HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats (original) (raw)
. Author manuscript; available in PMC: 2009 Aug 21.
Abstract
Traumatic brain injury (TBI) produces a rapid and robust inflammatory response in the brain characterized in part by activation of microglia. A novel histone deacetylase (HDAC) inhibitor, 4-dimethylamino-_N_-[5-(2-mercaptoacetylamino)pentyl]benzamide (DMA-PB), was administered (0, 0.25, 2.5, 25 mg/kg) systemically immediately after lateral fluid percussion TBI in rats. Hippocampal CA2/3 tissue was processed for acetyl-histone H3 immunolocalization, OX-42 immunolocalization (for microglia), and Fluoro-Jade B histofluorescence (for degenerating neurons) at 24 h after injury. Vehicle-treated TBI rats exhibited a significant reduction in acetyl-histone H3 immunostaining in the ipsilateral CA2/3 hippocampus compared to the sham TBI group (p<0.05). The reduction in acetyl-histone H3 immunostaining was attenuated by each of the DMA-PB dosage treatment groups. Vehicle-treated TBI rats exhibited a high density of phagocytic microglia in the ipsilateral CA2/3 hippocampus compared to sham TBI in which none were observed. All doses of DMA-PB significantly reduced the density of phagocytic microglia (p<0.05). There was a trend for DMA-PB to reduce the number of degenerating neurons in the ipsilateral CA2/3 hippocampus (p = 0.076). We conclude that the HDAC inhibitor DMA-PB is a potential novel therapeutic for inhibiting neuroinflammation associated with TBI.
Keywords: traumatic brain injury, microglia, inflammation, histone deacetylase, fluid percussion
1. Introduction
Traumatic brain injury (TBI) is a serious and complex injury that occurs in approximately 1.4 million people each year in the United States (Langlois et al., 2006). TBI is associated with a broad spectrum of symptoms and disabilities, including a risk factor for developing neurodegenerative disorders, such as Alzheimer’s disease (Jellinger et al., 2001; Nemetz et al., 1999; Van Den Heuvel et al., 2007). TBI produces a number of pathologies including axonal injury, cell death, contusions, and inflammation (Teasdale & Graham, 1998). The inflammatory cascade is characterized by proinflammatory cytokines (Dietrich et al., 2004; Morganti-Kossmann et al., 2002) and activation of microglia (Carbonell and Grady, 1999; Morganti-Kossmann et al., 2007) which can exacerbate other pathologies. Although the role of inflammation in experimental TBI is well established, no truly efficacious and approved anti-nflammatory therapies are currently available for the treatment of TBI.
Considerable research activity has focused on histone deacetylase (HDAC)1 inhibitors as novel therapeutics in models of ischemic stroke (Faraco et al., 2006; Kim et al., 2007; Ren et al., 2004), multiple sclerosis (Camelo et al., 2005) and Huntington’s diseases (Ferrante et al., 2003; Gardian et al., 2005). However no data have been reported regarding the effects of HDAC inhibitors on TBI. The post-translational acetylation status of chromatin is determined by the activities of two classes of enzymes, histone acetyltransferases (HATs) and HDACs, which compete for control of the acetylation of lysine residues making up the histones. In general, HATs function to acetylate lysine groups in nuclear histones, resulting in neutralization of the charges on the histones and a more open, transcriptionally active chromatin structure. In contrast, HDACs function to deacetylate and suppress transcription. Therefore the action of HDAC inhibitors is to reactivate silenced genes by modulating the condensation status of DNA. Any shift in the balance of acetylation on chromatin may result in changes in the regulation of patterns of gene expression (Butler and Kozikowski, 2008; Kozikowski et al., 2007). Additionally, HDAC inhibitors may also have anti-inflammatory actions acting through acetylation of non-histone proteins (Adcock, 2007).
A recent study in experimental pediatric TBI reported a decrease in hippocampal CA3 histone H3 acetylation lasting hours to days after injury (Gao et al., 2006). These changes were attributed to documented upstream excitotoxic and stress cascades associated with TBI. A novel HDAC inhibitor, 4-dimethylamino-_N_-[5-(2-mercaptoacetylamino)pentyl]benzamide (DMA-PB), belonging to the family of mercaptoacetamides was recently developed which exhibits some degree of HDAC6 selectivity (Kozikowski et al., 2007). In vitro studies demonstrated that DMA-PB exhibited robust neuroprotective effects when tested in cortical neurons using the homocysteic acid model of oxidative stress with little toxicity compared to the hydroxamate-based HDAC inhibitors such as suberoyanilide hydroxamic acid (Kozikowski et al., 2007). The current investigation tested the efficacy of DMA-PB when applied acutely after TBI. Outcome was assessed using immunohistochemistry and histofluorescent techniques to examine histone H3 acetylation, inflammation, and neurodegeneration in the brains of adult rats following experimental TBI.
2. Results
There were no significant differences between groups in mean injury magnitude, righting time, rectal temperature, or temporalis muscle temperature values (Table 1).
Table 1.
Fluid percussion injury magnitude, righting times, and temperatures (mean ± SEM)
| Treatment group | ATM | Rightingtime (min) | Temperature (°C) | ||
|---|---|---|---|---|---|
| Rectal(during surgery) | Rectal(post TBI) | Temporalis muscle(post TBI) | |||
| Vehicle | 2.13 ± 0.01 | 14.1 ± 1.1 | 37.0 ± 0.07 | 36.9 ± 0.11 | 35.6 ± 0.04 |
| DMA-PB (0.25mg/kg) | 2.13 ± 0.01 | 13.2 ± 0.8 | 37.0 ± 0.08 | 37.0 ± 0.12 | 35.7 ± 0.11 |
| DMA-PB (2.5mg/kg) | 2.13 ± 0.01 | 13.6 ± 1.0 | 37.0 ± 0.04 | 37.0 ± 0.08 | 35.7 ± 0.09 |
| DMA-PB (25mg/kg) | 2.12 ± 0.01 | 13.6 ± 0.8 | 37.0 ± 0.08 | 37.0 ± 0.10 | 35.7 ± 0.06 |
2.1. Histone acetylation
Immunohistochemistry was performed to investigate the effect of the HDAC inhibitor on histone acetylation in the hippocampal CA2/3 region. Immunolabeled products were visible as blue/gray staining localized in the nucleus though staining intensity measured by optical density (OD) varied between cells (Fig. 2). The OD measurements included all cells (e.g., neurons, microglia and astrocytes) in the region of interest. However, the OD measurements most likely reflect changes in neuronal nuclei since the stratum pyramidale is predominantly populated with neurons. In general, lower OD was observed in the hemisphere ipsilateral to fluid percussion injury (Fig. 2). In order to control for differences in staining intensity between subjects, relative OD was calculated by dividing ipsilateral OD by contralateral OD. Quantitative analysis of relative OD (average of two sections) revealed significantly differences between groups (F (4, 35) = 2.894, p=0.036). Dunnett’s post hoc comparisons revealed the vehicle group had a significant reduction (p=0.015) in relative OD compared to the sham group (Fig. 3). The relative OD among DMA-PB dosage treatment groups was not significantly different from the sham group. No immunostaining was detected in the minus primary antibody control condition.
