Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma (original) (raw)
. Author manuscript; available in PMC: 2009 Aug 23.
Published in final edited form as: Cancer Cell. 2007 Aug;12(2):115–130. doi: 10.1016/j.ccr.2007.07.004
Summary
Mechanisms of constitutive NF-κB signaling in multiple myeloma are unknown. An inhibitor of IκB kinase beta (IKKβ), targeting the classical NF-κB pathway, was lethal to many myeloma cell lines. Several cell lines had elevated expression of NIK due to genomic alterations or protein stabilization while others had inactivating mutations of TRAF3; both kinds of abnormality triggered the classical and alternative NF-κB pathways. A majority of primary myeloma patient samples and cell lines had elevated NF-κB target gene expression, often associated with genetic or epigenetic alteration of NIK, TRAF3, CYLD, BIRC2/BIRC3, CD40, NFKB1, or NFKB2. These data demonstrate that addiction to the NF-κB pathway is frequent in myeloma and suggest that IKKβ inhibitors hold promise for the treatment of this disease.
Significance
Here we show the importance of classical NF-κB signaling in multiple myeloma. We discovered diverse genetic and epigenetic mechanisms leading to NF-κB activity in myeloma cell lines and patient samples. Targeted disruption of classical NF-κB signaling with a small molecule inhibitor of IKKβ blocked myeloma cell proliferation and induced cell death. Most primary MM patient samples had evidence of NF-κB pathway activation, suggesting that therapeutic strategies targeting the classical NF-κB pathway should be pursued.
Keywords: multiple myeloma, NF-κB, NIK, TRAF3, IκB kinase, CYLD, CD40, c-IAP1, c-IAP2
Introduction
Multiple myeloma (MM), a malignancy of plasma cells (PCs), was diagnosed in over 16,000 people in the US in 2005 and contributed to over 11,000 deaths (Jemal et al., 2006). The current standard treatment of MM includes high dose chemotherapy and stem cell transplantation. New agents with broad or undefined molecular specificity, such as the proteasome inhibitor bortezomib and the thalidomide analogue lenalidomide (Barlogie et al., 2004; Mitsiades et al., 2004), can prolong survival but have associated toxicities. There is a clear need for new therapeutic strategies targeting defined pathogenetic events in MM.
Previous studies suggested the importance of NF-κB signaling in MM, by the nuclear presence of NF-κB in MM cells and the sensitivity of some MM cell lines to NF-κB inhibition (Hideshima et al., 2002; Hideshima et al., 2006). Within the bone marrow microenvironment, NF-κB signaling in stromal cells can lead to production of IL6, BAFF or APRIL, known growth factors for and activators of NF-κB in MM (Hideshima et al., 2002; Marsters et al., 2000; Moreaux et al., 2005).
Five subunits combine into hetero- and homo-dimers to create the NF-κB transcription factor family (Ghosh and Karin, 2002) yielding two general pathways of activation (Figure 1A). In the classical pathway, IKKβ phosphorylates the inhibitory subunits IκBα, IκBβ or IκBε leading to their degradation in the proteasome. As a result, the NF-κB heterodimers p50/p65 and c-rel/p65 accumulate in the nucleus. In the alternative pathway, IKKα homodimers phosphorylate p100/NFKB2, resulting in proteasomal removal of an inhibitory C-terminal domain and generating the NF-κB p52 subunit (Senftleben et al., 2001; Xiao et al., 2001). Consequently, the p52/RelB heterodimers preferentially accumulate in the nucleus.
Figure 1.
