Methylation Status of CpG Islands Flanking a CRE Motif on the Protein Phosphatase 2Acα Promoter Determines CREB Binding and Activity (original) (raw)
. Author manuscript; available in PMC: 2010 Feb 1.
Abstract
Protein phosphatase 2A (PP2A) is a major serine-threonine protein phosphatase in eukaryotic cells and is involved in many essential aspects of cell function. The catalytic subunit of the enzyme (PP2Ac), a part of the core enzyme, has two isoforms, α (PP2Acα) and β (PP2Acβ), of which PP2Acα is the major form expressed in vivo. Deregulation of PP2A expression has been linked to several diseases, but the mechanisms which control the expression of this enzyme are still unclear. We conducted experiments to decipher molecular mechanisms involved in the regulation of the PP2Acα promoter in human primary T cells. After constructing serially truncated PP2Acα promoter luciferase constructs, we found that the region stretching around 240 bases upstream from the translation initiation site was of functional significance and included a cAMP response element (CRE) motif flanked by three GC- boxes. Shift assays revealed that CRE-binding protein (CREB)/phosphorylated CREB (pCREB) and stable protein 1 (Sp1) could bind to the region. Furthermore, we demonstrated that methylation of deoxycytosine (dC) in the CpG islands limited binding of pCREB and the activity of the PP2Acα promoter. In contrast, the binding of Sp1 to a GC-box within the core promoter region was not affected by DNA methylation. Primary T cells treated with 5-azacitidine (5-azaC), a DNA methyltransferase (DNMT) inhibitor, showed increased expression of PP2Acα mRNA. We propose that conditions associated with hypomethylation of CpG islands, such as drug- induced lupus and cancer, permit increased PP2Ac expression.
Keywords: Human, T Cells, Systemic Lupus Erythematosus, Protein Kinases/Phosphatases, Gene Regulation
Introduction
Protein phosphatase 2A (PP2A) is a major serine/threonine phosphatase with complex composition and is involved in many essential aspects of cell function (1–3). The heterodimeric PP2A core enzyme consists of a well-conserved 36 kDa catalytic subunit (C subunit; PP2Ac) and a 65 kDa scaffold subunit (A subunit) (4). To gain full activity toward specific substrates, the PP2A core enzyme associates with a variable regulatory subunit (B subunit) to form a heterotrimeric holoenzyme (1–3).
Molecular cloning has revealed the existence of two mammalian PP2Ac isoforms, α (PP2Acα) and β (PP2Acβ), which share 97% identity in their primary sequence (5). Both isoforms are ubiquitously expressed and these genes are composed of seven exons and six introns encoded by different genes, localized to human chromosome 5q23-q31 for α and to 8p12-p11.2 for β. The promoters of both genes are GC rich and lack the TATA and CCAAT sequences typical of many housekeeping genes. However, the 5′ upstream regions as well as the regions encoding the 5′ and 3′ untranslated sequences of each mRNA are quite different. The activity of the PP2Acα gene promoter is 7 ± 10-fold stronger than that of the PP2Acβ gene promoter, which may explain why mRNA level of PP2Acα is about 10 times higher than that of PP2Acβ (6). The expression of PP2Ac is tightly controlled through autoregulation to ensure the presence of relatively constant levels of PP2A (7), but many essential aspects of PP2Ac regulation remain poorly understood.
Abnormal expression of PP2A has been linked to many diseases such as cancer (8–9), Alzheimer’s disease (10) and Opitz BBB/G syndrome (11). We have reported that the message, protein, and enzymatic activity of PP2Ac are increased in the peripheral T cells from the patients with systemic lupus erythematosus (SLE) compared to normal T cells (12). Phosphorylated cAMP response element–binding protein (pCREB), which is an important transcription factor in the regulation of the expression of interleukin 2 (IL-2) (13) is known to be one of the substrates of the PP2Ac (14), and decreased production of IL-2 by SLE T cells is considered to be of pathogenic importance (15–20). Treatment of SLE T cells with PP2Ac-siRNA decreases the protein levels and activity of PP2Ac in a specific manner and increases the levels of pCREB and its binding to the IL-2 and c-fos promoters, as well as increases activator protein 1 (AP-1) activity, causing normalization of IL-2 production.
SLE is an autoimmune disease that affects multiple organs including the joints, skin, kidneys and brain. Several signaling abnormalities of the immune system have been described in SLE and are thought to be central in the pathophysiology of this disease. At the cellular level, T lymphocytes orchestrate the altered immune responses in SLE (21, 22). One of the well-known abnormalities of T cell signal transduction in SLE is the defective signaling via extracellular signal-regulated kinases (ERK-1/ERK-2) (23), which contribute to the DNA hypomethylation by the decreased expression of DNA methyltransferase (DNMT-1) (21–26). Because methylation of deoxycytosine (dC) in regulatory sequences can suppress transcription of the associated genes, abnormal hypomethylation may result in the overexpression of certain genes such as lymphocyte function-associated antigen 1 (LFA-1; CD11a) and CD70 in SLE T cells (27, 28).
Because PP2Ac mRNA was found to be increased in T cells from patients with SLE and its stability was not compromised (12), we have initiated studies to define the transcriptional control of the PP2Ac gene. We report here that the PP2Acα promoter defines a CRE site flanked by CpG motifs and that methylation controls the binding of pCREB and the activity of the promoter.
Materials and Methods
Cells and reagents
Primary T cells were purified from peripheral venous blood obtained from healthy volunteers. The blood was incubated for 30 minutes with a rosette T cell purification kit (Stem Cell Technologies Inc., Vancouver, Canada) which contained a tetrameric antibody mixture against CD14, CD16, CD19, CD56, and glyA that attaches non-T cells to erythrocytes. Ficoll containing Lymphoprep gradient (Greiner Bio-One, Monroe, NC) was subsequently used to separate these complexes from T cells. Using flow cytometry, we established that the purified cells were >98% positive for CD3.
T cells (up to 10 × 106/sample) were plated onto appropriate size plates in RPMI 1640 medium (Mediatech Inc., Manassas, VA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Quality Biological Inc., Gaithersburg, MD) and 2 mM L-glutamine and maintained in a humidified incubator (37°C, 5% CO2). For some experiments, T cells were preincubated with 200 U/ml of human recombinant IL-2 (R&D Systems, Inc., Minneapolis, MN) overnight, and treated with 0~10μM of 5-Azacytidine (5-azaC; Sigma- Aldrich, St. Louis, MO) for 24 or 48 hours. Studies were approved by the human use committee of our institution.
