Zebrafish colgate/hdac1 functions in the non-canonical Wnt pathway during axial extension and in Wnt-independent branchiomotor neuron migration (original) (raw)
. Author manuscript; available in PMC: 2009 Jun 25.
Published in final edited form as: Mech Dev. 2007 Jul 14;124(9-10):682–698. doi: 10.1016/j.mod.2007.07.003
Abstract
Vertebrate gastrulation involves the coordinated movements of populations of cells. These movements include cellular rearrangements in which cells polarize along their medio-lateral axes leading to cell intercalations that result in elongation of the body axis. Molecular analysis of this process has implicated the non-canonical Wnt/Frizzled signaling pathway that is similar to the planar cell polarity pathway (PCP) in Drosophila. Here we describe a zebrafish mutant, colgate (col), which displays defects in the extension of the body axis and the migration of branchiomotor neurons. Activation of the non-canonical Wnt/PCP pathway in these mutant embryos by overexpressing Δ_Ndishevelled, rho kinase2_ and van gogh-like protein 2 (vangl2) rescues the extension defects suggesting that col acts as a positive regulator of the non-canonical Wnt/PCP pathway. Further, we show that col normally regulates the caudal migration of nVII facial hindbrain branchiomotor neurons and that the mutant phenotype can be rescued by misexpression of vangl2 independent of the Wnt/PCP pathway. We cloned the col locus and found that it encodes histone deacetylase1 (hdac1). Our previous results and studies by others have implicated hdac1 in repressing the canonical Wnt pathway. Here, we demonstrate novel roles for zebrafish hdac1 in activating non-canonical Wnt/PCP signaling underlying axial extension and in promoting Wnt-independent caudal migration of a subset of hindbrain branchiomotor neurons.
Keywords: Wnt signaling, histone deacetylase, convergent extension, migration, PCP, zebrafish
Introduction
The basic vertebrate body plan consists of the three germ layers that emerge during gastrulation. Carefully orchestrated movement of groups of cells relative to each other culminates in the transformation of an unstructured mono-layered blastula into a gastrula with germ layers. Cell intercalations result in the elongation of the body axis. An important driving force for these cell movements is a process known as convergent-extension (CE). Studies suggest that CE in zebrafish has at least two distinct components (Kane and Warga, 1994; Solnica-Krezel et al., 1995; Wallingford et al., 2002). The first involves directed migration of cells towards the dorsal side of the gastrula, termed dorsal convergence. Convergence is a migratory event not involving cell rearrangements. This is followed by cellular rearrangements where cells converging at the dorsal midline become polarized along the medio-lateral axis resulting in cell intercalations and elongation of the body axis.
The dissociation of convergence and medio-lateral intercalation and extension is evident from zebrafish mutants affecting CE movements differently. For example, in silberblick (slb) mutants, both convergence and extension movements are defective (Heisenberg et al., 2000), whereas in no tail (ntl) and somitabun (sbn) mutants convergence is significantly affected with extension occurring almost normally (Myers et al., 2002; Solnica-Krezel et al., 1996). In knypek (kny) mutants, mediolateral intercalations that underlie CE movements are impaired (Topczewski et al., 2001).
The molecular basis for CE movements in vertebrates is incompletely understood. The polarization of cells within the plane of tissues undergoing CE in vertebrate embryos is akin to the polarization of epithelial cells in the insect cuticle. In Drosophila, the orientation of cells in a plane, planar cell polarity (PCP), is regulated by a non-canonical Wnt signaling cascade. As in the case of the canonical Wnt signaling pathway, this pathway also uses the Frizzled receptor and Dishevelled (Dsh). Other proteins, such as Inversin, (Simons et al., 2005), Diversin, (Schwarz-Romond, 2002), Naked and Casein Kinase 1 (Yan et al, 2001; McKay et al., 2001), all of which either interact directly with Dsh or with Dsh-associated proteins, have been shown to regulate both Wnt pathways. However, downstream of Dsh, PCP signaling recruits a different set of molecules including Van gogh-like protein 2, Prickle, and JNK (Shulman et al, 1998; Boutros and Mlodzik, 1999; Adler and Lee, 2001).
Recent studies have revealed that the orthologs of PCP pathway molecules control CE during gastrulation in Xenopus and zebrafish (Park and Moon, 2002; Kibar et al., 2001; Carreira-Barbosa et al., 2003). Mutant versions of Dsh have implicated the PCP signaling pathway as a regulator of CE movements in vertebrates (Heisenberg et al., 2000; Tada and Smith, 2000; Wallingford et al., 2000). A construct of Dsh that specifically disrupts PCP signaling in Drosophila, but does not affect the canonical Wnt pathway was able to block CE movements in both Xenopus and zebrafish (Wallingford et al., 2000; Heisenberg et al., 2000). Conversely, deletion constructs of Dsh that are unable to activate the canonical Wnt pathway were shown to rescue CE in silberblick, a zebrafish wnt11 mutant, as well as the overexpression of a dominant-negative form of wnt11 in Xenopus embryos (Tada and Smith, 2000; Heisenberg et al, 2000). In addition to dsh, other PCP genes also have homologs in vertebrates. For example, the zebrafish trilobite mutant is defective in the homolog of the van gogh-like protein 2 gene and is expressed in cells undergoing CE (Park and Moon, 2002). Two homologs of prickle that regulate gastrulation movements in zebrafish have been identified recently (Veeman et al., 2003; Carreira-Barbosa et al., 2003). Additionally, other CE genes specific to vertebrates have been isolated, including the formin morphology protein daam-1, knypek/glypican 4/6 and Wnt ligands wnt5/pipetail and wnt11/silberblick (Hammerschmidt et al., 1996; Heisenberg et al., 2000; Jessen et al., 2002; Kilian et al., 2003; Rauch et al., 1997; Solnica-Krezel et al., 1996; Topczewski et al., 2001).
Chromatin modifications play a key role in regulating eukaryotic gene expression (Jenuwein and Allis, 2001). Histones have numerous sites where post-translational modifications occur, and the pattern of modification encodes the expression status of a gene (Strahl and Allis, 2000; Rice and Allis, 2001). The silencing of gene expression has been found to be associated with deacetylation whereas acetylation of histones is associated with activation of gene expression (Allfrey, 1966). Histone deacetylases (HDACs) are primarily nuclear enzymes involved in removing acetyl groups from histone lysine tails (de Ruijter et al., 2003; Marks et al., 2003). A role for Hdac1 in repressing the expression of canonical Wnt target genes has been shown in Drosophila and vertebrates (Chen et al., 1999; Billin et al., 2000; Brantjes et al., 2001; Yamaguchi et al., 2005). Hdac1 has been shown to exert its repressive function via association with Groucho and LEF1 in the nucleus (Chen et al., 1999; Brantjes et al., 2001; Billin et al., 2000). Roles for zebrafish hdac1 in notch and sonic hedgehog signaling have also been reported (Yamaguchi et al., 2005; Cunliffe, 2004).
We have shown that the zebrafish mutant colgate (col) displays defects in early dorso-ventral and brain patterning (Nambiar and Henion, 2004) that can exclusively be rescued by overexpression of canonical Wnt pathway antagonists (Nambiar and Henion, 2004). Here, we show that col mutants also display defects both in axial extension and the migration of a subset of hindbrain branchiomotor neurons that can be selectively and differentially rescued by overexpressing molecules of the non-canonical Wnt/PCP signaling pathway. We have cloned the col locus and found that it encodes histone deacetylase 1 (hdac1). In this study we demonstrate novel roles for Hdac1 in the non-canonical Wnt/PCP pathway during axial extension as well as in Wnt/PCP-independent neuronal migration, functions not previously attributed to hdac1.
Materials and Methods
Fish strains
Adult zebrafish and embryos were maintained at 28.5 C and staged by hours post fertilization (hpf), days post fertilization (dpf) or morphological criteria (Kimmel et al., 1995). Mutant embryos (*AB and WIK background) were collected from pair-wise matings of heterozygous adults. All phenotypic analyses of col mutants were done using embryos homozygous for the col b382 allele (Henion et al., 1996).
Axis length, somite number and notochord diameter measurements
To compare axis length between col mutant and wildtype embryos (Table 1), images of wild-type and col/hdac1 mutant embryos were taken at identical positions under a Leica compound light microscope and then measurements were made using the Leica SPOT v 4.0 software measurement tools that were calibrated with a standard stage micrometer. 20 wild-type and 20 col/hdac1 mutants were used for analysis at 25 hpf and 10 each of wild-type and col/hdac1 mutant embryos were measured at 48 hpf and 72 hpf. The data obtained was analyzed using 2 way ANOVA and Bonferroni post hoc tests. Graph prism pad version 4.0 software was used to conduct statistical analysis and graph data.
Table 1.
Statistical analysis of mean length of wild-type and col/hdac1 embryos at 25 hpf, 48 hpf and 72 hpf.
| Hours post fertilization | Mean length of embryos in mm ± 1 SD | P value | |
|---|---|---|---|
| wild-type | col/hdac1 | ||
| 25 | 2.115± 0.085n=20 | 1.943± 0.058n=20 | P< 0.001 |
| 48 | 2.934± 0.054n=10 | 2.710± 0.052n=10 | P< 0.001 |
| 72 | 3.466± 0.067n=10 | 2.684± 0.184n=10 | P< 0.001 |
Quantification of somite numbers in time-matched (hpf) col mutant and wildtype embryos was performed at 16hpf, 27hpf and 48hpf (Table 2). Embryos were obtained from col heterozygous adults. Somite pairs of a clutch of live individual embryos at 16hpf were counted within 25 minutes and the embryos were allowed to develop to 27hpf when the col phenotype is readily apparent in order to assign genotype to individual embryos. For counts at 27hpf and 48hpf, embryos were anaesthetized with tricaine to immobilize them for counts.
Table 2.
Somite counts of wildtype and col mutants at 16, 27 and 48 hour post fertilization
| Hours post fertilization | Number of somites ± 1 SD | P value | |
|---|---|---|---|
| wild-type | col | ||
| 16 | 15.5± 0.93n= 24 | 14.9± 0.99n=20 | P>0.05 |
| 27 | 30.1± 0.88n=10 | 28.7± 1.25n=10 | P< 0.01 |
| 48 | 29.8± 0.63n=10 | 28.5± 0.85n=10 | P< 0.01 |
For notochord diameter quantification (Table 3),10 embryos of each type (wildtype uninjected, col uninjected, Δ_Ndsh_ injected col and wildtype, rok2 injected col and wildtype and vangl2 injected col and wildtype) at 48 hpf were fixed and labeled with f59 antibody to provide tissue contrast. Embryos were cryosectioned (16 μm) and 6–7 mid-trunk sections per embryo were imaged. The images were then transferred to a drawing program (Adobe Photoshop 7.0) and notochord measurements were made using the scale bar. The significance of differences from observed values was assessed using the Mann-Whitney U test. The increase in notochord diameter observed in col mutants compared to wildtype was found to be significant, p<0.002. Because no significant differences were observed between uninjected wildtype and all injected wildtype embryos, these data are not shown (see Results).