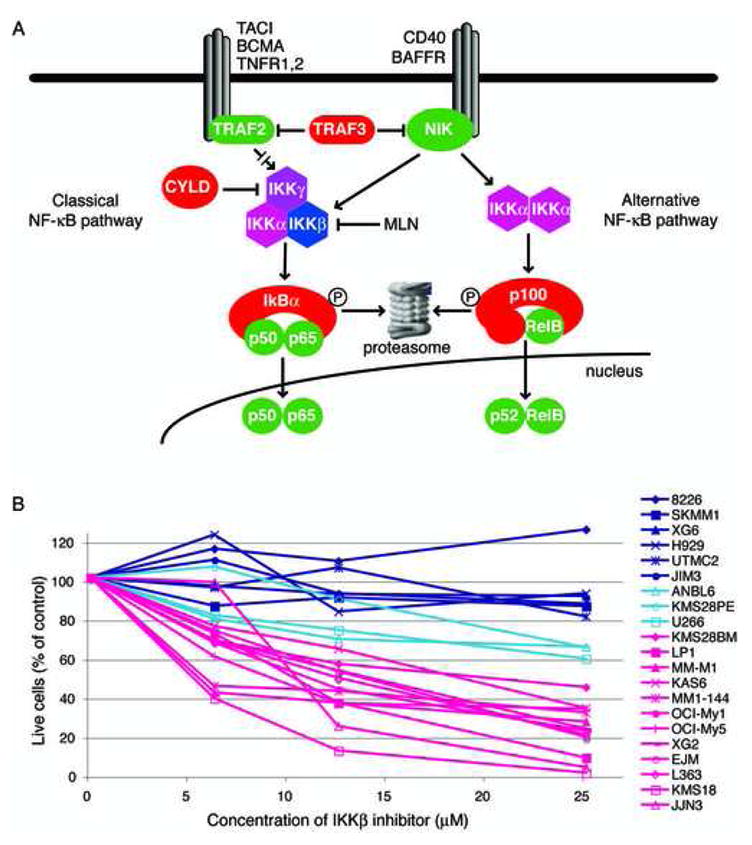
A. Schematic of NF-κB signaling. B. Growth inhibition of MM cell lines by the IKKβ inhibitor MLN120b (MLN). Cell lines were cultured in the presence of MLN (25 μM), and cells were enumerated by flow cytometry as described (Davis et al., 2001). After 12 days, cell numbers were determined and displayed relative to a control culture treated with the same volume of DMSO (solvent) alone.
Various cancer types utilize constitutive NF-κB signaling to block apoptosis (Basseres and Baldwin, 2006), but the molecular mechanisms leading to constitutive IKKβ activation in cancer are largely unknown. Cancer-causing mutations in the NF-κB pathway could lead to over-activation of positive inputs to NF-κB activation, or loss of negative regulators. The NF-κB-inducing kinase (NIK) falls into the former category. NIK is necessary for alternative NF-κB signaling initiated by various members of the TNF receptor (TNFR) superfamily (Claudio et al., 2002; Coope et al., 2002; Ramakrishnan et al., 2004; Yin et al., 2001). However, NIK is required for signaling to the classical pathway by certain TNF family members, such as CD40 ligand and BAFF (Ramakrishnan et al., 2004). When overexpressed, NIK can activate the classical pathway, with resultant IκB degradation and nuclear translocation of p50/p65 and/or c-rel/p65 (O’Mahony et al., 2000; Woronicz et al., 1997).
Negative regulators act on many steps in the NF-κB pathway. TRAF3, a putative ubiquitin ligase, can inhibit signaling to both the classical and alternative NF-κB pathways by TNFR family members (Hauer et al., 2005), and may exert this effect, in part, by mediating NIK protein degradation (Liao et al., 2004; Xiao and Sun, 2000). The tumor suppressor CYLD inhibits the NF-κB pathway at multiple steps by deubiquitinating the IKKγ subunit, TRAF2, TRAF6, and BCL3 (Brummelkamp et al., 2003; Kovalenko et al., 2003; Massoumi et al., 2006; Regamey et al., 2003; Trompouki et al., 2003). The ubiquitin ligase c-IAP1 can attenuate NF-κB signals via TNFR family members (Li et al., 2002; Rothe et al., 1995a).
In the present study, we discovered diverse genetic abnormalities causing constitutive NF-κB signaling in MM. Gene expression profiling revealed that the NF-κB pathway was activated in MM with surprising frequency. These genetic and functional data provide a molecular framework for the rational development of NF-κB pathway inhibitors for the therapy of multiple myeloma.
Results
Sensitivity of myeloma cell lines to IKKβ inhibition
We examined the toxicity of a small molecule inhibitor of IKKβ, MLN120b (MLN) (Nagashima et al., 2006), in 21 cell lines representing many different molecular subgroups of MM. Twelve cell lines had moderate to high sensitivity, 3 had intermediate sensitivity, and 6 were resistant (Figure 1B). Cell lines showed varying degrees of apoptosis and growth arrest in response to IKKβ inhibition (Figure S1).
The presence of nuclear NF-κB subunits in cell lines corresponded with their sensitivity to IKK inhibition. Nuclear p65 and p52 reflect activity of the classical and alternative NF-κB pathways, respectively. Both subunits were detected in nuclear extracts from 12 MLN-sensitive cell lines but were low or undetectable in nuclear extracts from 3 resistant lines (Figure 2A).
Figure 2.
