TORC-specific phosphorylation of mTOR: phospho-Ser2481 is a marker for intact mTORC2 (original) (raw)
. Author manuscript; available in PMC: 2010 Mar 1.
Abstract
The mammalian target of rapamycin (mTOR) Ser/Thr kinase is the catalytic component of two evolutionarily conserved signaling complexes. mTOR signaling complex 1 (mTORC1) is a key regulator of growth factor and nutrient signaling. S6 kinase (S6K) is the best characterized downstream effector of mTORC1. mTOR signaling complex 2 (mTORC2) has a role in regulating the actin cytoskeleton and activating Akt through S473 phosphorylation. Herein we demonstrate that mTOR is phosphorylated differentially when associated with mTORC1 and mTORC2 and that intact complexes are required for these mTORC-specific mTOR phosphorylations. Specifically, we find that mTORC1 contains mTOR phosphorylated predominantly on S2448 whereas mTORC2 contains mTOR phosphorylated predominantly on S2481. Using S2481 phosphorylation as a marker for mTORC2 sensitivity to rapamycin, we find that mTORC2 formation is in fact rapamycin-sensitive in several cancer cell lines in which it had been previously reported that mTORC2 assembly and function were rapamycin-insensitive. Thus phospho-S2481 on mTOR serves as a biomarker for intact mTORC2 and its sensitivity to rapamycin.
Keywords: Kinase, phosphorylation, biomarker, rapamycin, cell growth
Introduction
mTOR is a member of the PI-3 kinase-like kinase family (PIKKs) that plays an integral role in coordinating cell growth and division in response to growth factors, nutrients and the energy status of the cell. mTOR is found in two distinct signaling complexes that are evolutionarily conserved from yeast to mammals. These complexes have differing substrate specificity that is determined by the unique mTOR-interacting proteins that are found in each complex. The rapamycin-sensitive mTORC1 contains mTOR, Raptor, mLST8 and PRAS40 (1-4), and regulates cell growth and translation in part by phosphorylating S6K and the eIF-4E binding protein 1 (4E-BP1) (5). The rapamycin-insensitive mTORC2 contains mTOR, Rictor, mSin1, mLST8 and Protor (6-10). In select tumor cell lines, mTORC2 is sensitive to prolonged rapamycin treatment, which inhibits mTORC2 assembly and function (11). mTORC2 regulates organization of the actin cytoskeleton through the phosphorylation of PKCα, and also phosphorylates and activates Akt at the hydrophobic motif (HM) site, S473 (6, 12). Although several other kinases have been reported to phosphorylate Akt at S473, including the PIKK-family members DNA-PK and ATM (13-15), genetic evidence from Rictor, mSin1 and mLST8 knockout mice demonstrates that intact mTORC2 is necessary for maximal phosphorylation and activation of Akt in mouse embryos, suggesting it is the major S473 kinase under normal conditions (8, 16, 17). Nevertheless, DNA-PK may be an important regulator of S473 phosphorylation in response to genotoxic stress and DNA damage (15).
Upon activation, mTOR is phosphorylated on several residues, including T2446, S2448 and S2481. T2446 is phosphorylated in response to nutrient availability (18). Initially, S2448 was reported to be an Akt phosphorylation site because its phosphorylation is sensitive to PI-3 kinase (PI-3K) inhibition, which reduces Akt activity. However, more recent reports have shown that S6K is the S2448 kinase (19, 20). S2481 is a rapamycin-insensitive autophosphorylation site (21). All three phosphorylation sites are in a region lying between the catalytic domain and the FATC domain near the C-terminus of mTOR. Mutation of T2446 and S2448 to alanine has no discernible effect on the ability of mTOR to activate its downstream effectors. Nevertheless, the fact that internal deletion of residues 2430-2450 reportedly increases mTOR kinase activity suggests that this region is regulatory (22). At present, the functional significance of mTOR phosphorylation at these sites has not been fully explored and remains poorly understood.
