Mutation analysis of the hyperpolarization-activated cyclic nucleotide-gated channels HCN1 and HCN2 in idiopathic generalized epilepsy (original) (raw)
. Author manuscript; available in PMC: 2009 Jul 13.
Published in final edited form as: Neurobiol Dis. 2007 Aug 24;29(1):59–70. doi: 10.1016/j.nbd.2007.08.006
Abstract
Hyperpolarization-activated cyclic nucleotide-gated (HCN1-4) channels play an important role in the regulation of neuronal rhythmicity. In the present study we describe the mutation analysis of HCN1 and HCN2 in 84 unrelated patients with idiopathic generalized epilepsy (IGE). Several functional variants were identified including the amino acid substitution R527Q in HCN2 exon 5. HCN2 channels containing the R527Q variant demonstrated a trend towards a decreased slope of the conductance-voltage relation. We also identified a variant in the splice donor site of HCN2 exon 5 that results in the formation of a cryptic splice donor. In HCN1, the amino acid substitution A881T was identified in one sporadic IGE patient but was not observed in 510 controls. Seven variants were examined further in a case-control association study consisting of a larger cohort of IGE patients. Further studies are warranted to more clearly establish the contribution of HCN1 and HCN2 dysfunction to the genetic variance of common IGE syndromes.
Keywords: Hyperpolarization-activated cyclic nucleotide-gated channel, HCN1, HCN2, idiopathic generalized epilepsy, IGE, mutation analysis
Introduction
Idiopathic generalized epilepsies (IGEs) account for 15–20% of all epilepsies and affect approximately 0.2% of the general population (Jallon and Latour, 2005). IGEs encompass several syndromes that are characterized by age-related, recurrent, unprovoked generalized seizures in the absence of detectable brain lesions or metabolic abnormalities (International League Against Epilepsy, 1989). The most common IGE subtypes are childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), juvenile myoclonic epilepsy (JME), and epilepsy with generalized tonic-clonic seizures (EGTCS) (Nordli, 2005). The etiology of IGE is genetically determined, but the complex inheritance pattern suggests involvement of a large number of susceptibility genes in the majority of IGE patients (Greenberg et al., 1992; Ottman, 2005). Most of the genes currently identified in the rare, monogenic forms of idiopathic epilepsy encode voltage-gated or ligand-gated ion channels (Turnbull et al., 2005).
Generalized seizures can result from synchronized hyperexcitability of thalamocortical circuits facilitated by the nucleus reticularis thalami, which serves as the pacemaker in the generation of underlying rhythmic seizure activity (Avanzini et al., 1999; Crunelli and Leresche, 2002; Blumenfeld, 2005). The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in thalamocortical neurons contribute to the generation of this oscillatory activity (Pape, 1996) and are therefore good candidates for involvement in human epilepsy.
HCN channels are encoded by a gene family that consists of four members, HCN1, HCN2, HCN3, and HCN4 (Santoro et al., 1997; Ludwig et al., 1998), which are localized to human chromosomes 5p12, 19p13.3, 1q22, and 15q24–q25, respectively. HCN proteins share a topological arrangement consisting of six putative transmembrane segments (S1–S6), with a pore region (comprised of the S5, pore loop and S6) and a cyclic nucleotide-binding domain (CNBD) in the C-terminal region (Fig. 1A). Four HCN subunits assemble into a functional homomeric or heteromeric channel, and different subunit compositions lead to hyperpolarization-activated cation currents (Ih) with distinct biophysical properties (for review, see Santoro and Baram, 2003; Robinson and Siegelbaum, 2003; Frere et al., 2004; Herrmann et al., 2007).
Figure 1.
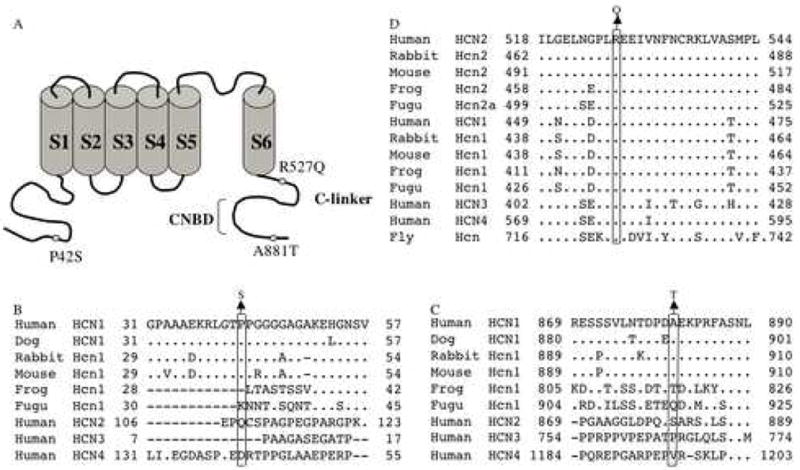
Location and evolutionary conservation of P42, A881, and R527. A. Schematic representation of HCN channels. The locations of the three amino acid substitutions identified in this study are shown. B. P42, located in the N-terminus of HCN1, is conserved in mammalian HCN1 channels. C. A881, located in the C-terminus of HCN1, is conserved in mammalian HCN1 channels. D. R527, located in the C-linker between the S6 segment and the CNBD, is invariant. Dots indicate identity to the top sequence. The alignments were generated using ClustalW1.82 (http://www.ebi.ac.uk/cgi-bin/clustalw).
HCN subunits differ in their activation kinetics and sensitivity to modulation by cAMP. cAMP favors channel activation, shifting the voltage dependence of activation to more depolarized potentials, speeding channel kinetics, and increasing the maximum current level (Craven and Zagotta, 2006). HCN1 exhibits the fastest activation kinetics and weak sensitivity to modulation by cAMP, whereas HCN2 displays slower activation kinetics and a more sensitive response to cAMP (Santoro et al., 1998; Ludwig et al., 1998, 1999; Moosmang et al., 2001). Differences have also been observed in the spatial and temporal expression patterns of HCN subtypes (Pape, 1996; Moosmang et al., 2001; Bender et al., 2001). HCN1 is primarily expressed in cortical, hippocampal, and cerebellar regions, while HCN4 is predominantly expressed in subcortical regions, such as the thalamus. HCN2 is broadly expressed in the brain with highest levels in thalamus and brainstem nuclei. HCN3 is expressed at low levels throughout the central nervous system (Moosmang et al., 1999; Santoro et al., 2000). During development of the rat hippocampus, HCN1 expression increases steadily with age, while HCN2 achieves maximum levels by post-natal day 11, and HCN4 progressively decreases after birth (Brewster et al., 2007).
While the causal relationship between HCN channel function and epilepsy is still unclear, there is increasing evidence that altered expression of HCN subtypes is associated with epileptogenesis and the chronic seizure state. Homozygous HCN2 knockout mice exhibit reduced locomotor activity and spontaneous absence seizures and provide the most direct evidence for the involvement of HCN channels in epilepsy (Ludwig et al., 2003). Reduced HCN1 protein levels, which precede the onset of seizures, have been observed in the cortex and hippocampus of the WAG/Rij rat model of absence epilepsy (Strauss et al., 2004; Kole et al., 2007). In contrast, decreased responsiveness of Ih to cAMP was observed in thalamocortical neurons of WAR/Rij and GAERS rats (Budde et al., 2005; Kuisle et al., 2006). This electrophysiological finding was attributed to increased HCN1 mRNA levels without changes in HCN2 and HCN4 (Budde et al., 2005). Brewster et al. (2005) demonstrated that experimental febrile seizures lead to reduced HCN1 expression and increased formation of hippocampal heteromeric HCN1/HCN2 channels. Increased HCN1 mRNA levels were also observed in an experimental rodent model of chronic hippocampal epilepsy, as well as in surviving dentate gyrus granule cells from patients with temporal lobe epilepsy and hippocampal sclerosis (Bender et al., 2003).
