnas.0705837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung KM, et al. Antibodies against West Nile virus non-structural (NS)-1 protein prevent lethal infection through Fc gamma receptor-dependent and independent mechanisms. J Virol. 2006;80:1340–1351. doi: 10.1128/JVI.80.3.1340-1351.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnston LJ, Halliday GM, King NJ. Phenotypic changes in Langerhans’ cells after infection with arboviruses: a role in the immune response to epidermally acquired viral infection? J Virol. 1996;70:4761–4766. doi: 10.1128/jvi.70.7.4761-4766.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnston LJ, Halliday GM, King NJ. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J Invest Dermatol. 2000;114:560–568. doi: 10.1046/j.1523-1747.2000.00904.x. [DOI] [PubMed] [Google Scholar]
- 30.Byrne SN, Halliday GM, Johnston LJ, King NJ. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J Invest Dermatol. 2001;117:702–709. doi: 10.1046/j.0022-202x.2001.01454.x. [DOI] [PubMed] [Google Scholar]
- 31.Daffis S, Samuel MA, Keller BC, Gale M, Jr, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and independent mechanisms. PLoS Pathog. 2007;3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arjona A, et al. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J Immunol. 2007;179:8403–8409. doi: 10.4049/jimmunol.179.12.8403. [DOI] [PubMed] [Google Scholar]
- 33.Silva MC, Guerrero-Plata A, Gilfoy FD, Garofalo RP, Mason PW. Differential activation of human monocyte-derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J Virol. 2007;81:13640–13648. doi: 10.1128/JVI.00857-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourne N, et al. Early production of type I interferon during West Nile virus infection: role for lymphoid tissues in IRF3-independent interferon production. J Virol. 2007;81:9100–9108. doi: 10.1128/JVI.00316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82:609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao SY, Guzman H, Zhang H, Travassos da Rosa AP, Tesh RB. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg Infect Dis. 2001;7:714–721. doi: 10.3201/eid0704.010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chambers TJ, Diamond MS. Pathogenesis of flavivirus encephalitis. In: Chambers TJ, Monath TP, editors. The Flaviviruses: Current Molecular Aspects of Evolution, Biology, and Disease Prevention. London: Academic Press; 2003. pp. 273–342. [Google Scholar]
- 38.Shrestha B, Gottlieb DI, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J Virol. 2003;77:13203–13213. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samuel MA, Morrey JD, Diamond MS. Caspase-3 dependent cell death of neurons contributes to the pathogenesis of West Nile virus encephalitis. J Virol. 2007;81:2614–2623. doi: 10.1128/JVI.02311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel MA, Diamond MS. Type I IFN protects against lethal West Nile Virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79:13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diamond MS, Sitati E, Friend L, Shrestha B, Higgs S, Engle M. Induced IgM protects against lethal West Nile Virus infection. J Exp Med. 2003;198:1–11. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehlhop E, Diamond MS. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med. 2006;203:1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS. Complement activation is required for the induction of a protective antibody response against West Nile virus infection. J Virol. 2005;79:7466–7477. doi: 10.1128/JVI.79.12.7466-7477.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 45.Arjona A, et al. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117:3059–3066. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunsperger EA, Roehrig JT. Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. J Neurovirol. 2006;12:129–139. doi: 10.1080/13550280600758341. [DOI] [PubMed] [Google Scholar]
- 47.Samuel MA, et al. PKR and RNAse L contribute to protection against lethal West Nile virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. RNase L plays a role in the antiviral response to West Nile virus. J Virol. 2006;80:2987–2999. doi: 10.1128/JVI.80.6.2987-2999.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilfoy FD, Mason PW. West Nile virus-induced IFN production is mediated by the double-stranded RNA-dependent protein kinase, PKR. J Virol. 2007;81:11148–11158. doi: 10.1128/JVI.00446-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller BC, Fredericksen BL, Samuel MA, Mock RE, Mason PW, Diamond MS, et al. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J Virol. 2006;80:9424–9434. doi: 10.1128/JVI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson JF, Rahal JJ. Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis. 2002;8:107–108. doi: 10.3201/eid0801.010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lucas M, et al. Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol Cell Biol. 2003;81:230–236. doi: 10.1046/j.1440-1711.2003.01166.x. [DOI] [PubMed] [Google Scholar]
