E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity (original) (raw)
. Author manuscript; available in PMC: 2009 Aug 10.
Published in final edited form as: Oncogene. 2008 Jul 28;27(51):6561–6570. doi: 10.1038/onc.2008.253
Abstract
The E2f transcription factors are key downstream targets of the retinoblastoma protein tumor suppressor that control cell proliferation. E2F3 has garnered particular attention because it is amplified in various human tumors. E2f3 mutant mice typically die around birth and E2f3-deficient cells have a proliferation defect that correlates with impaired E2f-target gene activation and also induction of p19Arf and p53. The E2f3 locus encodes two isoforms, E2f3a and E2f3b, which differ in their N-termini. However, it is unclear how E2f3a versus E2f3b contributes to E2f3's requirement in either proliferation or development. To address this, we use E2f3a- and E2f3b-specific knockouts. We show that inactivation of E2f3a results in a low penetrance proliferation defect in vitro whilst loss of E2f3b has no effect. This proliferation defect appears insufficient to disrupt normal development since E2f3a and E2f3b mutant mice are both fully viable and have no detectable defects. However, when combined with E2f1 mutation, inactivation of E2f3a, but not E2f3b, causes significant proliferation defects in vitro, neonatal lethality and also a striking cartilage defect. Thus, we conclude that E2f3a and E2f3b have largely overlapping functions in vivo and that E2f3a can fully substitute for E2f1 and E2f3 in most murine tissues.
Keywords: E2f3, Arf, cell cycle, E2f1
Introduction
The retinoblastoma protein (pRB) was the first identified tumor suppressor. pRB's tumor suppressive activity is largely dependent on its ability to regulate the E2f transcription factors. E2fs control the cell cycle dependent transcription of genes that encode key components of the cell cycle machinery (Attwooll et al., 2004; Dimova and Dyson, 2005). pRB, and its relatives p107 and p130, bind to E2fs in quiescent cells and this prevents transcription of E2f target genes via binding to the transactivation domain and blocking its function and by recruiting chromatin regulators which directly repress transcription (Attwooll et al., 2004; Dimova and Dyson, 2005; Blais and Dynlacht, 2007). Dissociation of E2f/pocket protein complexes is triggered by mitogen-induced phosphorylation of the pocket proteins by cyclin dependent kinases, Cdks, thus allowing activation of target gene transcription by E2fs (Trimarchi and Lees, 2002; Attwooll et al., 2004; Dimova and Dyson, 2005). The majority of human tumors carry mutations that either inactivate the retinoblastoma gene or activate the Cdks that mediate pocket protein phosphorylation. This strongly suggests that the pocket-protein associated E2fs, E2f1-5, play an important role in tumorigenesis.
The prevailing view is that individual E2f proteins are preferentially involved in either the activation or repression of E2f-responsive genes. E2f4 and E2f5 play a key role in gene repression whilst E2f1 and E2f2 play a key role in activating E2f-target genes (Trimarchi and Lees, 2002; Attwooll et al., 2004; Dimova and Dyson, 2005). E2f4 and E2f5 are expressed constitutively but E2f1 and 2 levels increase upon cell cycle re-entry because E2f1 and E2f2 are E2f-responsive genes. E2f3 is unusual in that it encodes two different proteins, E2f3a and E2f3b (He et al., 2000; Leone et al., 2000). These isoforms are generated by two separate promoters that govern the expression of alternative first exons that are spliced to a common second exon (Adams et al., 2000). E2f3a shares many of the properties of E2f1 and E2f2: it is inhibited by pRB in quiescent cells, recruits coactivators to E2f-responsive genes in G1 and its promoter is E2f-responsive. E2f3b is constitutively expressed, like E2f4 and E2f5, suggesting that it could function as a transcriptional repressor (Adams et al., 2000). However, in other regards, E2f3b more closely resembles the activating E2fs. It lacks the nuclear export signals that are characteristic of the repressive E2fs and thus remains in the nucleus after release from pocket proteins. E2f3 also associates specifically with pRB, and not p107 and p130, in vivo. Importantly, recent studies have linked E2F3 amplifications to the development of human bladder, lung and prostate tumors (Feber et al., 2004; Foster et al., 2004; Oeggerli et al., 2004; Cooper et al., 2006; Oeggerli et al., 2006; Hurst et al., 2007).
We previously generated an E2f3 mutant mouse strain that disrupts expression of both E2f3a and E2f3b. A large proportion of the E2f3-deficient mice die in utero or as neonates (Humbert et al., 2000; Cloud et al., 2002). Analysis of mouse embryonic fibroblasts (MEFs) shows that loss of E2f3 impairs both asynchronous proliferation and mitogen-induced cell cycle re-entry (Humbert et al., 2000). This latter defect correlated with two changes in gene regulation. First, there is a clear defect in the transcriptional activation of E2f-responsive genes (Humbert et al., 2000). This is consistent with the notion that E2f3a, and also possibly E2f3b, contribute to gene activation. Second, E2f3-loss is sufficient to derepress the Arf tumor suppressor, triggering activation of p53 and expression of the cdk-inhibitor p21Cip1 (Aslanian et al., 2004). In wildtype MEFs, the Arf promoter is specifically occupied by E2f3b, and not other E2fs, suggesting that E2f3b contributes to Arf repression in vivo (Aslanian et al., 2004). These observations offer two distinct mechanisms by which E2f3a and/or E2f3b could promote proliferation and tumorigenesis. Given these findings, we have generated E2f3a or E2f3b mutant mouse strains to determine how each of these isoforms contributes to E2f3's key roles in cellular proliferation and normal development.
