HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis (original) (raw)
Introduction
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) represent two enzyme classes that, respectively, catalyze forward and backward reaction kinetics of lysine residue acetylation in specific protein substrates. These substrates most importantly include nucleosomal histones and various transcription factors (TFs), which form part of the transcription initiation complex. Accordingly, HATs and HDACs are found embedded in large multiprotein complexes near euchromatic regions of the chromatin. HATs modify core histone tails by post-translational acetylation of specific lysine residues, thereby creating appropriate ‘histone code’ for chromatin modification and enhanced DNA accessibility of TFs. Moreover, TFs like RelA, E2F, p53 and GATA1 are also acetylated by HATs. In addition to enhancing protein stability, such acetylation, in most cases, enhance their transactivation potential by facilitating their interactions with DNA and other proteins of the transcription apparatus. Contrarily, HDACs attenuate transcription process at a particular site by deacetylating these targets. Taken together, the HAT–HDAC system, owing to their involvement in turnover of histone and transcription machinery regulation, is one of the ultimate regulatory switches of gene expression. In addition to transcriptional regulation, HAT–HDAC system is also postulated to modulate other chromatin-associated processes like replication, site-specific recombination and DNA repair, thereby playing a major role in modulating overall cellular fate.
The Acetylation Homeostasis
Identification of mammalian HDAC as a homolog of the yeast corepressor, reduced potassium deficiency3 (RPD3),1 gave rise to the hypothesis that regulated transcriptional events may involve alternative exchange of HDAC complexes with those containing acetylase activities. Kinetically, such exchange in steady-state cells suggests a stoichiometrical balance in HAT and HDAC enzymatic undertakings, which in turn confers stability to the cellular homeostasis by coordinating gene expression and repression on both temporal and spatial basis. We will refer to this balance in HAT and HDAC activity as ‘acetylation homeostasis’ to aver the importance of regulated acetylation in acceding cellular homeostasis. Acetylation homeostasis is indeed closely linked to cellular homeostasis. This concept is buttressed by studies in various cancer forms. Somatic mutations of cyclic adenosine monophosphate (cAMP)-response element binding (protein) (CREB) binding protein (CBP)/p300 result in either loss- or gain-of-function for these HATs leading to oncogenic orientations. Similarly, altered HDAC activity due to chromosomal translocation of HDAC-recruiting elements leads to various forms of hematological tumorigenesis. Thus, the well-tuned, sensitive balance between HATs and HDACs plays a major role in modulating overall cell fate, a fact further highlighted by their monoallelic compromised functionality in other noncancerous human disorders like Rubinstein–Tabi syndrome and fragile-X syndrome.2
In this review, we circumvent discussions on various HAT and HDAC families and mode of their function, which has been extensively covered elsewhere.3, 4 However, we will present a snap-shot of signaling pathways responsible for regulating HAT–HDAC activity prior to focusing extensively on the involvement of HAT–HDAC system in neurodegeneration, where we assemble emerging evidences to implicate the disproportionate presence of HATs and HDACs and loss of acetylation homeostasis as major reasons for neuronal dysfunction and toxicity. Although HATs like p300 and CBP are multifunctional proteins, currently we shall restrict our focus on their acetylation activity. Finally, we also attempt to justify the consideration of HAT–HDAC system as a potential epigenetic avenue to mitigate neurodegeneration.
Signals Regulating HAT and HDAC Activity
Consistent with their central importance in mediating overall cellular homeostasis, signal-induced activity of HATs and HDACs are regulated by various means. Despite dearth of in-depth knowledge about such signaling mechanisms, we now have few pieces of the puzzle to provide a general roadmap of signaling pathways regulating these enzymes.
Regulating HATs
In an interesting study with two different antibodies against CBP, it was found that one of the antibodies revealed a uniform distribution of the protein in nucleoplasm, while the other detected the protein in organized subnuclear compartments like promyelocytic leukemia (PML) bodies.5 This opposing set of observations, explainable by epitope masking, revealed that, depending on the transcriptional activity of the cell, only a subset of CBP is recruited to PML bodies where they are involved in a dynamic association with the basal transcription machinery and thereby remain in the mainframe of active transcription.
Is the above-mentioned subset of CBP specifically modified? It has been demonstrated at least once that the recruitment of CBP to transcriptional complex requires direct phosphorylation on its serine 436 residue.6 Subsequently, it is now becoming clear that, like most other signaling components of cellular response, CBP activity is also regulated by phosphorylation. CBP is modified by several kinases including, cyclin E–cyclin-dependent kinase (CDK)2, nonclassical protein kinase C, protein kinase A (PKA), calcium/calmodulin-dependent kinase (CaMK)-IV (CAMK-IV) and p42/p44 mitogen-activated protein kinase (MAPK). Similarly, phosphorylation-mediated regulation has also been reported for other HATs like p300, ATF-2 and Tip60. In addition to the recruitment of HATs to transcriptional complexes, phosphorylation in most cases enhances their acetyltransferase activity and facilitates transactivation of target promoters.
Neurons show no exception to phosphorylation-dependent activation of HATs like CBP. However, only few kinases are reported till date in neuronal HAT activation. As illustrated in Figure 1a, nerve growth factor (NGF) can activate CBP in PC12 neuronal cells via the p42/p44 MAPK.7 CBP directly associates with the p42/p44 MAPK enzymes both prior to and following their activation by NGF. However, this association bears a fruitful phosphorylation of CBP only after NGF treatment.8 Among four transactivation domains (TAD) of CBP, two of which are located at the N-terminus and remaining two in the C-terminus,9 this NGF response was found to be mediated by dual C-terminal TADs without involving any region from the N-terminus. NGF treatment has also been reported to stimulate the recruitment of S6 kinase pp90RSK to CBP and their complex formation.10 This interaction is dependent on the activation of Ras pathway (Figure 1a). Formation of this CBP–pp90RSK complex has been shown to induce Ras-responsive genes in PC-12 cells, while simultaneously inhibiting cAMP-response element (CRE)-mediated gene transcription.10 CRE-dependent responses involve CREB-dependent activation of numerous survival-associated genes.