In order to test the hypothesis that mutations in HCN genes may underlie some forms of human epilepsy, we screened HCN1 and HCN2 for mutations in 84 unrelated IGE patients. We observed five HCN1 variants, including two nonsynonymous substitutions and a polymorphic variation in the length of a glycine repeat in exon 1. We identified 36 variants in HCN2, including the substitution of the conserved residue R527 in the C-linker and the intron 5 variant c.1584+7C>T, which creates a cryptic splice donor site. Seven HCN2 variants were further evaluated in a population-based association study comprising larger groups of IGE patients and ethnically matched unaffected controls.
Materials and Methods
Subjects
Unrelated study participants of German origin were recruited by the Department of Neurology, Charité University Medicine in Berlin, and by the University Clinic of Epileptology in Bonn, Germany. The Institutional Review Boards of both clinical sites approved the study protocol, and informed consent was obtained in writing from all participants. IGE syndromes were diagnostically classified according to the revised “Classification of Epilepsies and Epileptic Syndromes” as proposed by the Commission on Classification and Terminology of the International League Against Epilepsy (International League Against Epilepsy, 1989; Nordli, 2005).
Mutation Screening Sample
Eighty-four unrelated IGE patients were examined. Of these, 41 IGE patients had a positive family history for epilepsy, and the remaining cases were sporadic. Epilepsy subtypes represented among the patients included childhood absence epilepsy (CAE, n=23), juvenile absence epilepsy (JAE, n=20), juvenile myoclonic epilepsy (JME, n=40), and epilepsy with generalized tonic-clonic seizures (EGTCS, n=1). For the initial evaluation of IGE-associated susceptibility alleles, 50 unaffected, ethnically matched control subjects were also examined. The controls were screened for neuropsychiatric disorders via standardized interview. Individuals with a history of epileptic seizures or major neuropsychiatric disorders were excluded from the control sample.
Case-Control Sample
The IGE sample included 459 patients of German descent with the following IGE subtypes: childhood (CAE, n=127) and juvenile (JAE, n=73) absence epilepsy, juvenile myoclonic epilepsy (JME, n=197), and epilepsy with generalized tonic clonic seizures (EGTCS: age of-onset <25 years, EEG: generalized spike-wave discharge; n=62). The control sample consisted of 462 unaffected individuals of German decent. To test for potential population substructures in these samples, we applied a novel genomic control approach that assesses genomic similarities among unrelated individuals using 24 unlinked, highly polymorphic microsatellite polymorphisms, which were chosen as ethnicity markers based on normative data from various European populations (Stassen et al., 2004). Adaptive cluster analysis showed that there were no discernible differences in the population substructures between the IGE patients and controls. A recent study by the German Genome Research Network also demonstrated a lack of substantial genetic substructures in the German population (Steffens et al., 2006). SNP-based analysis (212 SNPs) of genetic substructures in the German population indicated a low degree of population heterogeneity between three subsamples collected in the Southwest and Northwest regions of Germany from >700 individuals with German-born grandparents. Thus, even if cryptic substructures do exist in the present study sample, the effect of undetected population stratification will be predicted to be small.
Mutation Detection
Genomic DNA was extracted from blood or lymphoblastoid cell lines using routine protocols (Miller et al., 1988). The coding regions and exon/intron boundaries of HCN1 and HCN2 were PCR-amplified from the genomic DNA of each patient. Primer sequences are available on request. PCR conditions for the amplification of all exons of HCN1 and HCN2, with the exception of HCN2 exon 8, were 94°C for 2 minutes, followed by 32 cycles of 94°C for 45 seconds, 55–62°C for 45 seconds, 72°C for 45 seconds, and a final extension of 72°C for 5 minutes. HCN2 exon 8 was amplified using the slowdown PCR technique that was developed for the amplification of GC-rich templates (Bachmann et al., 2003) with the modification that an extension time of 90 seconds was used. PCR products were incubated at 99°C for 10 minutes and then 68°C for 30 minutes to generate heteroduplex products. Samples were analyzed by conformation-sensitive gel electrophoresis (CSGE) and visualized by ethidium bromide staining as previously described (Escayg et al., 2000). PCR products that generated mobility variants on the CSGE gel were purified using QIAquick PCR Purification Kit (Qiagen) and were sequenced using an ABI 3100 automated sequencer (Applied Biosystems). Despite numerous attempts, we were unable to obtain satisfactory PCR amplification of HCN2 exon 1 which has a GC content of 83.5%. As a result, this exon was not analyzed. All novel variants identified in this study were submitted to the SNP database (http://www.ncbi.nlm.nih.gov/SNP/snpblastByChr.html).
Genotyping of Coding Sequence Variants
HCN1 coding variants, c.124C>T and c.2641G>A, and the HCN2 exon 2 variant, c.858T>C, were genotyped using TaqMan nuclease assays (Livak, 1999). TaqMan MGB probes and primers were synthesized by the Assays-by-Design service from Applied Biosystems (Applied Biosystems, Foster City, CA, USA). The HCN2 c.1580G>A variant was genotyped by PCR-RFLP analysis, based on an alternative PstI restriction site.
Genotypes of the polymorphic HCN1 exon 1 polyglycine repeat were assessed by fragment length analysis using PCR with flanking fluorescent primers. PCR fragments were separated on an ABI 3730 DNA Analyzer, and genotypes were scored using GeneMapper software version 3.0 (Applied Biosystems, Foster City, CA, USA).
Statistical Analyses
Allele and genotype frequencies, χ2-tests, and the test for Hardy-Weinberg equilibrium were calculated using the SAS computer program (SAS, 1988). A two-tailed type I error rate of 0.05 was chosen for the analyses. Power analysis for a presumed risk factor frequency of 5% in the entire case-control sample indicated a statistical likelihood of >90% to detect a predisposing effect, with an attributable relative risk (RR) of 2 and a likelihood of >50% for RR=1.5.
Splice Assay
Human genomic DNA was PCR-amplified from one individual who was heterozygous for the HCN2 intron 5 c.1584+7C>T variant and from the proband of Family 22, who was heterozygous for the linked variants, c.1584+7C>T and c.1580G>A, using the primer pair F1 (CGGGGTACCTGGGCCACAGAGCGAGACAGTG), R1 (CGGGGTACCACAGACCCAGGCAGTTCTGCTA). The 5′ end of each primer included a KpnI restriction site for subsequent cloning. The 771-bp PCR product consisted of the last 385 bp of intron 4, exon 5 and the first 239 bp of intron 5. The PCR product was digested with KpnI and cloned into the KpnI site in the Exontrap vector (MoBiTec). COS-7 cells were transfected with 1 μg of each plasmid in 6-well plates using FuGENE 6 Transfection Reagent (Roche). Cells were harvested two days later, and RNA was isolated using Trizol Reagent (Invitrogen) and purified using the RNeasy MinElute Cleanup Kit with DNase I treatment (Qiagen). First-strand cDNA was synthesized from 5 μg RNA using random hexamers and Superscript III following the manufacturer’s instructions (Invitrogen). Transcripts containing the longer version of exon 5, due to use of the cryptic splice donor site, were identified by PCR amplification of the cDNA using the primer pair F2 (GCCCCTGCGGGAGGTGAG), R2 (CCACGATGCCGCGCTTCT). PCR products were visualized on a 2.0% agarose gel, preparative grade for small fragments (Promega). Transcripts containing the longer version of exon 5 were confirmed by RT-PCR using the primer pair F3 (GAGGGATCCGCTTCCTGCCCC), R3 (CTCCCGGGCCACCTCCAGTGCC) that annealed to exons contained in the ExonTrap vector that flanked our sequence of interest. PCR products, predicted to comprise of both wild-type exon 5 and the longer version of exon 5, were cloned into the TOPO vector using the TOPO TA Cloning Kit (Invitrogen). To quantify the use of the cryptic splice donor site, the inserts from 100 clones were PCR amplified with primer pair F3, R3 and sequenced.