Results
E2f3b binding to the Arf promoter is not required for Arf repression
To create E2f3a- and E2f3b-specific mutants we used recombineering to replace the relevant ATG translation start codon with a single loxP sequence (Figure 1a and Supplementary Information). ES cell lines shown to be correctly targeted by Southern blotting (Figure 1b) were used to produce E2f3a and E2f3b mutant mouse strains in both mixed (C57BL/6 × 129S/v) and pure 129S/v genetic backgrounds. Our first goal was to verify the specificity of our mutation strategy. To this end, we generated MEFs from mutant and wildtype littermates and conducted western blotting using a pan-E2F3 antibody, LLF3#2G2, which recognizes a C-terminal sequence common to both E2f3 isoforms (Parisi et al., 2007). This analysis confirmed that the introduced mutations specifically abolished the expression of either E2f3a or E2f3b (Figure 1c). Moreover, there was no detectable change in the levels of the remaining E2f3 isoform (Figure 1c) or in the levels of E2f1 or E2f4 (data not shown).
Figure 1.

Mutation of E2f3a or E2f3b by gene targeting. (a) Schematic of the targeting construct and the endogenous E2f3 locus. The E2f3a and E2f3b exons are represented by the shaded boxes. The common second exon is 65kb from these exons. DTa represents the diphtheria toxin negative selection cassette and loxPneo the loxP site flanked PGKEM7neobpA positive selection cassette. The two loci after the predicted homologous recombination event are shown for E2f3a and E2f3b. The two lines underneath the E2f3 locus represent the 5' and 3' probe sequences used for Southern analysis of the homologous recombination event. (b) The predicted sizes of the genomic DNA fragments identified by these probes are shown along with Southern analysis of representative targeted clones. In each case one targeted clone and one non-targeted clone is shown. (c) Western blotting analysis of protein extracts derived from mouse embryo fibroblasts (MEFs) of the indicated genotypes. In wildtype MEFs, both E2f3a and E2f3b are detected. Both of these isoforms run as doublets. In E2f3 mutant MEFs both isoforms are absent. Mutation of E2f3a or E2f3b leads to specific loss of only the mutated isoform indicating that the targeting strategy successfully disrupted expression of each isoform separately.
Our previous studies showed that E2f3 mutation lead to the induction of Arf in MEFs, and implicated the E2f3b isoform as a direct transcriptional repressor of Arf (Aslanian et al., 2004). Thus, we wanted to establish how the loss of either E2f3a or E2f3b affects the regulation of Arf. First, we performed chromatin immunoprecipitation (ChIP) experiments to examine E2f binding to the Arf promoter in MEFs derived from E2f3a-/- or E2f3b-/- embryos alongside their wildtype littermate controls (Figure 2). We used a pan-E2f3 antibody for these studies to allow direct comparison of the binding of E2f3a versus E2f3b. Consistent with our previous studies, we found that Arf is specifically bound by E2f3 in wildtype MEFs. This is in clear contrast to a classic E2f-responsive gene, p107, which shows significant occupancy by both E2f3 and E2f4. As expected, mutation of E2f3a did not alter the anti-E2f3 ChIP signal detected at Arf (Figure 2a). This supported our prior conclusion that E2f3b is the major E2f bound to Arf in MEFs but it does not preclude the possibility that both E2f3a and E2f3b can occupy the Arf promoter in wildtype MEFs. In the absence of E2f3b, we now observed ChIP signals with antibodies to E2f3, E2f4 and, to a lesser extent E2f1, at Arf (Figure 2b). Similar results were observed in three other isoform specific mutant lines analyzed (data not shown). Thus, we conclude that E2f3a, and also other E2fs, are able to bind to the Arf promoter in place of E2f3b in E2f3b-/- MEFs.
Figure 2.

Arf promoter regulation, p19Arf and p21Cip1 expression and asynchronous proliferation properties of E2f3a- and E2f3b-deficient MEFs. (a) Chromatin immunoprecipitation (ChIP) was performed using asynchronously proliferating wildtype and E2f3a-/- littermate MEFs, or (b) wildtype and E2f3b-/- littermate MEFs. Sonicated cross-linked chromatin was immunoprecipitated with antibodies to E2f3, E2f1, E2f4, or control IgG. The purified DNA was analyzed by PCR with primers specific for the p107 or Arf promoters, or a control sequence lacking E2f binding sites (1kb upstream of a control promoter). Input, 0.5% of chromatin in IP reaction was analyzed by PCR. These analyses show that in the absence of E2f3b, E2f3a is detected bound to the Arf promoter. (c) The majority of E2f3a mutant MEFs show little or no increase in p19Arf and p21Cip1 levels relative to wildtype controls, as illustrated by western blot analysis of two representative sets of serum arrested MEFs. Gapdh is shown as a loading control. (d) No increase in p19Arf or p21Cip1 levels are observed in E2f3b mutant MEFs relative to wildtype littermate controls. (e) E2f3a-/-, E2f3a+/- and E2f3a+/+ MEFs or (f) E2f3b-/-, E2f3b+/-, and E2f3b+/+ MEFs were assayed for asynchronous proliferation. Cells were plated in duplicate at 6×104/3cm dish and their growth monitored by daily counting for six days. No significant growth defect was observed in the isoform specific mutant cells.