Figure 2. Acetyl-Histone H3 (Lys9) immunoreactivity in CA2/3 region.
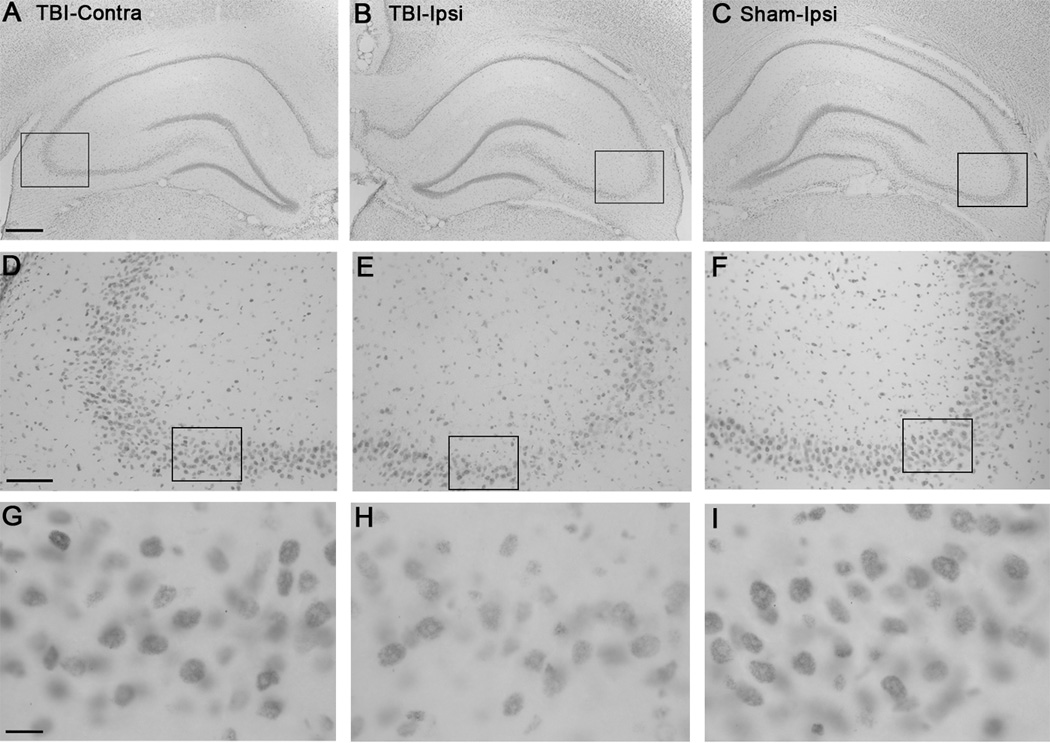
A) Contralateral hippocampus at 24 h post-TBI. Note the OD of immunostaining in CA2/3 region compared to B. B) Ipsilateral hippocampus at 24 h post TBI with lower OD in CA2/3 region. C) Ipsilateral hippocampus 24 h post sham TBI. Note that the OD is similar to A. D, E, F) Higher-magnification of the boxed highlighted regions of A, B, and C. Note the weaker immunostaining in the ipsilateral TBI section compared to the contralateral TBI section or the sham ipsilateral section. G, H, I) Higher-magnification of the boxed highlighted regions of D, E, and F. Note the lower OD of the neuronal nuclei of pyramidal neurons in H (ipsilateral TBI) compared to G (contralateral TBI) or I (sham). Calibration bar for A, B, and C, 400 µm; for D, E, and F, 100 µm; for G, H, and I, 20 µm.
Figure 3. Quantification of histone H3 acetylation immunoreactivity.
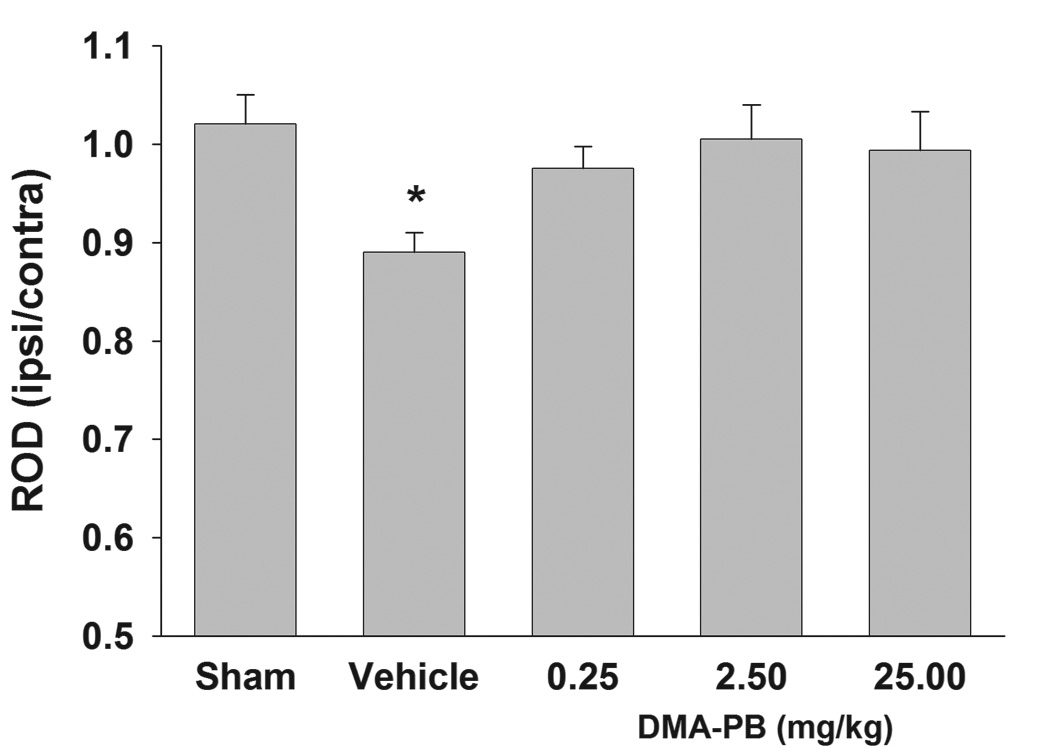
Vehicle-treated TBI rats exhibited a significant decrease in relative OD of histone H3 acetylation immunoreactivity in the ipsilateral CA2/3 at 24 h after injury. Treatment with all doses of DMA-PB (0.25, 2.5, 25 mg/kg) increased the immunoreactivity which was not significantly different from the sham group. The data are group means + SEM. * p<0.05 compared to the sham group.