- 53.Fredericksen BL, Smith M, Katze MG, Shi PY, Gale M. The host response to West Nile virus infection limits spread through the activation of the interferon regulatory factor 3 pathway. J Virol. 2004;78:7737–7747. doi: 10.1128/JVI.78.14.7737-7747.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA. Inhibition of interferon signaling by the New York 99 strain and kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J Virol. 2005;79:1934–1942. doi: 10.1128/JVI.79.3.1934-1942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T, et al. IFN-γ-producing γδ T cells help control murine West Nile virus infection. J Immunol. 2003;171:2524–2531. doi: 10.4049/jimmunol.171.5.2524. [DOI] [PubMed] [Google Scholar]
- 56.Shrestha B, et al. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol. 2006;80:5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perelygin AA, Scherbik SV, Zhulin IB, Stockman BM, Li Y, Brinton MA. Positional cloning of the murine flavivirus resistance gene. Proc Natl Acad Sci USA. 2002;99:9322–9327. doi: 10.1073/pnas.142287799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mashimo T, et al. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc Natl Acad Sci USA. 2002;99:11311–11316. doi: 10.1073/pnas.172195399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shrestha B, Diamond MS. The role of CD8+ T cells in the control of West Nile virus infection. J Virol. 2004;78:8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Lobigs M, Lee E, Mullbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003;77:13323–13334. doi: 10.1128/JVI.77.24.13323-13334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, Lobigs M, Lee E, Mullbacher A. Exocytosis and Fas mediated cytolytic mechanisms exert protection from West Nile virus induced encephalitis in mice. Immunol Cell Biol. 2004;82:170–173. doi: 10.1046/j.0818-9641.2004.01227.x. [DOI] [PubMed] [Google Scholar]
- 63.Shrestha B, Diamond MS. Fas Ligand interactions contribute to CD8+ T cell-mediated control of West Nile virus infection in the central nervous system. J Virol. 2007;81:11749–11757. doi: 10.1128/JVI.01136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 65.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. The envelope glycoprotein from tick-borne encephalitis virus at 2 Angstrom resolution. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 66.Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanai R, et al. Crystal structure of west nile virus envelope glycoprotein reveals viral surface epitopes. J Virol. 2006;80:11000–11008. doi: 10.1128/JVI.01735-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nybakken GE, Nelson CA, Chen BR, Diamond MS, Fremont DH. Crystal structure of the West Nile virus envelope glycoprotein. J Virol. 2006;80:11467–11474. doi: 10.1128/JVI.01125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allison SL, Schalich J, Stiasny K, Mandl CW, Heinz FX. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J Virol. 2001;75:4268–4275. doi: 10.1128/JVI.75.9.4268-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Modis Y, Ogata S, Clements D, Harrison SC. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 71.Bressanelli S, et al. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004;23:728–738. doi: 10.1038/sj.emboj.7600064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhardwaj S, Holbrook M, Shope RE, Barrett AD, Watowich SJ. Biophysical characterization and vector-specific antagonist activity of domain III of the tick-borne flavivirus envelope protein. J Virol. 2001;75:4002–4007. doi: 10.1128/JVI.75.8.4002-4007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu JJ, Rajamanonmani R, Li J, Bhuvanakantham R, Lescar J, Ng ML. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. J Gen Virol. 2005;86:405–412. doi: 10.1099/vir.0.80411-0. [DOI] [PubMed] [Google Scholar]
- 74.Chu JJ, Ng ML. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J Biol Chem. 2004;279:54533–54541. doi: 10.1074/jbc.M410208200. [DOI] [PubMed] [Google Scholar]
- 75.Lee JW, Chu JJ, Ng ML. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J Biol Chem. 2006;281:1352–1360. doi: 10.1074/jbc.M506614200. [DOI] [PubMed] [Google Scholar]