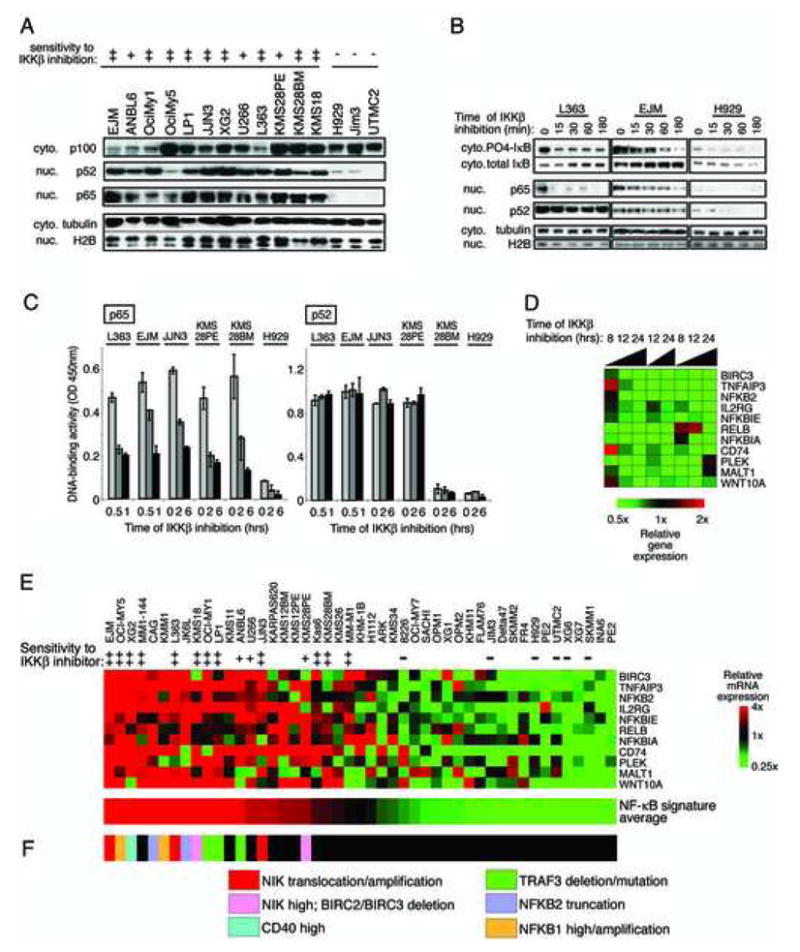
Effect of IKKβ inhibition on NF-κB signaling in MM cells. A: Steady state levels of NF-κB subunits in cytoplasmic or nuclear enriched protein fractions from cell lines. B: Effect of IKKβ inhibition by MLN on the abundance of NF-κB subunits and of total and phosphorylated IkBα. C: Effect of IKKβ inhibition on the nuclear DNA-binding activity of the NF-κB p65 and p52 subunits. Binding to an oligonucleotide containing the NF-κB consensus sequence was measured in nuclear extracts prepared from MM cells treated with MLN for the indicated times. DNA-binding activity was quantified by colorimetry (mean +/− SD). D: NF-κB target genes in MM. L363 cells were treated with the IKKβ inhibitor MLN for the indicated times and gene expression changes were assessed using DNA microarrays and depicted according to the color scale shown. E: Affymetrix U133plus2.0 microarray data from 47 MM cell lines were ordered using the average of the 11 NF-κB target genes determined in D, median centered, and depicted according to the color scale shown. F: Abnormalities of NF-κB pathway components and regulators in the indicated cell lines.
The level of IκBα phosphorylation decreased with IKKβ inhibition over time in two MLN-sensitive cell lines (Figure 2B). Nuclear p65 also decreased with MLN treatment, but nuclear p52 remained unchanged. NF-κB motif DNA-binding activity was measured in nuclear extracts from 5 sensitive cell lines and one resistant cell line. DNA binding by complexes containing p65 decreased after IKKβ inhibition while p52 DNA binding was unaffected (Figure 2C). This result is consistent with the specificity of MLN for IKKβ and the classical NF-κB pathway (Nagashima et al., 2006).
We defined an NF-κB target gene signature by profiling gene expression changes with MLN treatment of L363 cells (Figure 2D). Eleven genes were consistently downregulated by IKKβ inhibition and correlated in expression across 47 cell lines (Figure 2E). These same genes were downregulated in MM cell line EJM upon retroviral expression of the IkBα super-repressor, a specific inhibitor of classical NF-kB signaling (Davis et al., 2001), which was toxic to both L363 and EJM cell lines (data not shown). The expression levels of these NF-κB target genes were combined to create an NF-κB signature average, which was used to rank the cell lines. Notably, all MLN-sensitive cell lines had higher NF-κB signature expression than the MLN-insensitive cell lines (Figure 2E).