In order to determine if the reduction in acetylated histone immunoreactivity was due merely to neuronal cell loss, OD measurements of a sample of individual surviving neurons were made in representative animals using 100X oil objective (Figure 2G–I). The mean OD value (0.0268±0.0048 SD) of neuronal nuclei of surviving neurons in the CA3 stratum pyramidale from the ipsilateral hemisphere of a vehicle-treated TBI rat was reduced by ~30 percent compared to the corresponding region of contralateral hemisphere (0.0385 ± 0.0055 SD) or to that of a sham TBI rat (0.0395 ± 0.0038 SD). These data indicate that the changes in relative OD quantification in Figure 3 are not likely due to neuronal cell loss.
2.2. Microglia
The effects of HDAC inhibition on inflammatory processes after TBI were assessed by quantification of the activation of resident microglia and/or recruitment of peripheral macrophages in the CA2/3 region at 24 h after injury (Fig. 4 A–F). The immunolabeled products were visible as blue/gray stained cytoplasm under a light microscope. OX-42 immunoreactive cells exhibited four distinct morphologies: resting microglia with small cell body and multiple thin, long processes (Fig. 4G), hypertrophic microglia with thickened and shorter processes (Fig. 4H), activated microglia with enlarged amoeboid morphology and much fewer and shorter processes (Fig. 4I), and phagocytic microglia or macrophages with amoeboid morphology and no processes (Fig. 4J). Each of the four classifications of microglia was quantified in the CA2/3 region of the ipsilateral hippocampus and expressed as cell density (cells/mm2). The mean density of resting microglia was significantly different between groups (F (4, 42) =235.06, p<0.001). The sham group had a preponderance of resting microglia density (254±11.7) that was significantly greater (p<0.001) than any of the injury groups (range from 4.0±1.3 to 24±9.1 for vehicle and 0.25mg/kg DMA-PB, respectively) (Fig. 5A). The mean density of hypertrophic microglia was significantly different between groups (F (4, 42) =2.954, p=0.031). A Dunnett’s post-hoc analysis determined that the 0.25 and 2.5mg/kg DMA-PB groups had significantly higher densities of hypertrophic microglia (p=0.023 and p=0.043, respectively) compared to sham (Fig. 5B). No activated or phagocytic microglia were detected in the sham group. Thus, to provide a more conservative data interpretation, the analysis of variance (ANOVA) was limited to the four TBI groups. There were no significant differences in mean density of activated microglia between the injury groups (F (3, 36) = 0.421, p=0.739) (Fig. 5C). The density of phagocytic microglia was significantly different between injury groups (F (3, 36) =4.308, p=0.011) (Fig. 5D). The 0.25, 2.5, and 25 mg/kg DMA-PB treatments had significantly lower density of phagocytic microglia (p=0.015; 0.011; 0.023, respectively) compared to the vehicle-treated TBI group. The mean area (±SEM) for the hippocampal CA2/3 ranged from 0.427 (± 0.027) to 0.471 (± 0.028) mm2 and was not significantly different. No immunostaining was detected in the minus primary antibody control condition.
Figure 4. OX-42 immunoreactivity of microglia in CA2/3 region.
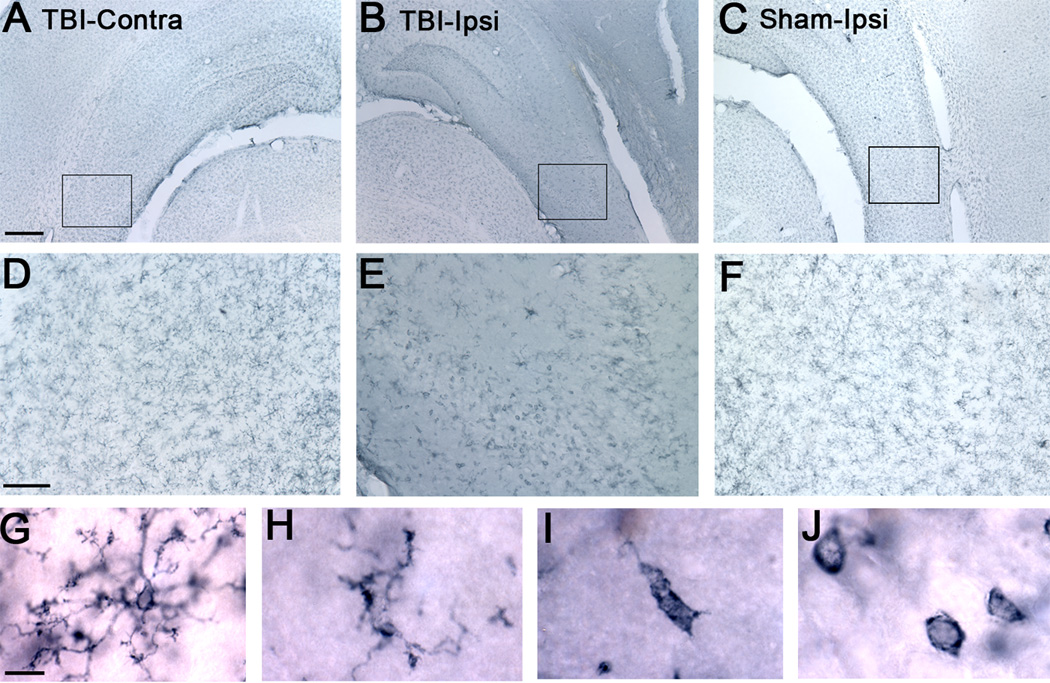
A) Contralateral hippocampus at 24 h post-TBI. Note the resting microglia in CA2/3 region compared to B. B) Ipsilateral TBI hippocampus with many activated and phagocytic microglia in CA2/3 region at 24 h post-TBI. C) Ipsilateral sham TBI with a predominance of resting microglia. D, E, F) Higher-magnification of the boxed highlighted regions of A, B, and C. Note the abundance of activated and phagocytic microglia in the ipsilateral TBI section compared to contralateral TBI and ipsilateral sham sections which are predominantly populated with resting microglia. G, H, I. J) Distinct morphologies of OX-42 immunoreactive microglia detected in CA2/3 region under high-magnification including resting microglia with small cell body and multiple thin, long processes (G), hypertrophic microglia with thickened and shorter processes (H), activated microglia with enlarged amoeboid morphology and much fewer, shorter processes (I), and phagocytic microglia or macrophage with amoeboid morphology and no processes (J). Calibration bar for A, B, and C, 400 µm; for D, E, and F, 100 µm; for G, H, I, and J, 10 µm.
Figure 5. Quantification of microglia types.