To determine whether the loss of E2f3b or E2f3a increases the levels of p19Arf and p21Cip1 as observed in E2f3 and E2f1;E2f2;E2f3 mutant MEFs (Wu et al., 2001; Aslanian et al., 2004; Sharma et al., 2006; Timmers et al., 2007), we analyzed the levels of these proteins in serum starved mutant and wildtype MEFs generated from littermate embryos. We conducted these studies using both mixed (C57BL/6 × 129Sv) and pure 129Sv background MEFs, since the E2f3 mutant phenotype is always stronger in the pure 129Sv background (Cloud et al., 2002). Irrespective of the genetic background, the levels of p19Arf or p21Cip1 were not elevated in the E2f3b mutant cells (n=8 independent mutant cell lines) relative to wildtype littermate controls (Figure 2d). The mixed (C57BL/6 × 129Sv) background E2f3a-/- MEFs did not display any detectable defect in this, or any other, assay (data not shown). In contrast, the pure 129Sv background E2f3a-/- MEFs showed somewhat variable phenotypes. Five of the six lines tested showed either no upregulation of either p19Arf or p21Cip1, or they had slightly elevated levels of just one of these proteins (Figure 2c, data not shown). The remaining line had increased levels of both Arf and p21 (Supplementary Figure 1). These changes are subtle. However, the E2f3a-deficient MEFs seem to be more predisposed to upregulate p19Arf and/or p21Cip1 than wildtype controls. Taken together, these data show that the robust activation of the p19Arf-p53-p21Cip1 network that occurs in E2f3 mutant MEFs cannot be reproduced by the loss of either E2f3b or E2f3a. Since E2f3b is the predominant Arf promoter binder in wildtype cells but E2f3a takes its place in E2f3b-deficient MEFs and Arf and p21 regulation is subtly impaired in E2f3a-deficient MEFs, we conclude that E2f3a and E2f3b can play overlapping roles in the appropriate regulation of the p19Arf-p53-p21Cip1 network.
E2f3a and E2f3b play overlapping roles in controlling asynchronous proliferation and cell cycle entry
E2f3-loss impairs the proliferation properties of MEFs and this correlates with both the derepression of Arf and the failure to appropriately induce classic E2f-responsive genes (Humbert et al., 2000; Aslanian et al., 2004). Having shown that loss of E2f3a or E2f3b results in occasional or no increase in p19Arf and p21Cip1levels respectively, we wished to assess whether cell proliferation was compromised. To test this we compared the properties of mutant versus wildtype MEFs in standard proliferation assays, and also assayed their ability to re-enter the cell cycle from a serum starvation-induced quiescent state. The vast majority of the E2f3b mutant MEF lines had proliferation properties that were indistinguishable from the controls irrespective of whether they were generated in the mixed (6/7 lines tested) or pure 129Sv (6/7 lines tested) genetic backgrounds (Figure 2f shows representative lines). Moreover, none of the three of E2f3b mutant lines tested in cell cycle re-entry experiments showed any detectable defect in the kinetics of cell cycle progression, as judged by analysis of BrdU incorporation (Figure 3a shows a representative line). In tandem with this analysis, we also assessed mRNA expression levels of three E2f target genes, Cyclin E, Cyclin A and p107 as well as Arf and p21Cip1. Consistent with lack of proliferation and re-entry defects, we saw no significant difference in levels of these mRNAs in control versus E2f3b-/- MEFs using two independent sets of lines (Figure 3b and data not shown). Thus, we conclude that E2f3b-loss has no dramatic affect on either Arf levels or cell cycle regulation.
Figure 3.
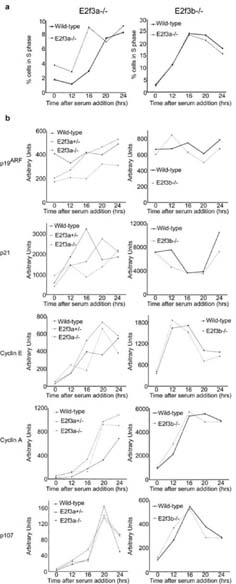
Cell cycle re-entry properties of E2f3a- and E2f3b-deficient MEFs. E2f3a-/- and E2f3b-/- MEFs were serum starved (T0) and induced to enter into the cell cycle with serum alongside wildtype littermate controls. (a) Entry into the cell cycle was analyzed by BrdU incorporation at the indicated times (hours) followed by propidium iodide staining and FACS analysis, the percentage of cells in S-phase (BrdU labeled) is plotted. (b) Quantitative PCR analyses of the mRNA levels of Arf and p21 as well as the E2f target genes Cyclin E, Cyclin A and p107 during the cell cycle re-entry experiment.