NF-κB activity and genetic abnormalities in primary MM cases
We analyzed the activity of the NF-κB pathway in gene expression profiles of 451 purified MM samples from newly diagnosed patients (Shaughnessy et al., 2007; Zhan et al., 2006). Each of the 11 NF-κB signature genes was significantly correlated in expression with the others in the primary MM samples (r-value range 0.63 – 0.87; Figure 3A). Since these genes were identified solely based on activity of the NF-κB pathway in myeloma cell lines, their co-regulation in primary patient samples suggests that they are surrogates for NF-κB pathway activity. Indeed, the proportion of MM cells with immunohistochemical evidence of nuclear p65 correlated with the NF-κB signature in patient samples (r=0.71; p<0.001) (Figure S2).
Figure 3.
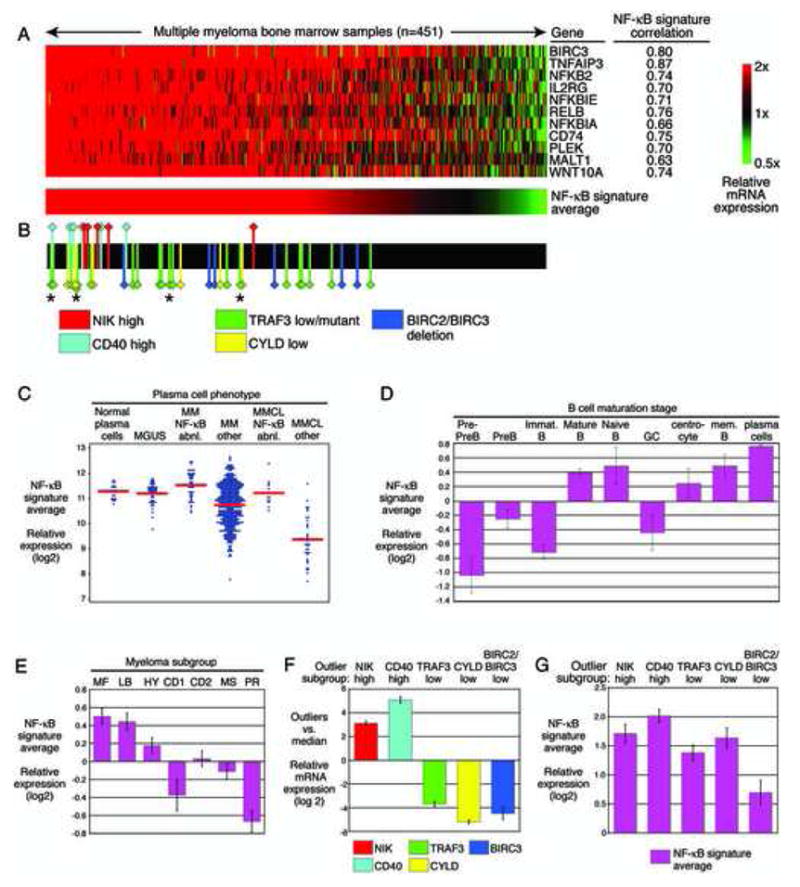
Multiple molecular mechanisms activate NF-κB in bone marrow PC from untreated MM patients. A: Expression of NF-κB target genes in primary MM patient samples. Affymetrix U133plus2.0 gene expression profiling data from 451 purified bone marrow plasma cell populations derived from untreated patients with MM (Zhan et al., 2006). Samples are ranked according to the average expression of the 11 NF-κB target genes. Expression was centered based on the median value in the MM cell lines (Figure 2E). B: MM samples with outlier gene expression and/or TRAF3 mutations. Cases with high NIK or CD40 expression and cases with low TRAF3, CYLD or BIRC2/BIRC3 expression are indicated with colored markers. The remaining cases are indicated in black. The asterisks indicate cases with inactivating TRAF3 mutations. C: NF-κB signature expression in normal and malignant plasma cell types. Data are taken from refs (Zhan et al., 2007; Zhan et al., 2006). D: NF-κB signature expression at different stages of human B cell differentiation. Shown are individual samples and the mean expression in each group. E: Expression of the NF-κB signature in MM gene expression subgroups. F: Expression of the indicated genes in the cases with outlier expression vs. other MM cases. G: Expression of the NF-κB signature in outliers. Shown is the NF-κB signature expression in outliers relative to the minimum expression in MM cell lines sensitive to IKKβ inhibition. In D – G, the mean value +/− S.E. is depicted.
For each MM sample, the average of the 11 NF-κB signature genes was calculated and used to rank the samples (Figure 3A). Among the MM patient samples, 368 (82%) had levels greater than that of MM-M1, the NF-κB-dependent cell lines with the lowest NF-κB signature expression. These data suggest that a majority of MM cases have functionally significant levels of the NF-κB signature, comparable to those in NF-κB-dependent cell lines.