We set out to analyze whether mTOR phosphorylation regulates the formation of either mTORC1 or mTORC2. We observed that the mTOR associated with mTORC1 and mTORC2 isolated from insulin-stimulated cells is phosphorylated differentially, with mTORC1 predominantly containing mTOR phosphorylated on S2448 and mTORC2 predominantly containing mTOR phosphorylated on S2481. We went on to show that S2481, which is specific for mTORC2, can be used as a marker to determine the rapamycin sensitivity of mTORC2 formation in several cancer cell lines that were reported to be insensitive to prolonged rapamycin treatment as deduced using phosphorylation of S473 of Akt as a marker. Our data demonstrate that S2481 phosphorylation of mTOR can serve as a biomarker for the prediction of rapamycin-induced mTORC2 suppression in particular cancer cell types. Because regulation of Akt S473 phosphorylation is rather complicated and not yet fully understood, we propose that phosphorylation of S2481 in mTOR is a better marker for intact mTORC2 and its sensitivity to rapamycin.
Materials and Methods
Antibodies and reagents
The following antibodies were purchased from Cell Signaling Technology: phospho-mTOR (S2448 and S2481), Akt and phospho-Akt (S473) rabbit polyclonal antibodies. Phospho-S6K (T389) and mTOR (mTab1) rabbit polyclonal antibodies were purchased from Millipore. Rictor and mSin1 rabbit polyclonal antibodies were purchased from Bethyl Laboratories. The anti-Raptor rabbit antiserum was developed with the antibody service from Invitrogen utilizing the peptide PHSHQFPRTRKMFDKG, amino acid sequence 918-933 of human Raptor. Rapamycin was purchased from Sigma. Insulin and IGF-1 were purchased from Research Diagnostics, Inc.
Lentivirus-mediated gene knockdown
We obtained pLKO.1 based short-hairpin constructs specific for mTOR (Addgene plasmid #1855), Raptor (Addgene plasmid #1857), Rictor (Addgene plasmid number #1853), and mSin1 (Addgene plasmid #13483), as well as a scrambled control sequence (Addgene plasmid #1864) from the plasmid repository at Addgene. They have been described previously (7, 12).
Plasmids were co-transfected together with the lentiviral packaging (pMDL), envelope (CMV-VSVG) and rev-expressing (RSV-REV) constructs into actively growing HEK293T cells using the Effectene transfection reagent (Qiagen) per manufacturer's protocol. Virus-containing supernatants were collected 48 hr. after transfection. Cells were infected twice in the presence of 1 μg/ml polybrene, selected for puromycin resistance and analyzed 48-72 hr. post-infection.
Cell culture, cell lysis and immunoprecipitation
All cells were cultured in DMEM/10% FCS supplemented with penicillin, streptomycin and ciprofloxacin at 37°C. Where applicable, cells were cultured in serum-free DMEM for 24 hr. prior to growth factor stimulation.
Cells were rinsed 2 × with cold PBS and lysed in 1 ml 50 mM Hepes pH 7.5, 150 mM NaCl, 10% glycerol, 0.3% Chaps (v/v), 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 500 μM sodium orthovanadate, 10 μg/ml aprotinin, and 10 μg/ml leupeptin per 10 cm tissue culture dish. Lysates were rotated end over end at 4°C for 20 min. and clarified by centrifugation at 12,000 rpm for 10 min at 4°C. Protein concentrations were determined using the Bio-Rad DC Protein Assay kit according to manufacturer's protocol. Lysates were either mixed v/v with 2X SDS sample buffer or subjected to immunoprecipitation.
For immunoprecipitation, lysates were incubated with the appropriate antibody while being mixed end over end for 1 hr at 4°C. Protein A-Sepharose was added and the samples were mixed for an additional hr. at 4°C. Immune complexes were isolated by centrifugation and washed 4X with ice-cold lysis buffer. Samples were boiled for 5 min. at 100°C in 1X SDS sample buffer.
Immunoblot analysis
Immunoprecipitates or whole-cell lysates normalized for total protein concentration were resolved by SDS-PAGE and proteins were electrotransferred to PVDF membranes. Immunoblotting was performed per manufacturer's protocol, and reactive proteins were visualized by ECL.