Figure 1
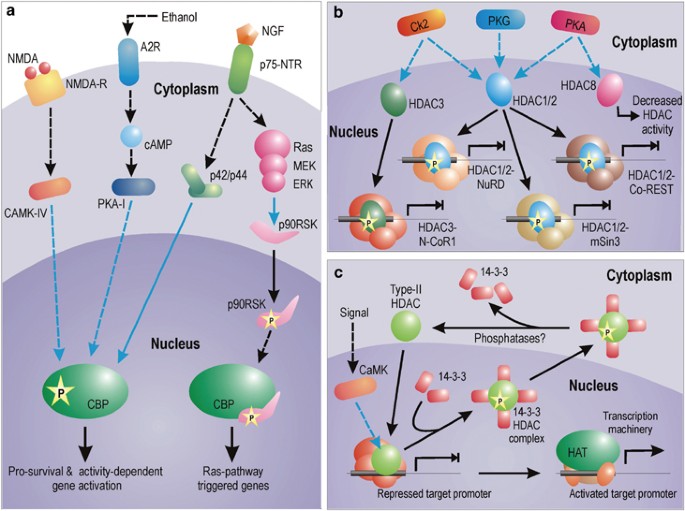
Signaling mechanisms for the activation of HAT and HDAC. (a) Pathways for the activation of CBP HAT in neurons. Activity-dependent activation of CBP depends on CAMK-IV-dependent phosphorylation on Ser 301. CBP is also activated by elevation in cAMP level. Ethanol, for example, phosphorylates adenosine A2 receptor by blocking adenosine uptake and its buildup. Subsequently, cAMP level is increased by adenylyl cyclase. cAMP-mediated activation of CBP is dependent on type-I PKA-mediated phosphorylation. Growth factors (like NGF) can trigger CBP phosphorylation via p42/p44 MAPK, which has been shown to interact directly with CBP. NGF can also trigger Ras pathway to mediate phosphorylation of 90 kDa ribosomal S6 kinase (RSK), which binds to CBP and canalizes expression of genes dependent on Ras pathway. However, this interaction inhibits expression of genes with CRE elements in PC12 cells. (b and c) Different modus operandi of class-I and class-II HDACs. HDAC1, HDAC2 and HDAC3 are phosphorylated by a distinct set of kinases. Once phosphorylated, they assemble into greater repressive complexes showing greater enzymatic activity and inhibit gene expression. In contrast, phosphorylation of HDAC8 decreases its enzymatic activity. Among all the represented HDAC complexes, the HDAC1/2-Co-REST complex is neuron specific. On the other hand, as shown in myoblasts, several type-II HDACs, once phosphorylated, are exported from the nucleus in complex with 14-3-3 proteins. The binding of HDACs with 14-3-3 proteins results in their release from the repression complex, allowing expression of the gene. Whether similar mechanisms are applicable to neurons is yet to be verified. Blue connectors represent kinase activity
Interestingly, the Ras-dependent formation of CBP–pp90RSK simultaneously inhibits the activity of yet another CBP activation pathway that is dependent on elevation of cAMP. This signal is transduced by a separate pathway involving cAMP-dependent PKA (type-I). In addition to responding to NGF, the C-terminus domain of CBP also responds to signaling via cAMP. Although phosphorylation of CBP is dependent on type-I PKA-I, however, owing to restricted nuclear entry of the kinase, the presence of an intermediate kinase(s) connecting PKA to CBP is strongly predicted. Interestingly, treatment of PC-12 cells with PKA increases the transcriptional activity of N-terminal CBP-TAD by 17-folds,9 despite its effect on CBP C-terminus. This observation was cell type specific and was not recorded in non-neuronal F9 or COS-7 cells.9 Probably cAMP-induced phosphorylation of CBP at its C-terminus via PKA increases the transcriptional ability of the N-terminal CBP-TAD domain by some molecular manipulation. It must also be asserted in this context that cytokines do not phosphorylate the C-terminus domain of CBP,7 suggesting that extracellular growth signals (like NGF) specifically utilize differential phosphorylation of CBP at C-terminus domains to transactivate specific gene subset.
During activity-dependent CBP engagement, the HAT is phosphorylated by yet another distinct pathway involving CAMK-IV. Impey et al.11 have shown that _N_-methyl-D-aspartate (NMDA) treatment induces phosphorylation of CBP serine 301 in a CAMK-IV-dependent manner. Mutation of this site attenuates NMDA-induced transcription. This finding is consistent with a previous study12 that had proposed the existence of a CAMK-IV-responsive element within the N-terminus of CBP in the range of 227–460 amino acids.
Activating HDACs
Regulation of HDACs may broadly be divided into two categories involving each of the two classes of mammalian HDACs. A general mode of regulation for HDAC class-I proteins (HDAC 1, 2, 3 and 8) involves association with proteins that modulate their deacetylase activity and recruitment to genomic arena (Figure 1b). However, class-II HDACs (HDAC 4, HDAC5, HDAC6, HDAC7 and HDAC9) are regulated by subcellular compartmentalization, where active nucleocytoplasmic trafficking delimits the availability of these enzymes for epigenetic utilization (Figure 1c). Both these processes are dependent on signal-induced phosphorylation.
HDAC1 and HDAC2 are best characterized among all HDACs. They are often found associated with three large multiunit protein complexes, called Sin3, Mi2/NuRD, and CoREST. Similarly, HDAC3 forms the N-CoR1 repressor complex. These large complexes demonstrate better deacetylase activity than HDAC components alone. Although extensive phosphorylation due to inhibition of phosphatase leads to disruption of the complex formation, yet specific phosphorylation of HDAC1 and HDAC2 is desired for complex stabilization and stimulation of deacetylase activity. Both HDAC1 and HDAC2 are primarily phosphorylated in this regard by casein kinase 2 (CK2). CK2 also phosphorylates HDAC3 but not HDAC8. In addition to CK2, cAMP-dependent PKA and cGMP-dependent protein kinase G also phosphorylates HDAC1. Furthermore, PKA also phosphorylates HDAC8 with an opposite outcome. In contrast with phosphorylation-mediated gain of function for all HDACs addressed so far, phosphorylation of HDAC8 by PKA leads to the inhibition of its deacetylation activity.13
Class-II mammalian HDACs are regulated differently by phosphorylation than class-I HDACs. Phosphorylated class-II HDACs bind 14-3-3 proteins leading to their nuclear expulsion. This kind of regulation plays a key role during myogenesis. In myoblasts, CAMK-I/IV phosphorylates two conserved serine residues on HDAC4, HDAC5 and HDAC7. Phosphorylation of these serines generate a docking site for the intracellular chaperone protein 14-3-3 and activation of a cryptic nuclear export signal in the HDAC leading to cytoplasmic translocation of the HDAC : 14-3-3 protein complex (Figure 1c). Recently, similar nucleocytoplasmic shuttling has also been reported for HDAC4 and HDAC5 in neurons in response to spontaneous electrical activity and calcium flux through NMDA receptors.14 However, as of now, we do not have any report on HDAC class-1 regulation in neurons.
Coupled signaling in HAT and HDAC regulation?
A closer look at regulatory modes of HATs and HDACs reveals few signaling events that apparently couple HAT activation to HDAC deactivation. While signal-triggered CAMK phosphorylates CBP and enhances its activity, nuclear expulsion of class-II HDACs are now known to be similarly dependent on the same kinase. Furthermore, while CBP acetyltransferase activity is enhanced by cAMP-dependent PKA, the same kinase is known to attenuate HDAC8 functioning. Are these events mutually exclusive or should we expect any contingency and corelation among them? Although we do not have any experimental data to support our speculation currently, yet logically, the coupling of HAT activation to HDAC inhibition by the same signal, at least in few instances, appears highly probable in light of cellular parsimony in organizing signal transduction pathways.
HAT–HDAC in Neurons: Acetylation Homeostasis and its Impairment during Neurodegeneration
Balanced acetylation status in normal neurons
During normal conditions, protein concentration (availability) and enzymatic activity of HATs (like CBP and p300) and HDACs remain in a highly harmonized state of balance where adequate active molecules from either group are present to effectively regulate chromatin and TF acetylation in a controlled manner (Figure 2a). Such equilibrium manifests neuronal homeostasis and is responsible for regulated gene expression leading to normal neurophysiological outputs like long-term potentiation, learning and memory. This equilibrium is maintained very stringently and any amount of tinkering with the HAT or HDAC dose and/or activity is not appreciated by cellular homeostasis machinery. Treatment of neurons with HDAC inhibitors like trichostatin A (TSA) in normal conditions induces neuronal apoptosis.15 Similarly, overexpression of CBP in resting CGC neurons under prosurvival conditions also leads to chromatin condensation and cell death.16 However, overexpression of an HAT domain-deleted CBP mutant did not generate similar results. No wonder, overexpression of CBP conveys the same fate in normal neurons as is scripted by blocking HDAC activity as both measures alter the precious HAT : HDAC balance towards enhanced histone acetylation, thereby opening chromatin gates for genes that were otherwise supposed to stay quiescent. These studies, therefore, strongly support the maintenance of precise balance between HATs and HDACs as a prerequisite of neuronal survival in normal conditions.