We next compared the NF-κB signature levels in MM with those in normal PCs and in monoclonal gammapathy of undetermined significance (MGUS), a benign condition that can progress to MM. Normal PCs had levels of the NF-κB signature that were comparable, on average, to those in MGUS and MM (Figure 3C). Among normal human B cell subpopulations, PCs had the highest expression of the NF-κB signature (Figure 3D), consistent with their dependence on signals in the bone marrow microenvironment that trigger NF-κB (O’Connor et al., 2004). The high expression of the NF-κB signature in many MM cases may therefore reflect a similar dependence on the microenvironment. However, not all molecular subgroups of MM had equivalent expression of the NF-κB signature: the MF and LB subgroups had relatively high expression while a proliferative (PR) subgroup had low expression (Figure 3E). Thus, the importance of the NF-κB pathway may be contingent upon other molecular features of these MM subgroups (Shaughnessy et al., 2006; Zhan et al., 2006).
To search for oncogenic abnormalities that might confer cell-autonomous NF-κB pathway activation in MM, we evaluated the expression of known regulators of NF-κB in patient samples. Genes with an “outlier” expression profile are those with a dramatically high or low expression in a subset of samples, which might reflect oncogenic events such as translocations, amplifications, or genomic deletions. Recently, a method to identify outlier genes was described, which relies on a scaling transformation of the expression data to accentuate extreme values (Tomlins et al., 2005). A similar method identified MM cases with FGFR3 and Cyclin D1 translocations (Zhan et al., 2002). We modified this approach to account for gene expression differences between known MM subgroups, attempting to focus on outliers associated with NF-κB activation in several MM subgroups (see Methods). We chose a high cutoff for outlier gene expression to focus on cases most likely to have genetic abnormalities affecting mRNA expression levels.
Four candidate NF-κB regulators- NIK, CD40, TRAF3 and CYLD- had outlier mRNA expression values in at least 5 MM cases. For NIK and CD40, outlier cases highly overexpressed the corresponding mRNA whereas for TRAF3 and CYLD, outlier cases had exceedingly low mRNA levels (Figure 3F). In addition, 6 cases with high NF-κB signature expression had exceedingly low expression of BIRC3 and the neighboring gene BIRC2, which was notable since BIRC3 is in the NF-κB signature. Cases with outlier levels of these NF-κB regulators were significantly skewed towards high NF-κB signature expression (Figure 3B). In many cases, the altered expression of the outlier genes was due to genetic abnormalities (see below).
Aberrations of the NIK genomic locus in MM
We first investigated the molecular basis for outlier expression of NIK, a kinase capable of activating both the alternative and classical NF-κB pathways (Claudio et al., 2002; Coope et al., 2002; O’Mahony et al., 2000; Ramakrishnan et al., 2004; Woronicz et al., 1997; Yin et al., 2001). Five patient samples were clear outliers, with average expression that was 8.8-fold above the median (Figures 3B, 3F, S3). NIK outliers had high expression of the NF-κB signature (Figures, 3B, G).
By FISH, three NIK outlier cases had chromosomal translocations between the NIK locus and the IGH locus (2 cases) or the IGL locus (1 case) (Figure 4A). Since chromosomal translocations or high-level amplifications lead to mono-allelic mRNA expression, we developed RT-PCR assays to distinguish two common NIK alleles that differ by a single nucleotide polymorphism (SNP). Three NIK outlier cases had mono-allelic NIK mRNA expression, one of which was confirmed to have a translocation of NIK by FISH (Figure 4A). Among 4 cases with intermediate (but not outlier) NIK expression, NIK mRNA was expressed from both alleles, consistent with transcriptional overexpression (Figure 4A).
Figure 4.