Plasmids
The PP2Acα gene promoter region was amplified using GoTaq DNA Polymerase (Promega, Madison, WI) and human genomic DNA template (Roche, Basel, Switzerland) with primers containing NheI, XhoI or HindIII restriction sites at the 5′ ends. The PCR primers used in these amplification reactions were: F1 (−1104): 5′-GCGCTAGCGACAGGTGCATCCATCTTCTCT-3′; F2 (−781): 5′-GGGCTAGCGGTGGGGGTGGTTAATCCAA-3′; F3 (−468): 5′-CCGCTAGCATGCTCCAGCTCCATCCTTC-3′; F4(−368): 5′-GCCTCGAGTGCGCTTTGACCCCCAGTTT-3′; F5 (−280): 5′-GCCTCGAGTTTTCCCCTCCGCTCCCC-3′; F6 (−218): 5′-CGGCTAGCGCCATTACAGAGAGCCGAGCT-3′; R (−83): 5′-CCAAGCTTGCCGGTTCCTCGTGTACTTCT-3′. The PCR-generated DNA fragments were then gel purified and ligated with the pGL3-basic luciferase constructs (Promega). The QuickChange Lightning Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used to mutate the CREB or stable protein 1 (Sp1) binding site in the construct including 386bp length of PP2Acα promoter region. Each primer that we used for mutagenesis is as below. Δ-468-MtCREB; F: 5′-CCTCCTGACGCCGGGCGTGCGTACACCACGCCCGG-3′, R: 5′-CCGGGCGTGGTGTACGCACGCCCGGCGTCAGGAGG-3′, Δ-468-MtSp1; F: 5′-GGGCGTGACGTCAATGTGCCCGGCGGCC-3′, R: 5′-GGCCGCCGGGCACATTGACGTCACGCCC-3′ (mutated bases are underlined). Plasmids were sequenced to detect any errors introduced by PCR.
20 μg of Δ-468 construct or empty vector were methylated using 16U of CpG Methyltransferase (_M. Sss_I) and S-adenosyl methionine (SAM, New England Biolabs Inc., Ipswich, MA) at 37°C for 16 hours, with subsequent inactivation of enzyme at 65°C for 20 minutes. Mock-methylation reactions were also performed in the absence of _M. Sss_I and SAM. The methylated or mock-methylated constructs were purified using QIAquick PCR purification kit (Qiagen, Valencia, CA) and the methylation status of each construct was determined by methylation senstive restriction enzyme AatII (New England Biolabs Inc.) for Δ-468 construct or SalI (New England Biolabs Inc.) for empty vector.
Cell transfection
The transfection of plasmids into human primary T cells was done using the Amaxa nucleoporator (Amaxa Inc., Gaithersburg, MD) according to the manufacturer’s instructions. In some experiments, a plasmid encoding CREB (Clontech, Mountain View, CA), Sp1 (InvivoGen, San Diego, CA) or corresponding empty vector (pCMV or pORF9) were cotransfected into primary T cells with PP2Acα-PGL3 basic construct. 0.25μg of pRL-TK (Promega) was cotransfected as an internal control for transfection efficiency in all cases. The total amount of plasmid used in each sample was up to 5 μg.
Following transfection, 3 × 106 T cells were cultured in 12-well plates and harvested after 24 hours incubation. Cytoplasmic extracts were prepared using a luciferase assay kit (Promega), and luminescence was measured immediately for 30 seconds using AutoLumat LB 953 (EG&G Berthold, Bad Wildbad, Germany). The luciferase activity was normalized using Renilla readings.
Electrophoresis mobility shift assay (EMSA)
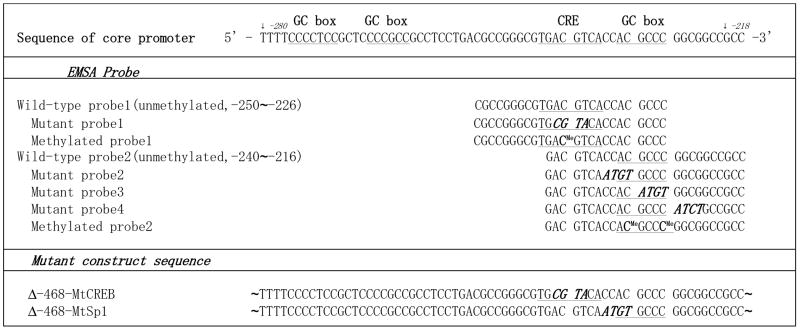
The purification of nuclear proteins and the conduction of EMSA have been described previously with minor modification (12). Briefly, the nuclear extracts of normal T cells were obtained by treatment with lysis buffer (20 mM HEPES (pH 7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM NaF, 1 mM Na3VO4, 1 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride, 2 mM aprotinin, and 1 mM leupeptin) after the collection of cytoplasmic extract. Oligonucleotides and methylated oligonucleotides were purchased from Operon Biotechnologies Inc. (Huntsville, AL). The sequences of the oligonucleotides used are shown in Table 1. All listed oligonucleotides included antiparallel partner oligonucleotides. In case of the methylated oligonucleotides, the antisense oligonucleotides pair was also methylated at the corresponding cytosine as the sense strand. Each pair of these complementary oligonucleotides were annealed and used as probe or competitor. The nuclear extracts (1μg) were incubated with1 μg of polydeoxyinosinic-deoxycytidylic acid (poly(dI-dC)) in the binding buffer (20mM Hepes (pH 7.5), 50mM KCL, 5mM MgCl2, 10μM ZnCl2, 4% Glycerol, and 100μg of BSA per ml) for 15 minutes at room temperature (29). For some samples, unlabeled double-stranded oligonucleotides were added as competitors at 10-fold moler excess. For supershift assays, the nuclear proteins were incubated with 1μg of anti-CREB antibody, anti- pCREB antibody (Upstate Biotechnology Inc., Termecula, CA), anti-cAMP response element modulator (CREM) antibody (Aviva system biology, San Diego, CA) or anti-Sp1 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA) at this time. After the incubation, the radiolabeled DNA probe was added and incubated for 20 minutes at room temperature. The reaction mixture was then subjected to separation in a 6% nondenaturing gel (Invitrogen Life Technologies, Carlsbad, CA). The dried gel was then autoradiographed.
Table 1.