HCN2 Mutagenesis
The mouse HCN2 cDNA (a gift from S.A. Siegelbaum) was subcloned into the pGEMHE expression vector (a gift from E.R. Liman (Liman et al., 1992)). Primers were designed to generate the amino acid substitution R500Q, which is orthologous to the human substitution R527Q. Mutagenesis was carried out with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). The construct was then sequenced to confirm the absence of unwanted substitutions. The cDNA was linearized, and cRNA was transcribed in vitro using the mMESSAGE mMACHINE Kit (Ambion).
Electrophysiology
Xenopus laevis oocytes were defolliculated and injected with cRNA as previously described (Zagotta et al., 1989). Recordings were made in the excised inside-out patch configuration (Hamill et al., 1981) using an Axopatch 200A patch-clamp amplifier (Axon Instruments) and a RSC-100 rapid solution changer (Biologic) for internal solution application. Data were acquired with PULSE acquisition software (HEKA Elektronik). Patch pipettes were pulled from borosilicate glass and their resistances were 0.25–1 MΩ after fire polishing. The recording solutions were as follows: pipette (external) solution: 130 mM KCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2; bath (internal) solution: 130 mM KCl, 3 mM HEPES, 0.2 mM EDTA, pH 7.2, with either no cyclic nucleotides or 4 mM cAMP.
Electrophysiology Data Analysis
Conductance-Voltage Relations
To obtain GV curves, peak tail current amplitudes at −40 mV, either with or without saturating cAMP, were normalized by the largest peak tail current amplitude. These normalized data were then plotted against the test voltage potential and fit with Boltzmann curves:
G/Gmax=a+b/{1+exp[(V−Vhalf)/s]}
where a=normalized leak current, b=maximal normalized tail current amplitude, V=test voltage (mV), Vhalf=midpoint activation voltage (mV), and s=slope of relation (mV).
Statistical Analysis
All numerical values indicate mean±SEM. Statistical significance was estimated by Student’s t-test, unless otherwise indicated, and P values under 0.05 were considered significant.
Results
HCN1 Mutation Screening
Nonsynonymous Substitutions
Two nonsynonymous HCN1 sequence variants (exon 1: c.124C>T, p.P42S; exon 8: c.2641G>A, p.A881T) were identified in two unrelated, sporadic patients (Fig. 1A). P42 and A881, located in the N-terminus and C-terminus, respectively, are both conserved in the orthologous HCN1 protein from human, dog, rabbit, and mouse (Fig.1B and C). To further investigate a possible role for these variants in epilepsy, a larger sample that consisted of 260 IGE patients and 510 unaffected controls was screened for each variant. Neither substitution was observed in the IGE patient cohort; however, one control sample was found to carry the P42S substitution (Table 1).
Table 1.
Variants Identified in the Human HCN1 Gene
| Location | Nucleotide | Amino acid | dbSNP | Frequency of minor allele in: | |
|---|---|---|---|---|---|
| Patients n=82 | Controls n=50 | ||||
| Nonsynonymous | |||||
| Exon 1 | c.124C>T | p.P42S | ss73757979 | 0.21 | 0.13 |
| c.187_195del | p.G63_G65del | ss73757931 | 2.4 | ND | |
| c.193_201del | p.G65_G67del | ss73757936 | 2.4 | ND | |
| c.202_210del | p.G68_G70del | ss73757939 | 1.2 | ND | |
| c.202_207del | p.G68_G69del | ss73757941 | 1.2 | ND | |
| Exon 8 | c.2641G>A | p.A881T | ss73757981 | 0.21 | 0.03 |
| Synonymous and intronic variants | |||||
| Exon 6 | c.1521C>T | p.A507A | ss73757962 | 0.62 | ND |
| Intron 7 | c.1783+7dupT | NA | ss73757966 | 1.22 | 0.0 |
We also found polymorphic variation in the number of trinucleotides that code for glycine in a polyglycine stretch of HCN1 exon 1 (Table 1). Genotyping of the trinucleotide repeat polymorphism in a larger sample of 446 IGE patients revealed that the most common allele consisted of 12 glycine (G12) residues; however, minor alleles consisting of G9 and G10 residues were also observed, with allele frequencies of 1.6% and 0.8%, respectively. Association analysis of the trinucleotide polymorphism showed similar allele frequencies in 446 IGE patients and 454 unaffected controls (Table 2). Two rare alleles that consisted of G13 and G15 residues were also found in the controls, but not the patients (Table 2). The G15 allele was also present in a three-generation family with six members affected by IGE; this family has been investigated previously in a genome-wide linkage scan (Hempelmann et al., 2006). The G15 allele, however, did not cosegregate with the IGE trait in this family.
Table 2.
Allele Frequencies of the Glycine Repeat Polymorphism in HCN1 Exon1
| Samples | Number | Repeat no./Allele freq. | ||||
|---|---|---|---|---|---|---|
| (G)9 | (G)10 | (G)12 | (G)13 | (G)15 | ||
| Controls | 454 | 0.014 | 0.009 | 0.974 | 0.002 | 0.001 |
| IGE | 446 | 0.016 | 0.008 | 0.976 | — | — |
| CAE | 119 | 0.021 | 0.004 | 0.975 | — | — |
| IAE | 194 | 0.021 | 0.005 | 0.974 | — | — |
| JME | 190 | 0.016 | 0.013 | 0.971 | — | — |
Synonymous and Intronic Variants
The single-base duplication in intron 7 (c.1783+7dupT), which is in close proximity to the exon 7 splice donor site, was identified in two patients. One patient was the proband of a small pedigree consisting of two unaffected parents and one affected sibling. Mutation analysis of the additional family members revealed that the variant was inherited from the unaffected mother, but was absent in the affected sibling. The second patient was a sporadic case. This variant was not observed in 50 control samples (Table 1). None of the identified HCN1 variants were present in the SNP database (http://www.ncbi.nlm.nih.gov/SNP/snpblastByChr.html).
HCN2 Mutation Screening
Nonsynonymous Substitutions
The missense variant (c.1580G>A, p.R527Q) was identified in HCN2 exon 5 in the proband of a small IGE pedigree (Family 22) consisting of two unaffected parents and two affected siblings. R527 is located in the C-linker, the region between the S6 segment and the CNBD (Fig. 1A and Fig. 4) and is evolutionarily conserved in the orthologous HCN2 protein in mammals, frog and fugu as well as the paralogous HCN subtypes (Fig. 1D). Analysis of exon 5 in the remaining family members revealed that the R527Q substitution was present in the unaffected mother, but absent in the other affected sibling. R527Q was not observed on the analysis of a larger sample that consisted of 93 IGE patients and 307 unaffected controls.
Figure 4.
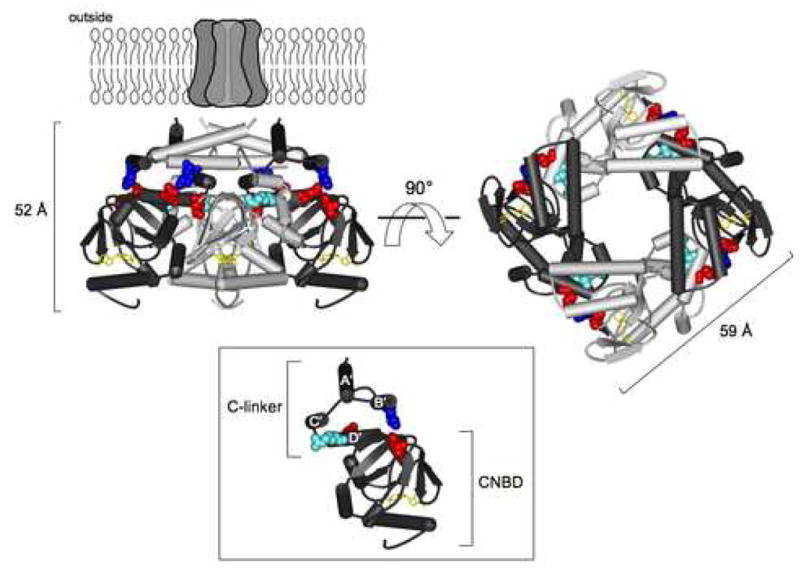
HCN2 C-terminal region crystal structure. Structure of the HCN2 C-terminal region (Zagotta et al., 2003) viewed from the side (left) and from the membrane (right). The structure is positioned below the membrane-spanning portion of the channel, as it is thought to be in vivo. The structure contains four subunits, two in dark gray and two in light gray, with the C-linkers making up the top half of the structure and the CNBDs the bottom half. cAMP (yellow) is bound in the CNBD of each subunit. Residues at position 500 (orthologous to position 527 in human sequence) and the salt bridge residues on the B′ and D′ helices, as well as the β-roll of the CNBDs, are shown in CPK format: R500 (light blue), K472 (blue), E502 and D542 (both in red). Side view of one subunit (inset). The A′-D′ helices of the C-linker are labeled, and the R500 residue can be seen projecting towards the hole down the center of the structure.