Figure 2
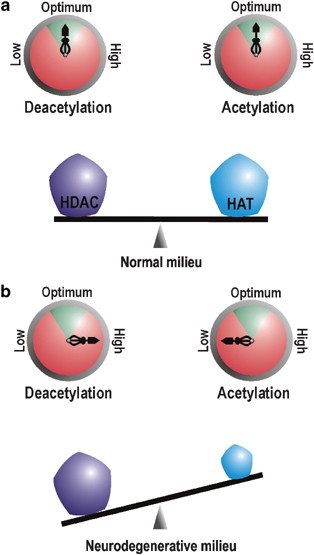
Neuronal acetylation homeostasis. Pentagons on the balance beam represent the protein level (dose) of HATs and HDACs, while compasses represent their activity. Enzymatic activity within the green arc of the compass is physiologically optimum. (a) In neurons under normal conditions, the dose and activity of HATs and HDACs are poised in a fine balance where they counteract each other to ensure physiological homeostasis. (b) During neurodegeneration, critical loss of HAT protein level ensue a rebated HAT dose and activity. This reclines the acetylation balance towards excessive deacetylation of target moieties
Loss of acetylation homeostasis during neurodegenerative maladies
Parameters of acetylation homeostasis alter significantly during neurodegenerative circumstances when histone acetylation level in neuron decreases globally,16 reflecting a malfunctioning acetylation apparatus. Such malfunction, as suggested by several observations, is manifested by comprehensive loss of HATs like CBP and p300 during various neurodegenerative challenges.16, 17 As reflected by antecedent deacetylation of histones in apoptotic conditions, this loss is an early event during apoptosis16 and is specific for affected neurons only. Immunocytological detection of CBP in a study with oxidative stress-challenged murine cortical neurons revealed conspicuous presence of CBP only in the nucleus of neurons surviving the hypoxic stress, whereas condensed or fragmented nuclei, representing neurons undergoing apoptosis, were not cofluorescent for the HAT.18 Furthermore, the critical nature of the loss of HAT is illuminated by several overexpression studies of CBP, all of which demonstrate enhanced neuronal viability in response to various challenges.16, 19, 20, 21 It may be mentioned here that, among all HATs, loss of CBP appears to be pivotal in facilitating neurodegenerative cascade of events. We will expand on this topic in the Therapeutics section.
Once the balance is disturbed by the loss of HAT dose, the HAT : HDAC ratio tilts in favor of HDAC in terms of availability and enzymatic functionality (Figure 2b), a fact highlighted by amelioration of several neurodegenerative conditions by various HDAC inhibitors. Oxidative stress, reported to be associated with several neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease, Huntington's disease (HD), stroke, multiple sclerosis and Friedreich's ataxia, fail to induce neuronal apoptosis when cells are treated with HDAC inhibitors.22 Similarly, HDAC inhibitors like TSA and suberoylanilide hydroxamic acid (SAHA) arrest polyglutamine toxicity in mouse motor neuron–neuroblastoma fusion cell line20and in Drosophila models of polyglutamine diseases.23 All these evidences strongly point towards a loss of neuronal acetylation homeostasis during neurodegeneration.
Mechanisms mediating loss of acetylation homeostasis during neurodegeneration
Neurodegeneration-coupled HAT loss is the main molecular event underpinning the forfeiture of neuronal acetylation homeostasis. Various mechanisms are now known to reduce CBP HAT availability in several models of neuronal insult. Nuclear translocation of expanded polyglutamine-containing neurotoxins (like mutated huntingtin protein), implicated in at least nine neurodegenerative diseases, selectively enhance ubiquitination and degradation of CBP by proteosomal pathway.17 Additionally, during AD progression, Presenilin-1-dependent epsilon-cleavage product N-Cad/CTF2 binds to CBP and facilitates its proteosomal degradation.24 Alternatively, CBP is redistributed from their normal nuclear location into Huntington aggregates, which compromises their availability for normal functions.19 Furthermore, caspase-6-dependent CBP proteolysis has been reported in low K+ shock model of neurodegeneration.16 Active caspase-6 is also reported in neuropil threads, neuritic plaques and neurofibrillary tracts of Alzheimer brain, suggesting that CBP may be lost in AD by caspase cleavage. Like CBP, p300 protein level also decreases during neurodegenerative conditions.16 However, we do not have adequate observations to explain the mechanism behind such loss.
The neurodegeneration-associated tilt in HAT : HDAC balance does not include augmentation of HDAC protein level. For example, total protein amount and activity of various HDACs is not altered by mutant huntingtin protein expression in primary cortical neurons.25 Despite no such alteration, the overall HDAC activity increases in a dying neuron due to loss of the counter-balancing effect of basal HAT activity. Effectiveness of HDAC blockers in ameliorating neurodegenerative diseases most probably rely on this tilt in acetylation balance. Knowing that HDAC inhibitors singularly block enzymatic activity of these enzymes and do not interfere with their protein turnover rate, we now have a picture where at the cost of HATs’ loss-of-availability HDACs attain a facilitated gain-of-function, thereby unsettling the acetylation homeostasis.
Fazed Acetylation Homeostasis: Molecular Inroads to Neuronal Dysfunction and Degeneration
How can impaired acetylation homeostasis lead to neuronal loss of function? The answer to this question revolves around the theory of ‘transcriptional dysfunction’, which has been implicated as the prime cause in pathosis of several neurodegenerative disorders including, HD and AD.26, 27 This theory attributes degenerative fate of neurons to an altered transcription profile, which differs sharply from activity-dependent normal transcription pattern in accentuating more proapoptotic genes and less prosurvival genes. The reader may note that the situation presents a riddle here. While the expression of survival-associated genes is being attenuated by altered acetylation, the same situation stimulates expression of death-inducing genes. In the following lines, few facts and theories are presented to explain this contradictory situation under two separate sections.
Relinquishing survival molecules
Bolted histone gates
Discounted HAT availability along with preponderant HDAC activity in a degenerating atmosphere is expected to confer deacetylation of histones. This, in turn, can potentially collapse the chromatin structure, thereby hindering the expression of vital genes (Figure 3). Indeed, this general mechanism of alteration in transcriptional output is supported by observations of global deacetylation of histones H3 and H4 during the onset of neuronal apoptosis in low potassium-induced neurodegeneration.16 Similar global histone deacetylation is also reported during CBP sequestration by polyglutamine huntingtin21, 23 and may represent the principle mechanism of neurotrophic gene repression. However, it remains to be elucidated that how proapoptotic genes, upregulated during initial stages of neurodegeneration, may be transcribed under similar conditions of global chromatin deacetylation. Light may be shed on such ambiguity by quantifying histone acetylation level in promoter regions of survival and apoptotic genes exclusively and separately. However, such studies are yet to be performed.