Sequence of oligonucleotides used in EMSA and mutant constructs
Reverse transcription and real-time PCR
RNA extracted from T cells (3 × 106) (RNEasy Mini Kit; Qiagen) was treated with DNase I (Qiagen). Total RNA (300 ng) was transcribed in cDNA in a conventional thermocycler using AMV reverse transcriptase and oligo-dT primer (RT-PCR kit; Promega). Real-time RT-PCR was performed in triplicate for every sample with a LightCycler® 480 System by adding SYBR green (Roche) to the reaction mixture. Primers used were: PP2Acα forward 5′-TCCGAGTCCCAGGTCAAGAG -3′, reverse 5′-GCTACAAGCAGTGTAACTGTTTCA -3′; and Glyceraldehyde phosphate dehydrogenase (GAPDH) forward 5′-CAACTACATGGT TTACATGTTCC -3′, reverse 5′-GGACTGTGGTCATGAGTCCT -3′. The averaged cycle threshold (CT) values of each reaction derived from the target gene, determined with LightCycler® 480 System software, were normalized to GAPDH levels. ΔCT was calculated using the following equation: ΔCT = CT of target − CT of GAPDH.
Methylation-specific PCR
Genomic DNA (1 μg) from primary T cells treated with 5-azaC for 48 hours was purified and treated with the methylation sensitive enzyme, Aat II (New England Biolabs Inc.). After DNA repurification, 50 ng of DNA were used as a template for PCR. To distinguish between methylated and non-methylated status within the CRE motif in the PP2Acα promoter, two sets of primers were used. F3 (−468) primer was used as forward primer of both control and methylation-sensitive products as described above. Reverse primer for methylation-sensitive product was the same as R (−83). Reverse primer for control product, R (−286) was the following oligonucletotide 5′-CGAAGCTTATGCCACCCGCCCCAG-3′. The Aat II recognition site was contained in the fragments generated by F3 and R (−83), but not in those by F3 and R (−286). PCR products were electrophoresed on 1% agarose gels, visualized by ethidium bromide staining and semiquantified with QuantityOne software.
Chromatin immunoprecipitation assays (ChIP)
ChIP analysis was done using ChIP assay kit (Upstate Biotechnology Inc.) according to the manufacturer’s instructions. T cells (3 × 106) with or without 5-azaC treatment were used per immunoprecipitating antibody. The cells were fixed with 1% formaldehyde for 10 minutes, lysed and sonicated. The DNA-protein complexes were immunoprecipitated with anti-pCREB antibody or its control antibody (Rabbit normal IgG; Santa cruz biotechnology Inc.) and after a series of washing steps, the cross-linking was reversed and the protein was digested with proteinase K (Qiagen). The DNA was extracted by QIAquick PCR purification kit (Qiagen). Immunoprecipitated and purified DNA was analyzed by PCR using the primers F3 (−468) and R (−83) described as above, which is specific for human PP2Acα promoter and were used to amplify a promoter fragment containing CRE motif. The corresponding nonimmunoprecipitated DNA (Input DNA) was also analyzed by same method for the control. PCR products were semiquantified using the same method as above in the methylation-specific PCR.
Statistics
Data are presented as mean value ± S.D. The paired two-tailed Student’s t-test and the Pearson product moment correlation coefficient were used for statistical analysis. Statistical significance was defined as P < 0.05.
Results
Identification of regions within the PP2Acα promoter responsible for the control of its activity in normal human T cells
To investigate the transcriptional mechanisms responsible for regulating PP2Acα expression, we first identified the PP2Acα promoter sequence via a BLAST search of the human genome against the cDNA sequence of the PP2Acα gene. Using human genomic DNA as a template, we amplified a 1023bp fragment containing the PP2Acα 5′-flanking region, upstream of the translation initiation start site (ATG), defined as +0. As shown in Figure 1A and 1B, the region was characterized by a high GC content and revealed the existence of several potential Sp1 transcription factor sites and a complete CRE motif. The major transcription start site is located at −211 position which was identified first using the primer extension method (6) and was confirmed by information from GenBank database (accession number; AK097599) (30).
FIGURE 1. Functional analysis and nucleotide sequence of the PP2Acα promoter.

A. Nucleotide sequence and putative regulatory elements within bp-1104/+2 of the 5′-flanking region of the human PP2Acα gene. The translation initiation codon, ATG, is indicated as +0. The major transcription factor is located at −211 position. The potential Sp1 transcription factor sites are highlighted by underlining and a complete CRE motif is boxed. The starting points of each primer used for the amplification of the fragments which were cloned into the pGL3-basic vector are indicated with arrows. The DNA sequence comparison between rat and human revealed that the genomic DNA is almost identical and contained a set of CRE and GC box immediately preceding each major transcription start site. B. Schematic representation of 5′-flanking region of PP2Acα and its CpG islands. Prediction of putative CpG islands was performed using CpG finder at the European Bioinformatics Institute (EMBL; http://www.ebi.ac.uk/). The parameters defined were: CpG length >200 bp, G+C >50% and a ‘CpG value’ (the ratio of observed to expected frequencies of the CpG dinucleotide) of at least 0.6. With the exception of the area between −630 and −592 the whole region qualifies as a CpG island. C. Activity of different parts of PP2Acα promoter in normal human T cells. Each construct or empty vector (5μg) were transfected into normal human T cells with pRL-TK plasmid as an intrinsic control of transfection efficacy. Luciferase activity was normalized to Renilla activity and is relative to pGL3-basic (empty vector), which was set as 1.0. Compared with the basal activity of empty vector (shown as a white bar), the promoter activity of all constructs was increased at statistically significant levels (P value of Δ-218 is 0.0232. P value of all other constructs is less than 0.01.). Compared with the activity of the shortest length construct (shown as a hatched bar), all constructs showed statististically significant higher activity (P < 0.02). The results represent the mean ± S.D. of 6 independent experiments.
To determine whether this fragment (nt –1104 to -83) defined an active promoter we cloned it into pGL3-basic luciferase construct and transiently transfected it into human normal T cells using the empty vector and a Renilla construct as controls. We also tested the promoter activity of several constructs which had different 3′ ends. Although the longer construct (nt -1104 to -2) had similar levels of promoter activity, the shorter one (nt -1104 to -180) displayed reduced activity compared to the Δ-1104 construct (nt -1104 to -83) (data not shown). Subsequently, we generated several truncated constructs by progressive deletion of nucleotides from the 5′ end and placed these fragments in the luciferase reporter construct to define the minimal sequences required for the transcriptional initiation of PP2Acα expression.
Compared to the basal activity of the empty vector, the promoter activity of all constructs was higher (p<0.05) and among them construct Δ-468 (nt -468 to -83) always displayed the highest activity (Fig. 1C). Moreover, the region between −280 and −218 was determined to be sufficient to support the majority of the promoter activity because the construct without this region showed significantly reduced promoter activity. Within this region there is a complete CRE motif surrounded by three potential Sp1 binding sequences immediately preceding the major transcription start site. When compared the human sequence of the region preceding the transcription start site to that of the rat, we found that this region is evolutionarily well-preserved and therefore of importance. (Fig.1A).