Table 3.
Wnt/PCP components are able to rescue the notochord phenotype in col mutant embryos.
| Embryos | Notochord diameter-mid trunk (μm) |
|---|---|
| Uninjected wildtype | 40.5±2.5 |
| Uninjected col mutants | 46.6±2.3 |
| Δ_Ndsh_ injected col mutants | 39.7±3.1 |
| rok2 injected col mutants | 39.5±3.3 |
| vangl2 injected col mutants | 40.2±1.6 |
Trichostatin A treatment
Trichostatin A (TSA; Biovision Research Products) was dissolved in DMSO at a concentration of 1mg/ml. This stock solution was then diluted in fish water to the concentrations indicated.
Genetic mapping and cloning
For linkage analysis, AB background heterozygous col individuals were crossed to a polymorphic WIK strain and mutant and wild type embryos were used. Genomic DNA was prepared from 1534 embryos and PCR was performed using Simple Sequence Length Polymorphic markers (SSLP; Knapik et al., 1996). Primer sequences for SSLP markers were obtained from the MGH zebrafish database. The ck2b, hdac1 and hey1 coding sequences were amplified from col b382 by RT-PCR and then directly sequenced. A single nucleotide polymorphism (T to A) at position 583 in the ck2b cDNA sequence was used to identify recombinants. The CHORI211 BAC library was screened by PCR using the ck2b primers to identify positive BAC clones. BAC end sequences were obtained from the Sanger Institute database (http://trace.ensembl.org) and PCR primers were designed to amplify regions of these sequences from mutant and wildtype embryos. Polymorphisms were identified in these amplicons and used to check for recombinants. The splice site lesion was identified from several independently amplified fragments from col and wildtype genomic DNA using PCR primers (5′-TAACGTAGGGGAGGATTGTC- 3′) and (5′-CAGCTCCAGAATGGCCAGTAC- 3′) that amplify across intron 4–5. Mutant and wildtype spice variants were examined using RT-PCR.
Plasmid constructs
The full-length hdac1 gene cloned into pBS SK+ was a gift from I. Masai (RIKEN, Japan). Other constructs used in this study were vangl2/tri (Jessen et al., 2002), rho kinase 2(Jessen et al., 2002), Δ_N dsh_ (Heisenberg et al., 2000). In situ probes for hdac1 were generated using the first 720 bp of the hdac1 cDNA and cloning into the TOPO TA vector. Probes were synthesized by digesting with Pvu I followed by transcription using T7 polymerase.
mRNA and morpholino injections
mRNA was synthesized using Ambion’s T7, T3 or SP6 mMessage mMachine kit (Ambion). Following transcription, the mRNA was extracted using phenol/chloroform and concentrated in Microcon YM-50 (Amicon) microconcentrator filter devices. RNA quality was assayed using gel electrophoresis. mRNA was diluted in 1% phenol red and pressure injected into the YSL of 1 to 8-cell stage embryos. The concentration of RNA injected into each embryo was approximately 50–500pg depending on the RNA used.
The antisense hdac1 morpholino was targeted to the translational initiation site (5′-TTGTTCCTTGAGAACTCAGCGCCAT -3′) and a modified morpholino was also used as a specificity control (5′-TTGcTCCcTGAGAtCTCAGgGCCAT-3′). The sequence for the MO targeting vangl2 was the same as Park and Moon, 2002. All morpholinos were obtained from Gene Tools. The morpholinos were diluted with phenol red/0.2M KCl (1:6) prior to injection and 2–8 ng was injected per embryo. Morpholinos were pressure injected into the YSL of 1 to 8-cell stage embryos.
In situ hybridization, immunohistochemistry and genotyping
In situ hybridization was performed using standard protocols. The following probes were used: no tail (Schulte-Merker et al., 1992), foxd3 (Odenthal and Nusslein-Volhard, 1998), vangl2 (Park and Moon, 2002), myod (Weinberg et al., 1996), islet1 (Korzh et al., 1993), dlx2 (Akimenko et al., 1994), wnt5a (Rauch et al., 1997), krox20 (Wilkinson et al., 1989), val (Moens et al., 1996), huC (Kim et al., 1996), hoxb3 (Prince et al., 1998), wnt11 (Heisenberg et al., 2000), wnt8 (Kelly et al., 1995). Probes were synthesized using T7, T3 or SP6 RNA polymerases and DIG labeled rNTPs as appropriate. For in situ hybridizations on embryos older than 24 hpf, the embryos were raised in 0.03g/l 1-phenyl-2-thiourea (PTU) to prevent melanin synthesis which allowed clear analysis of gene expression patterns without interference from pigmented melanophores.
Immunohistochemistry was performed according to Henion et al., 1996. The antibodies used were F59 (Crow and Stockdale, 1986), zn12 (Trevarrow et al., 1990), acetylated tubulin (Sigma), RMO44 (Zymed) and actin (Abcam).
To determine the genotype of embryos used in experiments before a readily apparent phenotype is seen, DNA from individual embryos was obtained and PCR was performed on genomic DNA using the closely linked SSLP marker Z7235.
Results
Defects in axial extension contribute to the col mutant phenotype
col mutant embryos are shorter (Table 1) and have a downward curved body compared to wildtype embryos. The somites of 48 hpf col mutants appear rounded, unlike chevron-shaped wildtype somites (Fig. 1A, B). There are also fewer somite pairs in col mutants than in wildtype by 27hpf, although no significant difference is apparent at 16hpf (Table 2). The 1–2 somite pair deficit in col mutants at 27hpf persists at 48hpf (Table 2). Compared to wildtype embryos, col mutants also have abnormally wide and stunted notochords (Fig. 1A-D) with defects being prominent by 30 hpf. Quantification of notochord diameter in transverse sections of the trunk of col mutants and wildtype siblings at 48 hpf revealed that notochord diameter was significantly larger (15%; Table 3; see Materials and Methods) in mutants (Fig.1C, D). Notochord specification and early somite development, however, appear overtly normal in col mutants as expression of ntl and myoD in embryos at late gastrulation (Fig. 1E, F; not shown) and at early somitogenesis (not shown) is similar to that of wildtype embryos. Likewise, neurulation and lengthening of the neural keel as evidenced by the expression of foxd3 (Fig. 1G, H) and huC (not shown) during early somitogenesis appears unaffected. It is nevertheless possible that subtle early defects are present and become accentuated over time during morphogenesis. This temporal pattern of phenotypic changes has been documented previously for anteroposterior brain patterning in col mutants (Nambiar and Henion, 2004).
Figure 1. _col_mutant embryos display a late axial extension phenotype.
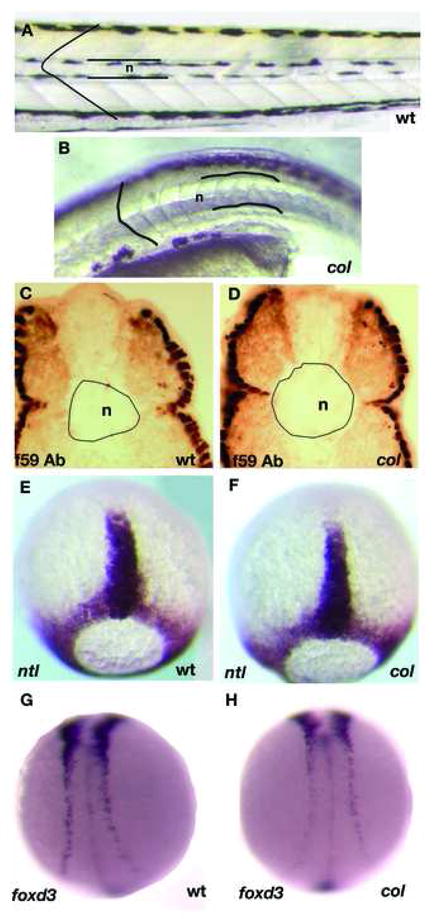
Live col mutants at 4 dpf (B) have wider notochords (n) than their wildtype siblings (A). Larger notochord diameter is observed in cross-sections of the trunk of 2 dpf col mutants stained with antibody f59 (D) as compared to wildtype (C). col mutants also display rounded somite morphology (B) unlike chevron-shaped wildtype somites (A). In contrast, col mutants do not display prominent early CE defects. For example, ntl expression in the notochord anlage at late gastrulation of col embryos (F) is indistinguishable from wildtype embryos (E). The degree of convergence of the neural plate border at the 12-somite stage identified by foxd3 expression also appears normal in col mutants (C, D).
The components of the axis extension phenotype of col mutants described above are reminiscent of, although much less pronounced than, those observed in the zebrafish Wnt/PCP mutants silberblick and trilobite as well as prickle morphants (Heisenberg et al., 2000; Sepich et al., 2000; Carreira-Barbosa et al., 2003), suggestive of a defect in extension movements as a result of the col mutation.
col mutant embryos display defects in the migration of hindbrain branchiomotor neurons
The branchiomotor neurons are born in specific rhombomeres in the hindbrain and innervate muscles of the pharyngeal arches (Noden, 1983; Chandrasekhar et al., 1997). In zebrafish the facial (nVII) and the glossopharyngeal (nIX) motor neurons migrate tangentially to their final destinations (Chandrasekhar et al., 1997). The trigeminal or nV neurons are specified as discrete clusters in r2 and r3. These two clusters of neurons are functionally distinct that can be attributed to the segmental origin of the motor neurons (Higashijima et al., 2000). At 21 hpf most of the facial (nVII) neurons are localized in r4 and r5, as judged by comparison to the otocyst. In the next 15 hrs the nVII motor neurons migrate tangentially such that by 36 hpf most of these neurons are located in r6 and r7.