As with our analysis of Arf regulation, the properties of the E2f3a-/- MEFs were more variable. The majority (4/6) of the pure background E2f3a-/- lines showed no detectable defect in asynchronous proliferation (Figure 2e shows two representative lines). Accordingly, their kinetics of cell cycle re-entry and regulation of Cyclin E, Cyclin A, p107, Arf and p21 mRNAs was similar to that of wildtype littermate controls (Figure 3; data not shown). In contrast, two of the six E2f3a-/- lines grew more slowly than the wildtype littermate lines in asynchronous proliferation assays (Supplementary Figure 1). They also had a significant defect in cell cycle re-entry and this was associated with a failure to appropriately induce the transcription of classic E2f-responsive targets (Supplementary Figure 1). Importantly, one of these cells lines had an associated upregulation of Arf and p21 but the other showed little change in these mRNAs (Supplementary Figure 1). Given these findings, we conclude that E2f3a-/- MEFs have a partially penetrant phenotype that can affect both the p19Arf-p53-p21Cip1 network and cell cycle regulation. In one E2f3a-/- cell line, the defects in E2f target induction and cell cycle progression appeared to be independent of changes in Arf and p21 expression, suggesting that these events can be uncoupled. Finally, our data show that the loss of either E2f3a or E2f3b has far less impact on cell cycle regulation than the combined loss of both E2f3 isoforms (Humbert et al., 2000), indicating that these two proteins, or other members of the E2f family, can largely substitute for one another in the regulation of cell cycle genes.
Neither E2f3a nor E2f3b are required for viability
To assess the role of E2f3a in normal development, E2f3a+/- animals were intercrossed in either a mixed (C57BL/6×129Sv) or 129Sv background. A similar strategy was used to test the consequences of mutating the E2f3b allele. In each case the frequency of mutant animals was not statistically significantly different from the expected Mendelian frequency (Table 1 and 2). This is in contrast with the mutation of E2f3 (both a+b isoforms) which results in reduced viability in a mixed background and embryonic lethality in a 129Sv background (Humbert et al., 2000; Cloud et al., 2002). In addition, histological analyses of E2f3a-/- adults and E2f3b-/- adults (n=8 mutant animals of each isoform, 4 of each sex) in comparison with wildtype littermates on either background did not identify any distinct pathology. Additionally, no distinct phenotype associated with either E2f3 isoform mutation has been observed in aging colonies (data not shown).
Table 1.
Mutation of E2f3a does not cause a significant reduction in viability
| Background | _E2f3a_-/- observed | χ2 test p value |
|---|---|---|
| E2f3a Mix (C57BL/6 × 129Sv) | 24% (23/94) | 0.534 |
| E2f3a Pure (129Sv) | 23% (60/257) | 0.615 |
Table 2.
Mutation of E2f3b does not cause a significant reduction in viability
| Background | _E2f3b_-/- observed | χ2 test p value |
|---|---|---|
| E2f3b Mix (C57BL/6 × 129Sv) | 27% (47/176) | 0.248 |
| E2f3b Pure (129Sv) | 33% (28/85) | 0.256 |
E2f3a but not E2f3b is required for viability and proliferation control in the absence of E2f1
We have previously shown that the additional mutation of one or both E2f1 alleles causes E2f3 mutant embryos to die at progressively earlier timepoints, indicating significant functional overlap between E2f1 and E2f3 (Cloud et al., 2002). Thus, to further test the relative roles of the two E2f3 isoforms, we intercrossed the E2f3a and E2f3b mutants with E2f1 mutant mice to determine the phenotypes of compound mutant mice in a mixed (C57BL/6×129Sv) background. Remarkably, we found that E2f1-/-;E2f3b-/- animals were present at the expected Mendelian frequency when weaned at three weeks of age (Table 3). Moreover, these mice were viable and fertile, and histological analyses of E2f1-/-;E2f3b-/- (n=5) versus E2f1+/-;E2f3b+/- (n=3) littermate controls showed that the compound mutants did not display any defects beyond those previously reported in the E2f1 mutant animals (data not shown). Thus, E2f3b-loss does not exacerbate the phenotypic consequences of E2f1-deficiency. In contrast, E2f3a mutation had a dramatic effect. First, the E2f1-/-;E2f3a-/- mice were greatly under-represented (p=0.021) at three weeks of age (Table 4). Moreover, the two surviving double mutants weighed less than one-third of their E2f1-/-;E2f3a+/- littermates (data not shown). To determine the time of death, we conducted E2f1-/-;E2f3a+/- intercrosses and recovered the pups at P1 for genotyping and also histological analysis. At P1, the observed number of living E2f1-/-;E2f3a-/- pups was not statistically different from the expected number (Table 4). This, along with daily monitoring of other litters, showed that the E2f1;E2f3a compound mutants die as neonates.
Table 3.
E2f1;E2f3b double mutant animals arise at the expected frequency
| Cross conducted | _E2f1_-/-;_E2f3b_-/- observed | Expected |
|---|---|---|
| E2f1+/-;_E2f3b_-/- _x E2f1_-/-;E2f3b+/- | 25% (6/25) | 25% |
| _E2f1_-/-;E2f3b+/- x E2f1+/-;E2f3b+/- | 12.5% (2/16) | 12.5% |
| E2f1+/-;_E2f3b_-/- x E2f1+/-;E2f3b+/- | 16.7% (3/18) | 12.5% |
| _E2f1_-/-;_E2f3b_-/- x E2f1+/-;E2f3b+/- | 23% (3/13) | 25% |
| _E2f1_-/-;_E2f3b_-/- x E2f1+/-;_E2f3b_-/- | 40% (2/5) | 50% |
| χ2 test p value (sum) | 0.98 |
Table 4.