Results
mTOR has several phosphorylation sites in the 60 amino acid region beyond the catalytic domain in the C-terminal tail, and these sites are conserved in all vertebrates but not in invertebrates (Fig. 1A). In fact, the entire 60 residue region containing these sites is highly conserved among vertebrate species, suggesting it could be a vertebrate-specific regulatory element (Fig. 1A). Because the regulation of mTORC1 and mTORC2 formation is poorly understood, we set out to analyze whether mTOR phosphorylation has any effect on either complex formation. Rictor and Raptor immunoprecipitates (IPs) from untreated serum-starved HEK293 cells and cells treated with 200 nM insulin for 5 min. were analyzed by immunoblotting with antibodies specific for either total mTOR, mTOR phosphorylated on S2448 or mTOR phosphorylated on S2481. Whole cell lysates were analyzed as controls to insure that insulin stimulation led to increased S2448 and S2481 phosphorylation. We were surprised to observe that mTOR phosphorylated on S2448 was mainly associated with Raptor, whereas mTOR phosphorylated on S2481 was predominantly associated with Rictor in HEK293 cells (Fig. 1B). The amount of mTOR associated with either Raptor or Rictor did not change as a result of insulin stimulation and concomitant mTOR phosphorylation. Rictor and Raptor IPs from actively growing U2OS cells were also analyzed to confirm that this result was not specific to HEK293 cells. As in HEK293 cells, mTOR phosphorylated on S2448 was associated with Raptor and mTOR phosphorylated on S2481 was associated with Rictor in U2OS cells (Fig. 1C). However, because there was a low level of S2448 phosphorylation associated with mTORC2 in HEK293 cells that was not observed in actively growing U2OS cells (Fig. 1B and C), it is possible that phospho-S2448 is not an mTORC1 specific phosphorylation site in all cell types under all conditions. Although it has been demonstrated that Rictor and Raptor binding to mTOR are mutually exclusive events (6), we wanted to confirm that the reason we detected a minor amount of mTOR phosphorylated on S2448 associated with Rictor was not due to a small fraction of mTORC1 bound to mTORC2. We performed Rictor and Raptor IPs using lysates from several cell lines and blotted for the presence of both proteins. We failed to detect any Rictor in Raptor IPs or any Raptor in Rictor IPs, confirming that mTORC1 and mTORC2 do not associate (Fig. S1).
Fig. 1. mTORC1 and mTORC2 contain differentially phosphorylated mTOR.

(A) Schematic of mTOR and multiple sequence alignment of the C-terminus of selected vertebrate and invertebrate TORs. Invertebrate species are Ciona intestinalis and C. savignyi (Cint and Csav); Drosophila melanogaster and D. virilis (Dmel and Dvir); Caenorhabditis elegans and C. briggsae (Cele and Cbrig); and Saccharomyces cerevisiae (TOR1 and TOR2). The region between the kinase domain (KD) and an N-terminal extension of the FATC domain (N-FATC) is conserved among vertebrates, including the marked phosphorylation sites S2448 and S2481. Asterisks indicate residues completely conserved in vertebrates. (B) HEK293 cells were serum-starved overnight. The indicated cells were stimulated with 200 nM insulin for 5 min at 37° C. Rictor and Raptor immunoprecipitates (IPs) from control and growth factor-stimulated cells were analyzed by immunoblotting with antibodies specific for mTOR phosphorylated on S2448 or S2481, or total mTOR. Whole cell lysates (WCL) were included as controls for total input. (C) Samples from actively growing U2OS cells were analyzed as in (B). Results are representative of multiple independent experiments.