In trilobite/vangl mutants, nVII and nIX neurons do not migrate into caudal rhombomeres following induction in r4 and r6 respectively (Bingham et al., 2002). Defects in the migration of these motor neurons are not a consequence of defective hindbrain patterning or widespread cell migration defects. Although tri/vangl mutants also display abnormal CE movements during gastrulation, the neuronal migration defect is not a consequence of gastrulation-associated cell movement abnormalities (Bingham et al., 2002). Similar defects are also observed in _pk1_morphants (Carreira-Barbosa et al., 2003).
We have found that col mutants display a branchiomotor neuron migration phenotype reminiscent of tri/vangl mutants and pk1 morphants. Specifically, the facial (nVII) neurons identified by _islet-1_mRNA and Islet1 protein expression fail to migrate caudally and remain in r4 (Fig. 2). Like in the case of vangl2 mutants, we observed that the general development of the hindbrain in col mutants is largely unaffected. The expression of the markers krox20, valentino and hoxb3 that are diagnostic for r3, 5 and 6 development, respectively, were comparable between wildtype and col mutant embryos (Fig. 3A-F). Also, the patterning of other hindbrain neuronal populations such as hindbrain commissural and reticulo-spinal neurons, as identified by staining with acetylated tubulin (not shown) and RMO44 antibodies respectively, was generally unaffected in col mutants (Fig. 3G, H). However, the number of neurons appeared to be reduced in number in col. These results suggest that the failure of facial (nVII) neuronal migration is specific to these neurons and not due to a general defect in hindbrain patterning.
Figure 2. Facial (nVII) hindbrain neurons in col mutants do not migrate tangentially.

Hindbrain neurons are marked by islet1 expression (A, B) at 56 hpf. In col mutant,, nVII neurons fail to migrate into r6 and r7 (B) as in wildtype siblings (A). In contrast, nV neurons are positioned correctly in mutants.
Figure 3. Gross patterning of the hindbrain is unperturbed in col mutants.
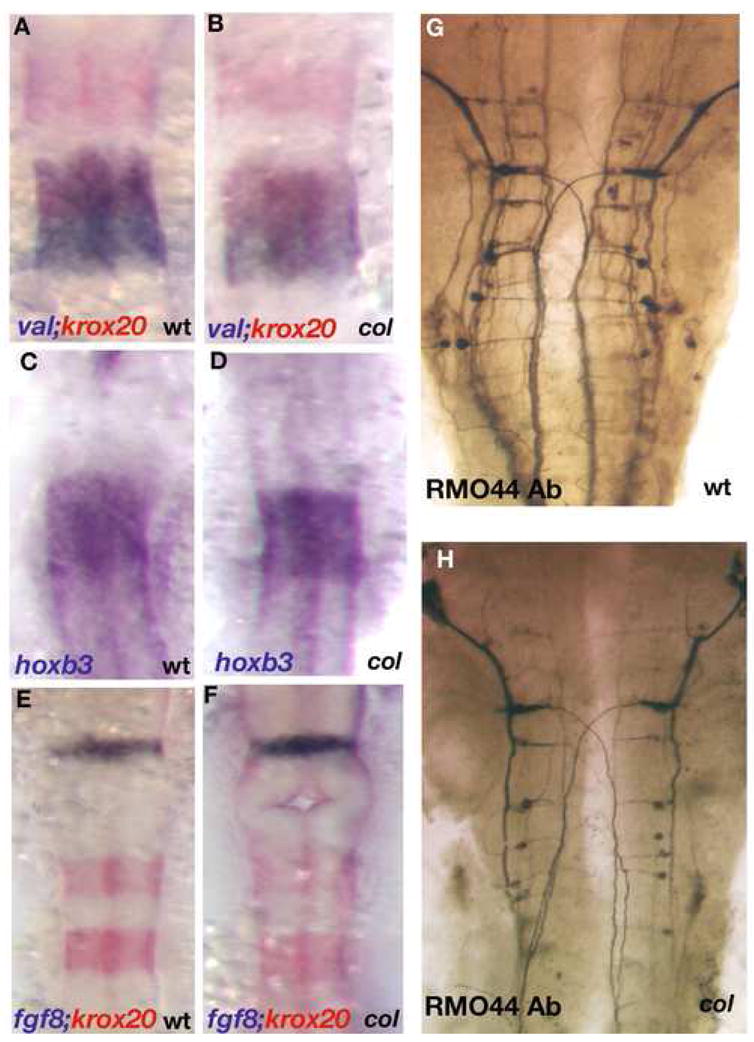
Hindbrain rhombomere patterning appears undisturbed in col mutant embryos (B, D, F) as compared to wild-type (A, C, E). Krox20 (red) labels r3 and r5 and valentino (blue) marks r5 and r6 (A, B). hoxb3 also marks r5 and r6 (C, D). fgf8 (blue) labels the mid-hindbrain boundary and krox20 (red) marks r3 and r5 showing that gross development between the mid-hindbrain boundary and r3 in the hindbrain also appears normal in col mutant embryos (F as compared to E). RMO44 labels reticulospinal neurons (G, H). The patterning of this neuronal population is relatively unperturbed in col mutants although overall neuronal numbers appear to be reduced (see Results).
Canonical and non-canonical Wnt pathway phenotypes of col mutants are genetically distinguishable
The embryonic patterning (Nambiar and Henion, 2004) and extension defects in col mutant embryos suggest a role for col in both canonical Wnt and non-canonical Wnt/PCP signaling. Therefore, we sought to determine the role of col in both arms of the Wnt signaling cascade. Injection of antagonists of the canonical pathway including dkk1, gsk3β and wnt8 MO, all resulted in the rescue of canonical pathway phenotypes of col mutants (Nambiar and Henion, 2004). In contrast, these canonical pathway antagonists failed to rescue the extension defects in col mutants (Nambiar and Henion, 2004; not shown). In addition, injection of the BMP inhibitor chordin mRNA and XFD, an FGF pathway antagonist also failed to rescue the extension defects in col embryos Nambiar and Henion, 2004; not shown). The persistence of extension defects in col mutants in which the patterning phenotypes were rescued is consistent with a role for col in regulating non-canonical as well as canonical Wnt signaling.
In order to delineate a potential role of col in the Wnt/PCP pathway, we first examined the expression of two wnt genes, wnt11 and wnt5a, both of which are known to play roles in regulating CE movements in vertebrates, including zebrafish (Heisenberg et al., 2000; Rauch et al., 1997). The overall patterns of embryonic expression of both genes were indistinguishable between col mutants (identified by genotyping with flanking marker Z9059) and wildtype embryos, except for an increase in wnt5a expression in the tailbud (not shown). This increased expression can be attributed to the accumulation of cells in the tailbud region as revealed by the expression of other markers such as ntl and wnt8 (Nambiar and Henion, 2004). Although we cannot rule out the existence of subtle changes, the generally unaltered expression of wnt5a and wnt11 in col mutants is consistent with a potential role of col in regulating the Wnt/PCP pathway independently of wnt5a and/or wnt11.
To further test whether col functions in the Wnt/PCP pathway, misexpression experiments were performed with regulatory components of the vertebrate Wnt/PCP pathway. dsh_Δ_N, a dsh construct lacking the N-terminal end that has been shown to activate the Wnt/PCP pathway and rescue CE defects in slb mutants (Heisenberg et al., 2000) and rho kinase2 RNA, which also activates the Wnt/PCP pathway (Marlow et al., 2002), were injected into embryos at 1–8 cell stage. We injected each of these RNAs independently into wildtype embryos and found that in the vast majority of individual embryos misexpression did not result in an abnormal phenotype, although a small number of injected embryos displayed variable defects in the morphogenesis of the prechordal plate and notochord (not shown). Misexpression of either RNA construct in homozygous col mutant embryos resulted in qualitatively equivalent suppression of the axial extension defects present in uninjected mutants (Fig. 4A-F). Injected mutant embryos (dsh_Δ_N 90%, n=50; rok2 90%, n=82; Table 4) displayed significantly longer, extended body axes (Fig. 4A-C) and their notochords were thinner (Table 3) and longer compared to uninjected mutants (Fig. 4D-F). dsh_Δ_N and rok2 injected col mutants retained defects in the canonical Wnt signaling pathway as evidenced by persistent brain patterning defects including, for example, reduced dlx2 expression in the anterior forebrain (Fig. 4G-I and not shown). Homozygous col mutants in all cases were identified by genotyping with flanking marker Z9059. These results strongly suggest that col functions in the regulation of the non-canonical/PCP pathway as well as in the canonical Wnt signaling pathway.
Figure 4. Regulators of the Wnt/PCP pathway are able to partially rescue col mutants.

Δ_Ndsh_ (B, F) and rok2 (C, E) were able to abolish the short body axis (B, C) and short wide notochords (E, F) observed in col mutants at 3 dpf (A, D). However, canonical Wnt signaling regulated defects in col mutants still remain in injected embryos. dlx2 expression in the telencephalon (asterisk) remains reduced compared to wildtype (G) in rock2 injected col mutant embryos (I) as compared to uninjected col mutants (H). The persistence of this phenotype is also observed in Δ_Ndsh_ injected col mutants (not shown).
Table 4.
Components of the non-canonical Wnt/PCP pathway can rescue aspects of the col mutant phenotype.
| RNA constructs | Rescue of body extension | Percentage (%) | Rescue of neuronal migration | Percentage (%) | Total (n) |
|---|---|---|---|---|---|
| Δ_Ndsh_ | 45±5 | 90 | 0 | 0 | 50 |
| rok2 | 73.8±6 | 90 | 0 | 0 | 82 |
| vangl2 | 40.5±5 | 90 | 36±4 | 80 | 45 |
The non-canonical Wnt/PCP pathway regulator vangl2 rescues defects in extension as well as branchiomotor neuron migration in col mutants
Although overexpression of dsh_Δ_N and rho kinase2 RNAs rescue axial extension defects in col mutants, branchiomotor neuron migration remains defective in injected mutants with cell bodies remaining in r4 in injected col mutants (not shown). However, another Wnt/PCP pathway regulator, van gogh-like protein 2 (vangl2), is known to be required for both CE movements in the gastrula and branchiomotor neuron migration (Bingham et al., 2002). We therefore tested the effects of vangl2 misexpression in col mutants on both axis extension and branchiomotor neuron migration. Injection of vangl2 RNA into col mutant embryos resulted in embryos with extended body axes and longer, thinner notochords when compared to uninjected mutants (Fig. 5A-D; Tables 3 and 4). Additionally, in vangl2 injected col mutants, facial branchiomotor neurons identified by islet-1 expression migrate from r4 to r6 and r7 in a pattern indistinguishable from wildtype embryos in 80% of injected mutants (Fig. 5A-C; Table 4). As was the case upon overexpression of dsh_Δ_N and rho kinase2 RNAs in col mutants (Fig. 4G-I), canonical Wnt signaling-dependent neural patterning defects remain defective in vangl2 injected col mutants (not shown). Phenotypic rescue of the extension defects in col mutants by vangl2 misexpression provides further evidence for the involvement of col in the regulation of the non-canonical/PCP pathway. In contrast, the rescue of branchiomotor neuron migration in vangl2 injected col mutants demonstrates an additional genetic interaction between col and vangl2 and indicates a novel role for col that is independent of Wnt signaling pathways.