Mutation of E2f1 and E2f3a significantly reduces viability
| Age | _E2f1_-/-;E2f3a+/+ | _E2f1_-/-;E2f3a+/- | _E2f1_-/-;_E2f3a_-/- | χ2 test p value |
|---|---|---|---|---|
| 1 Day old | 16 | 20 | 9 | 0.615 |
| 3 Weeks old | 51 | 89 | 2 | 0.021 |
The histological analyses of P1 E2f1-/-;E2f3a-/- pups (n=6) and paired littermate controls did not identify any tissue specific defects that could obviously account for the death of the E2f1-/-;E2f3a-/- neonates (data not shown). Instead, we believe that this results from their failure to thrive because of their small size. Histological analysis did, however, reveal a striking defect in cartilage morphology in the E2f1;E2f3a compound mutants. This was observed in various bones, including the spine, but was most apparent in the long bones (Figure 4; data not shown). Thus, we analyzed the defect in this setting. The E2f1-/-;E2f3a-/- femurs stained appropriately with Alcian Blue, indicating that mature cartilage was formed (data not shown), but the constituent chondrocytes were disorganized and/or displayed abnormal morphologies (Figure 4a). First, chondrocytes within the columnar layer of E2f1;E2f3a compound mutant epiphyseal plates did not form stacked columns typical of chondrocytes in this region, and many of the E2f1-/-;E2f3a-/- cells lack the condensed “bean shape” cytoplasm that is characteristic of their wildtype counterparts but instead had a more diffuse cytoplasm resembling prehypertrophic chondrocytes (Figure 4a). Second, chondrocytes within the resting, columnar and hypertrophic zones of the E2f1;E2f3a compound mutant femurs appear considerably larger than those in control E2f1-/-;E2f3a+/+ embryos (Figure 4a). To further investigate this defect, we measured chondrocyte sizes in comparable zones of E2f1-/-;E2f3a-/- versus littermate control E2f1-/-;E2f3a+/+ femurs from two different P1 litters (Figure 4b). In each of the three zones we detected two clear differences between the two genotypes; the E2f1-/-;E2f3a-/- chondrocytes showed a much greater range of cell sizes than their E2f1-/- littermate controls and they displayed a statistically significant 1.5-2.0 fold increase in their mean surface area (p<0.001 or <0.0001). Analyses of 18.5dpc embryos gave identical results (Fig 4c, Supplementary Figure 2). Age-matched E2f3a mutant embryos showed a negligible increase in cell size (1.08 fold) only in the hypertrophic chondrocytes whilst E2f1 mutant embryos showed a modest 1.2-1.4 fold increase in cell size in all of the chondrocyte zones (Fig 4c, Supplementary Figure 2). These results show that E2f1 mutation causes a subtle chondrocyte defect and this is exacerbated by the mutation of E2f3a. Collagen II and collagen X, markers of chondrocytes and hypertrophic chondrocytes respectively, were both expressed in the appropriate regions of the mutant embryos (Supplementary Figure 3). Thus, the increase in chondrocyte size following loss of E2f activity does not appear to result from premature activation of the hypertrophic program.
Figure 4.

Mutation of E2f1 and E2f3a results in abnormal cartilage morphology. (a) Hematoxylin and eosin stained sections of hind leg femoral epiphyses from E2f1-/- and E2f1-/-;E2f3a-/- mutant littermate P1 pups. The zones of resting, columnar and prehypertrophic/hypertrophic chondrocytes are shown. Cells in all three zones are larger in E2f1;E2f3a mutants in comparison with control E2f1 mutants, in addition cells within the columnar zone are disorganized in the E2f1;E2f3a mutants in comparison with control E2f1 mutants and don't form the typical stacked columns. (b) From photographs of E2f1 mutant and littermate E2f1;E2f3a mutant femurs chondrocyte sizes in each zone were measured and the data displayed as box plots for two representative P1 litters illustrating the universal increase in cell size. The data was also analyzed using a student's t-Test and, in all zones, cells in the E2f1;E2f3a mutants were statistically significantly larger than those in the same zones of the E2f1 mutants (p values indicated). (c) Box plot analyses of chondrocyte size quantification from three or more E2f1;E2f3a mutant embryos and littermate controls of the indicated genotypes at 18.5dpc show a similar phenotype (left panel). A weaker but statistically significant increase in cell size is observed in 18.5dpc E2f1-/- embryos relative to wildtype controls (middle panel). A minor cell size increase is also seen in 18.5dpc E2f3a-/- embryos versus wildtype controls only in the hypertrophic chondrocytes (right panel), p values derived from Student's t-test analysis of the mean cell sizes are shown. (d) Quantification of proliferation markers in resting and columnar chondrocytes at 18.5 dpc. Immunohistochemistry was used to label cells that had incorporated BrdU or expressed Ki67. No significant difference in BrdU labeling was detected but Ki67 was detected in a smaller percentage of cells within the columnar layer in E2f1;E2f3a mutants relative to E2f1 mutants (mean +/- s.d. and p values indicated).