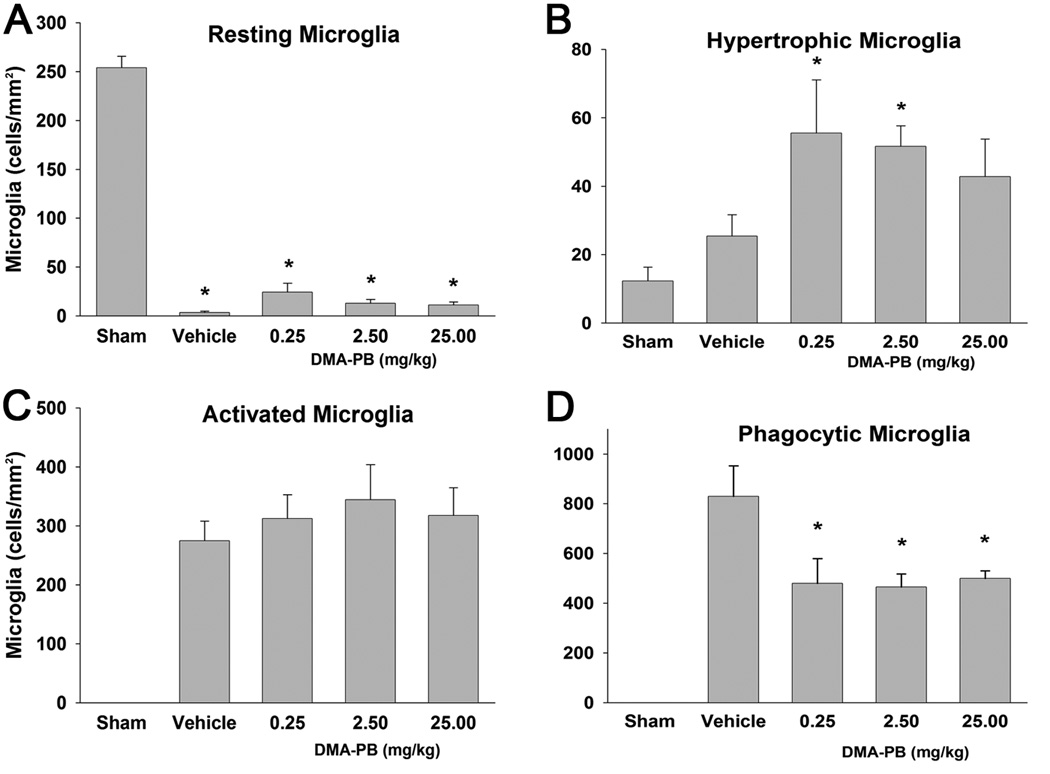
A) TBI resulted in a significant decrease in the density of resting microglia. B) The density of hypertrophic microglia were significantly elevated in the 0.25 and 2.5 mg/kg DMA-PB dose groups. C) There were no detectable activated microglia in the sham group. Injury produced a large increase in the density of activated microglia but there were no differences between any of the TBI groups. D) There were no detectable phagocytic microglia in the sham group. Injury produced a larger increase in density of phagocytic microglia that was attenuated by each of the DMA-PB treatment groups. The data are group means ± SEM of cell density. * p<0.05 compared to the sham group in A and B; * p<0.05 compared to the Vehicle group in D.
2.3. Neuronal degeneration
Neuronal degeneration was quantified in the stratum pyramidal of the ipsilateral CA2/3 region with Fluoro-Jade B at 24 h after TBI. Positive pyramidal cells fluoresced bright and somas appeared large with extensive dendritic arborization into the stratum radiatum (Fig. 6A,B). There was a trend for the DMA-PB treatment to reduce the number of ipsilateral degeneration neurons compared to vehicle (F(3,36)=2.491, p = 0.076) (Fig. 6C). Visual inspection at 200× failed to detect any Fluoro-Jade B positive neurons in the hippocampus contralateral to the fluid percussion injury in any groups examined.
Figure 6. Fluoro-Jade histofluorescence of degenerating neurons.


A) Degenerating neurons are detected by brightly fluorescing cell bodies in the stratum pyramidale of the CA2/3 and dendritic processes in the stratum radiatum. B) higher magnification of Fluoro-Jade positive cells in the stratum pyramidale. C) Quantification of Fluoro-Jade positive cells in the hippocampus CA2/3 at 24 h after injury. There was a trend (ANOVA: p = 0.076) for DMA-PB treatment to reduce the number of degenerating neurons. The data are group means ± SEM of total cell counts from two sections per animal. Calibration bar for A, 300 µm; for B, 100 µm. SO stratum oriens; SP stratum pyramidale; SR stratum radiatum.
3. Discussion
In the present study, DMA-PB, a new HDAC inhibitor, was used to test the hypothesis that HDAC inhibition represents a novel strategy for reducing inflammation following TBI. The results demonstrate that DMA-PB treatment, at all doses examined, significantly inhibited microglia transformation to phagocytes in CA2/3 at 24 h after lateral fluid percussion TBI in adult rats. These anti-inflammatory effects coincided in time (24 h after TBI) and region (dorsal hippocampus CA2/3) with DMA-PB effects on acetylation of H3 histone proteins. Administration of DMA-PB also resulted in a trend for reduced neuronal degeneration in the CA2/3 at 24 h after TBI. Thus, HDAC inhibitors may represent a new therapeutic approach to secondary brain pathology after TBI.
The observed decrease in acetylated histone levels in the ipsilateral CA2/3 region of the vehicle-treated TBI rats was similar to that reported in a study performed in immature rats following controlled cortical impact (Gao et al., 2006). In contrast to the present study, Gao and colleagues did not observe CA2/3 neuronal cell death in immature rats following TBI. In order to deal with the possibility that the reduction in acetylated histone immunoreactivity in the present study was due merely to neuronal cell loss, OD measurements were performed on nuclei of surviving neurons in samples from representative animals using much higher magnification. Since the mean OD of nuclei of individual surviving neurons in the ipsilateral CA3 of TBI rats was reduced compared to neurons in the contralateral TBI or sham TBI rats, the changes in relative OD reported in Figure 3 are most likely unrelated to changes in neuronal cell loss. DMA-PB, in comparison with the commercially available hydroxamate-based HDAC inhibitors, is a mercaptoacetamide-based HDAC inhibitor that owes its action to the ability of the thiol and carbonyl groups to interact with the zinc atom present in the catalytic gorge of the HDACs (Chen et al., 2005). Previous studies with DMA-PB demonstrated an ability to protect cortical neurons in culture from oxidative stress-induced death greater than hydroxamate-based inhibitors and with no detectable cytotoxicity (Kozikowski et al., 2007). In the present study, treatment with DMA-PB increased the expression of histone H3 acetylation, an effect not previously reported. Kozikowski and colleagues reported that DMA-PB was predominantly HDAC6-selective and did not modulate histone acetylation levels in cultured neurons (Kozikowski et al., 2007). Thus, the present data showing increased histone acetylation in the CA2/3 with DMA-PB treatment after TBI may represent an indirect action related to the drug’s effects on reducing microglia activation and the trend for reduced neuronal degeneration. There is an increasing focus on HDAC inhibitor-induced protection in a variety of inflammatory models. A series of HDAC inhibitors have shown anti-inflammatory effects in peripheral inflammatory diseases (Chung et al., 2003; Glauben et al., 2006; Lin et al., 2007; Nishida et al., 2004) and central nervous system (CNS) injuries (Faraco et al., 2006; Kim et al., 2007; Sinn et al., 2007). The results from the present study further characterize HDAC inhibitors as anti-inflammatory agents.