We also identified a 9-bp deletion in HCN2 exon 8 in one sporadic patient. The deletion occurred within a 21-bp “CCG” repeat that codes for seven proline residues in the common allele. The rare allele, present in the patient, contains four proline residues. This variant was observed in 2/50 controls, indicating that it is unlikely to be disease causing.
Synonymous and Intronic Variants
We identified 14 silent substitutions, 14 intronic variants, and 6 3′ UTR variants from our analysis of _HCN_2 (Table 3). We found that 27 of these 34 variants (79%) were not listed in the dbSNP database. Six of the silent substitutions were identified in exon 2. Of these, the minor alleles of the exon 2 SNPs, namely c.858T>C, c.915C>T, and c.963C>T, were always observed together in the same individuals, suggesting that they form part of a common haplotype.
Table 3.
Variants Identified in the Human HCN2 Gene
| Location | Nucleotide | Amino acid | dbSNP | Frequency of minor allele in: | |
|---|---|---|---|---|---|
| Patients n=84 | Controls n=50 | ||||
| Nonsynonymous | |||||
| Exon 5 | c.1580G>A | p.R527Q | ss73757987 | 0.61 | 0.02 |
| Exon 8 | c.2155_2163delCCGCC | p.P719_P72 | ss73758064 | 0.6 | 2.0 |
| GCCG | 1del | ||||
| Synonymous and intronic variants | |||||
| Exon 2 | c.714T>C | p.D238D | ss73757943 | 15.23 | 13.0 |
| c.723T>C | p.T241T | ss73757945 | 15.23 | 13.0 | |
| c.[858T>C; 915C>T; c.963C>T] | p.[Y286Y; F305F; R321R] | ss73757984; ss73757947; ss73757951 | 15.93 | 12.0 | |
| c.921C>T | p.I307I | ss73757949 | 12.23 | 9.4 | |
| Intron 2 | c.1056+55G>A | NA | rs10409268 | 49.43 | 48.0 |
| c.1056+80C>T | NA | rs12973067 | 3.13 | 8.3 | |
| Exon 3 | c.1086C>T | p.S362S | ss73757968 | 5.9 | ND |
| c.1089G>A | p.A363A | ss73757970 | 5.9 | ND | |
| c.1167T>C | p.P389P | rs12981860 | 33.0 | ND | |
| Intron 3 | c.1218+33C>T | NA | rs12977789 | 24.1 | ND |
| c.1219-23C>T | NA | ss73757972 | 0.6 | ND | |
| Exon 4 | c.1239G>C | p.L413L | rs3752158 | 5.9 | ND |
| Intron 4 | c.1438-16C>G | NA | ss73757974 | 11.2 | ND |
| Exon 5 | c.1452G>A | p.E484E | ss73757976 | 11.8 | ND |
| Intron 5 | c.1584+7C>T | NA | ss73757990 | 2.9 | 0 |
| Exon 6 | c.1644C>T | p.A548A | rs2301778 | 22.4 | ND |
| Intron 6 | c.1825+3G>A | NA | ss73757959 | 2.4 | 4.0 |
| c.1825+22_1825+42del CTGGAGGGGGAGGGGGCACGC | NA | ss73758050 | 19.4 | 14.0 | |
| c.1826-17delC | NA | ss73757957 | 32.93 | ND | |
| c.1826-18C>G | NA | ss73757953 | 0.63 | ND | |
| c.1826-20C>G | NA | ss73757955 | 0.63 | ND | |
| Exon 7 | c.1872C>T | p.A624A | rs1054786 | 31.0 | ND |
| Intron 7 | c.1990+24_1990+57dup GGGGCGGGTGCCCT GGCGGGGGAGGGG CGTGGCC | NA | ss73758057 | 48.8 | 40.0 |
| c.1991-25C>T | NA | ss73758060 | 1.2 | ND | |
| c.1991-4G>T | NA | ss73758062 | 0.6 | ND | |
| Exon 8 | c.2253G>C | p.A751A | ss73758066 | 19.6 | ND |
| 3′ UTR | c.2670+35G>A | NA | ss73758068 | 11.3 | ND |
| c.2670+94C>T | NA | ss73758070 | 0.6 | ND | |
| c.2670+137G>A | NA | ss73758072 | 0.6 | ND | |
| c.2670+197C>T | NA | ss73758074 | 4.8 | ND | |
| c.2670+214G>A | NA | ss73758076 | 17.3 | ND | |
| c.2670+227G>A | NA | ss73758078 | 17.3 | ND |
The intron 5 variant (c.1584+7C>T) was identified in three individuals with family histories of epilepsy and in two sporadic patients. Analysis of a larger sample comprising 452 IGE patients and 459 unaffected controls revealed similar frequencies of this minor allele in both groups, which makes this variant an unlikely candidate as a cause of epilepsy. Nonetheless, the close sequence similarity of the exon 5 splice donor site to the sequence created by the c.1584+7C>T substitution (Fig. 2A) raised the possibility that this variant could lead to the formation of a cryptic splice donor site. Interestingly, this variant was also identified in the proband of Family 22. Analysis of additional members of this family revealed that the c.1584+7C>T variant was present in the unaffected mother, but absent from the other affected sibling, which is reminiscent of the R527Q substitution. Therefore, the HCN2 variants, Q527 and c.1584+7T, appear to be on the same haplotype in Family 22.
Figure 2.
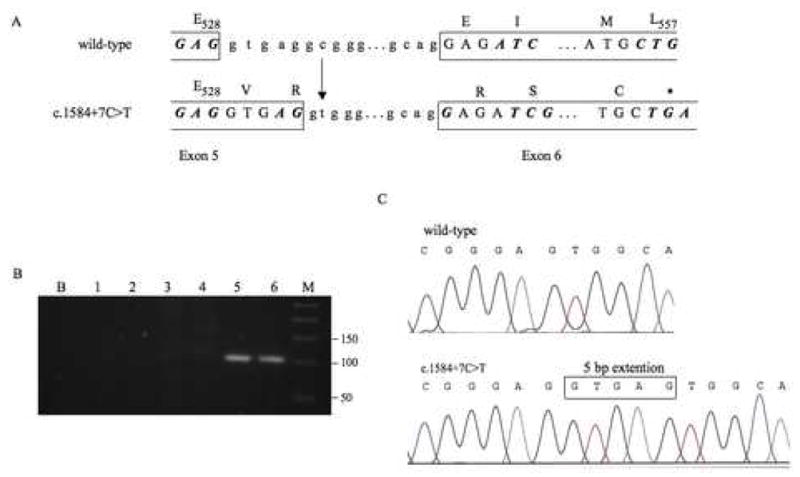
Location and functional effect of the HCN2 intron 5 variant c.1584+7C>T. A. The substitution of the cytosine residue (c) at c.1584+7 with a thymine (t) residue generates a potential cryptic splice donor site that closely matches the sequence of the wild-type splice donor site. Use of the cryptic donor site will result in the extension of exon 5 (boxed) by five additional nucleotides. Transcripts containing the longer version of exon 5 will result in a premature stop codon (asterisk) in exon 6. B. Transcripts containing the longer version of exon 5 were detected by RT-PCR analysis and visualized in 2% agarose gels. No PCR amplification was observed from samples containing either the wild-type sequence (Lane 1) or the c.1584+7C>T variant (Lane 2) when reverse transcriptase was omitted from the cDNA synthesis. No PCR amplification was observed from samples containing only the wild-type splice donor site (Lanes 3 and 4). The expected 101-bp PCR product was observed from samples that contained the c.1584+7C>T variant (Lanes 5 and 6). Lane B represents the negative control containing no cDNA in the PCR reaction; Lane M represents the 50-bp molecular weight ladder (Invitrogen). C. Transcripts in which exon 5 was longer by 5 bp were confirmed by sequence analysis.