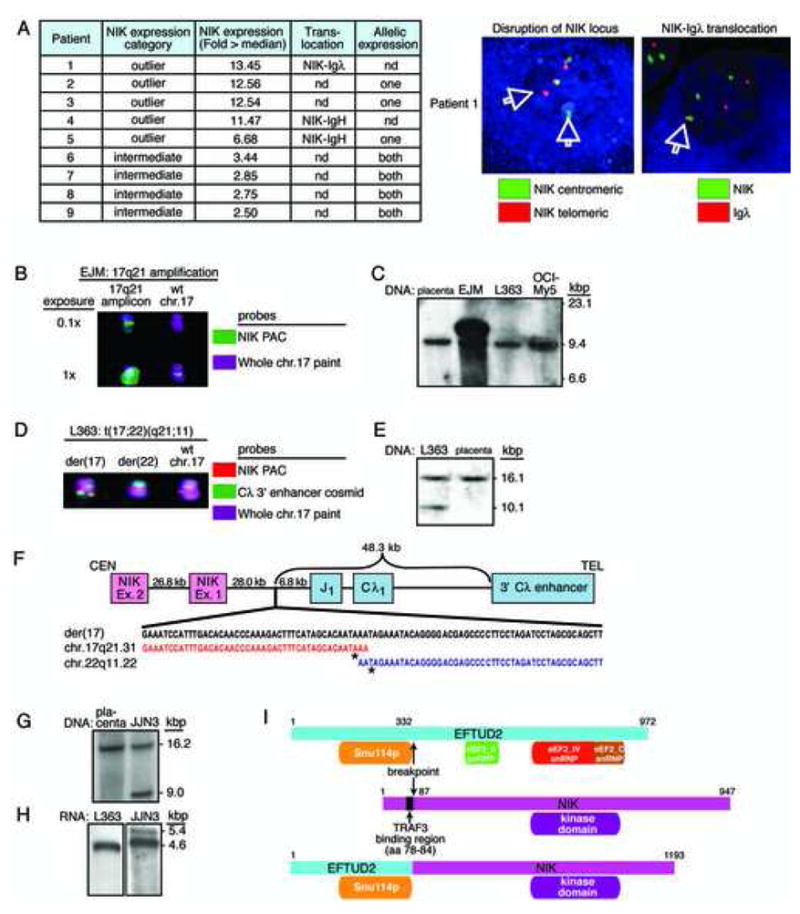
Amplification or translocation of NIK in MM cell lines. A: Chromosomal translocations of NIK in primary MM patient samples. Three samples with NIK translocated to either the IGH locus or the IGL locus were documented by FISH (one representative sample shown at right). Other cases had monoallelic expression of NIK, consistent with a chromosomal translocation or other cis-acting event (see text for details). B: FISH analysis of NIK amplification in the EJM cell line. Two exposures are shown to illustrate the high level NIK amplicon on 17q21. C: Southern blot of NIK amplification in the EJM cell line. Genomic DNAs from the indicated cell lines were digested with HindIII and hybridized with a radiolabeled 568 bp PCR fragment amplified from NIK exon 15. D: FISH analysis of NIK translocation to the IGL locus in the L363 cell line. The signal from the 92 kb NIK PAC probe is split between the der(17) and der(22) chromosomes, and is also present on the wild type chr 17. The der(17) contains NIK sequences juxtaposed to the IGL enhancer. E: Southern blot of the NIK translocation in the L363 cell line. HpaI digest of genomic DNA hybridized with a NIK probe (Chr17:40,769,557–40,770,000 (build35)) shows a 16.1 kb wild type fragment in placental DNA and a 10.1 kb fragment in L363. F: Structure of the NIK translocation breakpoint in L363. CEN, centromeric end; TEL, telomeric end. The breakpoint on chr 17 is bp 40,778,164 (asterisk) and on chr 22 is bp 21,553,824 (asterisk) (NCBI build 35). G: Southern blot of the NIK translocation in JJN3 cells. HpaI digest of genomic DNA hybridized with NIK exon 5 probe shows the wild type locus as a 16.2 kb fragment. The translocated locus is a 9.0 kb fragment. H: Northern blot of NIK mRNA species. mRNA from L363 or JJN3 cells was hybridized with a radiolabeled cDNA probe derived from the 3′ end of the NIK mRNA. A wild type 4.6 kb mRNA species was detected in L363 cells (17 hr exposure). JJN3 cells (5 d exposure) show the wild type mRNA and an additional 5.4 kb mRNA, presumably from the translocated allele. I: Schematic of the EFTUD2-NIK fusion protein in JJN3 (see text for details).
Two NF-κB-dependent cell lines (EJM, L363) had NIK mRNA levels equivalent to those in the NIK outlier patient samples (not shown). In the EJM cell line, the NIK locus was highly amplified, as judged by both FISH and Southern blot (Figure 4B,C). In the L363 cell line, FISH revealed a chromosomal translocation juxtaposing the NIK and the IGL locus (Figure 4D,E). The breakpoint occurred 28kb 5′ of NIK, bringing the NIK promoter in proximity to IGL enhancer elements (Figure 4F).
In the JJN3 cell line, one NIK allele generated an aberrant restriction fragment that hybridized with a probe derived from NIK exon 5 (Figure 4G). These cells expressed a 5.4kb NIK mRNA instead of the wild type 4.6kb species (Figure 4H). Cloning of the aberrant NIK allele identified a translocation fusing a 5′ fragment of EFTUD2 to a 3′ portion of NIK, with the breakpoint between exons 11–12 of EFTUD2 and between exons 2–3 of NIK (Figure S4). The translocation is predicted to generate a 5152 bp mRNA encoding a 132kD in-frame EFTUD2-NIK fusion protein (Figure 4H,I), and these cells indeed expressed an aberrant high molecular weight NIK protein (Figure 6A). The EFTUD2-NIK fusion protein consists of the N-terminal region of EFTUD2, containing a conserved domain of unknown function, fused to the C-terminal NIK kinase domain (Figure 4I).