E2f3 and E2f1 have both been linked to the regulation of proliferation and cell death. Thus, we examined the state of both processes by screening matched femoral sections by immunohistochemical staining for Ki67, a proliferation marker and known E2f-responsive gene, and incorporated BrdU, to detect replicating cells, and TUNEL or cleaved caspase 3 staining to detect apoptotic cells. Given the mild chondrocyte defect in the E2f1-/- mice, we first compared femurs from E2f1-/- embryos at 18.5dpc with those of wildtype littermate controls. In these two genotypes, we saw no statistically significant difference in BrdU or Ki67 labeling of the cells in the resting and columnar zones (data not shown). We could not assess cells in the hypertrophic zone, since these typically lose their nuclei. We next compared femurs from E2f1-/-;E2f3a-/- versus E2f1-/-;E2f3a+/+ littermates at 18.5dpc. There was no detectable difference in the level of apoptosis between these two genotypes (data not shown). Similarly, there was no statistically significant difference in the percentage of BrdU-positive cells in either the resting or columnar zones of E2f1-/-;E2f3a-/- versus E2f1-/-;E2f3a+/+ femurs at 18.5dpc (Figure 4d). In contrast, analysis of adjacent sections showed the percentage of Ki67 labeled cells was modestly decreased in the resting zone (p=0.058), and significantly decreased in the columnar zone (p=0.015), of the E2f1-/-;E2f3a-/- versus E2f1-/-;E2f3a+/+ embryos (Figure 4d). We therefore conclude that the combined loss of E2f1 and E2f3a impairs the expression of at least one E2f-target gene, Ki67, in chondrocytes. Although there is no detectable impairment of DNA replication at this timepoint, these data suggest that loss of E2f1 and E2f3a somehow impairs cell cycle progression.
To further explore this possibility, we generated MEFs from E2f1-/-;E2f3a-/- embryos and compared their properties to E2f1-/- littermate controls, since it is well established that E2f1-deficiency does not significantly impair MEFs (Field et al., 1996; Humbert et al., 2000). Three of the four E2f1-/-;E2f3a-/- MEF lines analyzed were impaired in both asynchronous proliferation and cell cycle re-entry (Figure 5a,b; data not shown). Accordingly, we observed an upregulation in both Arf and p21 mRNA and protein levels (Figures 5c,d), and poor induction of the classic E2f-responsive genes Cyclin E, Cyclin A and p107 following cell cycle entry (Figure 5e). This spectrum of defects is strikingly similar to that seen in E2f3-/- MEFs, lacking both E2f3a and E2f3b (Humbert et al., 2000; Aslanian et al., 2004). Consistent with the viability of E2f1-/-;E2f3b-/- mice, preliminary studies indicate that the E2f1-/-;E2f3b-/- MEF lines don't have a proliferation defect (data not shown). Taken together, these data suggest that there is significant overlap in the functions of E2f1, E2f3a and E2f3b. At least in the context of MEFs and E2f1-deficiency, E2f3a plays a more important role than E2f3b.
Figure 5.

E2f1;E2f3a MEF lines show defects in proliferation, cell cycle re-entry and E2f target gene induction. (a) E2f1-/-;E2f3a-/- MEFs were assayed for asynchronous proliferation alongside E2f1-/- littermate controls. Cells were plated in duplicate at 6×104/3cm dish and their growth monitored by daily counting for six days. (b) E2f1-/- and E2f1-/-;E2f3a-/- littermate MEFs were serum starved (T0) and induced to enter into the cell cycle with serum. Entry into the cell cycle was analyzed by BrdU incorporation at the indicated times (hours) followed by propidium iodide staining and FACS analysis, the percentage of cells in S-phase (BrdU labeled) is plotted. (c) Quantitative PCR analyses of the mRNA expression levels of Arf and p21 during cell cycle re-entry. (d) Western blotting for p19Arf, p21Cip1 and Gapdh (as a loading control) in serum starved MEFs. (e) Quantitative PCR analyses of the mRNA expression levels of the E2f target genes Cyclin E, Cyclin A and p107 in E2f1-/- and E2f1-/-;E2f3a-/- littermate MEFs during the cell cycle re-entry experiment. Both E2f1-/-;E2f3a-/- MEF lines showed reduced proliferation, reduced S-phase entry, increased p19Arf and p21Cip1 expression and lower levels of E2f target gene expression.
Discussion
The E2f transcription factors are key downstream targets of the pRB tumor suppressor. Considerable attention has focused on E2F3 because it is amplified in a variety of human tumors including bladder, lung and prostate tumors (Feber et al., 2004; Foster et al., 2004; Oeggerli et al., 2004; Cooper et al., 2006; Oeggerli et al., 2006; Orlic et al., 2006; Hurst et al., 2007). We, and others, have previously generated E2f3 mutant mouse strains that eliminate expression of both E2f3a and E2f3b (Humbert et al., 2000; Wu et al., 2001). Analyses of these models show that E2f3 is of central importance. First, E2f3 promotes the development of various tumor types (Ziebold et al., 2003; Parisi et al., 2007). Second, it is the only E2f knockout that leads to embryonic lethality or to a profound defect in cellular proliferation (Humbert et al., 2000; Wu et al., 2001; Cloud et al., 2002). Notably, this proliferation defect correlates with two distinct changes: there is an impaired activation of E2f responsive genes and also a derepression of Arf and a consequent activation of the p53-p21Cip1 anti-proliferative response (Aslanian et al., 2004). Our prior analysis strongly suggested that this latter defect reflects a specific role for E2f3b in the transcriptional repression of Arf (Aslanian et al., 2004). However, it was unclear to what extent E2f3a versus E2f3b contributes to either the cellular or developmental requirements for E2f3. In this study, we address this question through the generation and analyses of E2f3a and E2f3b-specific mutant mouse strains. These analyses show that the two isoforms have largely overlapping functions. First, the loss of E2f3a has a low penetrance effect on the asynchronous proliferation and cell cycle re-entry properties of MEFs. In contrast, we found that E2f3b is not required for the appropriate repression of Arf, asynchronous proliferation, or cell cycle re-entry in MEFs. Second, in stark contrast to the high frequency of late stage embryonic or early neonatal lethality that results from E2f3-inactivation, we find that the E2f3a-/- and E2f3b-/- mice are born at the expected frequency and they live into adulthood without any significant pathology. Thus, these data show that the presence of either E2f3a or E2f3b is largely sufficient to fulfill the essential role of the E2f3 locus in both the control of cellular proliferation and normal development.