Another potentially pathogenic HCN2 variant was c.1825+3G>A, located in the splice donor site of exon 6. This variant, however, occurred at a higher frequency in controls (4/50) than in patients (3/84), suggesting that it, too, is unlikely to be disease-causing. In addition to the identification of a large number of SNPs, we also identified a 21-bp deletion (c.1825+22_1825+42del) and a 34-bp duplication (c.1990+24_1990+57dup) in HCN2 intron 6 and intron 7, respectively. Both variants were present at similar frequencies in patients and controls (Table 3).
Case-Control Study
Seven HCN2 variants were tested in a case-control study consisting of at least 383 IGE patients and 275 unaffected controls (Table 4). The variants examined were c.858T>C, c.1239G>C, c.1584+7C>T, c.1825+22_1825+42del, c.1990+24_1990+57dup, and two SNPs, rs10408159 and rs4919872, selected from the dbSNP database. No evidence of an allelic or genotypic association between the controls and the IGE groups was found, indicating that these variants are unlikely to be associated with common IGE syndromes (Table 4).
Table 4.
Association Analysis of Seven HCN2 Variants
| c.858T>C | Number | C/C | C/T | T/T | f(C) | P*(df1) | P**(df2) |
|---|---|---|---|---|---|---|---|
| Controls | 455 | 0.004 | 0.185 | 0.811 | 0.097 | ||
| IGE | 450 | 0.007 | 0.191 | 0.802 | 0.102 | 0.695 | 0.868 |
| CAE | 122 | 0.000 | 0.189 | 0.811 | 0.094 | 0.909 | 0.762 |
| IAE | 196 | 0.005 | 0.214 | 0.781 | 0.112 | 0.394 | 0.672 |
| JME | 193 | 0.010 | 0.171 | 0.819 | 0.096 | 0.962 | 0.628 |
| c.1239G>C | Number | G/G | G/C | C/C | f(G) | P*(df1) | P**(df2) |
| Controls | 275 | 0.000 | 0.131 | 0.869 | 0.065 | ||
| IGE | 393 | 0.005 | 0.094 | 0.901 | 0.052 | 0.305 | 0.167 |
| CAE | 96 | 0.000 | 0.115 | 0.885 | 0.057 | 0.689 | |
| IAE | 163 | 0.000 | 0.086 | 0.914 | 0.043 | 0.165 | |
| JME | 175 | 0.011 | 0.114 | 0.874 | 0.069 | 0.855 | 0.184 |
| Exon 5 c.1584+7C>T | Number | T/T | C/T | C/C | f(T) | P*(df1) | P**(df2) |
| Controls | 459 | 0.000 | 0.052 | 0.948 | 0.026 | ||
| IGE | 452 | 0.000 | 0.035 | 0.965 | 0.018 | 0.219 | |
| CAE | 122 | 0.000 | 0.041 | 0.959 | 0.020 | 0.615 | |
| IAE | 197 | 0.000 | 0.036 | 0.964 | 0.018 | 0.360 | |
| JME | 193 | 0.000 | 0.047 | 0.953 | 0.023 | 0.767 | |
| Intron 6 c.1825+22_1825+42del21 | Number | 171/171 | 171/192 | 192/192 | f(171) | P*(df1) | P**(df2) |
| Controls | 460 | 0.026 | 0.313 | 0.661 | 0.183 | ||
| IGE | 453 | 0.024 | 0.292 | 0.684 | 0.184 | 0.924 | 0.971 |
| CAE | 124 | 0.022 | 0.258 | 0.713 | 0.190 | 0.803 | 0.688 |
| IAE | 197 | 0.027 | 0.284 | 0.689 | 0.180 | 0.917 | 0.907 |
| JME | 195 | 0.021 | 0.305 | 0.674 | 0.192 | 0.680 | 0.878 |
| Intron 7 c.1990+24_1990+57dup34 | Number | 200/200 | 200/234 | 234/234 | f(200) | P*(df1) | P**(df2) |
| Controls | 460 | 0.252 | 0.506 | 0.241 | 0.505 | ||
| IGE | 456 | 0.265 | 0.493 | 0.242 | 0.505 | 0.998 | 0.991 |
| CAE | 124 | 0.251 | 0.508 | 0.235 | 0.480 | 0.474 | 0.741 |
| IAE | 198 | 0.253 | 0.506 | 0.242 | 0.503 | 0.923 | 0.983 |
| JME | 196 | 0.272 | 0.473 | 0.255 | 0.500 | 0.857 | 0.910 |
| rs10408159 (c.1218+663C>T) | Number | T/T | C/T | C/C | f(T) | P*(df1) | P**(df2) |
| Controls | 275 | 0.015 | 0.218 | 0.767 | 0.124 | ||
| IGE | 393 | 0.015 | 0.219 | 0.766 | 0.125 | 0.954 | 0.997 |
| CAE | 95 | 0.011 | 0.211 | 0.779 | 0.116 | 0.775 | 0.944 |
| IAE | 163 | 0.026 | 0.215 | 0.761 | 0.132 | 0.772 | 0.752 |
| JME | 175 | 0.000 | 0.217 | 0.783 | 0.109 | 0.494 | 0.275 |
| rs4919872 (c.1584+985C>T) | Number | T/T | C/T | C/C | f(T) | P*(df1) | P**(df2) |
| Controls | 275 | 0.244 | 0.462 | 0.295 | 0.475 | ||
| IGE | 383 | 0.232 | 0.457 | 0.311 | 0.461 | 0.623 | 0.891 |
| CAE | 93 | 0.237 | 0.484 | 0.280 | 0.478 | 0.926 | 0.932 |
| IAE | 159 | 0.245 | 0.453 | 0.302 | 0.472 | 0.936 | 0.982 |
| JME | 169 | 0.249 | 0.420 | 0.331 | 0.459 | 0.643 | 0.645 |
c.1584+7C>T Creates a Cryptic Splice Donor Site
To determine whether the HCN2 intron 5 variant c.1584+7C>T generated a novel splice donor site, a 771-bp fragment consisting of exon 5 and flanking sequences from intron 4 and intron 5 was PCR-amplified from the genomic DNA of an individual who carried the variant. The PCR product was cloned into the Exontrap vector and expressed in COS-7 cells. Use of the cryptic splice donor site would be expected to generate a mutant mRNA in which exon 5 is longer by five nucleotides (Fig. 2A). RT-PCR analysis using a forward primer that was specific for the longer version of exon 5 generated the expected 101-bp product from cells containing the c.1584+7C>T variant, but not from cells expressing the wild-type HCN2 sequence, demonstrating use of the cryptic splice donor site (Fig. 2B).
To determine the relative use of the wild-type and mutant splice donor sites, RT-PCR analysis was repeated using a primer pair with homology to flanking exons of the Exontrap vector, and the PCR products were cloned into the TOPO vector. Sequence analysis of 100 individual clones revealed three clones (3%) containing the longer version of exon 5 and 97 clones (97%) containing the wild-type exon 5 (Fig. 2C). We obtained the same result on analysis of the allele containing both the c.1584+7C>T and c.1580G>A (R527Q) variants, indicating that the c.1580G>A substitution did not influence the use of the cryptic splice donor.