Figure 3
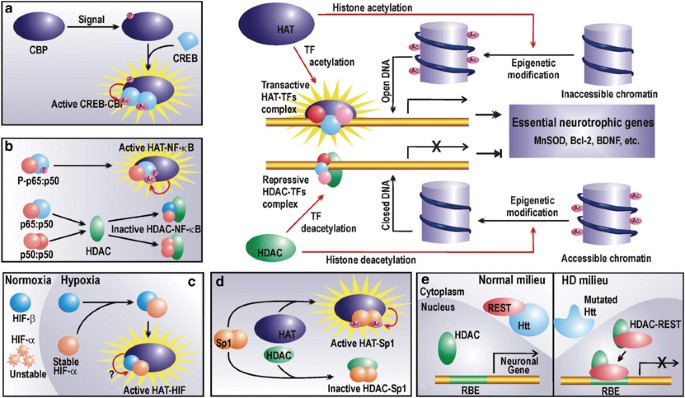
Regulation of chromatin and prosurvival TFs by HATs and HDACs. HATs acetylate lysine residues on nucleosomal histones to render the chromatin accessible to the transcription machinery, which then upregulates the expression of an array of neurotrophic factors. HDACs reverse the process by histone deacetylation. Furthermore, HATs and HDACs also regulate the activity of certain prosurvival TFs (a–d). (a) CREB activity depends on its acetylation status. Phosphorylated CBP can acetylate CREB, which then undergoes conformational modification and forms a transactive dimer in association with the HAT. (b) The time and strength of NF-_κ_B p65 : p50 is mediated by acetylation of p65 by HATs. However, if unphosphorylated, NF-_κ_B p65 : p50 can interact with HDACs to form nontransactive complexes. Furthermore, p50 : p50 can form repressive complex with HDACs. (c) Hypoxic responses of HIF require steric interaction of HATs with HIF heterodimers. However, the probable enzymatic HAT activity is not yet reported. (d) Sp1 may conditionally interact with HATs or HDACs. Transactivation by Sp1 is accrued by its interaction with HATs and subsequent acetylation. (e) Nonepigenetic repression by HDAC. During HD, HDAC represses the expression of vital neuronal genes by forming a repressor complex with REST, which otherwise in normal conditions is arrested in the cytoplasm by wild huntingtin protein (Htt). Refer to the text for abbreviations
Incapacitation of prosurvival TFs
In addition to gene repression by deacetylation of histones, transcriptional dysfunction may also result from anomalistic deacetylation of certain TFs like CREB, RelA (nuclear factor kappaB (NF-_κ_B)), hypoxia-induced factor (HIF) and specificity protein 1 (Sp1), which have well-recognized roles in regulating the expression of prosurvival elements (Table 1). Manipulation of their acetylation status may therefore lead to inhibition of vital gene activation (Figure 3), subsequently negotiating the loss of cellular homeostasis. These possibilities are expanded in the following lines.
Table 1 Role of acetylation balance in augmenting neuronal survival
cAMP response element binding protein
Several studies have now revealed CREB as a bona fide regulator of neuronal homeostasis,27 where it mediates neurotrophic responses to neurotrophins like NGF and BDNF.28 Loss of CREB function contributes to neuronal dysfunction and death during oxidative stress. CREB-mediated gene activation is highly dependent on its association with CBP as is revealed in a study with ataxia-telangiectasia (A-T)-mutated (ATM) gene, where ATM phosphorylates CREB and subsequently contributes to A-T neurodegeneration by inhibiting its interaction with CBP.29 Normally however, activated CBP binds to CREB and acetylates the latter at three putative acetylation sites within its activation domain. It has been proposed that during resting conditions, these nonacetylated lysine residues restrain CREB-mediated transactivation by maintaining the molecule in an inactive conformation. However, once acetylated by the bound HAT and consequential charge neutralization at the lysine residues, CREB attains the active state and dimerizes to bind CRE found in promoters of many survival genes in response to, most emphatically, cAMP-mediated signals (Figure 3, inset a). Table 1 summarizes a list of prosurvival molecules regulated by CREB. In few cases, as revealed by independent studies, CBP has also been implicated in the CREB-mediated upregulation of gene products like MnSOD and Bcl-2. In other instances represented in the table, involvement of CBP is still subject to experimental verification.
Furthermore, CBP–CREB association is required for acetylation and binding of histone binding protein RbAp48 to the CBP–CREB complex in order to allow the interaction of the complex with core histones and mononucleosomes.30 Thus, CREB-dependent gene expression is heavily contingent on the presence and enzymatic activity of CBP and may thus fall short of the threshold level during neurodegenerative conditions when the quantity and functionality of HATs are severely compromised. Indeed, the expression of BDNF, a well-known CREB target gene, is impaired during HD.31 Mutant huntingtin-mediated sequestration of CBP away from transcription pockets may be recalled in this respect.
Nuclear factor kappaB
NF-_κ_B is yet another TF accredited with well-defined roles in neuronal survival during neurodegenerative conditions. Survival promoting role of NF-_κ_B is manifested by augmented transactivation of several antiapoptotic genes like _bcl_-2 (Table 1). This dimeric TF is usually composed of two subunits, p65 and p50, both of which undergo signal-coupled acetylation in the nucleus by p300/CBP or p300/CBP-associated factor (P/CAF).32 Such post-translational acetylation is contingent on binding of the HAT with phosphorylated p6532 (Figure 3, inset b). Acetylation enhances DNA binding and transcriptional activity of p65 : p50 NF-_κ_B heterodimer. However, p65 acetylation is reversible and is attenuated by HDAC3. Deacetylation of lysine 221 of p65 is required for its nuclear export by interacting with its newly synthesized arrestor molecule, inhibitory kappaB (I_κ_B).33 This molecular mechanism regulates the strength and duration of NF-_κ_B activity in the nucleus. Thus, in the deacetylation-prone neurodegenerative atmosphere, both strength and duration of this antiapoptotic transactivator are expected to be retarded. Additionally, HDACs may interact with unphosphorylated p65 : p50 heterodimer or p50 homodimers (another NF-_κ_B dimer) to form a repressive complex32 that may inhibit expression of vital genes (Figure 3, inset b).
Hypoxia-induced factor
HIF is part of the cellular oxygen-sensing system that intercedes upregulation of genes encoding essential growth factors or enzymes during hypoxic conditions.34 HIF acts as a heterodimer consisting of constitutively present HIF-β and O2-sensing HIF-α, which has several isoforms. HIF-1_α_, the most characterized HIF-α, is degraded during normoxia via ubiquitin–proteasome pathway thereby keeping the HIF system in an inoperative state. Alternatively during hypoxia, HIF-1_α_ becomes stable and forms the active dimer capable of transactivating target genes. In neuronal cells, which are extremely anoxia-sensitive, the exact role of HIF is dependent on the degree and duration of hypoxic stress. Prolonged hypoxic shock induces coordinated activities of p53 and HIF-1_α_ in driving ischemia-induced delayed neuronal death. However, HIF triggered by transient or moderate ischemia elicits neuronal preconditioning by upregulation of genes like erythropoietin and vascular endothelial growth factor (VEGF) that play an important role in neuronal oxygen delivery. Moreover, these HIF-induced gene products also mediate survival of amyloid-_β_-resistant neurons in AD. These survivor neurons exhibited greater glucose influx, which presumably requires several HIF-1 target products involved in glucose uptake and metabolism35 (Table 1).
Hypoxia-responsive activity of HIF is dependent on its interaction with p300/CBP coactivators (Figure 3, inset c). Interaction of HIF-1_α_ with p300/CBP has been recorded using a region of p300 as probe that differs from its CREB binding domain.36 Subsequently, p300/CBP and HIF-1_α_ were colocalized in a hypoxia-induced DNA-bound complex that was responsible for transactivating several hypoxia-activated genes. Such interaction of HIF with its coactivators is hypoxic event specific and does not occur during normoxia. Whether this association employs acetyltransferase activity of CBP/p300 or is limited to physical protein–protein interaction in order to achieve facilitated DNA accessibility is still not elucidated. However, the interaction of HIF with HAT coactivators is nevertheless a prerequisite for HIF-mediated gene activation and may therefore be severely compromised during scarcity of the HAT molecules during neurodegeneration.