While our data demonstrated that S2448 phosphorylation of mTOR is associated with mTORC1 and that S2481 phosphorylation of mTOR is associated with mTORC2, it was unclear if intact mTORC1 and mTORC2 complexes are required for these mTOR phosphorylations. To investigate this, short hairpin RNA (shRNA) sequences that specifically deplete endogenous mTOR, Rictor or Raptor were expressed in HEK293 cells via lentiviral infection. Three days after infection, cells were serum-starved overnight, control and insulin-stimulated cells were lysed, and whole cell lysates were analyzed by immunoblotting with antibodies that recognize either phospho-S2448 or phospho-S2481. In cells in which Raptor levels were significantly reduced, we found complete ablation of insulin-stimulated S2448 phosphorylation without any effect on S2481 phosphorylation. Conversely, in cells in which Rictor had been depleted, insulin-stimulated S2481 phosphorylation was abolished without any reduction in S2448 phosphorylation. The data demonstrate that intact mTORC1 is necessary for S2448 phosphorylation and that intact mTORC2 is necessary for S2481 phosphorylation, further underscoring the specificity of these mTOR phosphorylation sites for the different mTOR signaling complexes (Fig. 2A). Although the depletion of mTOR, Raptor and Rictor was not as efficient in U2OS cells as in HEK293 cells, decreased Raptor expression led to diminished S2448 phosphorylation, and decreased Rictor expression led to diminished S2481 phosphorylation in actively growing U2OS cells (data not shown). To confirm that mTORC2 is necessary for S2481 phosphorylation we utilized shRNA knockdown of the other major mTORC2 specific component, mSin1 in HEK293 cells. We found that depletion of mSin1 also reduced the level of Rictor, as previously reported (7) (Fig. 2B). In cells with reduced levels of both mSin1 and Rictor, the insulin-induced phosphorylation of mTOR on S2481 was completely abolished, confirming that intact mTORC2 is necessary for S2481 phosphorylation (Fig. 2B). To definitively prove that S2481 phosphorylation requires intact mTORC2, we analyzed mTOR phosphorylation in Sin1-/- mouse embryo fibroblasts (MEFs) (8). Consistent with the shRNA results, the basal and growth factor-induced phosphorylation of S2481 was severely diminished in Sin1-/- MEFs as compared to WT (Fig. 2C). Genetic ablation of Sin1 appears to have less of an effect on S2481 phosphorylation than does shRNA-mediated knockdown of mSin1 (Fig. 2B and C). This is most likely due to the presence of a low level of Rictor in Sin1-/-MEFs [(8) and Fig. 2C].
Fig. 2. Intact mTORC1 and mTORC2 are necessary for mTOR phosphorylation.

(A) HEK293 cells were infected with lentiviruses expressing a control shRNA or shRNAs targeting mTOR, Raptor or Rictor. Cells were selected with puromycin 24 hr. after infection and then serum-starved overnight 2 days post-selection. The indicated cells were stimulated with 200 nM insulin for 5 min. at 37°C. WCLs were normalized for total protein concentration and were analyzed by immunoblotting with antibodies specific for mTOR phosphorylated on S2448 or S2481. Blots for total mTOR, Rictor and Raptor were included as controls. (B) HEK293 cells were infected with lentiviruses expressing a control shRNA or shRNAs targeting Rictor or mSin1. Cells were treated as in (A) and analyzed by immunoblotting for mTOR phosphorylated on S2448 or S2481. Blots for total mTOR, Rictor and mSin1 were included as controls. (C) Wild type (WT) and Sin1-/- mouse embryo fibroblasts (MEFs) were serum starved overnight. The indicated cells were stimulated with 200 nM insulin for 5 min. at 37°C. WCLs were normalized for total protein concentration and were analyzed by immunoblotting with antibodies specific for mTOR phosphorylated on S2481, total mTOR and Rictor. (D) HEK293 cells were infected with lentiviruses and were treated as in (A). Rictor and Raptor IPs from control and growth factor- stimulated cells were analyzed by immunoblotting for bound mTOR. WCLs were analyzed by immunoblotting with antibodies specific for mTOR phosphorylated on S2448 or S2481. Results are representative of multiple independent experiments.
Although insulin-stimulated S2481 phosphorylation remained unchanged in Raptor-depleted cells, basal levels of S2481 phosphorylation were higher (Fig. 2A). Careful examination of the data from several independent experiments show that in some cases S2481 phosphorylation was less effectively abolished by serum depletion than S2448 phosphorylation, and this was independent of Raptor knockdown (for example, compare the insulin-induced S2481 phosphorylation shown in Figs. 2A, 2B, 2D and 3A). However, others have reported findings that indirectly suggest that Raptor knockdown may have an effect on mTORC2, possibly as a result of freeing up more mTOR to interact with Rictor (9, 12). To test this, we performed Rictor IPs from cells in which Raptor was depleted and Raptor IPs from cells in which Rictor was depleted and compared the level of mTOR associated with each protein. Although there was a significant decrease in the level of S2448 phosphorylation, which indicates efficient Raptor depletion, there was no discernible change in the amount of mTOR associated with Rictor in cells with decreased Raptor expression (Fig. 2D). In Rictor-depleted cells, S2481 phosphorylation was completely abolished, yet the levels of mTOR associated with Raptor were unchanged (Fig. 2D). These data provide direct evidence that the relative amount of intact mTORC1 has no effect on the relative amounts of intact mTORC2 and vice versa.