Functional Analysis of R527Q
To determine whether the HCN2 substitution R527Q affects channel function, we introduced this variant into the orthologous mouse channel, which is 91% identical in amino acid sequence to the human channel. HCN2 channels are activated by both membrane hyperpolarization and binding of cAMP to the intracellular cyclic nucleotide-binding domain (CNBD). Homomeric wild-type and mutant HCN2 channels containing the R500Q substitution (orthologous to R527Q in the human HCN2 channel) were expressed in Xenopus laevis oocytes, and currents were recorded using the patch-clamp technique in the inside-out configuration. Figure 3A shows currents from a representative patch of wild-type and a representative patch of mutant HCN2 channels. These currents were evoked by voltage steps to potentials ranging from −70 mV to −150 mV. In response to this hyperpolarization, wild-type and mutant HCN2 channels opened with a predominantly exponential time course following an initial lag. Normalized conductance (the ease with which ions pass through the ion channel) with respect to the voltage of the membrane (i.e., the conductance-voltage, or GV) curves indicate the steady-state activation of the channels and are shown in Figure 3B. Upon compilation and statistical analysis of these data, there were no statistical differences between the midpoints of these GV curves (Vhalf for wild-type (−126±1.9 mV) and mutant (−124±2.1 mV) channels) (Table 5). Although the slope values were not significantly different, (3.56±0.30 mV for wild-type and 4.23±0.31 mV for mutant channels, P = 0.16) (Table 5), there was a trend towards a shallower slope for the mutant channels. Saturating concentrations (4 mM) of cAMP increased the rates of activation and the steady-state current levels at hyperpolarizing membrane potentials, as well as led to a shift in the voltage dependence of activation to more depolarized potentials, for both wild-type and mutant channels (Figure 3 and Table 5). Neither the fold-increase in activation rate due to cAMP (wild-type: 2.67±0.62 and mutant: 3.34±0.64) nor the depolarizing shift in Vhalf due to cAMP (wild-type: −110±2.2 mV and mutant: −108±2.0) were significantly different between wild-type and mutant channels (Table 5). cAMP therefore stabilizes the closed-to-open equilibrium for mutant as well as wild-type HCN2 channels, thereby enabling activation with less hyperpolarization and more complete activation at hyperpolarized voltages. The difference between mutant and wild-type HCN2 channels in the conductance-voltage relations was not statistically significant either with or without cAMP.
Figure 3.
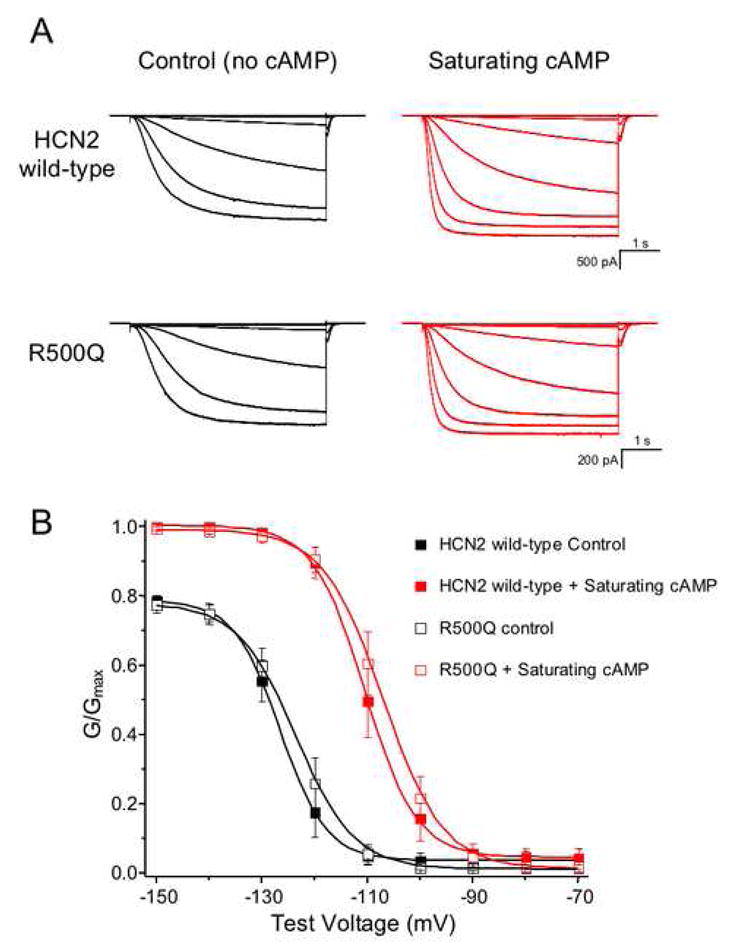
Functional analysis of homomeric mouse HCN2 channels, consisting of either wild-type or R500Q mutant subunits. (A) Currents from one representative patch of each channel construct are shown in the absence (black) and presence (red) of saturating (4 mM) cAMP. The currents were recorded in response to voltage pulses from a holding potential of 0 mV to test potentials between −70 mV and −150 mV, returning to a tail potential of −40 mV. (B) Conductance-voltage relations were obtained from normalized tail currents, either for HCN2 wild-type (filled symbols) or mutant (open symbols). The data shown are mean normalized values ± SEM and are fit with a Boltzmann relation.
Table 5.
Ih Properties of Wild-type and Mutant HCN2
| HCN2 channel | Control | Saturating cAMP | ΔVhalf (mV) | Rate _cAMP/_Rate Control | ||
|---|---|---|---|---|---|---|
| Vhalf (mV) | Slope (mV) | Vhalf (mV) | Slope (mV) | |||
| Wild-type (n=5) | −126±1.9 | 3.56±0.30 | −110±2.2* | 4.02±0.36 | 15.9±0.51 | 2.670.62 |
| Mutant (n=6) | −124±2.1 | 4.23±0.31 | −108±2.0* | 4.73±0.45 | 15.8±0.28 | 3.340.64 |
Discussion
In our analysis of 84 IGE patients, we identified five HCN1 and 36 HCN2 variants. There are likely additional HCN2 variants that remain to be identified, since we were unable to screen exon 1, due its high GC content. We did not consider HCN3 to be a likely candidate gene for epilepsy due to its low level of expression in the brain. HCN4 is the dominant HCN isoform in the adult sinoatrial node (SAN), and mutations have been identified in patients with SAN dysfunction, suggesting that it may play a more important role in the heart (Moosmang et al., 2001; Schulze-Bahr et al., 2003; Ueda et al., 2004; Milanesi et al., 2006). However, HCN4 should be considered a candidate for neonatal forms of epilepsy due to its high expression in neonatal hippocampal regions (Brewster et al., 2007). Patients with such forms of epilepsy were not included in this study and, as a result, HCN4 was not included in the analysis.
Two nonsynonymous substitutions, P42S and A881T, were detected in the N- and C-termini of HCN1 respectively. Since both variants were identified in sporadic patients, cosegregation with disease could not be confirmed. The N-terminus of HCNs is believed to play an essential role in the formation of functional homomeric or heteromeric channels (Proenza et al., 2002). Tran et al. (2002) demonstrated that the deletion of HCN1 residues 2–130, which includes P42, resulted in reduced currents via a number of possible mechanisms, including a decrease in the number of functional HCN channels or the disruption of the tertiary structure. Thus it is possible that the substitution of a nonpolar proline residue with a polar serine residue in the N-terminus of HCN1 could result in altered channel function or the ability to form functional homomeric or heteromeric channels. P42S was not observed in the larger sample of 260 IGE patients; however, it was detected in 1/510 unaffected controls and may therefore represent a rare nonpathogenic variant.
A881T was not observed in our analysis of 510 control samples, raising the possibility that this variant might be disease-causing. A881 is located seven nucleotides upstream of the conserved amino acid sequence (SNL) in the C-termini of HCNs. The SNL sequence is believed to be involved in the binding of a brain-specific protein, TRIP8b, that facilitates the transport of HCN channels to the dendrites (Santoro et al., 2004). The A881T substitution may alter the conformation of HCN1, thereby affecting its binding affinity to TRIP8b. Functional analysis of the A881T substitution will be necessary in order to determine whether it affects the biophysical properties or localization of HCN1.