Neuroinflammatory responses which are mediated in part by activated microglia are considered to have important implications for neuronal survival and brain function. Microglia are the resident immune cell population of the CNS (Kreutzberg, 1996). Under normal conditions microglia are in a resting state, characterized by a small cell body with fine, ramified processes and low expression of surface antigens. CNS injury triggers rapid changes in the morphology and function of microglia. Hypertrophic microglia with thickened and slightly shorter processes are in a reactive state which suggests a largely passive response to injury. In contrast, activated microglia and phagocytic microglia (or macrophages) have a more aggressive role which, depending upon the circumstances, can produce deleterious effects to the CNS through secretion of various inflammatory molecules (Garden and Moller, 2006; Streit, 2002). Activated and phagocytic microglia release pro-inflammatory cytokines, cytotoxic proteases and reactive oxygen species, which in turn can exacerbate the degree of blood brain barrier disruption, cerebral edema, excitotoxicity, and neuronal dysfunction (Morganti-Kossmann et al., 2007). Pathological activation of phagocytic microglia (or macrophages) is especially involved in mediating tissue damage both by release of harmful mediators and phagocytosis (Gehrmann and Kreutzberg, 1995). Pharmacological therapy that can modulate pro-inflammatory cytokines (Atkins et al., 2007) or microglia inflammatory responses could have potential efficacy in CNS injury and disease in which inflammation is thought to play a prominent role in neurotoxicity (Garden and Moller, 2006). Additional studies reported that treatment with drugs that interfere with macrophage reactions improved the neurological status in animals with spinal cord ischemia (Giulian and Robertson, 1990) and that depletion of macrophages prior to the onset of experimental autoimmune encephalomyelitis attenuated the disease (Huitinga et al., 1990). Popovich and colleagues reported that depletion of macrophages promoted partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury (Popovich et al., 1999). Thus, mounting evidence points toward modulation of inflammatory responses after CNS injury as a promising avenue for therapeutic intervention.
Residential microglia activation and infiltration of macrophages from the peripheral blood is well known to contribute to post-injury inflammation after TBI (Aihara et al., 1995; Kreutzberg, 1996). Such secondary injuries often occur following the initial TBI insult and contribute to further brain pathology and neurological impairment (McIntosh et al., 1998; Thompson et al., 2005). The present results quantifying hippocampal microglia at 24 h following TBI showed a notable increase in total microglia numbers and very different morphology (most of the microglia were macrophages or activated microglia) compared to the sham group which was populated predominately by resting microglia. DMA-PB treatment significantly reduced the number of phagocytic microglia or macrophages, indicating that DMA-PB had significant effects on microglia inflammatory responses after TBI. These promising results prompted us to do further studies to determine if DMA-PB could reduce acute neuronal degeneration following TBI.
Consistent with previous studies (Hallam et al., 2004; Lyeth et al., 2001; Zhong et al., 2005), TBI produced robust Fluoro-Jade positive staining of pyramidal neurons in the ipsilateral CA2/3 region at 24 h post-injury. Fluoro-Jade is a novel anionic fluorochrome with specific affinity for neurons undergoing degeneration (Schmued et al., 1997; Schmued and Hopkins, 2000). It stains those degenerating neurons in their entirety, including cell bodies, distal dendrites, axons, and terminals and is used as a marker for neuronal degeneration in experimental TBI (Hallam et al., 2004; Wang et al., 2007; Zhao et al., 2003; Zhong et al., 2005). There was a trend for DMA-PB treatment groups to reduce neuronal degeneration (p = 0.076) compared to vehicle-treated group, with the highest dose (25 mg/kg) producing the largest reduction in neurodegeneration. The less that optimal effects on neuronal degeneration may have been due to the fact that in addition to HDAC6 there are also high levels of HDAC5 and HDAC10 (de Ruijter et al., 2003) in the brain, which, may not be modulated by the HDAC6 inhibitor, DMA-PB, employed in this study. The trend for reduced neuronal degeneration coupled with the significant recovery of histone acetylation levels with DMA-PB treatment also suggests a potential link between these processes after TBI. However, the concomitant significant reduction in phagocytes with the DMA-PB treatment opens the distinct possibility of an indirect effect of DMA-PB on neuronal degeneration through reduction in the inflammatory response.
It is generally accepted that microglia play a predominant role in the initiation and progression of neurodegeneration in neurodegenerative disorders (Liu and Hong, 2003; McGeer and McGeer, 1998). Previous studies reported the immunomodulatory effects of HDAC inhibitors on microglia (Chen et al., 2007; Huuskonen et al., 2004) in addition to their protective effects in neurodegenerative diseases (Ferrante et al., 2003; Gardian et al., 2005; Petri et al., 2006;; Saha and Pahan, 2006) and cerebral ischemia (Ren et al., 2004). Although the temporal relationship between microglia activation and neurodegeneration following TBI has not been thoroughly investigated, it appears that neurodegeneration can induce microglia activation and microglia activation can aggravate neurodegeneration in various neuropathological conditions (Garden and Moller, 2006; Popovich et al., 2002; Zhou et al., 2005). This illustrates the complex relationship of microglia and the potential benefits and or consequences depending upon whether activation is physiological or pathological. Would further reduction in microglia activation spare more neurons or is neuronal death what is triggering activation of microglia? Future studies examining differing severities of injury and timing of drug administration may begin to parse out the relationship between activated microglia and cell death processes. However, it should be emphasized that in the present study by far the most robust (and significant) effects of DMA-PB were on microglia activation. This suggests that the primary effects of the HDAC inhibitor were on reducing the inflammatory response to injury.
A limited number of studies to date examining the function of HDAC6 have produced mixed results with some studies reporting that HDAC6 provides cellular protection qualities by recognizing, binding, and clearing the cell of cytotoxic misfolded protein aggregates (Kawaguchi et al., 2003; Pandey et al., 2007) while others report that inhibition of HDAC6 rescues intracellular transport in the pathological situation associated with Huntington’s disease (Dompierre et al., 2007). In the present study, DMA-PB reduced markers of inflammation and attenuated the TBI-induced reduction in histone H3 acetylation while producing a trend for reduced neuronal degeneration. Further studies are required to understand the relationships and interactions between histone acetylation, inflammation, and neuronal degeneration following TBI.
In conclusion, the present results provided further evidence for the deacetylation of histone H3 by TBI and demonstrated that the novel HDAC6 inhibitor, DMA-PB, reversed the deacetylation measured in the hippocampus at 24 h after injury likely through an indirect action. The HDAC inhibitor also robustly attenuated the inflammatory microglia response and produced a trend toward reduced neuronal degeneration in the hippocampus. Further studies of HDAC inhibitors appear warranted for clarification of the underlying mechanisms of action and evaluation of chronic endpoints and delayed administration for potential development of therapeutics for patients suffering from TBI.