Figure 6.
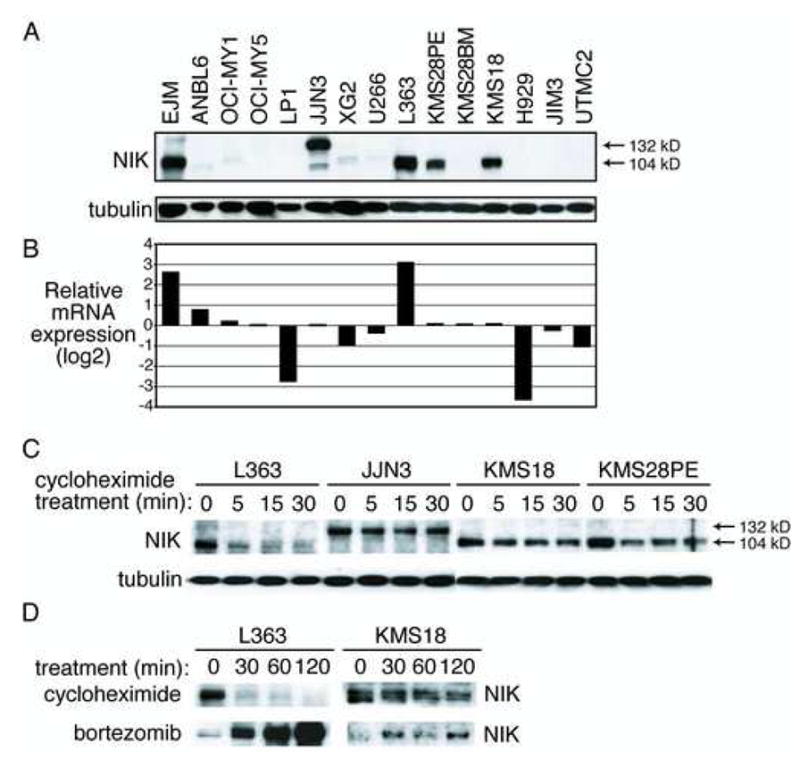
NIK protein overexpression in MM cell lines. A: Western blot of NIK protein levels in cytoplasmic extracts from the indicated cell lines. The wild type NIK protein is a 104 kD protein whereas the EFTUD2-NIK fusion protein in JJN3 is 132 kD. B: Relative NIK mRNA levels in cell lines as assessed by gene expression profiling on Affymetrix U133plus2.0 arrays (probe ID 205192_at). C: Enhanced NIK protein stability in a subset of myelomas. Western blot of NIK and tubulin levels before and after protein synthesis inhibition with cycloheximide. D: Proteasomal degradation of NIK varies in MM cell lines. L363 or KMS18 cells were treated with cycloheximide (20 μg/ml) or the proteasome inhibitor bortezomib (250 nM) for the indicated times and assayed for NIK levels.
Mutations, deletions and silencing of TRAF3 in MM
By the outlier approach, 17 MM cases had low expression of TRAF3, a negative regulator of NIK (Liao et al., 2004; Xiao and Sun, 2000). On average, outliers expressed TRAF3 mRNA at levels 13-fold below the median (Figures 3B, 3F, S3). TRAF3 outliers had high expression of the NF-κB signature (Figures, 3B, G).
In the NF-κB-dependent cell line OCI-My1, which does not express TRAF3 mRNA, the TRAF3 gene could not be amplified by PCR from genomic DNA (data not shown) and array-based comparative genomic hybridization (aCGH) analysis confirmed a homozygous TRAF3 deletion (Figure 5A). We next constructed a quantitative PCR (qPCR) assay for TRAF3 genomic copy number. Of the 6 TRAF3 outliers analyzed, 2 had bi-allelic deletion, 3 had mono-allelic TRAF3 deletion (one of which had a TRAF3 mutation on the remaining allele), and 1 had a normal copy number (Figure 5B). In these cases, epigenetic silencing or decreased mRNA stability due to nonsense mediated decay may contribute to the low TRAF3 mRNA expression; indeed, case P993 had a frameshift mutation of TRAF3 that resulted in early termination of the protein (see below). Homozygous deletions of TRAF3 were not present in other “NF-κB-high” cases (NF-κB signature above the median; n=12) nor in NF-κB-low cases (NF-κB signature in the lowest quintile; n=12).