We have previously demonstrated a strong synergy between E2f3 and E2f1 by showing that the combined mutation of these genes causes lethality between day 10 and 12 of gestation (Cloud et al., 2002). Given this observation, we further probed the relative roles of E2f3a and E2F3b by intercrossing the isoform-specific knockouts with E2f1 mutant mice. Remarkably, we found that the E2f1-/-;E2f3b-/- double mutant mice were fully viable and had only the limited spectrum of developmental defects that are characteristic of the E2f1-deficient animals. Since E2f1-/-;E2f3-/- embryos die in midgestation (Cloud et al., 2002), this shows that E2f3a can fully substitute for E2f1 and E2f3 genes in the vast majority of murine tissues. This does not assume that E2f3a is acting alone to mediate all of the functions of E2f1, E2f3a and E2f3b, but that the combination of E2f3a and the other endogenous E2f family members gives sufficient total E2f activity to allow near normal development. Given these observations, we conclude that the E2f network has a significant degree of redundancy and robustness, reminiscent of that seen for other core cell cycle regulators including the cyclins and cdks (Berthet and Kaldis, 2007). In contrast to the E2f1-/-;E2f3b-/- animals, we found that the E2f1-/-;E2f3a-/- double mutants arise at expected frequency but die within the first few weeks of life. Thus, at least in the context of E2f1 loss, E2f3a plays a more important role than E2f3b in vivo. A similar result was observed in vitro: E2f1-/-;E2f3a-/- double mutant MEFs exhibit a reduced level of proliferation, are impaired in their ability to re-enter the cell cycle and fail to appropriately regulate E2f-target genes. In contrast, preliminary analysis suggests that the E2f1-/-;E2f3b-/- double mutant lines are essentially normal, again indicating a more important role for E2f3a. It is formally possible that E2f3a has one, or more, function(s) that are specifically shared by E2f1 and not other E2fs. However, we favor the hypothesis that this reflects some difference in the relative levels, or timing of expression, of the two isoforms such that E2f3a, but not E2f3b, places the total E2f activity above a critical threshold. Notably, our analysis of the E2f1;E2f3a mutant neonates also reveals an essential role for E2f1 plus E2f3a in cartilage development. Specifically, we find that the E2f1-/-;E2f3a-/- chondrocytes have an abnormal morphology, are not appropriately organized within the epiphysis and are significantly larger than normal. This appears to be an exacerbation of a milder phenotype in E2f1 mutant embryos. Further experiments will be required to establish the precise cause of these defects. However, our analysis suggests that the chondrocyte phenotype could be due to reduced E2f target gene expression. One intriguing possibility is that these cells have increased their size in preparation for cell division but have trouble proceeding through the cell cycle because there is insufficient accumulation of E2f target gene products, many of which are rate limiting for DNA replication and mitosis.
Materials and Methods
Generation of mutant mouse strains
see Supplementary Information.
MEF generation and analyses
Passage 4 MEF lines were prepared as described (Humbert et al., 2000). Mutant MEFs were always compared with littermate controls. Proliferation assays and cell cycle re-entry experiments were performed essentially as described (Aslanian et al., 2004) apart from the determination of S-phase progression which was monitored by FACS based detection of incorporated BrdU using a FITC conjugated anti-BrdU antibody (347583 BD Biosciences, San Jose, CA, USA) as described (Janumyan et al., 2003), using a BD FACScan. For Western blotting, protein extracts were prepared in 20mM Tris pH7.5, 250mM NaCl, 5mM NaF, 1mM EDTA, 1mM EGTA, 1% Triton as described using antibodies against E2f3 (LLF3#2G2; Parisi et al., 2007), p19Arf (sc32748, Santa Cruz Biotechnology, Santa Cruz, USA), p21Cip1 (sc6246, Santa Cruz Biotechnology) and Gapdh (4300, Ambion, Austin, TX, USA). ChIP was performed as described (Aslanian et al., 2004). For quantitative PCR, RNA was collected from cells at 0, 12, 16, 20, and 24 hours after re-entry and RNA was processed as described (Courel et al., 2008). qPCR primers are listed in the Supplementary Information.