Sp1 and specificity protein 3 (Sp3)
Zinc-finger TF Sp1 and Sp3 are ubiquitously expressed members of SP family of TFs. In neurons, these TFs have been associated with neuronal vitality. Owing to its glutamine-rich domains, Sp1 is sequestered in protein aggregates of intranuclear inclusions that develop during expanded glutamine diseases. This in turn, as seen in the mutant huntingtin model, disrupts normal pattern of Sp1-dependent gene expression by prohibiting interaction of Sp1 with its coactivator TAFII130, thereby promoting HD progression. Overexpression of Sp1 reversed neuronal vulnerability to mutant huntingtin neurotoxicity, suggesting a prosurvival role for this transactivator. Indeed, subsequent studies have revealed Sp1 and Sp3 as oxidative stress-inducible neuroprotective TFs.37 Sp1 needs to be hyperacetylated to mediate oxidative stress adaptive responses and has been shown to be acetylated by p300.22 Appropriately acetylated Sp1 binds target DNA more avidly (Figure 3, inset d) and facilitates recruitment of coactivator TAFII130 to the transcription machinery in promoters of vital genes like superoxide dismutase (Table 1). During altered acetylation status of neurons during neurodegenerative stress, paucity of survival-enhancing molecules may result from lack of Sp1 acetylation. Indeed, HDAC inhibitors abrogate oxidative stress-induced neuronal death by augmenting Sp1 acetylation.22
Gene silencing with repressor element TF (REST)
The above-discussed shift in catalytic activity of HAT–HDAC system towards deacetylation of histones and several prosurvival TFs, however, is not the exclusive molecular mechanism to explain transcriptional dysregulation. Preponderant availability of HDAC may lead to its interaction with certain repressors during neurodegenerative conditions and physical repression of vital gene. To illustrate the point, we may consider interaction of HDAC with the REST/neuron restrictive silencer factor. The HDAC : REST complex mediates repression of neuron-specific genes in non-neuronal cells under normal conditions. These suppressed genes are, however, expressed in neurons where they encode a wide array of products involved in neuronal functioning and survival. Such cell-specific gene expression in neuron is in part facilitated by cytosolic association of REST with wild huntingtin protein, which prohibits formation of HDAC : REST in the nucleus, thereby permitting transcription of these genes in neurons (Figure 3, inset e). However, during neuropathogenesis of CAG triplet repeat disorders like HD, the mutated huntingtin fails to arrest REST in cytosol leading to the formation of HDAC : REST complex in the nucleus (Figure 3, inset e). This situation represents the scenario in a non-neuronal cell where the repressor complex dampens expression of vital genes like BDNF. Given that REST silencing activity occurs by HDAC-dependent chromatin reorganization,38 we now have yet another mechanism where HDAC predominance may corrupt prosurvival transcriptional profile required for neuronal homeostasis.
Derepression of lethal molecules
A cyclic way to die
In addition to repression of prosurvival molecules, altered transcriptional profile of a dying neuron involves derepression of several proapoptotic gene products. Neuronal death is distinguished from general apoptotic machinery of dividing cells by the expression and involvement of cell-cycle regulators. Indeed, in the past decade, much evidence has accrued to corelate expression of mammalian cell cycle molecules and neuronal death.39 Normally, differentiated adult neurons do not undergo cellular division and remain in a quiescent state. During neurodegeneration, uncoordinated ectopic re-entry of neurons into the cell cycle proves abortive and subsequently leads to apoptosis. In case of AD, it is speculated that cell cycle arrest prior to mitosis leads to the dysregulation of mitochondrial function and apoptosis is caused by oxidative stress generated subsequently.
Neuronal mitotic catastrophe resulting due to anomalous and deregulated re-entry into the cell cycle is mainly mediated by upregulation of CDK and their regulatory subunits, the cyclins. Cyclin D1, cyclin E, cyclin B, CDK4/6, CDK1 and CDK2 are aberrantly expressed in various diseases and models of neurodegeneration.39 Is this facilitated by altered acetylation status of dying neurons? At present, no experiments have been conducted in neurons to validate this corelation. However, we may pick cues from other cell types with customary caution of cell-specific eventualities. CBP and p300 antisense vector-mediated depletion in nontransformed breast epithelial cells initiates emergence of these cells from quiescence even in the absence of growth factors.40, 41 These p300/CBP-depleted cells, however, do not complete normal cell cycle and a significant proportion of these cells die upon serum restimulation. This observation is apparently similar with neuronal fate after their nonobligatory cell cycle entry. Furthermore, antisense-treated epithelial cells demonstrate a dramatic reduction in CDK inhibitor p21, a well-known negative regulator of cell cycle. A similar downregulation of another cell-cycle inhibitor, p27, is observed during cerebral ischemia and neuronal apoptosis due to reduced potassium.42 As a matter of fact, loss of cell-cycle inhibitors has been proposed as an early indicator of striatal neurodegeneration during cerebral ischemia.42 Is this downregulation a functional output of the loss of acetylation homeostasis? In a study with neuro2a cells, treatment with HDAC inhibitors blocked cell cycle progression that was associated with concomitant increase in p21 acetylation. Thus, there is hint that acetylation homeostasis plays a part in neuronal quiescence. Further support for this theory is obtainable from studies in normal (nontransformed) breast epithelial cells where HDAC inhibitors decreased expression of cyclin D1 and increased expression of p21.43 However, the effect of HDAC inhibitors on neuronal cell-cycle inhibitors during neurodegenerative stress still remains untested.
Leasing and unleashing proapoptotic TFs
Neuronal apoptosis requires de novo transcription whereby, in addition to cell cycle regulators, several apoptotic factors like APAF-1, SIVA and PUMA are expressed in response to neurotoxic insults. The expression of these molecules is dependent on the activation and functioning of a set of proapoptotic TFs including, p53, E2F, c-Jun and FOXO family members.39 Do these TFs oblige the death process independent of acetylation control or do they temporally precede the events of HAT loss and thus have a chance to recruit them prior to their deficit? As a matter of fact, expression of several above-mentioned apoptotic molecules is identified as early warnings of neuronal death. May be, transcription of these genes begin before neurodegenerative loss of CBP/p300. Thus, facilitation of lethal gene promoter activation by HATs prior to their loss cannot be ruled out.
An alternative hypothesis suggests that these TFs may be regulated by HAT coactivators other than the ones that are depleted during the onset of neurodegeneration. For example, p53 is known to promote transcription by interacting with individual HATs like CBP, P/CAF, p300 or multiprotein acetyltransferase complexes like transactivation/transformation-domain-associated protein (TRRAP) and Ada3, which potentially contain acetyltransferases like P/CAF, general control nonderepressible5 (GCN5) or Tip60. It is contextual at this point to recall observations of Rouaux et al.,16 where they have detected a decline of dose and acetylation activity of only p300 and CBP during apoptosis. In contrast, level of other HATs like TAF II p250 and P/CAF remained unaltered. Functional redundancy among available HATs may pave a way for p53-dependent transcription in neurons even in the absence of CBP/p300. In another instance, E2F1, a major proapoptotic TF related to neuronal cell-cycle re-entry,39 is stabilized in response to DNA damage by acetylation activity of P/CAF but not p300.44 Along same lines, E2F may remain active in apoptotic neurons even in the absence of CBP or p300.