Figure 5. _tri/vangl2_is able to rescue extension defects and tangential migration of hindbrain branchiomotor neurons in col mutant embryos.
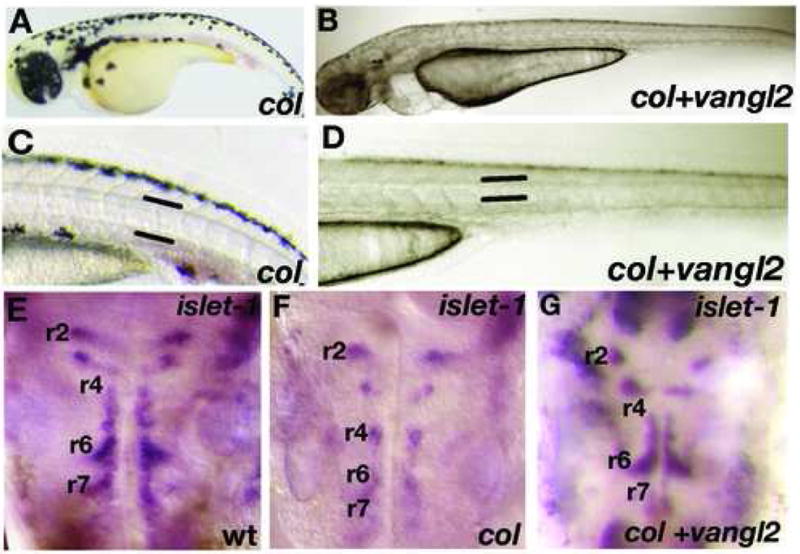
Overexpression of tri/vangl2 in col mutants (B, D) is able to rescue the stunted trunk development and broad notochords observed in uninjected col mutants (A, C). Facial (nVII) hindbrain motor neurons labeled by islet1 expression are positioned ectopically in r4 in col mutants at 56 hpf (F) compared to wildtype (E). Overexpression of tri/vangl2 restores tangential migration of these neurons into r6 and r7 (G) as in wildtype siblings (E). nV neurons are positioned correctly in r2 in tri/vangl2 injected (G) and uninjected (F) col mutants.
The col locus encodes histone deacetylase 1
The colb382 mutation was mapped to LG19 (Nambiar and Henion, 2004). Using SSLP markers it was placed approximately 0.6cM south of Z9059 (6 recombinants out of 1004) and 1.6cM north of Z22532 (17 recombinants out of 1004). An examination of the annotated genes between Z9059 and Z22532 revealed that the zebrafish orthologs of casein kinase 2b (ck2b), histone deacetylase1 (hdac1) and hairy/enhancer-of-split related with YRPW motif1 (hey1) lie within this interval (Fig. 6A). The roles of ck2b and hdac1 as negative regulators of canonical Wnt signaling have been established (Dominguez et al., 2004; Willert et al., 1997; Billin et al., 2000; Yamaguchi et al., 2005) prompting us to sequence the cDNAs of these genes. We also sequenced cDNA of the hey1 gene that lies within the same critical region. The sequencing of ck2b and hey1 cDNAs revealed no lesions. In order to completely rule out ck2b, we used a SNP in the gene to identify recombinants. We found two recombinants out of 1024 genomes. Using the ck2b primers we screened a bacterial artificial chromosome (BAC) library. We identified SNPs in the end sequences of two of the BACs, K206D2 and C261D24, and used them as additional markers. Sequencing of the hdac1 cDNA revealed a 9bp insertion at position 405 resulting in the insertion of 3 amino acids, glutamate, phenylalanine and serine in a conserved motif potentially compromising the structural integrity of the Hdac1 protein. Sequence comparison of col mutant and wildtype hdac1 cDNAs revealed that the wildtype form is absent in col mutants (Fig. 6D).
Figure 6. The_col_ locus encodes hdac1.
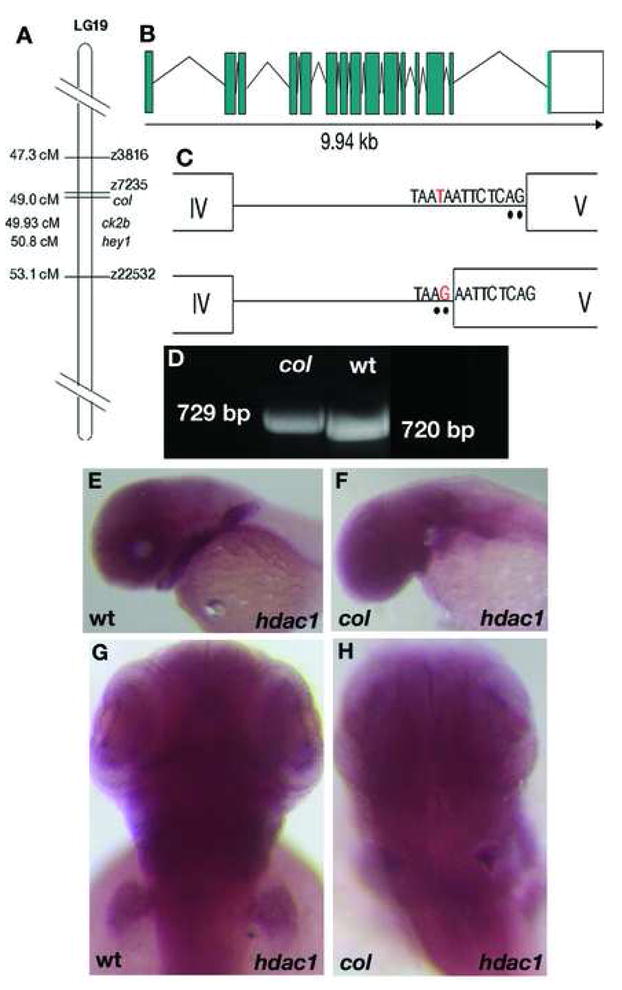
col maps close to the zebrafish hdac1 locus on LG 19 (A). A schematic showing the genomic organization of the zebrafish hdac1 gene (B). The zebrafish hdac1 gene has 15 exons and 14 introns spanning 9.94 kb of the genome and a T to G transversion in the intronic sequence adjacent to exon 5 causes aberrant splicing between exons 4 and 5 in col mutants (C). Aberrantly spliced hdac1 cDNA fragment in col with an additional 9 bp running in lane 1 can be distinguished from the wildtype product running in lane 2 (D). hdac1 expression is unaffected in 2 dpf col mutants (F, H) compared to wildtype (E, G).
Since a 9 bp insertion is unlikely to result from ENU mutagenesis, we reasoned that the defect could be the result of a point mutation that causes a splicing defect. In order to test this possibility we sequenced hdac1 from mutant genomic DNA and found a T-to-G transversion in the intron sequence flanking exon 5 that creates a new splice acceptor site (Fig. 6B, C). This results in the addition of 9 bases from the adjacent intron to the 5′ end of exon 5 in col mutants.
To determine if the expression pattern of hdac1 correlates with the development of the col mutant phenotype, we analyzed hdac1 RNA distribution. The hdac1 gene is ubiquitously expressed from the one cell stage until 18 hpf, suggesting a maternal contribution (not shown). At 24 hpf expression predominates in the brain and eyes and at 2 dpf hdac1 expression is also seen in the pectoral fin bud, branchial arches and hindbrain (Fig. 6E, G). Our results are consistent to observations by Cunliffe (2004) and Pillai et al (2004). We have shown selective and prominent developmental defects in col mutants in all of these regions (Nambiar and Henion, 2004). Overall, col mutants do not display a noticeable reduction in the levels of hdac1 expression (Fig. 6F, H), suggesting that the col mutation likely results in impaired Hdac1 protein function due to defective RNA processing
To further test whether the hdac1 gene is defective in col mutants, we assessed the ability of hdac1 RNA to rescue col mutant phenotypes. We found that all characterized phenotypic defects observed in col mutants are rescued by hdac1 RNA injection (85% n=125 Fig. 7; Table 5; Rescued embryos identified by genotyping, see Methods). For example, extension movements are rescued in injected embryos, leading to the development of normally elongated notochords and regular chevron shaped somites (Fig. 7A, C). The forebrain is specified and patterned normally based on the pattern of dlx2 expression compared to col mutants (Fig. 7D, F). Another aspect of phenotypic rescue by hdac1 RNA injections is the restored migration of branchiomotor neurons and melanophores. Hindbrain branchiomotor facial (nVII) neurons visualized by islet-1 expression migrate normally into r7 by 33 hpf in injected col mutant embryos whereas they fail to migrate out of r4 in uninjected col mutants (Fig. 7G, I). Injected col mutant embryos displayed the absence of the characteristic cluster of melanophores just posterior to the otic vesicle, a prominent feature of the col mutant phenotype (Fig. 7A, C). Lastly, we injected colb382hdac1 RNA into wildtype embryos and observed no obvious phenotypic consequences (not shown), inconsistent with the possibility of the col mutant form of hdac1 having a dominant-negative activity. We also noted the effects of hdac1 RNA overexpression in wildtype embryos. Injection of moderate concentrations (400 pg) resulted in a mild reduction of trunk tissue, partial loss of eyes and strikingly, an enlargement of the forebrain (Supplemental Fig. 2A, B). Injection of higher doses (1 ng; 17 0f 20 injected embryos=85%) caused a significant loss of trunk tissue and eyes and a loss of forebrain (Supplemental Fig. 2C, D) and enlargement of midbrain (Supplemental Fig. 2E, F) consistent with our previous data demonstrating that col is required for forebrain and midbrain patterning (Nambiar and Henion, 2004). Together, these results suggest that the col locus encodes hdac1 and that the colb382 mutation results in a reduction in or loss of hdac1 activity.
Figure 7. hdac1 morphants phenocopy col mutants and hdac1 RNA rescues col mutant phenotypes.