We also observed polymorphic variation in the number of glycine residues (G9–15) in a polyglycine stretch of HCN1 exon 1. The most common allele consisted of 12 glycine residues; however, alleles containing nine and ten glycine residues were also identified at similar frequencies in both patients and controls. Rare longer alleles consisting of 13 and 15 glycines were also detected in control samples. Based on the comparable frequencies of these alleles in IGE patients and controls, it seems unlikely that they are associated with IGE. Nonetheless, we cannot rule out the possibility that the length of the polyglycine stretch may influence the biophysical properties of HCN1 channels, contributing to genotype-dependent differences in neuronal excitability. Such variation could influence seizure thresholds or contribute to other disorders. Polymorphic glycine repeats have previously been associated with disease (Brito et al., 2005; Werner et al., 2006), hence further analysis of the correlation between the number of glycine residues and HCN1 function is warranted.
In contrast to HCN1, we observed a high degree of sequence variation at the HCN2 locus. However, of the 36 HCN2 variants identified, only c.1580G>A (p.R527Q) and c.2155_2163del (p.P719_P721del) altered the amino acid sequence of the HCN2 protein. R527 is located in a highly conserved segment of the C-linker of HCN2. The C-linker plays a critical role in the activation gating of HCN channels (Ishii et al., 2001). It is composed of six α–helices (A′-F′) connected with short loops, which participate in several interactions, including the formation of salt bridges (Zagotta et al., 2003). The crystal structure of the HCN2 C-terminal region (Fig. 4) indicates that a positively charged lysine in the B′ helix interacts with both a negatively charged glutamic acid in the D′ helix and a negatively charged aspartic acid in the β-roll site of the CNBD to form one intersubunit and one intrasubunit salt bridge, respectively (Craven and Zagotta, 2004). Mutations in the residues forming salt bridges disrupt the salt bridges and favor channel opening. R500, the residue homologous to human R527, is located in the D′ helix and is two amino acids upstream of the negatively charged glutamic acid residue forming the intersubunit salt bridge (Fig. 4). The R527Q substitution may therefore alter the tertiary structure, thereby disrupting salt bridge formation or other hydrophobic interactions or hydrogen bonds.
Biophysical analysis of channels containing the orthologous R527Q substitution confirmed that they were still activated by hyperpolarization and modulated by cAMP, just like wild-type HCN2 channels. The mutant channels do show a trend for a shallower GV slope, indicating that these channels may respond to voltage differently than wild-type channels. However, cAMP modulation was not statistically different for wild-type (15.9±0.51) versus mutant (15.8±0.28) channels (Table 5), suggesting that the R500Q substitution did not affect the mechanism of cAMP modulation. Since the experiments conducted here examined the effect of saturating levels of cAMP, rather than a dose response relation, we cannot exclude the possibility that mutant channels may have a different binding affinity for cAMP. Furthermore, it is possible that mutant channels may gate differently than wild-type HCN2 channels in response to membrane voltage; however, as the exact voltage-sensing mechanism for hyperpolarization activation has yet to be elucidated, we cannot postulate how the mechanism may be altered by the R527Q substitution.
On the basis of structure/function analyses, several other mutations in the HCN2 C-terminal region (Craven and Zagotta, 2004; Decher et al., 2004; Zhou et al., 2004) and the C-termini of other HCN channel types (Mistrik and Torre, 2004) have been shown to result in shallower GV curves. Mutations in the S4–S5 linker of HCN2 also result in a shallower GV slope (Macri and Accili, 2004). Changes in the voltage sensitivity or voltage response of HCN channels may therefore be a common consequence of altered amino acid interactions or protein conformation.
Since the C-linker provides the predominant interface between channel subunits, it is possible that the R527Q mutation may alter the protein configuration of the HCN2 C-linker D′ helix, leading to a compromised intersubunit interaction in heteromeric channels. This will be predicted to alter subunit stoichiometry and channel function. Similar channel assembly mechanisms have been suggested for the related cyclic nucleotide-gated channels (Zheng et al., 2002; Zheng and Zagotta, 2004).
Functional analysis of the HCN2 intron 5 variant c.1584+7C>T demonstrated that it was likely to generate a cryptic splice donor site. Using the Splice Site Score Calculation (http://rulai.cshl.edu/new_alt_exon_db2/HTML/score.html), the wild-type exon 5 splice donor site and the cryptic splice donor site generated scores of 9.9 and 6.3, respectively. The mean score of the seven HCN2 splice donor sites is 6.6. Use of the cryptic splice donor site is predicted to disrupt the reading frame, resulting in premature termination of the HCN2 protein in exon 6. We observed 3% usage of the cryptic splice donor site in our in vitro assay; however, it is likely that the relative usage of the wild-type and cryptic splice donor sites would be different in vivo. Although the c.1584+7C>T substitution was observed at comparable frequencies in patients and controls, it is possible that this variant may influence seizure thresholds by reducing the level of functional HCN2 channels. Further studies to determine the in vivo effect of this substitution on HCN2 levels are therefore warranted.
We identified a total of 41 variants in this study. Functional analysis of two of these variants, R527Q and c.1584+7C>T, indicate that they may affect channel function. However, because R527Q did not cosegregate in the other affected family member and because c.1584+7C>T was observed at similar frequencies in patients and controls, a disease-causing role in epilepsy for these variants cannot be claimed. There are other variants, such as A881T, which was identified in HCN1 exon 8 and was not observed in 510 unaffected controls, which should also be explored as potential causes of disease. Results from the current study are consistent with the theory that common IGE subtypes arise from the complex interactions of several genes, each producing small to moderate effects. Our findings serve to underscore the difficulties posed by such complexity, highlighting the challenge of identifying the genes that lead to common forms of epilepsy. Further research into the HCN gene family will be necessary to more clearly establish their role in epilepsy.