On the flip side, we wonder, does acetylation homeostasis play a role in harnessing these proapoptotic TFs? Under peroxide stress, transcriptional activity of FOXO4 is repressed by CBP-mediated acetylation.45 Furthermore, activity of c-Jun, a monomer of proapoptotic TF AP-1, is also repressed under certain conditions by acetylation. As shown in the E1A model of c-Jun repression, acetylation of the c-Jun-Lys271 by p300 is required for suppression of c-Jun activity by E1A.46 Thus, a balanced acetylation status may maintain a lease on these TFs, which may be compromised during loss of CBP/p300. However, we need experimental ratification of this hypothesis before we attempt to extrapolate these findings to neurons. For now, we forward these theories as mere possibilities.
Reinstating Acetylation Homeostasis: Therapeutic Potentials
Despite differences in disease etiology and cellular properties of affected neurons in various forms of neurodegenerative disorders, loss of acetylation homeostasis appears to represent a critical and decisive mechanism commonly underlying catastrophic neuronal dysfunction and degeneration. General loss of acetylating agents along with parallel gain of HDAC activity represents the crux of the altered situation. Once this was realized, research in this field introduced itself to experimental therapeutics and the pipeline of drug development was established initially by HD researchers, who attempted to fine-tune the altered HAT–HDAC balance by using general HDAC inhibitors to re-establish the acetylation threshold in endangered neurons. Furthermore, proposals targeting HAT loss reversal have also been forwarded. In the following discussion, the efficacy of both modes of resetting acetylation homeostasis is evaluated.
Rescuing acetylation homeostasis by HDAC inhibitors
Taking a leaf out of cancer drug development schemes, several groups explored the possibility of addressing deranged acetylation in HD by blocking augmented HDAC activity with several well-known HDAC inhibitors. The logic behind using these inhibitors is based on the argument that transcriptional repression is the predominant reason of transcriptional dysregulation by global deacetylation. Thus, manipulative strategies aimed at re-establishing the acetylation homeostasis by rebating excess HDAC activity could remove repressive blocks from promoters of essential gene. Indeed, HDAC inhibitors like TSA, SAHA and sodium butyrate (NaBu) showed encouraging results in yeast,47 Drosophila,23 mammalian cells in culture20 and mouse model.48 Interestingly, the last study involving preclinical application of HDAC inhibitor, SAHA, showed encouraging results. SAHA increased acetylation globally in the brain and enhanced motor impairment significantly. Although SAHA does not appreciably penetrate brain barriers by itself, it does so when complexed with 2-hydroxypropyl-_β_-cyclodextrin in solution. Thus, a solution of this complex in drinking water can make SAHA a cost-effective pharmacological candidate that could be administered by unsophisticated means.
Among several tested HDAC inhibitors, butyrates are considered to be the most effective ones due to their ability to cross the blood–brain barrier. In a study with NaBu in R6/2 transgenic mouse model of HD, intraperitonial administration of the inhibitor enhanced acetylation status of histones and prosurvival TFs like Sp1.49 At 90 days of treatment, marked neuroprotection was observed in treated animals, which showed significant reduction in several neuropathological hallmarks like striatal neuronal atrophy. However, here the inhibitor was administered presymptomatically. In a different study, when administered postdisease onset, phenylbutyrate extended survival and showed neuroprotection in N171-82Q transgenic mouse model of HD.50 In addition to HD, NaBu has also been reported to be effective in treating spinal muscular atrophy (SMA),51 an autosomal recessive disease marked by the degeneration of anterior horn cells of spinal cord, and spinal and bulbar muscular atrophy,52 an inheritable motor neuron disease caused by an expanded polyQ tract within the androgen receptor. Finally, it has been shown very recently that NaBu treatment in transgenic amyotrophic lateral sclerosis mice resulted in significant enhancement of survival and improvement in clinical phenotypes of diseased mice treated with the inhibitor in comparison to untreated animals.53
Taken together, HDAC inhibitors are promising candidates for harnessing neuronal loss. Are these inhibitors the real wonder drug? Selection of a molecule for future drug development is based on detailed assessment of its biological potency, safety and pharmacotoxicity. It must be warned that mode of action for HDAC inhibitors is highly nonspecific and nontargeted. On long-term usage, this may reverse deacetylation-mediated blockage of undesired non-specific promoters leading to cytotoxicity. Indeed, TSA displays prominent toxic effects in normal neuronal cells by upregulating the E2F TF.15 Thus, in vivo, general administration of HDAC inhibitors may enhance survival potency of previously challenged neurons at the very high cost of endangering other normal brain cells in vicinity. Probably, this criticism will invite objection from studies demonstrating amelioration of detrimental symptoms in HD models. However, it must be taken into account that HDAC inhibitors affect expression of only 2% genes in mammals. This is a very small fraction to highlight any nonspecific nocuous outcome during short-term laboratory experimentation schedule, which may otherwise arise from long-term clinical usage of these inhibitors. Therefore, despite being able to ameliorate neuronal loss of function and homeostasis in mouse models of several diseases, HDAC inhibitors may not be the magic wand to treat neurodegeneration.
Rescuing acetylation homeostasis by enhancing CBP functionality
Among all the HATs known to be involved in regulating transcription kinetics and specificity, probably CBP has the most important role in mediating neuronal survival response during neurodegenerative agitations. Apoptosis induced by K+ deprivation in cerebellar granule neurons is coupled by significant and early loss of CBP and p300 to a lesser extent.16 Other HATs like P/CAF and TAF II p250 recorded no alteration in protein level. Loss of p300, however, does not appear to be as critical as that of CBP. Mutant polyQ proteins do not sequester p300 or manipulate p300-mediated transactivation, as this adapter protein, unlike CBP, lacks polyglutamine domain.17 Despite such unaltered availability of p300, CBP deracination by mutant polyQ proteins has been singularly implicated as a major cause of neuronal loss during polyQ diseases.54 Furthermore, p300 lacks any analogous CBP serine 301 site, which is phosphorylated by CAMK-IV during activity-dependent transcriptional activity.18 All these arguments identify loss of CBP as a major degenerative threat to neurons, thereby approbating the molecule as a strong target for neurodegenerative therapeutic intervention.54, 55 Indeed, calculated re-expression of CBP during catastrophic times may be the perfect molecular bandage to heal neuronal loss.
Quantitative reconstitution of CBP is possible by either overexpressing the protein, as has been done in several studies, or by blocking its degradation in the first place. This can be achieved by designing devices to arrest caspase-6 activity on CBP or blocking attachment of CBP to ubiquitin so as to subsequently deter its proteosomal degradation. Blocking interaction of the Presenilin-1-dependent epsilon cleavage product with CBP during AD progression is yet another viable anti-Alzheimer's therapeutic approach. Furthermore, several small molecule modulators (SMM) of p300 and P/CAF have been developed.56 Similar activating SMMs may also be developed for CBP.
However, restituting acetylation homeostasis via enhanced CBP dosage calls for the same concern as does utilization of HDAC inhibitors. Nonspecific encouragement of CBP activity is undesired as it may tilt the acetylation balance of adjacent normal brain cells towards hyperacetylation leading to further complications. Although we are being able to qualitatively appreciate acetylation homeostasis, further research need to be carried out in order to quantify HAT–HDAC doses in cells. Therapeutic attempts targeting CBP re-establishment, although promising, will have to wait for our enhanced understanding of physiological settings of acetylation homeostasis.