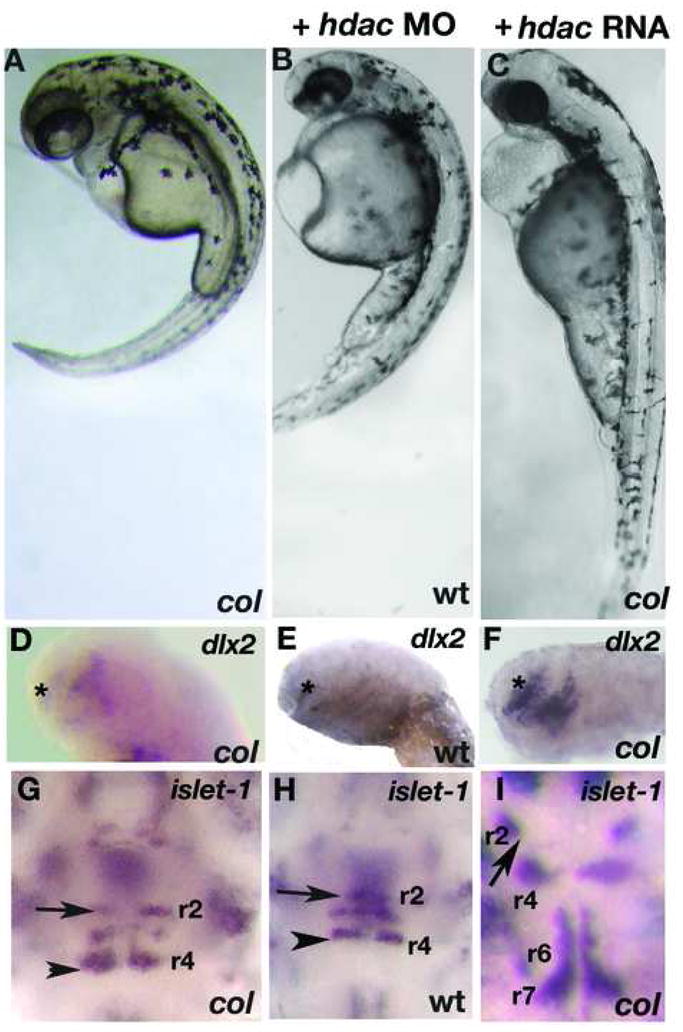
hdac1 MO injected wildtype embryos at 3 dpf (B) resemble uninjected col mutants (A). Reduced dlx2 expression (asterisk) and ectopically positioned facial hindbrain motor neurons (black arrowheads) in col mutants (D, G) are phenocopied in hdac1 morphants (E, H). The position of nV neurons in r2 are marked with arrows (G-I). hdac1 RNA is able to rescue axis extension and melanophore defects in col mutants (C). Telencephalon dlx2 expression and migration of facial hindbrain motorneurons is restored in col mutants injected with hdac1 RNA (F, I compared to D, G).
Table 5.
hdac1 RNA is able to rescue the col mutant phenotype while the hdac1 MO is able to phenocopy the mutant.
| Construct injected | Rescue of CE and neuronal migration defects | Percentage (%) | Mutant phenotype observed | Percentage (%) | *Abnormal | Percentage (%) | Total (n) |
|---|---|---|---|---|---|---|---|
| hdac1 RNA | 106.3±7 | 85 | 4±1 | 3.3 | 14.7±2 | 11.7 | 125 |
| hdac1 MO | 0 | 0 | 110±5 | 91.6 | 10±2 | 8.3 | 120 |
To determine whether hdac1 knockdown by translational interference in wildtype embryos phenocopies col mutants, we injected wildtype embryos with morpholino oligonucleotides (MO) directed to the translational start site of hdac1. A large number of embryos injected with the MO (91.6%; n=120; Table 5) very closely resembled col mutants (Fig. 7B, E, H). They were defective in extension movements evidenced by short, curved body axes and thick notochords (Fig. 7A, B). Like col embryos, they lacked pectoral fins, retained a cluster of melanophores posterior to the otocyst and displayed grossly truncated anterior brain development (Fig. 7A, B and not shown). These embryos also displayed the reduced domain of dlx2 expression in the telencephalon (Fig. 7D, E) and migration defect of facial branchiomotor hindbrain neurons expressing islet-1 (Fig. 7G, H), both characteristic phenotypes of col mutants. Embryos injected with the control MO did not show specific defects (not shown).
Taken together, since the hdac1 gene contains a lesion in col mutants, the col hdac1 gene produces an RNA that is incorrectly spliced, wildtype hdac1 RNA is able to rescue col mutant phenotypes and morpholino-based hdac1 gene knockdown phenocopies col mutants, we conclude that the col locus encodes hdac1.
col/hdac1 appears to function genetically upstream of vangl2
Because vangl2 misexpression rescues the axial extension and branchiomotor neuron migration phenotypes of col mutant embryos, we sought to establish a functional hierarchy between vangl2 and col/hdac1. To do so, we first injected wildtype embryos with tri/vangl2 MOs. These embryos developed highly stunted body axes with compressed somites and notochords, like tri mutants (Supplemental Fig. 1A; Jessen et al., 2002). We then coinjected hdac1 RNA with the tri/vangl2 MO in wildtype embryos. We observed that overexpression of hdac1 RNA was unable to rescue the phenotypes resulting from the injection of tri/vangl2 MOs (Supplemental Fig. 1B). This result suggests that hdac1 may be acting genetically upstream of vangl2 in the Wnt/PCP pathway, although a more detailed analysis is required to define this interaction.
Differential temporal requirements for histone deacetylase activity
To further understand the requirement of histone deacetylase activity in the manifestation of col mutant phenotypes, we attenuated HDAC activity in wildtype embryos using the pharmacological agent trichostatin A (TSA), a specific and potent inhibitor of histone deacetylases. TSA inhibits the activity of both type I and type II Hdac proteins by binding to their catalytic domains (Yoshida et al., 1995). We performed two different treatments of embryos with TSA. In the first, we treated wildtype embryos continuously with 50 nM TSA from 5 hpf and fixed embryos at multiple subsequent time points. These embryos displayed specific defects that were more severe than developmental defects in col mutant embryos. We observed CE defects in these embryos starting at early somitogenesis (Fig. 8A, B; Table 6). These embryos have shorter, thicker notochords marked by ntl expression at the 6-somite stage and 24 hpf (Fig. 8C-F) and severely shortened body axes with compressed somites marked by myod expression at 24 hpf (Fig. 8G, H). At 2 dpf these embryos have highly compressed body axes, thick notochords and narrow compressed somites (Fig. 9B; not shown). Like in col mutants, we also observe clustering of melanophores posterior to the otocysts (Fig. 9B). We also observed the ectopic positioning of the hindbrain facial branchiomotor neurons in r4 due to lack of caudal migration as seen in col mutants (Fig. 9F). These embryos additionally showed a severe reduction in islet-1 expression in these neuronal populations (Fig. 9F) as well as a drastic reduction in dlx2 expression in the forebrain (Fig. 9D). Exposing embryos to higher concentrations of TSA at 5 hpf resulted in high mortality by 24 hpf. In order to determine the effect of blocking HDAC activity later in development, we continuously exposed wildtype embryos to TSA beginning at 16 hpf (Table 6). Embryos at this stage required a higher concentration of TSA (800 nM) to show phenotypes resembling col mutants. These embryos partially phenocopied the col mutant phenotype. Treated embryos at 2 dpf developed stunted body axes, the melanophore migration defect and had pectoral fins (Fig. 9A; not shown). These embryos also displayed aberrant branchiomotor hindbrain neuronal migration (Fig. 9E). In contrast to untreated col mutant embryos, dlx2 expression in the forebrain is only slightly reduced in treated wildtype embryos (Fig. 9C) compared to untreated wildtype embryos. Early notochord development also appeared unaffected in treated embryos (not shown). Treatment of wildtype embryos with 1200nM TSA resulted in widespread cell death in treated embryos by 2 dpf. Together, these results suggest an early requirement for HDAC function in brain patterning and notochord development whereas extension of the body axis, tangential migration of hindbrain facial branchiomotor neurons, pectoral fin development and melanophore patterning, require or also require HDAC function at relatively later stages of development.
Figure 8. CE defects in early TSA-treated wildtype embryos resembles other zebrafish Wnt/PCP mutants.
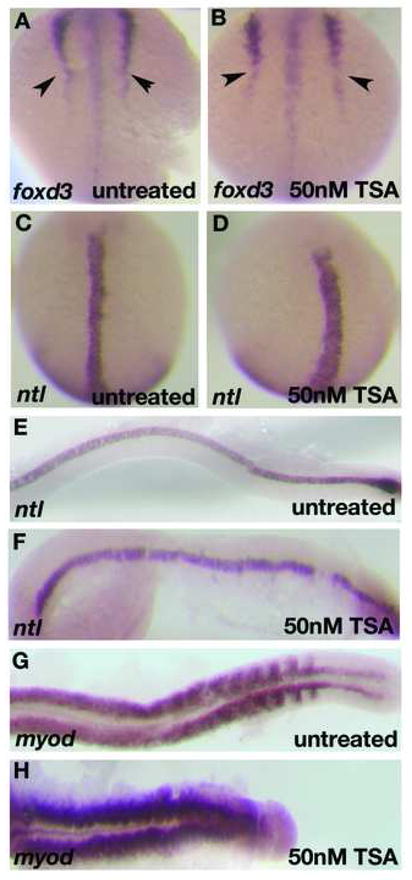
foxd3 expression at the 6-somite stage in TSA-treated embryos shows defects in convergence (arrowheads; B) as compared to wildtype (A). ntl expression at the 6-somite stage (C, D) and 22 hpf (E, F) reveals shorter, thicker notochord in TSA-treated embryos (D, F) than in wildtype embryos (C, E). myoD expression at 22 hpf shows compressed, laterally expanded somites in TSA-treated embryos (H) as compared to wildtype (G).
Table 6.
TSA treatment is able to phenocopy the col mutant phenotype.
| Conc of TSA (nM) | Defects in forebrain development, CE and neuronal migration observed | Percentage (%) | Defects in CE and neuronal migration only observed | Percentage (%) | Total (n) |
|---|---|---|---|---|---|
| 50 nM at 5 hpf | 105 | 100 | 0 | 0 | 105 |
| 800 nM at 16 hpf | 0 | 0 | 96 | 100 | 96 |
Figure 9. Wildtype embryos treated late with TSA phenocopy col mutants.