Binding of CREB and Sp1 proteins to the core promoter region of PP2Acα
In order to identify whether these _cis_-acting elements physically interacted with trans factors, we incubated nuclear protein from normal T cells with two labeled double-stranded oligonucleotides containing either the CRE motif or the Sp1 site at the center. The sequence of the oligonucleotides that we used in EMSA is shown in Table 1. Shift assays shown in Fig. 2A and 2B demonstrate the formation of one band (lane 1; indicated by asterisk). Although there are two candidates for Sp1 binding upstream of the CRE motif, no nuclear protein binding to the labeled probe was recorded (data not shown).
FIGURE 2. EMSA analysis of the core promoter region of the PP2Acα promoter.

A. Wild-type probe1 (W1), 32P-end labeled double-stranded oligonucleotides including the CRE (nt -250 to -226), was incubated with 1μg of nuclear protein from normal human T cells. For competition EMSA or supershift assays, excess amount of unlabeled W1 or mutant probe (Mt1) or 1μg of each antibody were preincubated with nuclear protein. The putative CREB/pCREB and DNA complexes marked with an asterisk whereas the arrow indicates the supershifted complexes. B. A similar experiment was conducted with wild-type probe2 (W2) probe including the GC box in the center (nt -240 to -216). Three different mutant oligonucleotides (Mt2-4) were used as competitors to determine the critical site for Sp1 binding. The sequences of all probes used in these experiments are shown in Table 1. The putative Sp1 and DNA complex is shown as an asterisk whereas the arrow indicates the supershifted complex.
We conducted competitive EMSA using unlabeled wild-type oligonucleotides or several oligonucleotides mutated around the consensus binding site to determine the exact binding requirements for nuclear protein. Both (W1 and W2) unlabeled wild-type probes out competed the binding between the labeled wild-type probe and nuclear protein and this confirmed the specificity of the bands (lane 2 and 3 in Fig.2A and lane 2 in Fig2B). The observation that unlabeled mutant oligonucleotides (Mt1 in CRE motif, Mt2 and Mt3 in Sp1 site) did not interrupt protein-oligonucleotide interaction supports the claim that the mutated region is critical for the binding of these proteins (Fig. 2A, lanes 4 & 5 and Fig.2B, lanes 3 & 4). The third Sp1 site (downstream of the CRE site, defined by the W2 probe) defined yet another GC region could also bind Sp1. We generated another mutated oligonucleotide (Mt4) which when used in competitive EMSA inhibited the binding of Sp1 suggesting that it is not important for the binding (Fig. 2B, lane 5). Supershift assays were performed at the same time to determine which transcription factors bind this sequence in a specific manner. CREB and its activated form, pCREB, could bind to the CRE motif (Fig 2A, lanes 6 to 8), but anti-CREM antibody did not affect the location and density of the band even though CREM can bind to the CRE motif (Fig.2A, lane 9). The presence of anti-Sp1 antibody in the sample containing nuclear protein and labeled W2 probe resulted in a clear shift of the band (Fig.2B, lane 6). Antibodies against other candidate transcription factors binding the W2 sequence such as CREM, transcription factor IIB (TFIIB) did not result in a similar band shift (data not shown).
CREB and Sp1 regulate the PP2Acα promoter activity
Because the competition shift assay shown in Fig. 2A and 2B suggested the presence of critical binding sites for CREB and Sp1, we cloned the PP2Acα promoter with the CRE motif or Sp1 site mutated upstream of the luciferase gene and compared their activity to that driven by the wild type promoter in human T cells. We chose the Δ-468 construct as a wild type because it displayed the most potent promoter activity (shown in Fig. 1C) and the GC box was highly represented within this region. The mutant constructs defined the same pattern of mutations as these designed for the mutant oligonuclotides which we used in EMSA (Table 1). As shown in Fig 3A, the promoter activity of each of the mutant constructs was suppressed compared to the activity of the wild-type construct.
FIGURE 3. CREB and Sp1 are essential for the PP2Acα promoter activity.

A. Mutagenesis within CRE or Sp1 sites of the PP2Acα core promoter region suppressed the promoter activity. Five μg of wild type construct (Δ-468; nt -468 50 -83) or its mutant constructs (Δ-468-MtCREB or Δ-468-MtSp1) were transfected into normal human T cells. Each sequence of mutant construct is shown in Table1. Luciferase activity was normalized to Renilla activity and normalized luciferase activity for the wild type promoter was set as 100%. Compared to the basal activity of wild type construct (shown as a white bar), the promoter activities of the mutated constructs were decreased significantly. The results represent the mean ± S.D. of 5 independent experiments. B. CREB and Sp1 overexpression increases the activity of the PP2Acα promoter. The pCMV-CREB, pORF9-Sp1 or its corresponding empty vector (2 μg) was cotransfected with the Δ-468 construct into normal human T cells. The normalized luciferase activity of the sample cotransfected with empty vector was set as 100% (shown as a white bar). The promoter activity increased in the presence of cotransfected plasmids encoding CREB or Sp1 gene. The results represent the mean ± S.D. of 3 independent experiments. C. CREB and Sp1 overexpression also induced increased PP2Acα mRNA expression levels. Three μg of pCMV-CREB, pORF9-Sp1, or its corresponding empty vector, were transfected into normal human T cells and cells were harvested 18 hours later. PP2Acα transcripts were measured and normalized to GAPDH by real-time RT-PCR. The relative ratio of PP2Acα RNA in T cells transfected with empty vector was set as 1.0 for a control (shown in white bar). The relative expression levels of PP2Acα mRNA were also increased following overexpression of each transcription factor (black bars). The results represent the mean ± S.D. of 4 independent experiments.
Next, we designed experiments to determine the effect of the identified transcription factors on the PP2Acα promoter activity. T cells were cotransfected with 2 μg of pCMV-CREB, pORF9-Sp1 or the empty vector (pCMV or pORF9) with 3 μg of Δ-468 PP2Acα promoter-luciferase fusion construct. Similar to other transfection experiments, 0.25μg of pRL-TK was also cotransfected as an internal control for transfection efficiency in all samples. Compared to transfected samples with empty vector, the level of relative promoter activity was increased in the samples in which pCMV-CREB or pORF9-Sp1 had been cotransfected (Fig. 3B). We also quantified the PP2Acα mRNA expression levels of primary T cells transfected with CREB or Sp1 encoding plasmids using real-time RT-PCR. Overexpression of these transcription factors up-regulated the expression of PP2Acα (Fig.3C). Taken together, CREB and Sp1 bind to the PP2Acα promoter and enhance its activity.