- 76.Davis CW, Nguyen HY, Hanna SL, Sanchez MD, Doms RW, Pierson TC. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol. 2006;80:1290–1301. doi: 10.1128/JVI.80.3.1290-1301.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davis CW, Mattei LM, Nguyen HY, Doms RW, Pierson TC. The location of N-linked glycans on West Nile virions controls their interactions with CD209. J Biol Chem. 2006;281:37183–37194. doi: 10.1074/jbc.M605429200. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Y, et al. Conformational changes of the flavivirus E glycoprotein. Structure (Camb) 2004;12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang W, et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ. Structure of West Nile virus. Science. 2003;302:248. doi: 10.1126/science.1089316. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y, Kaufmann B, Chipman PR, Kuhn RJ, Rossmann MG. Structure of immature West Nile virus. J Virol. 2007;81:6141–6145. doi: 10.1128/JVI.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heinz F, et al. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with the protein prM. Virology. 1994;198:109–117. doi: 10.1006/viro.1994.1013. [DOI] [PubMed] [Google Scholar]
- 83.Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 85.Colombage G, Hall R, Pavy M, Lobigs M. DNA-based and alphavirus-vectored immunisation with prM and E proteins elicits long-lived and protective immunity against the flavivirus, Murray Valley encephalitis virus. Virology. 1998;250:151–163. doi: 10.1006/viro.1998.9357. [DOI] [PubMed] [Google Scholar]
- 86.Pincus S, et al. Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology. 1992;187:290–297. doi: 10.1016/0042-6822(92)90317-i. [DOI] [PubMed] [Google Scholar]
- 87.Falconar AK. Identification of an epitope on the dengue virus membrane (M) protein defined by cross-protective monoclonal antibodies: design of an improved epitope sequence based on common determinants present in both envelope (E and M) proteins. Arch Virol. 1999;144:2313–2330. doi: 10.1007/s007050050646. [DOI] [PubMed] [Google Scholar]
- 88.Vazquez S, et al. Immune response to synthetic peptides of dengue prM protein. Vaccine. 2002;20:1823–1830. doi: 10.1016/s0264-410x(01)00515-1. [DOI] [PubMed] [Google Scholar]
- 89.Chung KM, Thompson BS, Fremont DH, Diamond MS. Antibody recognition of cell surface-associated NS1 triggers Fc-g receptor mediated phagocytosis and clearance of WNV infected cells. J Virol. 2007;81:9551–9555. doi: 10.1128/JVI.00879-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wong SJ, et al. Immunoassay targeting nonstructural protein 5 to differentiate West Nile virus infection from dengue and St. Louis encephalitis virus infections and from flavivirus vaccination. J Clin Microbiol. 2003;41:4217–4223. doi: 10.1128/JCM.41.9.4217-4223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kimura-Kuroda J, Yasui K. Protection of mice against Japanese encephalitis virus by passive administration with monoclonal antibodies. J Immunol. 1988;141:3606–3610. [PubMed] [Google Scholar]
- 92.Roehrig JT, Staudinger LA, Hunt AR, Mathews JH, Blair CD. Antibody prophylaxis and therapy for flaviviral encephalitis infections. Ann NY Acad Sci. 2001;951:286–297. doi: 10.1111/j.1749-6632.2001.tb02704.x. [DOI] [PubMed] [Google Scholar]
- 93.Schlesinger JJ, Walsh EE, Brandriss MW. Analysis of 17D yellow fever virus envelope protein epitopes using monoclonal antibodies. J Gen Virol. 1984;65:1637–1644. doi: 10.1099/0022-1317-65-10-1637. [DOI] [PubMed] [Google Scholar]
- 94.Roehrig JT, Mathews JH, Trent DW. Identification of epitopes on the E glycoprotein of Saint Louis encephalitis virus using monoclonal antibodies. Virology. 1983;128:118–126. doi: 10.1016/0042-6822(83)90323-9. [DOI] [PubMed] [Google Scholar]
- 95.Mathews JH, Roehrig JT. Elucidation of the topography and determination of the protective epitopes on the E glycoprotein of Saint Louis encephalitis virus by passive transfer with monoclonal antibodies. J Immunol. 1984;132:1533–1537. [PubMed] [Google Scholar]
- 96.Beasley DW, Barrett AD. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J Virol. 2002;76:13097–13100. doi: 10.1128/JVI.76.24.13097-13100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sanchez MD, et al. Characterization of neutralizing antibodies to West Nile virus. Virology. 2005;336:70–82. doi: 10.1016/j.virol.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 98.Oliphant T, et al. The Induction of Epitope-Specific Neutralizing Antibodies against West Nile virus. J Virol. 2007;81:11828–11839. doi: 10.1128/JVI.00643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanchez MD, et al. The neutralizing antibody response against West Nile virus in naturally infected horses. Virology. 2007;359:336–348. doi: 10.1016/j.virol.2006.08.047. [DOI] [PubMed] [Google Scholar]