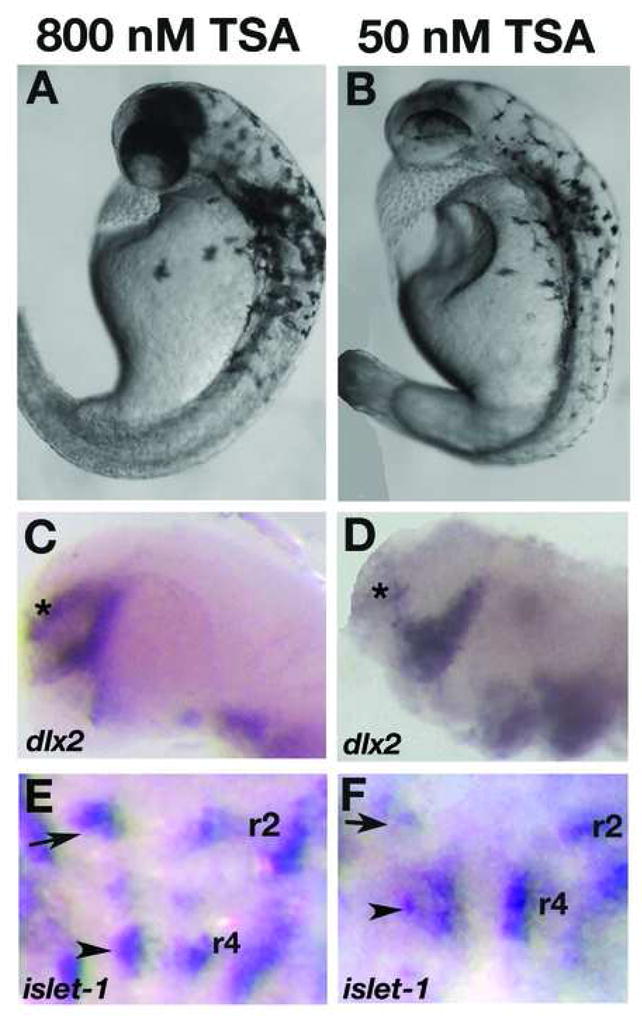
Live wildtype embryos at 2dpf treated with 800 nM TSA late (at 16 hpf) resemble col mutants (A, compare to Fig. 7A). dlx2 expression is unaffected in these embryos (C) while the facial hindbrain motorneurons remain in r4 (arrowhead; E) as in col mutants. The position of nV neurons are shown with black arrows. Phenotypes observed at 2dpf in wildtype embryos treated with 50 nM TSA early (5 hpf) appear more severe (B). The forebrain dlx2 expression domain is reduced (D), similar to col mutants, and facial hindbrain motorneurons remain in r4 (F) in these embryos.
Discussion
We have previously shown that the zebrafish mutant col displays specific defects in early DV patterning and AP patterning of the neuroectoderm (Nambiar and Henion, 2004). Our results indicated a function for Col as an inhibitor of canonical Wnt signaling (Nambiar and Henion, 2004). Our data also showed that while the early DV patterning and the neural AP patterning defects in col mutants were rescued by blocking excessive canonical Wnt signaling, these embryos still retained compressed body axes and notochords. Blocking of wnt8 function using wnt8 MOs was able to partially rescue body curvature (Nambiar and Henion, 2004) and this may be attributed to the activation of molecules such as Stat3 that are activated by the canonical Wnt pathway and regulate the initiation of CE (Sepich et al., 2005). These embryos, however, still are distinctly shorter than wildtype siblings.
In this study, we detail the defects in the extension of the body axis and notochord as well as the caudal migration of hindbrain nVII branchiomotor neurons in col mutants. We have identified col as zebrafish histone deacetylase 1. Our results indicate novel functions for col/hdac1 in non-canonical Wnt/PCP signaling during axial extension and in Wnt-indpendent hindbrain branchiomotor neuronal migration.
col encodes the histone deacetylase 1 gene
The hdac1 gene in col b382_mutants harbors a T to G tranversion in the intron flanking exon 5. This creates a new splice acceptor site resulting in the insertion of three additional amino acids in one of seven highly conserved motifs in the Hdac1 protein. Misexpression of hdac1 mRNA in col_−/− embryos rescues all aspects of the mutant phenotype and morpholino-mediated knockdown of the gene in wildtype embryos phenocopies col mutants. col mutant embryos very closely resemble other hdac1 mutants such as add and t24411 (Yamaguchi et al., 2005; Stadler et al., 2005), and correctly spliced hdac1 transcripts appear to be absent in col mutants, consistent with the possibility that col b382 locus may correspond to a null mutation of hdac1. However, we cannot rule out partial functionality of Col mutant protein or the presence of wildtype hdac1 transcripts in col mutants at levels below the detection limits of the methods we used. Thus, we cannot at present conclude that col represents a null hdac1 mutation (and see below).
Histone deacetylases play an important role in maintaining equilibrium between the acetylated and deacetylated states of chromatin and thus the tissue-specific expression status of genes and can also play a critical role in regulating the extracellular microenvironment (Whetstine et al., 2005). In zebrafish, hdac1 has been shown to regulate cell cycle exit and subsequent neurogenesis in the retina (Stadler et al., 2005; Yamaguchi et al., 2005) by antagonizing both the Notch and canonical Wnt signaling pathways (Yamaguchi et al., 2005). In addition to this, data from Cunliffe (2004) implicates hdac1 in promoting neuronal specification in the developing zebrafish brain by repressing Notch target gene expression. Our data provides the first evidence, in either vertebrates or invertebrates, for the functioning of hdac1 in regulating body extension and neuronal migration in the early embryo. Further, we show that hdac1 is involved in the regulation of these processes by means of the non-canonical Wnt/PCP pathway as well as independent of this pathway but involving genetic interaction with vangl2, respectively.
The role of Hdac1 in the non-canonical Wnt/PCP pathway
col mutants exhibit phenotypes characteristic of reduced non-canonical Wnt/PCP pathway signaling. By 2 dpf these mutant embryos typically develop stunted body axes, thick, short notochords and rounded somites. Similar phenotypes are observed in slb/wnt11 (Heisenberg et al., 2000), ppt/wnt5a (Rauch et al., 1997), tri/vangl2 (Sepich et al., 2000), kny/glp4 (Topczewski et al., 2001) mutants and pk morphants (Carreira-Barbosa et al., 2003). Consistent with this, activating the non-canonical pathway by overexpressing Δ_Ndsh_ (Heisenberg et al., 2000), rho kinase2 and vangl2 is able to completely rescue extension of the body axis and notochord and somite defects in col mutants. Our results, therefore, strongly suggest that col/hdac1 acts as a positive regulator of the non-canonical Wnt/PCP pathway. In contrast, analysis of the actin cytoskeleton of wildtype and col mutants during gastrulation revealed no discernable differences between col and wildtype embryos. This result is not consistent with compromised col/hdac1 function disrupting the cellular cytoskeleton generally and indirectly affecting cell polarity. However, it is important to note that this analysis of the cytoskeleton was not comprehensive and we cannot rule out subtle changes. It will be of considerable interest to examine potential defects in the subcellular localization of PCP pathway components during gastrulation in col mutants when reagents become available.
A major difference in the phenotypes displayed by other zebrafish Wnt/PCP mutants and col is that we do not observe an early manifestation of CE defects in col mutant embryos. Other Wnt/PCP mutants display defects in dorsal convergence and extension of the body axis starting at 9 hpf (Heisenberg et al., 2000; Rauch et al., 1997; Sepich et al., 2000; Carreira-Barbosa et al., 2003). In contrast, extension defects in col mutants become prominent only by 30 hpf. We have previously reported a similar late development of other mutant phenotypes in col such as defects in forebrain and midbrain patterning (Nambiar and Henion, 2004). Given the importance of Hdac1 as a co-repressor in yeast and mammalian systems, col mutants might be expected to display more severe patterning defects. It is possible that the persistence of maternal histone deacetylase and the activity of other Hdacs in col mutants may account for the lack of severe early CE defects in col mutants. Supporting this, we have shown that treating wildtype embryos with TSA, a potent inhibitor of all Hdacs, at different developmental time points results in different phenotypes. Early TSA treatment (5 hpf) produces embryos with CE defects similar to those observed in other zebrafish Wnt/PCP mutants and more severe than col mutants. Later treatment of embryos with TSA (16 hpf) produced embryos with milder defects that closely resembled col mutants. Since embryos exposed to TSA starting at the later time point displayed defects in extension of the body axis we suggest that persistent HDAC activity is required for extension movements during the later stages of development. Similar to col, Drosophila null mutants for rpd3, the hdac1 homolog, also display mild embryonic defects that have been attributed to possible redundancy among Drosophila histone deacetylases (Mannervik and Levine, 1999). Interestingly, _Hdac1_-null mouse ES cells show a marked compensatory increase in Hda_c_2 and Hdac3 expression (Lagger et al., 2002), lending further support to the possibility that functional redundancy among Hdacs and/or different maternal and zygotic functional contributions account for the phenotypes of col and other zebrafish hdac1 mutants. Nevertheless, we do observe an early DV patterning defect in col mutants attributable to disregulation of canonical Wnt signaling (Nambiar and Henion, 2004), which although not severe, suggests that neither maternal hdac1 function or the activities of other histone deacetylases can completely compensate for a requirement for early embryonic zygotic hdac1 function.
col/hdac1 is involved in mediating tangential migration of hindbrain facial (nVII) branchiomotor neurons
A number of genes that are required for both CE and neuronal migration have been identified so far. Amongst them are tri/vangl2 (Bingham et al., 2002; Jessen et al., 2002), pk1 (Carreira-Barbosa et al., 2003) and llk/scrb1 (Wada et al., 2005). Other signaling molecules in the non-canonical Wnt pathway such as slb/wnt11, ppt/wnt5a and kny/glypican4/6 regulate CE movements in the gastrula (Topczewski et al., 2001; Heisenberg et al., 2000; Kilian et al., 2003), but do not regulate neuronal migration (Bingham et al., 2002; Jessen et al., 2002). Also, branchiomotor neuronal migration is unaffected by the overexpression of a dominant negative form of Dsh which is able to suppress Wnt/PCP-mediated CE movements (Bingham et al., 2002; Jessen et al., 2002). These results therefore support the hypothesis that vangl2, pk1 and scrb1 regulate neuronal migration via a pathway independent of the Wnt/PCP signaling cascade.