Methylation of the PP2Acα promoter affects CREB binding to the CRE motif and suppresses its activity
The regulation of gene expression is a complex process that is achieved through the action of selective transcription factors, as well as via epigenetic regulatory mechanism, including DNA methylation and histone modification. Methylation of dC bases in the CpG dinucleotide promotes a repressive chromatin structure inaccessible to transcription factors, suppressing gene expression. Having detected the presence of concentrated CpG islands with the core PP2Acα promoter, we conducted experiments to determine the effect of DNA methylation on the regulation of its activity (Fig.1B).
The CRE motif has one CpG dinucleotide at the center. We constructed an oligonucleotide in which the dC base at −238 was converted to deoxymethylcytosine (dmC) with the complementary antisense oligonucleotides, which was also methylated at the corresponding dC as the sense strand (Table 1). Unmethylated or methylated pairs of the complementary oligonucleotides were annealed and labeled with 32P. Unlabeled double strand oligonucleotides were used as competitors. Supershift assays were performed at the same time to prove the specificity of the bound protein. Comparison of the bands present within lanes 1 and 5 (Fig.4A) demonstrated that methylation at −238 in the CRE motif inhibited protein binding. Usage of the methylated probe in competitive assays failed to inhibit the interaction between nuclear protein and labeled unmethylated probe (lanes 2 and 3).
FIGURE 4. Effect of DNA methylation on the PP2Acα promoter.

A. DNA methylation within specific transcription factor binding sites interfered with the binding of CREB but not of Sp1 to the PP2Acα promoter. EMSA analysis was performed to investigate the influence of the ability of each transcription factor to bind to the promoter after methylation. Unmethylated (U1 and U2, similar to W1 and W2 in Fig.2) and methylated probes (Me1 and Me2), which have only 1 or 2 bases changed from dC to dmC within each binding motif, were labeled with 32P after annealing. The complete sequences of all oligonucleotides are shown in Table 1. For competition EMSA, unlabeled probes were preincubated before the addition of labeled probe. Supershift assays were also conducted at the same time using CREB or Sp1 antibody. The specific transcription factor binding band is shown by an asterisk and the supershifted band is marked with an arrow. B. Methylation suppresses PP2Acα promoter activity. The PP2Acα-luciferase fusion construct (Δ-468) and its empty vector were treated with or without CpG Methyltransferase (_M. Sss_I) and its substrate SAM. The normalized luciferase activity for the mock-plasmid (unmethylated construct) was set as 100% (shown as a white bar). The promoter activity decreased significantly following DNA methylation. The results represent the mean ± S.D. of 3 independent experiments.
We synthesized an oligonucleotide in which the −230 and −226 dC bases within the Sp1 binding site were converted to dmC to determine the effect of methylation on Sp1 binding. We did not detect any difference in Sp1 binding to methylated or unmethylated oligonucleotides (lane 9 and 13). Both unmethylated and methylated competitors could interrupt the protein binding to the labeled probe. These results revealed that the methylation of CRE motif inhibited the interaction to CREB directly, whereas methylation of the Sp1 site did not affect protein binding.
Furthermore, the pGL-PP2Acα promoter construct (Δ-468) and pGL3-basic vector were methylated using _M. Sss_I and its substrate SAM to determine the effect of DNA methylation on the activity of the promoter. As shown in Figure 4B, the promoter activity displayed by the methylated construct was significantly suppressed compared to the mock methylated construct.
DNA methylation inhibitors induced increased pCREB binding to hypomethylated CRE motif in the promoter and higher expression of PP2Acα
Next, we induced DNA hypomethylation in primary T cells using well-known DNA methylation inhibitor 5-azaC in order to determine the biological significance of our findings. Because DNMT inhibitor acts during the S phase of cell division and alters the methylation status in daughter cells, we treated human T cells with IL-2 prior to treatment with 5-azaC.
At first, we determined the effect within the CREB binding site of the PP2Acα promoter following treatment of cells with the DNMT inhibitor. DNA was purified from T cells treated with or without 10μM of 5-azaC for 48 hours and incubated with the methylation-sensitive restriction enzyme Aat II, which recognizes only unmethylated CRE motifs. The product was subjected to PCR using primers as described in the Methods section. The presence of dmC in the CRE motif prevents digestion by Aat II and a strong band can be detected using PCR. As shown in Figure 5A, treatment of T cells with 5-azaC reduced the amount of methylated DNA within the CRE motif of the PP2Acα promoter as the intensity of the PCR bands was decreased. In contrast the intensity of the PCR products of an area of the promoter which does not define Aat II sensitive motifs, referred to as control band, did not change.
FIGURE 5. Treatment of T cells with a DNA methylation inhibitor induced increased pCREB binding to hypomethylated CRE motif in the promoter and higher expression of PP2Acα.

A. Normal human T cells preincubated with IL-2 were treated with or without 10μM of 5-azaC, a DNA methylation inhibitor, for 48 hours and DNA was purified to determine its effect on the CREB binding site of the PP2Acα promoter. We used the methylation sensitive restriction enzyme Aat II which recognizes only unmethylated CRE motifs, then PCR was conducted using two set of primers to distinguish unmethylated or methylated status within this region. The band can be detected only when dmC remains in CRE motif because it can not be digested by Aat II. The control bands were generated by another set of primers defining an area of the PP2Acα promoter which did not contain any Aat II sensitive motifs. Each OD value of the bands was measured by densitometry. The ratio of the methylation-sensitive product to the control was calculated for the semiquantification of DNA methylation status within CRE of the promoter and the relative ratio of the sample from untreated T cells was set as 1.0. 5-azaC reduced the amount of methylated DNA within CRE in PP2Acα promoter. The results represent the mean ± S.D. of 4 independent experiments. B. DNA binding of pCREB to the PP2Acα promoter was examined using ChIP assays. T cells were treated with or without 10μM of 5-azaC for 24 hours or 48 hours and harvested to perform ChIP assays. Cells were fixed with formalin and sonicated. The anti-pCREB antibody and its control antibody precipitates were subjected to PCR using PP2Acα promoter –specific primers and visualized on a 1% agarose gel. The corresponding non-immunoprecipitated DNA (input DNA) was also analyzed using the same method. Three independent experiments were performed and the results represent the mean ± S.D. normalized to the input DNA. C. The expression of PP2Acα mRNA was calculated following inhibition of DNA methylation in human normal T cells. RNA from T cells treated with or without 5-azaC for 48 hours at the indicated concentration was purified and PP2Acα transcripts were measured using real-time RT-PCR in untreated cells (shown in white bar) and treated cells (black bars) (normalized against GAPDH). The results represent the mean ± S.D. of 6 independent experiments and were normalized against the untreated control which was set as 1.0.