- 100.Sitati E, Diamond MS. CD4+ T cell responses are required for clearance of West Nile Virus from the central nervous system. J Virol. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sitati E, McCandless EE, Klein RS, Diamond MS. CD40-CD40 Ligand Interactions Promote Trafficking of CD8+ T Cells into the Brain and Protection against West Nile Virus Encephalitis. J Virol. 2007;81:9801–9811. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oliphant T, Diamond MS. The molecular basis of antibody-mediated neutralization of West Nile virus. Expert Opin Biol Ther. 2007;7:885–892. doi: 10.1517/14712598.7.6.885. [DOI] [PubMed] [Google Scholar]
- 103.Nybakken G, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. Structural basis for neutralization of a therapeutic antibody against West Nile virus. Nature. 2005;437:764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choi KS, Nah JJ, Ko YJ, Kim YJ, Joo YS. The DE loop of the domain III of the envelope protein appears to be associated with West Nile virus neutralization. Virus Res. 2007;123:216–218. doi: 10.1016/j.virusres.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 105.Crill WD, Chang GJ. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J Virol. 2004;78:13975–13986. doi: 10.1128/JVI.78.24.13975-13986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Crill WD, Trainor NB, Chang GJ. A detailed mutagenesis study of flavivirus cross-reactive epitopes using West Nile virus-like particles. J Gen Virol. 2007;88:1169–1174. doi: 10.1099/vir.0.82640-0. [DOI] [PubMed] [Google Scholar]
- 107.Stiasny K, Kiermayr S, Holzmann H, Heinz FX. Cryptic properties of a cluster of dominant flavivirus cross-reactive antigenic sites. J Virol. 2006;80:9557–9568. doi: 10.1128/JVI.00080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goncalvez AP, Purcell RH, Lai CJ. Epitope determinants of a chimpanzee Fab antibody that efficiently cross-neutralizes dengue type 1 and type 2 viruses map to inside and in close proximity to fusion loop of the dengue type 2 virus envelope glycoprotein. J Virol. 2004;78:12919–12928. doi: 10.1128/JVI.78.23.12919-12928.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Daffis S, Kontermann RE, Korimbocus J, Zeller H, Klenk HD, Ter Meulen J. Antibody responses against wild-type yellow fever virus and the 17D vaccine strain: characterization with human monoclonal antibody fragments and neutralization escape variants. Virology. 2005;337:262–272. doi: 10.1016/j.virol.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 110.Ryman KD, Ledger TN, Weir RC, Schlesinger JJ, Barrett AD. Yellow fever virus envelope protein has two discrete type-specific neutralizing epitopes. J Gen Virol. 1997;78:1353–1356. doi: 10.1099/0022-1317-78-6-1353. [DOI] [PubMed] [Google Scholar]
- 111.Lai CJ, et al. Epitope determinants of a chimpanzee dengue virus type 4 (DENV-4)-neutralizing antibody and protection against DENV-4 challenge in mice and rhesus monkeys by passively transferred humanized antibody. J Virol. 2007;81:12766–12774. doi: 10.1128/JVI.01420-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pierson TC, et al. The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host and Microbe. 2007;1:135–145. doi: 10.1016/j.chom.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burton DR, Saphire EO, Parren PW. A model for neutralization of viruses based on antibody coating of the virion surface. Curr Top Microbiol Immunol. 2001;260:109–143. doi: 10.1007/978-3-662-05783-4_7. [DOI] [PubMed] [Google Scholar]
- 114.Klasse PJ, Burton DR. Antibodies to West Nile virus: a double-edged sword. Cell Host Microbe. 2007;1:87–89. doi: 10.1016/j.chom.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 115.Klasse PJ, Sattentau QJ. Mechanisms of virus neutralization by antibody. Curr Top Microbiol Immunol. 2001;260:87–108. doi: 10.1007/978-3-662-05783-4_6. [DOI] [PubMed] [Google Scholar]
- 116.Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J Gen Virol. 2002;83:2091–2108. doi: 10.1099/0022-1317-83-9-2091. [DOI] [PubMed] [Google Scholar]