In this study, we have shown that col mutants, in addition to extension defects, display aberrant migration of facial (nVII) hindbrain branchiomotor neurons. These neurons are born in the correct positions in the rhombomere 4 (Higashijima et al., 2000), but while their wildtype counterparts migrate caudally into rhombomeres 6 and 7 (Chandrasekhar et al., 1997), these neurons persist in their original positions. We were unable to test if these neurons function in their ectopic positions because of embryonic lethality. However, in a previous study it has been shown that these ectopic neurons in hdac1_−/_− mutants form axonal projections (Cunliffe, 2004).
Our studies also reveal that this migration defect is not a result of a more general effect of hdac1 on hindbrain patterning. Rhombomere formation and the development of the patterning of other hindbrain neurons are unaffected in col. Previous studies by Cunliffe (2004) also revealed no defects in segmentation of the hindbrain of hdac1 mutant embryos although he did report perturbed segmental organization of hindbrain Hu-expressing neurons. We, however, did not observe any defects in the organization of two sets of neurons arranged segmentally in the hindbrain of col mutants - the reticulospinal neurons and the commissural neurons. We did observe a decrease in the number of these neurons that is consistent with fewer Hu-positive hindbrain neurons reported by Cunliffe in hdac1 mutants suggesting that hdac1 function is required for the specification of neurons in the hindbrain. Consistent with results published by Cunliffe we also observed fewer cell bodies in the neuronal cluster marked by islet-1 antibody making them very difficult to detect. The intensity of the signal in situ preparations from mutant and wild-type however were not considerably different.
We were able to restore normal migration of these neurons by overexpressing vangl2/tri. Consistent with previous data, other components of the Wnt/PCP pathway such as Δ_Ndsh_ and rhokinase2 were unable to rescue the neuronal migration defect. This suggests that this phenotype is not a consequence of the CE defects observed in col and raises the possibility that Hdac1 functions along with Vangl2 via an independent pathway to regulate the migration of these neurons. These defects in neuronal migration are also not a consequence of aberrant canonical Wnt signaling since overexpression of negative regulators of the canonical Wnt pathway are not able to restore normal migration of the r4 neurons (not shown). We also found that defects in general patterning of the hindbrain do not contribute to the aberrant migration of these neurons since the expression patterns of segmentally expressed genes krox20, val and hoxb3 and the patterning of other hindbrain neuronal populations such as the reticulo-spinal neurons and commissural neurons are normal.
We were also able to phenocopy this defect with injection of hdac1 MOs and treatment with TSA. Embryos treated with TSA during early development, in addition to defects in the migration of branchiomotor neurons, also displayed a reduction in the islet-1 expression in these neurons. Previous data from Cunliffe (2004) has implicated hdac1 in the specification of hindbrain branchiomotor neurons, raising the possibility that the reduction in islet-1 expression in these TSA treated embryos could be due to defects in specification of this subset of neurons. We also show that treatment of wildtype embryos with TSA during later stages (at 21 hpf and 24 hpf; not shown) resulted in aberrant neuronal migration suggesting that continued Hdac activity is required for caudal migration of the facial branchiomotor neurons.
Novel functions of Hdac1 in the Wnt/PCP pathway
Our studies of col mutants have revealed novel functions of Hdac1 in major signaling pathways regulating embryonic development. However, precisely how Hdac1 functions in these pathways is not fully understood. In the canonical Wnt pathway (see Nambiar and Henion, 2004), Hdac1 functions as a co-repressor with molecules such as Groucho and LEF1 in the nucleus (Brantjes et al., 2001; Chen et al., 1999; Billin et al., 2000). Studies in Drosophila and vertebrates have shown that Groucho, a canonical Wnt signaling pathway repressor, readily interacts with Hdac1 forming a repressor complex that remains tethered to the promoter of Wnt target genes (Brantjes et al., 2001; Chen et al., 1999). Data also indicates that the Wnt transcription factor LEF1 can act as a repressor in the presence of Hdac1 (Billin et al., 2000). Activation of LEF-dependent target genes occurs when the increasing level of β-catenin in the nucleus is able to dissociate Hdac1 from LEF1 and itself bind to LEF1 to form a dimeric activator (Billin et al., 2000). Thus, Hdac1 appears to maintain Wnt target genes in a repressed state until replaced by activators such as β-catenin (Billin et al., 2000).
In this study we have shown that col/hdac1 regulates both the non-canonical Wnt/PCP pathway that controls CE movements as well as the pathway that mediates the caudal migration of hindbrain facial motor neurons. There are a number of possible ways in which Hdac1 functions in these pathways. For example, since Hdac1 regulates both pathways, it is conceivable then that Col/Hdac1 could act by regulating the transcription of vangl2 or its interacting proteins. We examined vangl2 expression in col mutants and there appeared to be no significant difference compared to wildtype siblings (not shown). Another possible scenario for the functioning of Col/Hdac1 in this context could be via an interaction with Vangl2 and its interacting proteins such as Pk and Scribble that act at the common branchpoint. Another possibility is that Col/Hdac1 regulates the transcription of other components of the Wnt/PCP pathway and/or the targets of Wnt/PCP pathway genes. In the latter case, additional interactions of Hdac1 with Wnt/PCP signaling-independent genes or components of the pathway that also regulate branchiomotor neuron migration are possible. Further studies exploring the function of col should reveal the molecular mechanism by which col/hdac1 affects the activities of the genes involved in the morphogenetic events we have described.
Supplementary Material
01
Supplemental Figure 1. hdac1 RNA does not rescue defects in vangl2 morphants. vangl2 morphants injected with hdac1 RNA (B) still display axial extension defects observed in vangl2 morphants (A). The tangential migration defect of hindbrain nV11 branchiomotor neurons in vangl2 morphants is also not rescued by hdac1 misexpression (not shown).
Supplemental Figure 2 Overexpression of hdac1 in wildtype embryos causes brain and trunk defects. Injection of hdac1 RNA causes and enlargement of the forebrain (arrows) at moderate doses (B, compare to A). At high concentration hdac1 RNA causes loss of forebrain tissue and an expansion of midbrain (D, compare to C) accompanying severe loss of trunk tissues. wnt1 expression marks the posterior edge of the midbrain (arrow E) in uninjected wildtype embryos at 2 dpf. Embryos injected with hdac1 RNA display an antero-posterior expansion of wnt1 expression (arrows).
Acknowledgments
We thank our colleagues for providing numerous reagents and are particularly thankful to Will Talbot. This work was supported by NSF grant IBN0315765 with additional support from NIH grant P30-NS045758.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler PN, Lee H. Frizzled signaling and cell-cell interactions in planar polarity. Curr Opin Cell Biol. 2001;13:635–640. doi: 10.1016/s0955-0674(00)00263-5. [DOI] [PubMed] [Google Scholar]
- Akimenko MA, Ekker M, Wegner J, Lin W, Westerfield M. Combinatorial expression of three zebrafish genes related to distal-less: part of a homeobox gene code for the head. J Neurosci. 1994;14:3475–86. doi: 10.1523/JNEUROSCI.14-06-03475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG. Structural modifications of histones and their possible role in the regulation of ribonucleic acid synthesis. Proc Can Cancer Conf. 1966;6:313–35. [PubMed] [Google Scholar]
- Billin AN, Thirlwell H, Ayer DE. Beta-catenin-histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol Cell Biol. 2000;20:6882–90. doi: 10.1128/mcb.20.18.6882-6890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham S, Higashijima S, Okamoto H, Chandrasekhar A. The Zebrafish trilobite gene is essential for tangential migration of branchiomotor neurons. Dev Biol. 2002;242:149–60. doi: 10.1006/dbio.2001.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Mlodzik M. Dishevelled: at the crossroads of divergent intracellular signaling pathways. Mech Dev. 1999;83:27–37. doi: 10.1016/s0925-4773(99)00046-5. [DOI] [PubMed] [Google Scholar]
- Brantjes H, Roose J, van De Wetering M, Clevers H. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res. 2001;29:1410–9. doi: 10.1093/nar/29.7.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira-Barbosa F, Concha ML, Takeuchi M, Ueno N, Wilson SW, Tada M. Prickle 1 regulates cell movements during gastrulation and neuronal migration in zebrafish. Development. 2003;130:4037–46. doi: 10.1242/dev.00567. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A, Moens CB, Warren JT, Jr, Kimmel CB, Kuwada JY. Development of branchiomotor neurons in zebrafish. Development. 1997;124:2633–44. doi: 10.1242/dev.124.13.2633. [DOI] [PubMed] [Google Scholar]
- Chen G, Fernandez J, Mische S, Courey AJ. A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev. 1999;13:2218–30. doi: 10.1101/gad.13.17.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunliffe VT. Histone deacetylase 1 is required to repress Notch target gene expression during zebrafish neurogenesis and to maintain the production of motoneurones in response to hedgehog signalling. Development. 2004;131:2983–95. doi: 10.1242/dev.01166. [DOI] [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez I, Mizuno J, Wu H, Song DH, Symes K, Seldin DC. Protein kinase CK2 is required for dorsal axis formation in Xenopus embryos. Dev Biol. 2004;274:110–24. doi: 10.1016/j.ydbio.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Pelegri F, Mullins MC, Kane DA, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Haffter P, Heisenberg CP, Jiang YJ, Kelsh RN, Odenthal J, Warga RM, Nusslein-Volhard C. Mutations affecting morphogenesis during gastrulation and tail formation in the zebrafish, Danio rerio. Development. 1996;123:143–51. doi: 10.1242/dev.123.1.143. [DOI] [PubMed] [Google Scholar]
- Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, Stemple DL, Smith JC, Wilson SW. Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000;405:76–81. doi: 10.1038/35011068. [DOI] [PubMed] [Google Scholar]
- Henion PD, Raible DW, Beattie CE, Stoesser KL, Weston JA, Eisen JS. Screen for mutations affecting development of Zebrafish neural crest. Dev Genet. 1996;18:11–7. doi: 10.1002/(SICI)1520-6408(1996)18:1<11::AID-DVG2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci. 2000;20:206–18. doi: 10.1523/JNEUROSCI.20-01-00206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074– 80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jessen JR, Topczewski J, Bingham S, Sepich DS, Marlow F, Chandrasekhar A, Solnica-Krezel L. Zebrafish trilobite identifies new roles for Van gogh-like protein 2 in gastrulation and neuronal movements. Nat Cell Biol. 2002;4:610–5. doi: 10.1038/ncb828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane DA, Warga RM. Domains of movement in the zebrafish gastrula. Semin Dev Biol. 1994;5:101–109. [Google Scholar]
- Kelly GM, Greenstein P, Erezyilmaz DF, Moon RT. Zebrafish wnt8 and wnt8b share a common activity but are involved in distinct developmental pathways. Development. 1995;121:1787–99. doi: 10.1242/dev.121.6.1787. [DOI] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P. Ltap, a mammalian homolog of Drosophila Van gogh-like protein 2/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet. 2001;28:251–5. doi: 10.1038/90081. [DOI] [PubMed] [Google Scholar]
- Kilian B, Mansukoski H, Barbosa FC, Ulrich F, Tada M, Heisenberg CP. The role of Ppt/Wnt5 in regulating cell shape and movement during zebrafish gastrulation. Mech Dev. 2003;120:467–76. doi: 10.1016/s0925-4773(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Kim CH, Ueshima E, Muraoka O, Tanaka H, Yeo SY, Huh TL, Miki N. Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci Lett. 1996;216:109–12. doi: 10.1016/0304-3940(96)13021-4. [DOI] [PubMed] [Google Scholar]
- Knapik EW, Goodman A, Atkinson OS, Roberts CT, Shiozawa M, Sim CU, Weksler-Zangen S, Trolliet MR, Futrell C, Innes BA, Koike G, McLaughlin MG, Pierre L, Simon JS, Vilallonga E, Roy M, Chiang PW, Fishman MC, Driever W, Jacob HJ. A reference cross DNA panel for zebrafish (Danio rerio) anchored with simple sequence length polymorphisms. Development. 1996;123:451–60. doi: 10.1242/dev.123.1.451. [DOI] [PubMed] [Google Scholar]
- Korzh V, Edlund T, Thor S. Zebrafish primary neurons initiate expression of the LIM homeodomain protein Isl-1 at the end of gastrulation. Development. 1993;118:417–25. doi: 10.1242/dev.118.2.417. [DOI] [PubMed] [Google Scholar]
- Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol. 2003;3:344–51. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- Marlow F, Topczewski J, Sepich D, Solnica-Krezel L. Zebrafish Rho kinase 2 acts downstream of Wnt11 to mediate cell polarity and effective convergence and extension movements. Curr Biol. 2002;11:876–84. doi: 10.1016/s0960-9822(02)00864-3. [DOI] [PubMed] [Google Scholar]
- McKay RM, Peters JM, Graff JM. The casein kinase I family in Wnt signaling. Dev Biol. 2001;235:388–96. doi: 10.1006/dbio.2001.0308. [DOI] [PubMed] [Google Scholar]
- Moens CB, Yan YL, Appel B, Force AG, Kimmel CB. valentino: a zebrafish gene required for normal hindbrain segmentation. Development. 1996;122:3981–90. doi: 10.1242/dev.122.12.3981. [DOI] [PubMed] [Google Scholar]
- Myers DC, Sepich DS, Solnica-Krezel L. Convergence and extension in vertebrate gastrulae: cell movements according to or in search of identity? Trends Genet. 2002;18:447–55. doi: 10.1016/s0168-9525(02)02725-7. [DOI] [PubMed] [Google Scholar]
- Nambiar RM, Henion PD. Sequential antagonism of early and late Wnt-signaling by zebrafish colgate promotes dorsal and anterior fates. Dev Biol. 2004;267:165–80. doi: 10.1016/j.ydbio.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Odenthal J, Nusslein-Volhard C. fork head domain genes in zebrafish. Dev Genes Evol. 1998;208:245–58. doi: 10.1007/s004270050179. [DOI] [PubMed] [Google Scholar]
- Park M, Moon RT. The planar cell polarity gene vangl2 regulates cell behavior and cell fate in vertebrate embryos. Nat Cell Biol. 2002;4:20–25. doi: 10.1038/ncb716. [DOI] [PubMed] [Google Scholar]
- Prince VE, Moens CB, Kimmel CB, Ho RK. Zebrafish hox genes: expression in the hindbrain region of wild-type and mutants of the segmentation gene, valentino. Development. 1998;125:393–406. doi: 10.1242/dev.125.3.393. [DOI] [PubMed] [Google Scholar]
- Rauch GJ, Hammerschmidt M, Blader P, Schauerte HE, Strahle U, Ingham PW, McMahon AP, Haffter P. Wnt5 is required for tail formation in the zebrafish embryo. Cold Spring Harb Symp Quant Biol. 1997;62:227–34. [PubMed] [Google Scholar]
- Rice JC, Allis CD. Code of silence. Nature. 2001;414:258–61. doi: 10.1038/35104721. [DOI] [PubMed] [Google Scholar]
- Schulte-Merker S, Ho RK, Herrmann BG, Nusslein-Volhard C. The protein product of the zebrafish homologue of the mouse T gene is expressed in nuclei of the germ ring and the notochord of the early embryo. Development. 1992;116:1021–32. doi: 10.1242/dev.116.4.1021. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Asbrand C, Bakkers J, Kuhl M, Schaeffer HJ, Huelsken J, Behrens J, Hammerschmidt M, Birchmeier W. The ankyrin repeat protein Diversin recruits Casein kinase Iepsilon to the beta-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 2002;16:2073–84. doi: 10.1101/gad.230402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepich DS, Calmelet C, Kiskowski M, Solnica-Krezel L. Initiation of convergence and extension movements of lateral mesoderm during zebrafish gastrulation. Dev Dyn. 2005;234:279–92. doi: 10.1002/dvdy.20507. [DOI] [PubMed] [Google Scholar]
- Sepich DS, Myers DC, Short R, Topczewski J, Marlow F, Solnica-Krezel L. Role of the zebrafish trilobite locus in gastrulation movements of convergence and extension. Genesis. 2000;27:159–73. doi: 10.1002/1526-968x(200008)27:4<159::aid-gene50>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Shulman JM, Perrimon N, Axelrod JD. Frizzled signaling and the developmental control of cell polarity. Trends Genet. 1998;14:452–8. doi: 10.1016/s0168-9525(98)01584-4. [DOI] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–43. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solnica-Krezel L, Stemple DL, Driever W. Transparent things: cell fates and cell movements during early embryogenesis of zebrafish. Bioessays. 1995;17:931–9. doi: 10.1002/bies.950171106. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, Stemple DL, Mountcastle-Shah E, Rangini Z, Neuhauss SC, Malicki J, Schier AF, Stainier DY, Zwartkruis F, Abdelilah S, Driever W. Mutations affecting cell fates and cellular rearrangements during gastrulation in zebrafish. Development. 1996;123:67–80. doi: 10.1242/dev.123.1.67. [DOI] [PubMed] [Google Scholar]
- Stadler JA, Shkumatava A, Norton WH, Rau MJ, Geisler R, Fischer S, Neumann CJ. Histone deacetylase 1 is required for cell cycle exit and differentiation in the zebrafish retina. Dev Dyn. 2005;233:883–9. doi: 10.1002/dvdy.20427. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Tada M, Smith JC. Xwnt11 is a target of Xenopus Brachyury regulation of gastrulation movements via Dishevelled, but not through the canonical Wnt pathway. Development. 2000;127:2227–38. doi: 10.1242/dev.127.10.2227. [DOI] [PubMed] [Google Scholar]
- Topczewski J, Sepich DS, Myers DC, Walker C, Amores A, Lele Z, Hammerschmidt M, Postlethwait J, Solnica-Krezel L. The zebrafish glypican knypek controls cell polarity during gastrulation movements of convergent extension. Dev Cell. 2001;1:251–64. doi: 10.1016/s1534-5807(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Trevarrow B, Marks DL, Kimmel CB. Organization of hindbrain segments in the zebrafish embryo. Neuron. 1990;4:669–79. doi: 10.1016/0896-6273(90)90194-k. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–5. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Fraser SE, Harland RM. Convergent extension: the molecular control of polarized cell movement during embryonic development. Dev Cell. 2002;2:695–706. doi: 10.1016/s1534-5807(02)00197-1. [DOI] [PubMed] [Google Scholar]
- Weinberg ES, Allende ML, Kelly CS, Abdelhamid A, Murakami T, Andermann P, Doerre OG, Grunwald DJ, Riggleman B. Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development. 1996;122:271–80. doi: 10.1242/dev.122.1.271. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Ceron J, Ladd B, Dufourcq P, Reinke V, Shi Y. Regulation of tissue-specific and extracellular matrix-related genes by a class I histone deacetylase. Mol Cell. 2005;18:483–90. doi: 10.1016/j.molcel.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Bhatt S, Chavrier P, Bravo R, Charnay P. Segment-specific expression of a zinc-finger gene in the developing nervous system of the mouse. Nature. 1989;337:461–4. doi: 10.1038/337461a0. [DOI] [PubMed] [Google Scholar]
- Willert K, Brink M, Wodarz A, Varmus H, Nusse R. Casein kinase 2 associates with and phosphorylates dishevelled. EMBO J. 1997;16:3089–96. doi: 10.1093/emboj/16.11.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Tonou-Fujimori N, Komori A, Maeda R, Nojima Y, Li H, Okamoto H, Masai I. Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development. 2005;132:3027–43. doi: 10.1242/dev.01881. [DOI] [PubMed] [Google Scholar]
- Yan D, Wallingford JB, Sun TQ, Nelson AM, Sakanaka C, Reinhard C, Harland RM, Fantl WJ, Williams LT. Cell autonomous regulation of multiple Dishevelled-dependent pathways by mammalian Nkd. Proc Natl Acad Sci USA. 2001;98:3802–7. doi: 10.1073/pnas.071041898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. BioEssays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
Supplemental Figure 1. hdac1 RNA does not rescue defects in vangl2 morphants. vangl2 morphants injected with hdac1 RNA (B) still display axial extension defects observed in vangl2 morphants (A). The tangential migration defect of hindbrain nV11 branchiomotor neurons in vangl2 morphants is also not rescued by hdac1 misexpression (not shown).
Supplemental Figure 2 Overexpression of hdac1 in wildtype embryos causes brain and trunk defects. Injection of hdac1 RNA causes and enlargement of the forebrain (arrows) at moderate doses (B, compare to A). At high concentration hdac1 RNA causes loss of forebrain tissue and an expansion of midbrain (D, compare to C) accompanying severe loss of trunk tissues. wnt1 expression marks the posterior edge of the midbrain (arrow E) in uninjected wildtype embryos at 2 dpf. Embryos injected with hdac1 RNA display an antero-posterior expansion of wnt1 expression (arrows).