Subsequently, we investigated the effect of 5-azaC on pCREB binding to the PP2Acα promoter. ChIP assays revealed that pCREB bound to PP2Acα promoter more intensely when T cells had been treated with 5-azaC (Fig.5B). Sp1 binding was not affected by 5-azaC treatment (data not shown). Finally, PP2Acα transcripts were quantified by real-time RT-PCR after 5-azaC treatment for 48 hours. The mRNA expression levels of PP2Acα were increased in a dose-dependent manner (Fig.5C).
These results indicate that the binding of CREB/pCREB to hypomethylated CRE motif in the PP2Acα promoter plays an important role in the regulation of its promoter activity. Sp1 may also play an important role in the regulation of the PP2Acα promoter activity, but its effect does not depend on the methylation status of the promoter.
Discussion
In this communication we present the first evidence on the transcriptional regulation of PP2Acα. A core area around the −240 site which defines both CRE and Sp1 binding sites is sufficient for the complete promoter activity. More importantly, whereas methylation excludes the binding of CREB to the CRE site, it does not affect the binding of Sp1 to its cis site. As methylation of some gene’s promoters which code for proteins involved in human and murine SLE (21, 24, 28, 29, 37) has been proposed to be of pathogenic importance, our data provide further evidence on its role in the expression of PP2Ac which is involved in the control of the expression of IL-2 (12).
PP2A is a highly abundant and ubiquitously expressed serine-threonine protein phosphatase in eukaryotic cells with various important roles in embryonic development, cell cycle progression, differentiation, oncogenic transformation and signal transduction (1–3). In general, the expression and activity of PP2A are tightly controlled by associating with regulatory subunits, posttranslational modification, or through the interaction with cellular proteins (31, 32). Abnormalities in the expression and function of PP2A have been implicated in several human diseases such as cancer, Alzheimer’s disease and SLE (8–12, 31).
Mammalian cDNA clones encoding two different subunits, termed PP2Acα and PP2Acβ, have been identified. PP2Acα and PP2Acβ differ only by 8 amino acids all found within the first 30 amino acids encoded by the first exon (6). The specific function of these two subunits is still unknown, but PP2Acα is assumed to the predominant type because of the embryonic lethality of PP2A Cα−/− knockout mice and because PP2Acα is expressed at higher levels than PP2Acβ (31–34).
Having established that PP2Ac mRNA is elevated in T cells from patients with SLE we sought to determine how the activity of the promoter is regulated. After cloning the proximal area we conducted experiments aimed at the functional analysis of the PP2Acα promoter. We identified an area around the −240 site to be the essential region of this promoter. It included a complete CRE motif, flanked by three GC-rich areas. Activated CREB and Sp1 could bind to the PP2Acα promoter and significantly up-regulate its activity (Fig. 3B). The fact that the sequence around these binding sites preceding the transcription start site is completely conserved in the different species also supports our claim that this region is important in the regulation of the PP2Acα expression. (35). In general, TATA-less promoters with CRE elements tend to show less induction following stimulation with forskolin, a pCREB inducer, compared with promoters which contain a TATA box (36). However, several reports have revealed that forskolin can induce stronger promoter activity or higher expression of PP2Acα, suggesting that CREB is important in the expression of PP2Acα (35,37). A previous report by Heim et al also demonstrated that a CREB-specific siRNA knockdown prevented the induction of PP2Ac by hepatitis virus C (HCV) protein and this finding is in perfect agreement with our findings because it postulates that CREB is required for the up-regulation of PP2Ac (37). It should be noted that a complete CRE motif is defined by the PP2Acα promoter and not by the PP2Acβ promoter. This, among other factors, could explain the differential expression of the two isoforms of PP2Ac.
The fact that the CRE site is flanked by CpG islands strongly suggested to us that epigenetic mechanisms are involved in the regulation of the expression of the PP2Acα promoter (38). Methylation of DNA represents one of the major epigenetic mechanisms involved in the regulation of gene expression. It is known to stabilize chromatin in an inactive configuration and thus inhibits gene transcription. In mammalian cells, the term DNA methylation usually refers to the postsynthetic methylation of dC residues at the 5-position to form dmC. Nearly all dmC residues are found in the dCs that precede guanines in DNA strands (CpG dinucleotides), and approximately 60%–90% of all CpG sequences in the genome are methylated, while unmethylated CpG dinucleotides are mainly clustered in the CpG-rich sequences (CpG islands) of the promoter region of each gene. Normally, both the core promoter and the transcription initiation site are included within CpG islands, and gene expression is completely repressed when they become hypermethylated (24, 25, 38). Our data show that CREB could not bind to the CRE motif if the dC was converted to dmC and this led to decreased activity of PP2Acα promoter. In contrast, the binding of Sp1 to the core promoter region was not affected by the methylation status of dmC within its binding site. Several reports indicate that the presence of dmC within the promoter inhibits the binding of methylation-sensitive transcription factors including AP-2, c-Myc/Myn, E2F, NF-_κ_B and CREB (39, 40), whereas other transcription factors, such as Sp1 or CAAT-box-binding transcription factor (CTF), can bind to promoter sequences even when the binding site is CpG methylated (24, 41, 42). Binding of transcription factors may be directly inhibited if CpG is methylated or indirectly through the involvement of dmC binding protein 1 and 2 (MeCP-1 and 2) which interferes with the binding of the transcription factors (24, 42, 43). In our studies we did not determine whether the presence of such proteins interferes with the binding of CREB. Furthermore, additional studies may be required to determine whether similar methylation sensitive regulatory processes affect the expression of other components of the PP2A holoenzyme. It has been reported that hypomethylation of the regulatory subunit B (PR55, β) induced its expression and was found to be involved in the expression of estrogen receptors in breast cancer cell-lines (44, 45).
Several studies have suggested that DNA hypomethylation may account for several T cell abnormalities in patients with SLE and to be involved in the pathogenesis of the disease (21, 22, 24, 25). Ultraviolet light and certain drugs known to be associated with lupus-like symptoms (drug-induced lupus), such as procainamide and hydralazine, inhibit DNA methylation in a manner similar to that of 5-azaC. It has been shown that the levels of DNMT-1 which is responsible for the methylation for newly replicated daughter DNA strands during mitosis, were decreased in SLE T cells compared to normal T cells (26, 46). This abnormality may account for the decreased methylation of promoter regions of LFA-1 and CD70 and the increased expression of these genes in SLE patients (27, 28). Also, high levels of hypomethlated CpG are found in the circulation of SLE patients and they have been implicated in the induction of autoreactive T and B cells in these patients (25, 46). Taken together, our finding that CREB could bind only to hypomethylated CRE motifs within the PP2Acα core promoter identifies a mechanism whereby PP2Ac is expressed in high levels in SLE T cells.