4. Experimental Procedure
4.1. Animals
Forty eight male Sprague Dawley rats (Harlan, Indianapolis, IN,USA) weighing 300–325g were used for this study. The animals were housed in individual cages in a temperature (22°C) and humidity-controlled (50% relative) animal facility with a 12-hour light/dark cycle. Water was continually available. The Institutional Animal Care and Use Committee at the University of California at Davis approved all animal procedures in these experiments.
4.2. Drug preparation and administration
DMA-PB (Fig.1) is an HDAC6-selective inhibitor (IC50 = 114 nM) which was first designed and synthesized in the laboratories of Dr. Kozikowski and details on the synthesis and selectivity of this compound have been reported (Kozikowski et al., 2007). DMA-PB was dissolved in DMSO and 3 doses (0.25, 2.5, or 25 mg/kg) or an equal volume of DMSO vehicle were administered at a volume of 1ml/kg intraperitoneal (i.p.)(n=10/TBI group; n=8 sham TBI) by injection just after moderate lateral fluid percussion TBI in the rats.
Figure 1.
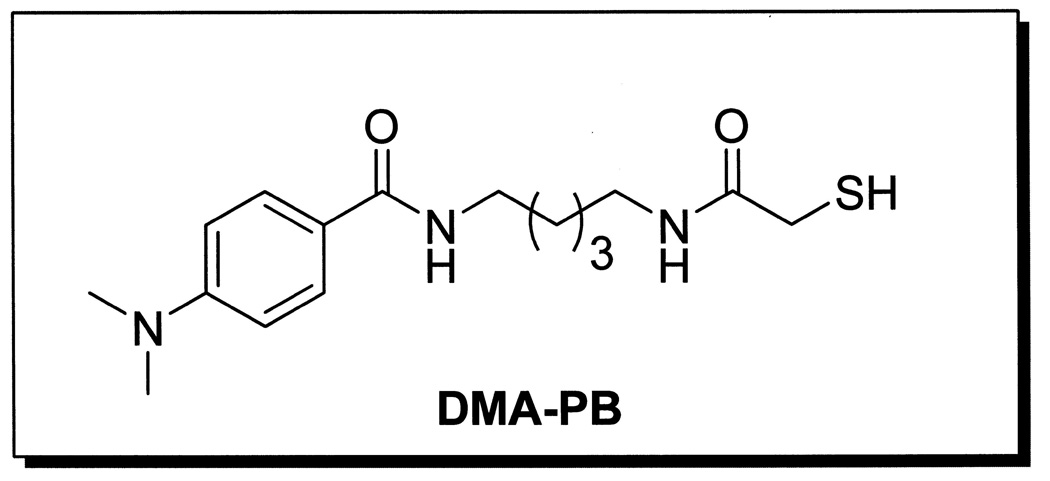
Structure of the HDAC inhibitor, 4-Dimethylamino-N-[5-(2- mercaptoacetylamino)pentyl]benzamide, (DMA-PB).
4.3. Surgery
Rats were anesthetized with 4% isoflurane in a 2:1 nitrous oxide/oxygen mixture, intubated, and mechanically normoventilated with a rodent volume ventilator (Harvard Apparatus model 683, Holliston, MA, USA) with isoflurane reduced to 2%. Sterile techniques were used during surgery. Rats were mounted in a stereotaxic frame, a scalp incision made along the midline, and a 4.8 mm diameter craniectomy was performed on the right parietal bone (centered at 4.5 mm Bregma and lambda, 3.0 mm right lateral). A rigid plastic injury tube (modified Luer-loc needle hub, 2.6 mm inside diameter) was secured over the exposed, intact dura with cyanoacrylate adhesive. Two skull screws (2.1 mm diameter, 6.0 mm length) were placed into burr holes, 1 mm rostral to Bregma and 1 mm caudal to lambda. The assembly was secured to the skull with cranioplastic cement (Plastics One, Roanoke, VA, USA). Rectal temperature was continuously monitored and maintained within normal ranges during surgical preparation by a feedback temperature controller pad (CWE model TC-1000, Ardmore, PA, USA). Temporalis muscle temperature was measured (Physitemp model TH-5, Clifton, NJ) by insertion of a needle temperature probe (Physitemp model MT-29/2, Clifton, NJ) between the skull and temporalis muscle (Jiang et al., 1991) and recorded (Table 1) just after injury.
4.4. Animal model
Experimental TBI was produced by a fluid percussion device (VCU Biomedical Engineering, Richmond, VA, USA) (Dixon et al., 1987) using the lateral orientation (Mcintosh et al., 1989). The device consists of a Plexiglas cylindrical reservoir filled with isotonic saline. One end of the reservoir has a Plexiglas piston mounted on O-rings and the opposite end has a transducer housing with a 2.6 mm inside diameter male Luer-loc opening. Injury was induced by the descent of a pendulum striking the piston, which injects a small volume of saline epidurally into the closed cranial cavity, producing a brief displacement and deformation of neural tissue. The resulting pressure pulse was measured in atmospheres (ATM) by an extracranial transducer (model EPN-0300 A*-100 A; Entran Devices Inc., Fairfield, NJ, USA) and recorded on a digital storage oscilloscope (model TDS 1002; Tektronix Inc., Beaverton, OR, USA).
The rat was disconnected from the ventilator, the injury tube connected to the fluid percussion cylinder, and a moderate fluid percussion pulse (range 2.12 – 2.13 ATM) was delivered within 10 seconds. Immediately after TBI the rat was again ventilated with a 2:1 nitrous oxide/oxygen mixture, followed by immediate injection of DMA-PB or vehicle. The plastic injury tube and skull screws were removed and the scalp incision was closed with 4.0 braided silk sutures. As soon as spontaneous breathing was observed the animal was disconnected from the ventilator. Assessment of the righting reflex was begun by placing the rat in a supine position at regular intervals (~30 sec) to test the rat’s ability to spontaneously recover to a prone position. The duration of suppression of the righting reflex was used as an indicator of traumatic unconsciousness and injury severity.
4.5. Tissue collection
Rats were euthanized at 24 h after TBI by deep sodium pentobarbital anesthesia (100 mg/kg intraperitoneal) followed by transcardially perfusion with 300 mL of 0.1 M sodium phosphate buffer (PBS, pH 7.4) followed by 300 mL of 4% paraformaldehyde (pH 7.4). Brains were removed and stored in 4% paraformaldehyde overnight at 4°C. Brains were cryoprotected in a 30% sucrose solution, frozen and coronal sections were cut at 45µm on a sliding microtome (American Optical, Model 860).