Fig. 3. Prolonged treatment of cells with rapamycin inhibits mTOR phosphorylation on S2448 and S2481.

(A) Serum-starved HEK293 cells were cultured in the presence or absence of 100 nM rapamycin for either 1 or 24 hr. The indicated cells were stimulated with 200 nM insulin for 5 min. at 37° C. WCLs were analyzed by immunoblotting with antibodies specific for mTOR phosphorylated on S2448 or S2481, or total mTOR. Rictor IPs were analyzed by immunoblotting with antibodies specific for Rictor, mTOR, and mTOR phosphorylated on S2481. (B) Actively growing U2OS cells were cultured in the presence or absence of 100 nM rapamycin for either 1 or 24 hr and analyzed as described as in (A). (C) Actively growing MDA 231, MDA 468, SKBR3 and A549 cells were treated as in (B). Rictor IPs were analyzed by immunoblotting with antibodies specific for Rictor and mTOR. WCLs were analyzed by immunoblotting for phospho-mTOR (S2481), phospho-Akt (S473), phospho-S6K (T389), total mTOR and total Akt. (D) Actively growing C2C12 myoblasts and HepG2 cells were treated as in (B) and WCLs were analyzed as in (C). Results are representative of multiple independent experiments.
The phosphorylation of mTOR on S2481 and the assembly and function of mTORC2 were initially reported to be rapamycin-insensitive (6, 21), but more recent studies indicate that prolonged rapamycin treatment inhibits both mTORC2 assembly and function (11). Because S2481 phosphorylation requires intact mTORC2, we tested whether prolonged rapamycin treatment had any effect on S2481 phosphorylation. Whole cell lysates from control and insulin-stimulated cells treated with 100 nM rapamycin for either one or 24 hr were analyzed for phosphorylation of mTOR on S2448 and S2481. As expected, we observed a marked reduction of S2448 phosphorylation in insulin-stimulated cells with either acute or prolonged treatment of rapamycin (Fig. 3A). In contrast, insulin-stimulated S2481 phosphorylation showed no discernible decrease after acute treatment with rapamycin but was completely absent after prolonged treatment (Fig. 3A). When Rictor IPs from these same cells were analyzed for bound mTOR, we observed a slight decrease in the amount of mTOR bound to Rictor after one hr of rapamycin treatment, indicating that acute rapamycin treatment may have a minor, yet reproducible, effect on mTORC2 formation. However, after 24 hr treatment, no detectible mTOR was bound to Rictor, indicating that prolonged rapamycin treatment inhibits the assembly of mTORC2 in HEK293 cells (Fig. 3A). In addition, both mTORC2 assembly and S2481 phosphorylation were inhibited by 24 hr but not one hr rapamycin treatment in actively growing U2OS cells (Fig. 3B).
Intriguingly, U2OS cells were reported to be insensitive to prolonged rapamycin treatment when phosphorylation of Akt at S473 was utilized as a marker for mTORC2 function (11). This led us to analyze S2481 phosphorylation and mTORC2 assembly in response to prolonged rapamycin treatment in several other cancer cell lines in which S473 phosphorylation is reported to be insensitive to rapamycin (11). Analysis of mTOR S2481 and Akt S473 phosphorylation in whole cell lysates of MDA-MB-231, MDA-MB-468, SKBR3 and A549 cells treated with 100 nM rapamycin for 24 hr showed that mTOR S2481 phosphorylation was greatly diminished (Fig. 3C) while Akt phosphorylation remained unchanged or increased, as reported [(11) and Fig. 3C]. However, when Rictor IPs were analyzed for bound mTOR in parallel, the amount of mTOR was dramatically reduced, if not completely abolished (Fig. 3C). The decreased amount of mTOR bound to Rictor paralleled the reduction in S2481 phosphorylation. As a control, we analyzed the phosphorylation of mTOR on S2481 and Akt on S473 in C2C12 myoblasts and HepG2 cells, two cell lines that were reported to be sensitive to prolonged rapamycin treatment (11). As expected, both S2481 and S473 phosphorylation were sensitive to rapamycin treatment in these cells (Fig. 3D). Our data demonstrate that phosphorylation of S2481 on mTOR is a more direct marker of intact mTORC2 than is phosphorylation of S473 of Akt. We assert that mTOR S2481 phosphorylation is a biomarker that can be used to analyze the sensitivity of mTORC2 to rapamycin treatment in various cancer types.