In conclusion, we have demonstrated that the activity of the PP2Acα promoter is controlled by the binding of pCREB and Sp1 around a CRE motif flanked by GC boxes and we have shown that pCREB can bind only if the motif is demethylated. Our findings provide novel information on the regulation of the PP2Ac promoter activity. Given that hypomethylation has been claimed to be involved in carcinogenesis (36, 47) and the pathogenesis of SLE (21–25), our studies bring PP2Ac on the forefront of effector molecules.
Footnotes
Disclosures
The authors have no financial conflict of interest.
1
This work was supported by PHS NIH grant RO1 AI068787.
3
Abbreviations used in this paper: PP2A, Protein phosphatase 2A; PP2Ac, PP2A catalytic subunit; PP2Acα, PP2Ac α isoform; PP2Acβ, PP2Ac β isoform; SLE, systemic lupus erythematosus; CRE, cAMP response element; pCREB, phosphorylated CREB; DNMT, DNA methyltransferase; dC, deoxycytosine; 5-azaC, 5-azacitidine; Sp1, stable protein 1; _M. Sss_I, CpG Methyltransferase; SAM, S-adenosyl methionine; CREM, cAMP response element modulator; CT, cycle threshold; ChIP, Chromatin immunoprecipitation assays; TFIIB, transcription factor IIB; dmC, deoxymethylcytosine; MeCP, dmC binding protein; HCV, hepatitis virus C.
References
- 1.Virshup DM. Protein phosphatase 2A: a panoply of enzymes. Current opinion in cell biology. 2000;12:180–185. doi: 10.1016/s0955-0674(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 2.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. The Biochemical journal. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta biochimica Polonica. 2001;48:921–933. [PubMed] [Google Scholar]
- 4.Orgad S, Brewis ND, Alphey L, Axton JM, Dudai Y, Cohen PT. The structure of protein phosphatase 2A is as highly conserved as that of protein phosphatase 1. FEBS letters. 1990;275:44–48. doi: 10.1016/0014-5793(90)81435-q. [DOI] [PubMed] [Google Scholar]
- 5.Stone SR, Hofsteenge J, Hemmings BA. Molecular cloning of cDNAs encoding two isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry. 1987;26:7215–7220. doi: 10.1021/bi00397a003. [DOI] [PubMed] [Google Scholar]
- 6.Khew-Goodall Y, Mayer RE, Maurer F, Stone SR, Hemmings BA. Structure and transcriptional regulation of protein phosphatase 2A catalytic subunit genes. Biochemistry. 1991;30:89–97. doi: 10.1021/bi00215a014. [DOI] [PubMed] [Google Scholar]
- 7.Baharians Z, Schonthal AH. Autoregulation of protein phosphatase type 2A expression. The Journal of biological chemistry. 1998;273:19019–19024. doi: 10.1074/jbc.273.30.19019. [DOI] [PubMed] [Google Scholar]
- 8.Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007;130:21–24. doi: 10.1016/j.cell.2007.06.034. [DOI] [PubMed] [Google Scholar]
- 9.Van Hoof C, Goris J. PP2A fulfills its promises as tumor suppressor: which subunits are important? Cancer cell. 2004;5:105–106. doi: 10.1016/s1535-6108(04)00027-3. [DOI] [PubMed] [Google Scholar]
- 10.Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neuro-Signals. 2002;11:262–269. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- 11.Schweiger S, Schneider R. The MID1/PP2A complex: a key to the pathogenesis of Opitz BBB/G syndrome. Bioessays. 2003;25:356–366. doi: 10.1002/bies.10256. [DOI] [PubMed] [Google Scholar]
- 12.Katsiari CG, V, Kyttaris C, Juang YT, Tsokos GC. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. The Journal of clinical investigation. 2005;115:3193–3204. doi: 10.1172/JCI24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothenberg EV, Ward SB. A dynamic assembly of diverse transcription factors integrates activation and cell-type information for interleukin 2 gene regulation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9358–9365. doi: 10.1073/pnas.93.18.9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wadzinski BE, Wheat WH, Jaspers S, Peruski LF, Jr, Lickteig RL, Johnson GL, Klemm DJ. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Molecular and cellular biology. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kracht M, Heiner A, Resch K, Szamel M. Interleukin-1-induced signaling in T-cells. Evidence for the involvement of phosphatases PP1 and PP2A in regulating protein kinase C-mediated protein phosphorylation and interleukin-2 synthesis. The Journal of biological chemistry. 1993;268:21066–21072. [PubMed] [Google Scholar]
- 16.Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, Gardner JP, Hambor JE, Neveu MJ, Thompson CB. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. 2000;13:313–322. doi: 10.1016/s1074-7613(00)00031-5. [DOI] [PubMed] [Google Scholar]
- 17.Linker-Israeli M, Bakke AC, Kitridou RC, Gendler S, Gillis S, Horwitz DA. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE) J Immunol. 1983;130:2651–2655. [PubMed] [Google Scholar]
- 18.Solomou EE, Juang YT, Gourley MF, Kammer GM, Tsokos GC. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J Immunol. 2001;166:4216–4222. doi: 10.4049/jimmunol.166.6.4216. [DOI] [PubMed] [Google Scholar]
- 19.Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, Kyttaris VC, Tsokos GC. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. The Journal of clinical investigation. 2005;115:996–1005. doi: 10.1172/JCI200522854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsiari CG, Tsokos GC. Transcriptional repression of interleukin-2 in human systemic lupus erythematosus. Autoimmunity reviews. 2006;5:118–121. doi: 10.1016/j.autrev.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Kammer GM, Perl A, Richardson BC, Tsokos GC. Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis and rheumatism. 2002;46:1139–1154. doi: 10.1002/art.10192. [DOI] [PubMed] [Google Scholar]
- 22.Crispin JC, V, Kyttaris C, Juang YT, Tsokos GC. How signaling and gene transcription aberrations dictate the systemic lupus erythematosus T cell phenotype. Trends in immunology. 2008;29:110–115. doi: 10.1016/j.it.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Cedeno S, Cifarelli DF, Blasini AM, Paris M, Placeres F, Alonso G, Rodriguez MA. Defective activity of ERK-1 and ERK-2 mitogen-activated protein kinases in peripheral blood T lymphocytes from patients with systemic lupus erythematosus: potential role of altered coupling of Ras guanine nucleotide exchange factor hSos to adapter protein Grb2 in lupus T cells. Clinical immunology (Orlando, Fla. 2003;106:41–49. doi: 10.1016/s1521-6616(02)00052-9. [DOI] [PubMed] [Google Scholar]