4.6. Acetyl-Histone H3 Immunohistochemistry
Acetyl-Histone H3 (Lys9) immunolocalization was carried out by using immunohistochemistry on 2 sections (−3.80 mm Bregma and −4.30 mm Bregma) from each animal. Free-floating tissue sections were washed two times (10 min each) in 0.1 M PBS with agitation followed by endogenous peroxidase blocking treatment. After three rinses (10 min each) in 0.1 M PBS, sections were incubated in 3% goat serum containing 0.3% Triton X-100, 3% BSA, and 8% Vector avidin D in 0.05M PBS for 40 mins at 37°C followed by 20 mins at room temperature. The sections were then transferred after a brief rinse in 0.1 M PBS and incubated with anti-acetyl-histone H3 antibody (polyclonal, rabbit, 1:500, #9671, Cell Signalling Technology, Inc., Danvers, MA, USA) in 0.1 M PBS which contained 8% Vector biotin for 48 hours at 4°C on a shaker. After six washes in 0.1 M PBS, the sections were then incubated in secondary antibody (goat anti-rabbit IgG, 1:500, Vector Laboratories, Burlingame, CA) for 2 hours at room temperature on a shaker. Sections were then washed six times in 0.1 M PBS and incubated with avidin-biotin-peroxidase complex (Vectastain Elite ABC kit PK6102, Vector Laboratories, Burlingame, CA) for 1 hour at room temperature on a shaker. Following four washes in 0.1 M PBS, the sections were visualized using Vector SG substrate kit for peroxidase (SK-4700, Vector Laboratories, Burlingame, CA). The reaction was stopped by transferring the sections into 0.1 M PBS, following by two 10-min washes in 0.1M PB with agitation. Finally, the sections were mounted on 1% gelatinized slides and allowed to dry in air, and then dehydrated serially in alcohol and coverslipped with Permount mounting medium. A control immunostaining experiment included identical immunohistochemistry procedures, but without the primary antibody. Image J software (National Institutes of Health) was used for OD step wedge calibration and subsequent image analysis.
In order to determine if neuronal cell loss after TBI was confounding the relative OD measurements in the region of interest, an additional analysis was performed on a limited number of tissue sections. Two sections (Bregma −3.80mm) per animal from one vehicle-treated TBI rat and one sham TBI rat were examined at high magnification (100X oil objective). The OD was measured in the nuclei of 10 neurons each at two aspects of the CA3 stratum pyramidale (0.5 and 1.2 mm away from the tip of the ventral blade of the dentate gyrus).
4.7. OX-42 Immunohistochemistry
OX-42 immunolocalization was carried out by using immunohistochemistry on one section (~4.30 mm Bregma) from each animal. Free-floating tissue sections were washed two times (10 min each) in 0.1 M PBS with agitation followed by endogenous peroxidase blocking treatment. After three washes (10 min each) in 0.1 M PBS, sections were incubated in 10% horse serum containing 0.3% Triton X-100, 3% BSA, and 8% Vector avidin D in 0.05 M PBS for 100 mins at room temperature. The sections were then transferred after a brief rinse in 0.1 M PBS and incubated with OX-42 antibody (monoclonal, mouse, 1:1000, CBL1512, Chemicon International) in 0.1 M PBS which contained 8% Vector biotin for 48 hours at 4°C on a shaker. After five washes (10 min each) in 0.1 M PBS, the sections were then incubated in secondary antibody (Horse anti-mouse IgG, 1:500, Vector Laboratories, Burlingame, CA) for 2 hours at room temperature on a shaker. Sections were then washed five times (10 min each) in 0.1 M PBS and incubated with avidin-biotin-peroxidase complex (Vectastain Elite ABC kit PK6102, Vector Laboratories, Burlingame, CA) for 1 hour at room temperature on a shaker. Following three washes in 0.1 M PBS, the sections were visualized using Vector SG substrate kit (SK-4700, Vector Laboratories, Burlingame, CA) for peroxidase. The reaction was stopped by transferring the sections into 0.1M PBS, following by two 10-min washes in 0.1 M PB with agitation. Finally, the sections were mounted on 1% gelatinized slides and allowed to dry in air, and then dehydrated serially in alcohol and coverslipped with Permount mounting medium. Control immunostaining experiment included identical immunohistochemistry procedures, but without the primary antibody. StereologerTM software (Systems Planning & Analysis, Inc., Alexandria, VA) was used for cell counting.
4.8. Fluoro-Jade histofluorescence
Fluoro-Jade B is a polyanionic fluorescein derivative which sensitively and specifically binds to degenerating neurons (Schmued et al., 1997; Schmued and Hopkins, 2000). Fluoro-Jade B fluorescence staining was carried out on 2 sections (−3.80 mm Bregma and −4.30 mm Bregma) from each animal. Briefly, tissue sections were mounted on 1% gelatinized slides in 0.05 M PB and air-dried overnight. The slide-mounted tissue sections were immersed subsequently in 100% alcohol (5 mins), 75% alcohol (5 mins), 50% alcohol (5 mins), 25% alcohol (5 mins), dH2O (3 mins), and 0.06% potassium permanganate (15 mins). Sections were rinsed in dH2O (2 mins), incubated in 0.0006% Fluoro-Jade B (Histo-Chem Inc., Pine Bluff, AK) staining solution in 0.1% acetic acid for 30 mins, rinsed again in dH2O (3 mins), and air-dried. Finally, the sections were immersed in xylene and coverslipped with DPX mounting medium (Electron Microscopy Sciences, Fort Washington, PA). Sections stained with Fluoro-Jade B were examined under UV light with a FITC fluorescence filter cube (Nikon B-2A, Tokyo). With this method neurons that undergo degeneration brightly fluoresce in comparison to the background (Schmued et al., 1997; Schmued and Hopkins, 2000). Stereologer™ software (Systems Planning & Analysis, Inc., Alexandria, VA) was used for cell counting.
4.9. Anatomical region of interest
Measurement of Acetyl-Histone H3 OD, Fluoro-Jade cell counts, and OX-42 positive microglia counts were made in the region of interest encompassing the stratum pyramidale of the hippocampus CA2 and CA3 bounded on one end by its entry into the dentate gyrus at the lateral tips of the dorsal and ventral blades of the granule cells and at the other end by the narrowing of the stratum pyramidale at the boundary of the CA2 to CA1.
4.10. Statistical analysis
All data are expressed as mean±SEM. Data analysis was performed using SPSS software, which adheres to a general linear model. Alpha level for Type I error was set at 0.05 for rejecting the null hypotheses. Acetyl-Histone H3 relative OD, microglia cell counts and degenerating neuron counts were each analyzed with one-way analysis ANOVA followed by Dunnett’s post hoc analysis. Fluid percussion injury magnitudes, righting times and temperature measurements were analyzed using separate one-way ANOVA between treatment groups.