Cells with rapamycin-insensitive Akt phosphorylation are reported to become sensitive to rapamycin treatment after partially reducing mTOR expression (11). One explanation is that even a small amount of intact mTORC2 can sustain robust Akt S473 phosphorylation in these cells, and that prolonged rapamycin treatment is not enough to decrease mTORC2 levels below the threshold necessary for Akt phosphorylation. Another possibility is that in certain cancer settings mTOR can phosphorylate Akt independently of its association with either Rictor or mSin1. To test this, we analyzed whether partial knockdown of either Rictor or mSin1 could render cells sensitive to rapamycin treatment. mTOR, Rictor or mSin1 expression was reduced by shRNA expression in MDA-MB-468 cells. Following rapamycin treatment for 24 hr, whole cell lysates were analyzed for mTOR S2481 phosphorylation and Akt S473 phosphorylation. As expected, a decrease in mTORC2, either by shRNA, rapamycin treatment, or both, led to a reduction in the amount of mTOR phosphorylated on S2481 (Fig. 4). Partial depletion of Rictor led to a mild, yet reproducible decrease in S473 phosphorylation upon treatment with rapamycin (Fig. 4). Partial depletion of mSin1 had a much more profound effect on S473 phosphorylation upon prolonged rapamycin treatment (Fig. 4). This is most likely due to a decrease in mSin1 protein levels leading to a concomitant decrease in Rictor protein levels, making mSin1 knockdown a more efficient way to diminish mTORC2 levels in the cell. Partial depletion of mTOR had the greatest effect on S473 phosphorylation upon prolonged rapamycin treatment. This makes sense, as mTOR is the catalytic component of the mTORC2 complex. These results indicate that rapamycin treatment alone is not enough to completely disrupt mTORC2 formation below levels that are necessary for S473 phosphorylation and that mTOR still requires Rictor/mSin1 in these cells to mediate S473 phosphorylation.
Fig. 4. Depletion of mTORC2 renders S473 phosphorylation of Akt sensitive to rapamycin.
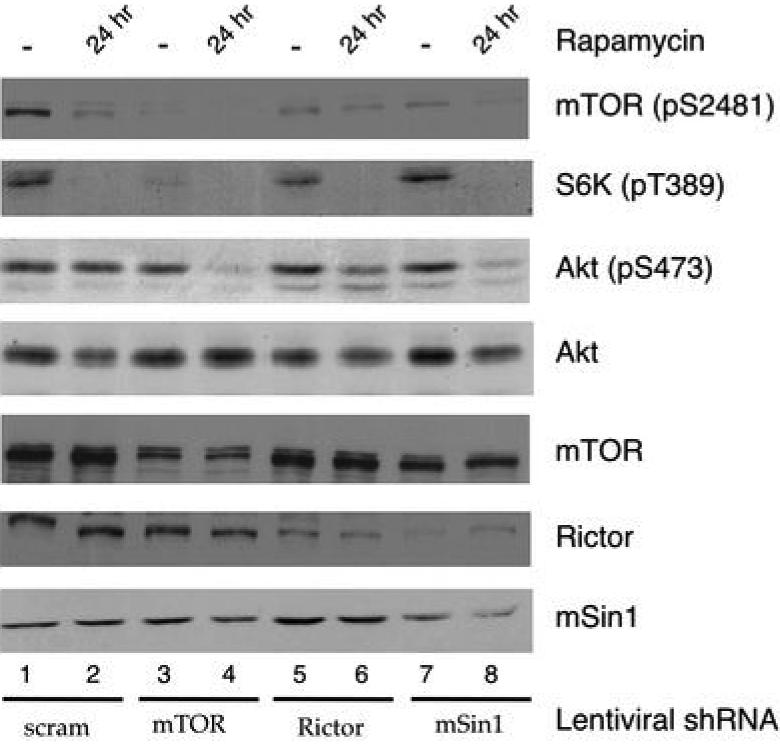
MDA-MB-468 cells were infected with lentiviruses expressing a control shRNA or shRNAs targeting mTOR, Rictor or mSin1. Cells were selected with puromycin 24 hr. after infection and the indicated cells were treated with 100 nM rapamycin for an additional 24 hr. WCLs were normalized for total protein concentration and were analyzed by immunoblotting for phospho-mTOR (S2481), phospho-Akt (S473), phospho-S6K (T389), and total Akt, mTOR Rictor and mSin1. Results are representative of multiple independent experiments.