Few Concluding Thoughts
Is the loss of acetylation homeostasis a cause or effect of neurodegeneration? A summary of our understanding of this question is represented in Figure 4. Loss of HATs by various means in a neuron primed to die during various forms of neurodegeneration represents a critical step in the apoptotic process. This loss may be thought of as one of the ‘last calls’ of an insulted neuron about to die. Once the acetylation homeostasis is lost at this phase, transcription profile is disrupted ensuring damage and death to the neuron. Thus, we see this loss as a critical cause of neurodegeneration, which represents a penultimate phase prior to the genetic response of the cell to apoptotic signal. Our argument is also positively supported by studies with HDAC inhibitors ameliorating neuronal loss. HDAC inhibitor-mediated neuronal fate reversal is possible only because a probable event can be logically avoided by manipulating its cause and not its effect.
Figure 4
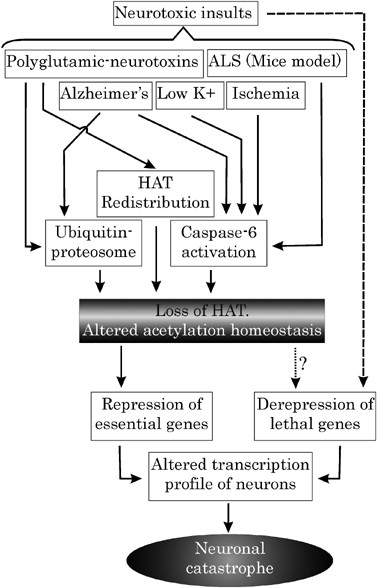
Proposed model depicting central role of acetylation homeostasis during neurodegeneration: Various models and diseases of neurodegeneration are marked by critical loss of HAT. This loss, leading to acetylation imbalance, may be manifested by various means, which are employed singularly or in combination during a particular disease. Loss of acetylation homeostasis forms the last common port where several neurotoxic insults converge to mediate a self-demolishing neuronal response. Such responses include repression of prosurvival genes on the one hand and derepression of lethal genes on the other
How appropriate is our attempt to project altered acetylation as a common factor in all these diseases? Various neurodegenerative diseases have their independent modes of inducing neuronal death. Moreover, different neuronal subsets are found to be susceptible to different degenerative disease etiology. Thus, it needs to be appreciated that although the loss of acetylation homeostasis may be critical for all modes of neurodegeneration, yet its underlying mechanism may vary among different diseases. For instance, CBP is lost in low K+-induced death by caspase activation, whereas in HD, it loses its efficacy due to redistribution in cellular compartments.
Currently, our attempt to recognize the importance of balanced acetylation in neuronal viability during neurodegeneration is severely crippled by dearth of experimental evidences. HAT–HDAC system research in nervous system is still in its juvenile days leaving us with more punctuations than statements. However, several lines of evidence provide us with enough incentive to consider this as a probable avenue of therapeutic intervention for neurodegenerative diseases. There is great hope that in-depth illumination of neuronal HAT–HDAC balancing act will help in designing restorative antineurodegenerative therapeutics in the near future.
Abbreviations
AD:
Alzheimer's disease
CaMK:
calcium/calmodulin-dependent kinase
CBP:
CREB binding protein
CDK:
cyclin-dependent kinase
CRE:
cAMP-response element
CREB:
cAMP-response element binding (protein)
HAT:
histone acetyltransferase
HD:
Huntington's disease
HDAC:
histone deacetylase
HIF:
hypoxia-induced factor
I_κ_B:
inhibitory kappaB
MAPK:
mitogen-activated protein kinase
NaBu:
sodium butyrate
NF-_κ_B:
nuclear factor kappaB
NGF:
nerve growth factor
NMDA:
_N_-methyl-D-aspartate
P/CAF:
p300/CBP-associated factor
PKA:
protein kinase A
PML:
promyelocytic leukemia
REST:
repressor element transcription factor
RPD3:
reduced potassium deficiency3
SAHA:
suberoylanilide hydroxamic acid
SMA:
spinal muscular atrophy
SMM:
small molecule modulators
SP:
specificity protein
TAD:
transactivation domain
TF:
transcription factor
TSA:
trichostatin A
References
- Taunton J, Hassig CA and Schreiber SL (1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272: 408–411
Article CAS PubMed Google Scholar - Timmermann S, Lehrmann H, Polesskaya A and Harel-Bellan A (2001) Histone acetylation and disease. Cell Mol. Life Sci. 58: 728–736
Article CAS PubMed Google Scholar - Marmorstein R and Roth SY (2001) Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 11: 155–161
Article CAS PubMed Google Scholar - Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A and Ponte JF (2003) Histone deacetylases: unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 983: 84–100
Article CAS PubMed Google Scholar - LaMorte VJ, Dyck JA, Ochs RL and Evans RM (1998) Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc. Natl. Acad. Sci. USA 95: 4991–4996
Article CAS PubMed PubMed Central Google Scholar - Zanger K, Radovick S and Wondisford FE (2001) CREB binding protein recruitment to the transcription complex requires growth factor-dependent phosphorylation of its GF box. Mol. Cell. 7: 551–558
Article CAS PubMed Google Scholar - Liu YZ, Chrivia JC and Latchman DS (1998) Nerve growth factor up-regulates the transcriptional activity of CBP through activation of the p42/p44 (MAPK) cascade. J. Biol. Chem. 273: 32400–32407
Article CAS PubMed Google Scholar - Liu YZ, Thomas NS and Latchman DS (1999) CBP associates with the p42/p44 MAPK enzymes and is phosphorylated following NGF treatment. Neuroreport 10: 1239–1243
Article CAS PubMed Google Scholar - Swope DL, Mueller CL and Chrivia JC (1996) CREB-binding protein activates transcription through multiple domains. J. Biol. Chem. 271: 28138–28145
Article CAS PubMed Google Scholar - Nakajima T, Fukamizu A, Takahashi J, Gage FH, Fisher T, Blenis J and Montminy MR (1996) The signal-dependent coactivator CBP is a nuclear target for pp90RSK . Cell 86: 465–474
Article CAS PubMed Google Scholar - Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR and Goodman RH (2002) Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron 34: 235–244
Article CAS PubMed Google Scholar - Hu SC, Chrivia J and Ghosh A (1999) Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron 22: 799–808
Article CAS PubMed Google Scholar - Lee H, Rezai-Zadeh N and Seto E (2004) Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 24: 765–773
Article CAS PubMed PubMed Central Google Scholar - Chawla S, Vanhoutte P, Arnold FJ, Huang CL and Bading H (2003) Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 85: 151–159
Article CAS PubMed Google Scholar - Boutillier AL, Trinh E and Loeffler JP (2003) Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J. Neurochem. 84: 814–828
Article CAS PubMed Google Scholar - Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP and Boutillier AL (2003) Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 22: 6537–6549
Article CAS PubMed PubMed Central Google Scholar - Jiang H, Nucifora Jr FC, Ross CA and DeFranco DB (2003) Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 12: 1–12
Article PubMed Google Scholar - Jin K, Mao XO, Simon RP and Greenberg DA (2001) Cyclic AMP response element binding protein (CREB) and CREB binding protein (CBP) in global cerebral ischemia. J. Mol. Neurosci. 16: 49–56
Article CAS PubMed Google Scholar - Nucifora Jr FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM and Ross CA (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423–2428
Article CAS PubMed Google Scholar - McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS and Fischbeck KH (2001) Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc. Natl. Acad. Sci. USA 98: 15179–15184
Article CAS PubMed PubMed Central Google Scholar - Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH and Min KT (2003) Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes. Dev. 17: 1463–1468
Article CAS PubMed PubMed Central Google Scholar - Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ and Ratan RR (2003) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc. Natl. Acad. Sci. USA 100: 4281–4286
Article CAS PubMed PubMed Central Google Scholar - Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL and Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413: 739–743
Article CAS PubMed Google Scholar - Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R and Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114: 635–645