Histological analyses and immunohistochemistry
Soft tissues were fixed in 3.7% formaldehyde in PBS overnight whilst adult bones were fixed in Bouin's fixative (Poly Scientific, Bat Shore, NY, USA) for 10 days. Paraffin sections were cut at 5μm, dewaxed and stained with hematoxylin and eosin. To assess proliferation, femur sections from a minimum of three pairs of control and mutant littermates were matched along the proximal-distal axis and immunohistochemistry was performed using antibodies raised against Ki67 and BrdU following BrdU labeling as described (Danielian et al., 2007). The percentage of positive nuclei was determined by counting 150-1000 nuclei within each zone in the femoral cartilage per section and the results analyzed by Student's t-Test. Cell area measurements were made using ImageJ software. A minimum of 35 cells of each chondrocyte type from each femoral head were measured and subject to the Student's t-Test and box-plot analyses. In all quantification studies at 18.5dpc between three and six pairs of control and mutant littermates embryos or pups were analyzed and both femurs were sectioned.
Supplementary Material
S1
S2
s3
sup. info
Acknowledgements
We thank Aurora Burds Connor and the MIT Fannie E. Rippel transgenic facility; the CCR Flow Cytometry facility for technical assistance; Donald Court, Neal Copeland, Nancy Jenkins, and Lili Yamasaki for reagents and mouse strains and Keara Lane, Daniel Garcia, GuangJun Zhang and Lees laboratory members for helpful suggestions. This work was supported by an NIH grant to J.A.L. (CA118757). J.A.L. is a Ludwig Scholar.
References
- Adams MR, Sears R, Nuckolls F, Leone G, Nevins JR. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol Cell Biol. 2000;20:3633–3639. doi: 10.1128/mcb.20.10.3633-3639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev. 2004;18:1413–1422. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. Embo J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Kaldis P. Cell-specific responses to loss of cyclin-dependent kinases. Oncogene. 2007;26:4469–4477. doi: 10.1038/sj.onc.1210243. [DOI] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud JE, Rogers C, Reza TL, Ziebold U, Stone JR, Picard MH, et al. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol Cell Biol. 2002;22:2663–2672. doi: 10.1128/MCB.22.8.2663-2672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CS, Nicholson AG, Foster C, Dodson A, Edwards S, Fletcher A, et al. Nuclear overexpression of the E2F3 transcription factor in human lung cancer. Lung Cancer. 2006;54:155–162. doi: 10.1016/j.lungcan.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Courel M, Friesenhahn L, Lees JA. E2f6 and Bmi1 cooperate in axial skeletal development. Dev Dyn. 2008;237:1232–1242. doi: 10.1002/dvdy.21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, Bender Kim CF, Caron AM, Vasile E, Bronson RT, Lees JA. E2f4 is required for normal development of the airway epithelium. Dev Biol. 2007;305:564–576. doi: 10.1016/j.ydbio.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- Feber A, Clark J, Goodwin G, Dodson AR, Smith PH, Fletcher A, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004;23:1627–1630. doi: 10.1038/sj.onc.1207274. [DOI] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr., Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Foster CS, Falconer A, Dodson AR, Norman AR, Dennis N, Fletcher A, et al. Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene. 2004;23:5871–5879. doi: 10.1038/sj.onc.1207800. [DOI] [PubMed] [Google Scholar]
- He Y, Armanious MK, Thomas MJ, Cress WD. Identification of E2F-3B, an alternative form of E2F-3 lacking a conserved N-terminal region. Oncogene. 2000;19:3422–3433. doi: 10.1038/sj.onc.1203682. [DOI] [PubMed] [Google Scholar]
- Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- Hurst CD, Tomlinson DC, Williams SV, Platt FM, Knowles MA. Inactivation of the Rb pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. Oncogene. 2007 doi: 10.1038/sj.onc.1210934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, et al. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. Embo J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, et al. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol. 2000;20:3626–3632. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeggerli M, Schraml P, Ruiz C, Bloch M, Novotny H, Mirlacher M, et al. E2F3 is the main target gene of the 6p22 amplicon with high specificity for human bladder cancer. Oncogene. 2006;25:6538–6543. doi: 10.1038/sj.onc.1209946. [DOI] [PubMed] [Google Scholar]
- Oeggerli M, Tomovska S, Schraml P, Calvano-Forte D, Schafroth S, Simon R, et al. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene. 2004;23:5616–5623. doi: 10.1038/sj.onc.1207749. [DOI] [PubMed] [Google Scholar]
- Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 2006;45:72–82. doi: 10.1002/gcc.20263. [DOI] [PubMed] [Google Scholar]
- Parisi T, Yuan TL, Faust AM, Caron AM, Bronson R, Lees JA. Selective requirements for E2f3 in the development and tumorigenicity of Rb-deficient chimeric tissues. Mol Cell Biol. 2007;27:2283–2293. doi: 10.1128/MCB.01854-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Timmers C, Trikha P, Saavedra HI, Obery A, Leone G. Control of the p53-p21CIP1 Axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and cellular transformation. J Biol Chem. 2006;281:36124–36131. doi: 10.1074/jbc.M604152200. [DOI] [PubMed] [Google Scholar]
- Timmers C, Sharma N, Opavsky R, Maiti B, Wu L, Wu J, et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27:65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol. 2003;23:6542–6552. doi: 10.1128/MCB.23.18.6542-6552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1
S2
s3
sup. info