Discussion
We have demonstrated that mTOR associated with mTORC1 or mTORC2 is phosphorylated on different sites. The rapamycin-sensitive mTORC1 complex contains phospho-S2448, which is consistent with S2448 phosphorylation being sensitive to acute rapamycin treatment. The rapamycin-insensitive mTORC2 complex contains phospho-S2481, which is consistent with S2481 being a rapamycin-insensitive autophosphorylation site. In all the cell lines we tested, the amount of mTOR recovered from Rictor IPs and the amount of S2481 phosphorylation of mTOR were reduced dramatically in response to prolonged rapamycin treatment. We have found a pharmacodynamic biomarker that directly monitors the effects of rapamycin on mTORC2 assembly and function. In several of the cancer cell lines tested the amount of Akt phosphorylated on S473 either increased or remained unchanged, as previously reported (11). Thus, S2481 phosphorylation of mTOR is a better marker for the amount of intact mTORC2 in the cell than is phospho-S473 Akt. Because Akt activation is downstream of both PI-3K and mTORC2, using S473 phosphorylation as a readout for mTORC2 does not differentiate between changes in PI-3K activity and changes in mTORC2 activity. Phospho-S2481 serves as a useful biomarker that distinguishes mTOR2 activity from PI-3K activity, which will make it an invaluable tool when evaluating inhibitors that are specific for mTORC2 only.
Because mTORC1-mediated activation of S6K can suppress Akt activation, long-term inhibition of mTORC1 by rapamycin can lead to increased Akt phosphorylation in some cell lines (23-25). We observed this in MDA-MB-231 cells (Fig. 3C). However, in all cell lines tested, rapamycin caused an almost complete disruption of the mTORC2 complex. Because the amount of S473 phosphorylation was either increased or unchanged in the presence of severely diminished levels of mTORC2, we looked for the presence of another S473 kinase in these cells. Following up on the report that DNA-PK, another PIKK, can serve as an S473 kinase under conditions of genotoxic stress (15), we have tested the effect of reducing DNA-PK expression in MDA-MB-468 cells and observed a significant reduction, but not complete ablation, of S473 phosphorylation (unpublished observations). We are currently investigating the role of DNA-PK in regulating Akt phosphorylation in these cells. The importance of understanding how Akt is regulated in cancer cells has been underscored by the recent finding that reducing Akt activity can restore sensitivity to rapamycin in cells that were resistant to the drug's cytostatic effects (26).
mTORC1 is required for the activation of S6K (1, 2). When we inhibited mTORC1 formation either by rapamycin or Raptor shRNA treatment, we observed diminished S2448 phosphorylation. Therefore, our data support the finding that S6K is the S2448 kinase rather than Akt (since S6K is downstream of mTORC1). S6K that is associated with Raptor via its TOS motif may mediate phosphorylation of mTOR at S2448 once it has been activated by mTOR phosphorylation at T389 in a possible feedback loop. However, it should be noted that S2448 phosphorylation, while predominantly associated with mTORC1, is not completely specific for mTOR in mTORC1 since we have observed some S2448-phosphorylated mTOR associated with Rictor in HEK293 cells and in some other cancer cell lines. S6K mediates the assembly of the translation preinitiation complex through a series of ordered phosphorylation events, and mTORC1 association with eIF3 increases upon insulin stimulation and is reduced upon rapamycin treatment, indicating that this association is phosphorylation dependent (27). Because phospho-S2448 is predominantly associated with mTORC1, we are currently analyzing whether it has a role in mTORC1 interacting with eIF3.