Article CAS PubMed Google Scholar - Hoshino M, Tagawa K, Okuda T, Murata M, Oyanagi K, Arai N, Mizutani T, Kanazawa I, Wanker EE and Okazawa H (2003) Histone deacetylase activity is retained in primary neurons expressing mutant huntingtin protein. J. Neurochem. 87: 257–267
Article CAS PubMed Google Scholar - Sugars KL and Rubinsztein DC (2003) Transcriptional abnormalities in Huntington disease. Trends Genet. 19: 233–238
Article CAS PubMed Google Scholar - Cha JH (2000) Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 23: 387–392
Article CAS PubMed Google Scholar - Riccio A, Ahn S, Davenport CM, Blendy JA and Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358–2361
Article CAS PubMed Google Scholar - Shi Y, Venkataraman SL, Dodson GE, Mabb AM, LeBlanc S and Tibbetts RS (2004) Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 101: 5898–5903
Article CAS PubMed PubMed Central Google Scholar - Zhang Q, Vo N and Goodman RH (2000) Histone binding protein RbAp48 interacts with a complex of CREB binding protein and phosphorylated CREB. Mol. Cell. Biol. 20: 4970–4978
Article CAS PubMed PubMed Central Google Scholar - Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S and Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493–498
Article CAS PubMed Google Scholar - Zhong H, May MJ, Jimi E and Ghosh S (2002) The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell. 9: 625–636
Article CAS PubMed Google Scholar - Chen LF and Greene WC (2004) Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell. Biol. 5: 392–401
Article CAS PubMed Google Scholar - Acker T and Acker H (2004) Cellular oxygen sensing need in CNS function: physiological and pathological implications. J. Exp. Biol. 207: 3171–3188
Article CAS PubMed Google Scholar - Semenza GL (2000) HIF-1 and human disease: one highly involved factor. Genes Dev. 14: 1983–1991
CAS PubMed Google Scholar - Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF and Livingston DM (1996) An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 93: 12969–12973
Article CAS PubMed PubMed Central Google Scholar - Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R and Ratan RR (2003) Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J. Neurosci. 23: 3597–3606
Article CAS PubMed PubMed Central Google Scholar - Huang Y, Myers SJ and Dingledine R (1999) Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 2: 867–872
Article CAS PubMed Google Scholar - Greene LA, Biswas SC and Liu DX (2004) Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 11: 49–60
Article CAS PubMed Google Scholar - Kolli S, Buchmann AM, Williams J, Weitzman S and Thimmapaya B (2001) Antisense-mediated depletion of p300 in human cells leads to premature G1 exit and up-regulation of c-MYC. Proc. Natl. Acad. Sci. USA 98: 4646–4651
Article CAS PubMed PubMed Central Google Scholar - Rajabi HN, Baluchamy S, Kolli S, Nag A, Srinivas R, Raychaudhuri P and Thimmapaya B (2005) Effects of depletion of CREB-binding protein on c-Myc regulation and cell cycle G1-S transition. J. Biol. Chem. 280: 361–374
Article CAS PubMed Google Scholar - Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG and Endres M (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J. Neurosci. 21: 5045–5053
Article CAS PubMed PubMed Central Google Scholar - Davis T, Kennedy C, Chiew YE, Clarke CL and DeFazio A (2000) Histone deacetylase inhibitors decrease proliferation and modulate cell cycle gene expression in normal mammary epithelial cells. Clin. Cancer Res. 6: 4334–4342
CAS PubMed Google Scholar - Ianari A, Gallo R, Palma M, Alesse E and Gulino A (2004) Specific role for p300/CREB-binding protein-associated factor activity in E2F1 stabilization in response to DNA damage. J. Biol. Chem. 279: 30830–30835
Article CAS PubMed Google Scholar - van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH and Burgering BM (2004) FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 279: 28873–28879
Article CAS PubMed Google Scholar - Vries RG, Prudenziati M, Zwartjes C, Verlaan M, Kalkhoven E and Zantema A (2001) A specific lysine in c-Jun is required for transcriptional repression by E1A and is acetylated by p300. EMBO J. 20: 6095–6103
Article CAS PubMed PubMed Central Google Scholar - Hughes RE, Lo RS, Davis C, Strand AD, Neal CL, Olson JM and Fields S (2001) Altered transcription in yeast expressing expanded polyglutamine. Proc. Natl. Acad. Sci. USA 98: 13201–13206
Article CAS PubMed PubMed Central Google Scholar - Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA and Bates GP (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA 100: 2041–2046
Article CAS PubMed PubMed Central Google Scholar - Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R and Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 23: 9418–9427
Article CAS PubMed PubMed Central Google Scholar - Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ and Beal MF (2005) Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J. Biol. Chem. 280: 556–563
Article CAS PubMed Google Scholar - Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH and Li H (2001) Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl. Acad. Sci. USA 98: 9808–9813
Article CAS PubMed PubMed Central Google Scholar - Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A and Sobue G (2004) Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 13: 1183–1192
Article CAS PubMed Google Scholar - Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH and Ferrante RJ (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93: 1087–1098
Article CAS PubMed Google Scholar - McCampbell A and Fischbeck KH (2001) Polyglutamine and CBP: fatal attraction? Nat. Med. 7: 528–530
Article CAS PubMed Google Scholar - Rouaux C, Loeffler JP and Boutillier AL (2004) Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem. Pharmacol. 68: 1157–1164
Article CAS PubMed Google Scholar - Varier RA, Swaminathan V, Balasubramanyam K and Kundu TK (2004) Implications of small molecule activators and inhibitors of histone acetyltransferases in chromatin therapy. Biochem. Pharmacol. 68: 1215–1220
Article CAS PubMed Google Scholar - Freeland K, Boxer LM and Latchman DS (2001) The cyclic AMP response element in the Bcl-2 promoter confers inducibility by hypoxia in neuronal cells. Brain Res. Mol. Brain Res. 92: 98–106
Article CAS PubMed Google Scholar - Guo Z, Boekhoudt GH and Boss JM (2003) Role of the intronic enhancer in tumor necrosis factor-mediated induction of manganous superoxide dismutase. J. Biol. Chem. 278: 23570–23578
Article CAS PubMed Google Scholar - Saura CA, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe OJ, Hellings P, Gorselink M, Heymans S, Theilmeyer G, Dewerchin M, Landenbach V, Vermylen P, Raat H, Acker T, Yleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D and Carmeliet P (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: 23–36
Article CAS PubMed Google Scholar - Sopher BL, Thomas Jr PS, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM and La Spada AR (2004) Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron 41: 687–699
Article CAS PubMed Google Scholar
Acknowledgements
This study was supported by grants from NIH (NS39940 and AG19487), National Multiple Sclerosis Society (RG3422A1/1), and Michael J Fox Foundation for Parkinson Research to KP and UNMC graduate assistantship to RNS. We thank V Yennu of MD Anderson, TX, for his support in literature research.
Author information
Authors and Affiliations
- Department of Oral Biology, Section of Neuroscience, University of Nebraska Medical Center, Lincoln, 68583-0740, NE, USA
R N Saha & K Pahan
Authors
- R N Saha
You can also search for this author inPubMed Google Scholar - K Pahan
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toK Pahan.
Additional information
Edited by L Greene
Rights and permissions
About this article
Cite this article
Saha, R., Pahan, K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis.Cell Death Differ 13, 539–550 (2006). https://doi.org/10.1038/sj.cdd.4401769
- Received: 28 April 2005
- Revised: 11 July 2005
- Accepted: 01 August 2005
- Published: 28 October 2005
- Issue Date: 01 April 2006
- DOI: https://doi.org/10.1038/sj.cdd.4401769