Acknowledgments
We are grateful to the individuals and families who participated in this study. We thank S.A. Siegelbaum for the HCN2 wild-type clone and Cheryl Strauss for critically reading the manuscript. This study was supported by grants from the NIH (NS051834 to A.E.), the National Eye Institute (EY010329 to William N. Zagotta), the Deutsche Forschungsgemeinschaft (SA434/4-1 to T.S.), the German Federal Ministry of Education and Research (BMBF/NGFN2: 01GS0479 to T.S.), and the European Integrated Project EPICURE (EC contract number: LSH-CT-2006-037315 to T.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avanzini G, de Curtis M, Pape HC, Spreafico R. Intrinsic properties of reticular thalamic neurons relevant to genetically determined spike-wave generation. Adv Neurol. 1999;79:297–309. [PubMed] [Google Scholar]
- Bachmann HS, Siffert W, Frey UH. Successful amplification of extremely GC-rich promoter regions using a novel ‘slowdown PCR’ technique. Pharmacogenetics. 2003;13:759–766. doi: 10.1097/00008571-200312000-00006. [DOI] [PubMed] [Google Scholar]
- Bender RA, Brewster A, Santoro B, Ludwig A, Hofmann F, Biel M, Baram TZ. Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1–4 suggests evolving roles in the developing rat hippocampus. Neuroscience. 2001;106:689–698. doi: 10.1016/s0306-4522(01)00314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H. Cellular and network mechanisms of spike-wave seizures. Epilepsia. 2005;46 (Suppl 9):21–33. doi: 10.1111/j.1528-1167.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- Brewster AL, Bernard JA, Gall CM, Baram TZ. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol Dis. 2005;19:200–207. doi: 10.1016/j.nbd.2004.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AL, Chen Y, Bender RA, Yeh A, Shigemoto R, Baram TZ. Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex. 2007;17:702–712. doi: 10.1093/cercor/bhk021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito M, Malta-Vacas J, Carmona B, Aires C, Costa P, Martins AP, Ramos S, Conde AR, Monteiro C. Polyglycine expansions in eRF3/GSPT1 are associated with gastric cancer susceptibility. Carcinogenesis. 2005;26:2046–2049. doi: 10.1093/carcin/bgi168. [DOI] [PubMed] [Google Scholar]
- Budde T, Caputi L, Kanyshkova T, Staak R, Abrahamczik C, Munsch T, Pape HC. Impaired regulation of thalamic pacemaker channels through an imbalance of subunit expression in absence epilepsy. J Neurosci. 2005;25:9871–9882. doi: 10.1523/JNEUROSCI.2590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels. J Gen Physiol. 2004;124:627–629. doi: 10.1085/jgp.200409178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. Review. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Decher N, Chen J, Sanguinetti MC. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: molecular coupling between the S4–S5 and C-linkers. J Biol Chem. 2004;279:13859–13865. doi: 10.1074/jbc.M313704200. [DOI] [PubMed] [Google Scholar]
- Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, Johnston J, Baloh R, Sander T, Meisler MH. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531–1539. doi: 10.1086/302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere SG, Kuisle M, Luthi A. Regulation of recombinant and native hyperpolarization-activated cation channels. Mol Neurobiol. 2004;30:279–305. doi: 10.1385/MN:30:3:279. Review. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Durner M, Delgado-Escueta AV. Evidence for multiple gene loci in the expression of the common generalized epilepsies. Neurology. 1992;42:56–62. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hempelmann A, Lenzen KP, Heils A, Lorenz S, Prud’Homme JF, Nabbout R, Dulac O, Rudolf G, Zara F, Bianchi A, Robinson R, Gardiner MR, Covanis A, Lindhout D, Stephani U, Elger CE, Weber YG, Lerche H, Kron KL, Scheffer IE, Mulley JC, Berkovic SF, Nürnberg P, Sander T. Exploration of the genetic architecture of idiopathic generalized epilepsies. Epilepsia. 2006;47:1682–1690. doi: 10.1111/j.1528-1167.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Stieber J, Ludwig A. Pathophysiology of HCN channels. Pflugers Arch. 2007;454:517–522. doi: 10.1007/s00424-007-0224-4. Review. [DOI] [PubMed] [Google Scholar]
- International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Takano M, Ohmori H. Determinants of activation kinetics in mammalian hyperpolarization-activated cation channels. J Physiol. 2001;537:93–100. doi: 10.1111/j.1469-7793.2001.0093k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallon P, Latour P. Epidemiology of idiopathic generalized epilepsies. Epilepsia. 2005;46 (Suppl 9):10–14. doi: 10.1111/j.1528-1167.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- Kole MH, Brauer AU, Stuart GJ. Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol. 2007;578 (Pt 2):507–525. doi: 10.1113/jphysiol.2006.122028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuisle M, Wanaverbecq N, Brewster AL, Frere SG, Pinault D, Baram TZ, Luthi A. Functional stabilization of weakened thalamic pacemaker channel regulation in rat absence epilepsy. J Physiol. 2006;575(Pt 1):83–100. doi: 10.1113/jphysiol.2006.110486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macri V, Accili EA. Structural elements of instantaneous and slow gating in hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem. 2004;279:16832–16846. doi: 10.1074/jbc.M400518200. [DOI] [PubMed] [Google Scholar]
- Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistrik P, Torre V. Histidine 518 in the S6-CNBD linker controls pH dependence and gating of HCN channel from sea-urchin sperm. Pflugers Arch. 2004;448:76–84. doi: 10.1007/s00424-003-1228-3. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol Chem. 1999;380:975–980. doi: 10.1515/BC.1999.121. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem. 2001;268:1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- Nordli DR., Jr Idiopathic generalized epilepsies recognized by the International League Against Epilepsy. Epilepsia. 2005;46 (Suppl 9):48–56. doi: 10.1111/j.1528-1167.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- Ottman R. Analysis of genetically complex epilepsies. Epilepsia 46 Suppl. 2005;10:7–14. doi: 10.1111/j.1528-1167.2005.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker:the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Proenza C, Tran N, Angoli D, Zahynacz K, Balcar P, Accili EA. Different roles for the cyclic nucleotide binding domain and amino terminus in assembly and expression of hyperpolarization-activated, cyclic nucleotide-gated channels. J Biol Chem. 2002;277:29634–29642. doi: 10.1074/jbc.M200504200. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. Review. [DOI] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarizationactivatedpacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Baram TZ. The multiple personalities of h-channels. Trends Neurosci. 2003;26:550–554. doi: 10.1016/j.tins.2003.08.003. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT User’s Guide, Release 6.03. Cary, NC: 1988. [Google Scholar]
- Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, Isbrandt D. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest. 2003;111:1537–1545. doi: 10.1172/JCI16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stassen HH, Bridler R, Hell D, Weisbrod M, Scharfetter C. Ethnicity-independent genetic basis of functional psychoses: a genotype-to-phenotype approach. Am J Med Genet B Neuropsychiatr Genet. 2004;124:101–112. doi: 10.1002/ajmg.b.20081. [DOI] [PubMed] [Google Scholar]
- Steffens M, Lamina C, Illig T, Bettecken T, Vogler R, Entz P, Suk EK, Toliat MR, Klopp N, Caliebe A, Konig IR, Kohler K, Ludemann J, Lacava AD, Fimmers R, Lichtner P, Ziegler A, Wolf A, Krawczak M, Nurnberg P, Hampe J, Schreiber S, Meitinger T, Wichmann HE, Roeder K, Wienker TF, Baur MP. SNP-based analysis of genetic substructure in the German population. Hum Hered. 2006;62:20–29. doi: 10.1159/000095850. [DOI] [PubMed] [Google Scholar]
- Strauss U, Kole MHP, Brauer AU, Pahnke J, Bajorat R, Rolfs A, Nitsch R, Deisz RA. An impaired neocortical Ih is associated with enhanced excitability and absence epilepsy. Eur J Neurosci. 2004;19:3048–3058. doi: 10.1111/j.0953-816X.2004.03392.x. [DOI] [PubMed] [Google Scholar]
- Tran N, Proenza C, Macri V, Petigara F, Sloan E, Samler S, Accili EA. A conserved domain in the NH2 terminus important for assembly and functional expression of pacemaker channels. J Biol Chem. 2002;277:43588–43592. doi: 10.1074/jbc.M208477200. [DOI] [PubMed] [Google Scholar]
- Turnbull J, Lohi H, Kearney JA, Rouleau GA, Delgado-Escueta AV, Meisler MH, Cossette P, Minassian BA. Sacred disease secrets revealed: the genetics of human epilepsy. Hum Mol Genet. 2005;14:2491–2500. doi: 10.1093/hmg/ddi250. [DOI] [PubMed] [Google Scholar]
- Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, Morita H, Higashiuesato Y, Hirano Y, Yasunami M, Takishita S, Yamashina A, Ohe T, Sunamori M, Hiraoka M, Kimura A. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004;279:27194–27198. doi: 10.1074/jbc.M311953200. [DOI] [PubMed] [Google Scholar]
- Werner R, Holterhus PM, Binder G, Schwarz HP, Morlot M, Struve D, Marschke C, Hiort O. The A645D mutation in the hinge region of the human androgen receptor (AR) gene modulates AR activity, depending on the context of the polymorphic glutamine and glycine repeats. J Clin Endocrinol Metab. 2006;91:3515–3520. doi: 10.1210/jc.2006-0372. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc Natl Acad Sci U S A. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zheng J, Trudeau MC, Zagotta WN. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 2002;36:891–896. doi: 10.1016/s0896-6273(02)01099-1. [DOI] [PubMed] [Google Scholar]
- Zheng J, Zagotta WN. Stoichiometry and assembly of olfactory cyclic nucleotide-gated channels. Neuron. 2004;42:411–421. doi: 10.1016/s0896-6273(04)00253-3. [DOI] [PubMed] [Google Scholar]
- Zhou L, Olivier NB, Yao H, Young EC, Siegelbaum SA. A conserved tripeptide in CNG and HCN channels regulates ligand gating by controlling C-terminal oligomerization. Neuron. 2004;44:823–834. doi: 10.1016/j.neuron.2004.11.012. [DOI] [PubMed] [Google Scholar]