It has been reported that S2481 is a rapamycin-insensitive autophosphorylation site (21). We have shown that phosphorylation of S2481 is dependent on intact mTORC2 and that this site is sensitive to prolonged rapamycin treatment. The sensitivity to rapamycin is most likely due to prolonged treatment inhibiting the assembly of mTORC2 (11). It is unclear why autophosphorylation of mTOR on S2481 requires the presence of Rictor and/or mSin1. These companion proteins may hold mTOR in a conformation that is amenable to autophosphorylation, perhaps in trans within an mTOR dimer. We are currently exploring why S2481 phosphorylation requires intact mTORC2.
While the functional significance of mTOR phosphorylation at S2448 and S2481 remains elusive, the fact that these sites are highly conserved across vertebrate species points to phosphorylation having a role in mTOR regulation (Fig. 1A). Alignment of multiple mTOR orthologs reveals that, while present in all vertebrate species analyzed, both phosphorylation sites analyzed here are absent in invertebrates (Fig. 1A). In fact, the entire region between the kinase and FATC domains is extremely well-conserved throughout vertebrate species but highly variable in other species, even closely related members of the same genus (Fig 1A). This high and selective conservation indicates that this region may function as a vertebrate-specific regulatory element. The deletion of amino acids 2430-2450 within this region leads to an elevated level of mTOR kinase activity, supporting the idea that it is involved in regulating mTOR function, possibly as a repressor domain (22). In the AGC family of protein kinases, the C-terminal tail has evolved as a regulatory module that is necessary for catalytic activity through various interactions with the catalytic domain (28). The C-terminal region of mTOR could modulate catalytic activity in a similar fashion. More work needs to be done to understand the role that this region plays in mTOR regulation.
Our data demonstrate the existence and the identity of mTORC-specific phosphorylation sites on mTOR, and that phospho-S2481 can be used as a specific marker to detect intact mTORC2 within the cell. Because S2448 and S2481 have evolved recently in vertebrate mTOR, they may regulate TOR activity in a manner not found in invertebrates. It is clear that mTOR phosphorylation is more complicated than previously thought, and further studies are needed to understand its role in regulating mTOR-mediated signaling.
Acknowledgements
The Sin1-/- MEFs were kindly provided by Dr. Bing Su. We thank Dr. Gray Pearson for many helpful discussions and Jill Meisenhelder for comments on the manuscript.
This work was supported by grants CA82683 and CA14195 from the NCI (to T.H.). J.C. was supported in part by the American Cancer Society, Illinois Division-Linda M. Campbell postdoctoral fellowship and NIH training grant T32 CA 09370. T.H. is a Frank and Else Schilling American Cancer Society Research Professor.
References
- 1.Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 2.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 3.Kim DH, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 4.Haar EV, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 5.Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 7.Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–70. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell. 2006;127:125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 9.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20:2820–32. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearce LR, Huang X, Boudeau J, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–22. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 13.Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–96. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- 14.Viniegra JG, Martinez N, Modirassari P, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem. 2005;280:4029–36. doi: 10.1074/jbc.M410344200. [DOI] [PubMed] [Google Scholar]
- 15.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–13. doi: 10.1016/j.molcel.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 16.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic Disruption of the rictor Gene in Mice Reveals that mTOR Complex 2 Is Essential for Fetal Growth and Viability. Dev Cell. 2006;11:583–9. doi: 10.1016/j.devcel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. J Biol Chem. 2004;279:15719–22. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 19.Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–90. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 20.Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280:26089–93. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- 21.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–23. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 22.Sekulic A, Hudson CC, Homme JL, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–13. [PubMed] [Google Scholar]
- 23.Harrington LS, Findlay GM, Gray A, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–6. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 25.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Yue P, Kim YA, et al. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–18. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 28.Kannan N, Haste N, Taylor SS, Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci U S A. 2007;104:1272–7. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]