- 24.Januchowski R, Prokop J, Jagodzinski PP. Role of epigenetic DNA alterations in the pathogenesis of systemic lupus erythematosus. Journal of applied genetics. 2004;45:237–248. [PubMed] [Google Scholar]
- 25.Sekigawa I, Kawasaki M, Ogasawara H, Kaneda K, Kaneko H, Takasaki Y, Ogawa H. DNA methylation: its contribution to systemic lupus erythematosus. Clinical and experimental medicine. 2006;6:99–106. doi: 10.1007/s10238-006-0103-x. [DOI] [PubMed] [Google Scholar]
- 26.Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, Hanash SM, Richardson BC. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis and rheumatism. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 27.Lu Q, Kaplan M, Ray D, Ray D, Zacharek S, Gutsch D, Richardson B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis and rheumatism. 2002;46:1282–1291. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 28.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol. 2005;174:6212–6219. doi: 10.4049/jimmunol.174.10.6212. [DOI] [PubMed] [Google Scholar]
- 29.Chang CC, Ye BH, Chaganti RS, Dalla-Favera R. BCL-6, a POZ/zinc-finger protein, is a sequence-specific transcriptional repressor. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6947–6952. doi: 10.1073/pnas.93.14.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, Kimura K, Makita H, Sekine M, Obayashi M, Nishi T, Shibahara T, Tanaka T, Ishii S, Yamamoto J, Saito K, Kawai Y, Isono Y, Nakamura Y, Nagahari K, Murakami K, Yasuda T, Iwayanagi T, Wagatsuma M, Shiratori A, Sudo H, Hosoiri T, Kaku Y, Kodaira H, Kondo H, Sugawara M, Takahashi M, Kanda K, Yokoi T, Furuya T, Kikkawa E, Omura Y, Abe K, Kamihara K, Katsuta N, Sato K, Tanikawa M, Yamazaki M, Ninomiya K, Ishibashi T, Yamashita H, Murakawa K, Fujimori K, Tanai H, Kimata M, Watanabe M, Hiraoka S, Chiba Y, Ishida S, Ono Y, Takiguchi S, Watanabe S, Yosida M, Hotuta T, Kusano J, Kanehori K, Takahashi-Fujii A, Hara H, Tanase TO, Nomura Y, Togiya S, Komai F, Hara R, Takeuchi K, Arita M, Imose N, Musashino K, Yuuki H, Oshima A, Sasaki N, Aotsuka S, Yoshikawa Y, Matsunawa H, Ichihara T, Shiohata N, Sano S, Moriya S, Momiyama H, Satoh N, Takami S, Terashima Y, Suzuki O, Nakagawa S, Senoh A, Mizoguchi H, Goto Y, Shimizu F, Wakebe H, Hishigaki H, Watanabe T, Sugiyama A, Takemoto M, Kawakami B, Yamazaki M, Watanabe K, Kumagai A, Itakura S, Fukuzumi Y, Fujimori Y, Komiyama M, Tashiro H, Tanigami A, Fujiwara T, Ono T, Yamada K, Fujii Y, Ozaki K, Hirao M, Ohmori Y, Kawabata A, Hikiji T, Kobatake N, Inagaki H, Ikema Y, Okamoto S, Okitani R, Kawakami T, Noguchi S, Itoh T, Shigeta K, Senba T, Matsumura K, Nakajima Y, Mizuno T, Morinaga M, Sasaki M, Togashi T, Oyama M, Hata H, Watanabe M, Komatsu T, Mizushima-Sugano J, Satoh T, Shirai Y, Takahashi Y, Nakagawa K, Okumura K, Nagase T, Nomura N, Kikuchi H, Masuho Y, Yamashita R, Nakai K, Yada T, Nakamura Y, Ohara O, Isogai T, Sugano S. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nature genetics. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- 31.Schild A, Ittner LM, Gotz J. Altered phosphorylation of cytoskeletal proteins in mutant protein phosphatase 2A transgenic mice. Biochemical and biophysical research communications. 2006;343:1171–1178. doi: 10.1016/j.bbrc.2006.03.066. [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Stock J. Protein phosphatase 2A catalytic subunit is methyl-esterified at its carboxyl terminus by a novel methyltransferase. The Journal of biological chemistry. 1993;268:19192–19195. [PubMed] [Google Scholar]
- 33.Gotz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12370–12375. doi: 10.1073/pnas.95.21.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gotz J, Schild A. Transgenic and knockout models of PP2A. Methods in enzymology. 2003;366:390–403. doi: 10.1016/s0076-6879(03)66029-5. [DOI] [PubMed] [Google Scholar]
- 35.Kitagawa Y, Shima H, Sasaki K, Nagao M. Identification of the promoter region of the rat protein phosphatase 2A alpha gene. Biochimica et biophysica acta. 1991;1089:339–344. doi: 10.1016/0167-4781(91)90174-k. [DOI] [PubMed] [Google Scholar]
- 36.Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Molecular cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- 37.Christen V, Treves S, Duong FH, Heim MH. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology (Baltimore, Md. 2007;46:558–565. doi: 10.1002/hep.21611. [DOI] [PubMed] [Google Scholar]
- 38.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochimica et biophysica acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Current opinion in genetics & development. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 40.Rountree MR, Selker EU. DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes & development. 1997;11:2383–2395. doi: 10.1101/gad.11.18.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holler M, Westin G, Jiricny J, Schaffner W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes & development. 1988;2:1127–1135. doi: 10.1101/gad.2.9.1127. [DOI] [PubMed] [Google Scholar]
- 42.Cross SH, Meehan RR, Nan X, Bird A. A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nature genetics. 1997;16:256–259. doi: 10.1038/ng0797-256. [DOI] [PubMed] [Google Scholar]
- 43.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science (New York, NY) 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keen JC, Garrett-Mayer E, Pettit C, Mack KM, Manning J, Herman JG, Davidson NE. Epigenetic regulation of protein phosphatase 2A (PP2A), lymphotactin (XCL1) and estrogen receptor alpha (ER) expression in human breast cancer cells. Cancer biology & therapy. 2004;3:1304–1312. doi: 10.4161/cbt.3.12.1458. [DOI] [PubMed] [Google Scholar]
- 45.Keen JC, Zhou Q, Park BH, Pettit C, Mack KM, Blair B, Brenner K, Davidson NE. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. The Journal of biological chemistry. 2005;280:29519–29524. doi: 10.1074/jbc.M505317200. [DOI] [PubMed] [Google Scholar]
- 46.Ogasawara H, Okada M, Kaneko H, Hishikawa T, Sekigawa I, Hashimoto H. Possible role of DNA hypomethylation in the induction of SLE: relationship to the transcription of human endogenous retroviruses. Clinical and experimental rheumatology. 2003;21:733–738. [PubMed] [Google Scholar]
- 47.Esteller M. Epigenetics in cancer. The New England journal of medicine. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]