Acknowledgements
This research was supported by NIH NS29995, NS45136 to BGL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1
Abbreviations used: HDAC histone deacetylase; HAT histone acetyltransferase; DMA-PB 4-dimethylamino-N-[5-(2-mercaptoacetylamino)pentyl]benzamide
References
- Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150:829–831. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aihara N, Hall JJ, Pitts LH, Fukuda K, Noble LJ. Altered immunoexpression of microglia and macrophages after mild head injury. J Neurotrauma. 1995;12:53–63. doi: 10.1089/neu.1995.12.53. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA, Jr., Alonso OF, Pearse DD, Bramlett HM, Dietrich WD. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol. 2007;208:145–158. doi: 10.1016/j.expneurol.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Grady MS. Regional and temporal characterization of neuronal, glial, and axonal response after traumatic brain injury in the mouse. Acta Neuropathol (Berl) 1999;98:396–406. doi: 10.1007/s004010051100. [DOI] [PubMed] [Google Scholar]
- Chen B, Petukhov PA, Jung M, Velena A, Eliseeva E, Dritschilo A, Kozikowski AP. Chemistry and biology of mercaptoacetamides as novel histone deacetylase inhibitors. Bioorg Med Chem Lett. 2005;15:1389–1392. doi: 10.1016/j.bmcl.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Chen PS, Wang CC, Bortner CD, Peng GS, Wu X, Pang H, Lu RB, Gean PW, Chuang DM, Hong JS. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience. 2007;149:203–212. doi: 10.1016/j.neuroscience.2007.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YL, Lee MY, Wang AJ, Yao LF. A therapeutic strategy uses histone deacetylase inhibitors to modulate the expression of genes involved in the pathogenesis of rheumatoid arthritis. Mol Ther. 2003;8:707–717. doi: 10.1016/s1525-0016(03)00235-1. [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich WD, Chatzipanteli K, Vitarbo E, Wada K, Kinoshita K. The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir Suppl. 2004;89:69–74. doi: 10.1007/978-3-7091-0603-7_9. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WM, Chadha MS, Kline AE, Clark RS, Kochanek PM, Dixon CE, Jenkins LW. Immunohistochemical analysis of histone H3 acetylation and methylation-Evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res. 2006;1070:31–34. doi: 10.1016/j.brainres.2005.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Kreutzberg GW. Microglia in experimental neuropathology. In: Kettenmann H, Ransom B, editors. Neuroglia. vol. New York: Oxford University Press; 1995. pp. 883–904. [Google Scholar]
- Giulian D, Robertson C. Inhibition of mononuclear phagocytes reduces ischemic injury in the spinal cord. Ann Neurol. 1990;27:33–42. doi: 10.1002/ana.410270107. [DOI] [PubMed] [Google Scholar]
- Glauben R, Batra A, Fedke I, Zeitz M, Lehr HA, Leoni F, Mascagni P, Fantuzzi G, Dinarello CA, Siegmund B. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- Hallam TM, Floyd CL, Folkerts MM, Lee LL, Gong QZ, Lyeth BG, Muizelaar JP, Berman RF. Comparison of behavioral deficits and acute neuronal degeneration in rat lateral fluid percussion and weight-drop brain injury models. J Neurotrauma. 2004;21:521–539. doi: 10.1089/089771504774129865. [DOI] [PubMed] [Google Scholar]
- Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. 1990;172:1025–1033. doi: 10.1084/jem.172.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huuskonen J, Suuronen T, Nuutinen T, Kyrylenko S, Salminen A. Regulation of microglial inflammatory response by sodium butyrate and short-chain fatty acids. Br J Pharmacol. 2004;141:874–880. doi: 10.1038/sj.bjp.0705682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Paulus W, Wrocklage C, Litvan I. Effects of closed traumatic brain injury and genetic factors on the development of Alzheimer's disease. Eur J Neurol. 2001;8:707–710. doi: 10.1046/j.1468-1331.2001.00322.x. [DOI] [PubMed] [Google Scholar]
- Jiang JY, Lyeth BG, Clifton GL, Jenkins LW, Hamm RJ, Hayes RL. Relationship between body and brain temperature in traumatically brain-injured rodents. J Neurosurg. 1991;74:492–496. doi: 10.3171/jns.1991.74.3.0492. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, Suto CM, Langley BC. Functional Differences in Epigenetic Modulators-Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies. J Med Chem. 2007 doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Langlois JA, Rutland-Brown W, Thomas KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalization, and Deaths. vol. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2006. [Google Scholar]
- Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L, Bastianelli E, Baron R, Rawadi G, Clement-Lacroix P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br J Pharmacol. 2007;150:862–872. doi: 10.1038/sj.bjp.0707165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Lyeth BG, Gong QZ, Shields S, Muizelaar JP, Berman RF. Group I metabotropic glutamate antagonist reduces acute neuronal degeneration and behavioral deficits after traumatic brain injury in rats. Exp Neurol. 2001;169:191–199. doi: 10.1006/exnr.2001.7643. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Dis Assoc Disord. 1998;12 Suppl 2:S1–S6. [PubMed] [Google Scholar]
- Mcintosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AI. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Juhler M, Wieloch T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury. J Neurotrauma. 1998;15:731–769. doi: 10.1089/neu.1998.15.731. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. American Journal of Epidemiology. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya Y, Inoue H, Asahara H. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 2004;50:3365–3376. doi: 10.1002/art.20709. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–645. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, McGaughy V, Fisher L, Hickey WF, Basso DM. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang D-M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. Journal of Neurochemistry. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker WJ. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Research. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade: novel fluorochromes for detecting toxicantinduced neuronal degeneration. Toxicologic Pathology. 2000;28:91–99. doi: 10.1177/019262330002800111. [DOI] [PubMed] [Google Scholar]
- Sinn DI, Kim SJ, Chu K, Jung KH, Lee ST, Song EC, Kim JM, Park DK, Kun Lee S, Kim M, Roh JK. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–472. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Teasdale GM, Graham DI. Craniocerebral trauma: protection and retrieval of the neuronal population after injury. Neurosurgery. 1998;43:723–737. doi: 10.1097/00006123-199810000-00001. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer's disease: a review. Prog Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- Wang H, Lynch JR, Song P, Yang HJ, Yates RB, Mace B, Warner DS, Guyton JR, Laskowitz DT. Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol. 2007;206:59–69. doi: 10.1016/j.expneurol.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG. Early loss of astrocytes after experimental traumatic brain injury. Glia. 2003;44:140–152. doi: 10.1002/glia.10283. [DOI] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Sarva J, Kozikowski A, Neale JH, Lyeth BG. NAAG peptidase inhibitor reduces acute neuronal degeneration and astrocyte damage following lateral fluid percussion TBI in rats. J Neurotrauma. 2005;22:266–276. doi: 10.1089/neu.2005.22.266. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Kovacs M, Jin J, Zhang J. Microglial activation induced by neurodegeneration: a proteomic analysis. Mol Cell Proteomics. 2005;4:1471–1479. doi: 10.1074/mcp.M500114-MCP200. [DOI] [PubMed] [Google Scholar]