- 117.Kauffman B, et al. West Nile virus in complex with a neutralizing monoclonal antibody. Proc Natl Acad Sci USA. 2006;103:12400–12404. doi: 10.1073/pnas.0603488103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mandl CW, Allison SL, Holzmann H, Meixner T, Heinz FX. Attenuation of tick-borne encephalitis virus by structure-based site-specific mutagenesis of a putative flavivirus receptor binding site. J Virol. 2000;74:9601–9609. doi: 10.1128/jvi.74.20.9601-9609.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holzmann H, Heinz FX, Mandl CW, Guirakhoo F, Kunz C. A single amino acid substitution in envelope protein E of tick-borne encephalitis virus leads to attenuation in the mouse model. J Virol. 1990;64:5156–5159. doi: 10.1128/jvi.64.10.5156-5159.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Se-Thoe SY, Ling AE, Ng MM. Alteration of virus entry mode: a neutralisation mechanism for Dengue-2 virus. J Med Virol. 2000;62:364–376. doi: 10.1002/1096-9071(200011)62:3<364::aid-jmv9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 121.Gollins SW, Porterfield JS. A new mechanism for the neutralization of enveloped viruses by antiviral antibody. Nature. 1986;321:244–246. doi: 10.1038/321244a0. [DOI] [PubMed] [Google Scholar]
- 122.Peiris JS, Porterfield JS. Antibody-mediated enhancement of Flavivirus replication in macrophage- like cell lines. Nature. 1979;282:509–511. doi: 10.1038/282509a0. [DOI] [PubMed] [Google Scholar]
- 123.Peiris JSM, Gordon S, Unkeless JC, Porterfield JS. Monoclonal anti-Fc receptor IgG blocks antibody-dependent enhancement of viral replication in macrophages. Nature. 1981;289:189–191. doi: 10.1038/289189a0. [DOI] [PubMed] [Google Scholar]
- 124.Halstead SB. In vivo enhancement of dengue virus infection in rhesus monkeys by passively transferred antibody. J Infect Dis. 1979;140:527–533. doi: 10.1093/infdis/140.4.527. [DOI] [PubMed] [Google Scholar]
- 125.Halstead SB. Observations related to pathogensis of dengue hemorrhagic fever. VI Hypotheses and discussion. Yale J Biol Med. 1970;42:350–362. [PMC free article] [PubMed] [Google Scholar]
- 126.Mehlhop E, et al. Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an IgG subclass-specific manner. Cell Host Microbe. 2007;2:417–426. doi: 10.1016/j.chom.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yamanaka A, Kosugi S, Konishi E. Infection-enhancing and -neutralizing activities of mouse monoclonal antibodies against dengue type 2 and 4 viruses are controlled by complement levels. J Virol. 2008;82:927–937. doi: 10.1128/JVI.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol. 1991;9:457–492. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 129.Shresta S, Sharar KL, Prigozhin DM, Beatty PR, Harris E. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J Virol. 2006;80:10208–10217. doi: 10.1128/JVI.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hamdan A, Green P, Mendelson E, Kramer MR, Pitlik S, Weinberger M. Possible benefit of intravenous immunoglobulin therapy in a lung transplant recipient with West Nile virus encephalitis. Transpl Infect Dis. 2002;4:160–162. doi: 10.1034/j.1399-3062.2002.01014.x. [DOI] [PubMed] [Google Scholar]
- 131.Shimoni Z, Niven MJ, Pitlick S, Bulvik S. Treatment of West Nile virus encephalitis with intravenous immunoglobulin. Emerg Infect Dis. 2001;7:759. doi: 10.3201/eid0704.010432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Morrey JD, et al. Defining limits of humanized neutralizing monoclonal antibody treatment for West Nile virus neurological infection in a hamster model. Antimicrob Agents Chemother. 2007;51:2396–2402. doi: 10.1128/AAC.00147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Berry DM, Almeida JD. The morphological and biological effects of various antisera on avian infectious bronchitis virus. J Gen Virol. 1968;3:97–102. doi: 10.1099/0022-1317-3-1-97. [DOI] [PubMed] [Google Scholar]
- 134.Cooper NR, Jensen FC, Welsh RM, Jr, Oldstone MB. Lysis of RNA tumor viruses by human serum: direct antibody-independent triggering of the classical complement pathway. J Exp Med. 1976;144:970–984. doi: 10.1084/jem.144.4.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jayasekera JP, Moseman EA, Carroll MC. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J Virol. 2007;81:3487–3494. doi: 10.1128/JVI.02128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 137.Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology. 2000;49:159–170. doi: 10.1016/s0162-3109(00)80301-x. [DOI] [PubMed] [Google Scholar]
- 138.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM. FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity. 2002;16:379–389. doi: 10.1016/s1074-7613(02)00287-x. [DOI] [PubMed] [Google Scholar]
- 139.Pierson TC, Diamond MS. Molecular mechanisms of antibody-mediated neutralization of flavivirus infection. Exp Rev Mol Med. 2008 doi: 10.1017/S1462399408000665. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]