Figure 5.
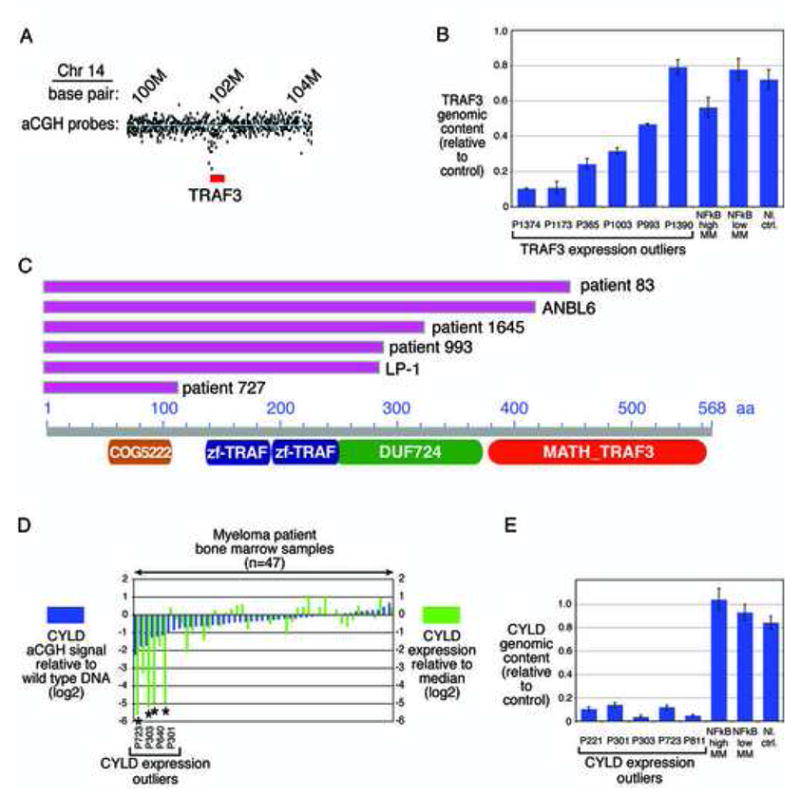
Genetic abnormalities of the negative NF-κB regulators TRAF3 and CYLD in MM. A: Genomic deletion of TRAF3 in the OCI-MY1 cell line. aCGH estimated copy number from the TRAF3 genomic region in OCI-MY1 versus a normal reference. Blue line indicates average ratio for probes within wild type regions. The area depicted is ~3 Mb on chr 14 (NCBI build 35). Probes within the region encompassed by TRAF3 (red) indicate genomic deletion of the TRAF3 gene. B: qPCR analysis of TRAF3 copy number. Copy number estimates of TRAF3 relative to a control locus in 6 primary MM cases. Also shown is average copy number (+/− S.E.) in 12 cases each with high or low NF-kB signature expression and in 10 normal control samples. C: Mutations in TRAF3 that truncate the protein. Base pairs are numbered based on the TRAF3 reference sequence NM_145725. Resequencing revealed nucleotide deletions or mutations in two NF-κB-positive MM cell lines (LP-1, ANBL-6) and in 4 patient samples. These changes produced early termination of the TRAF3 protein at the positions indicated in the diagram. Each mutation would delete all or part of the carboxy-terminal MATH domain required for binding to NIK and TNF receptor superfamily members. D: Comparison of aCGH and gene expression data for CYLD in MM samples. Cases are ranked according to their aCGH signals (Agilent probe 420863) relative to wild type, with negative values indicating deletion, and compared with CYLD gene expression levels. The 4 CYLD gene expression outliers for which array CGH data were available are indicated. E: qPCR analysis of CYLD copy number. Copy number estimates of CYLD relative to a control locus in 5 primary MM cases. Also shown is average copy number (+/− S.E.) in 12 cases each with high or low NF-κB signature expression and in 10 normal control samples.
Next, we investigated whether somatic mutations might inactivate TRAF3 in MM. We sequenced 11 exons of TRAF3 in genomic DNA from 10 cell lines and 47 samples from newly diagnosed MM patients, including 6 TRAF3 outliers, 29 NF-κB-high cases and 12 NF-κB-low cases. Two cell lines and 4 patient samples had mutations resulting in stop codons in exons 4, 9, 10 or 11 (Figure 5C). All of these had high NF-κB signature expression (Figures 2F, 3B). The predicted TRAF3 protein in the LP1 cell line would lack the C-terminal 283 amino acids, containing the entire MATH domain crucial for interaction of TRAF3 with NIK and with members of the TNFR superfamily (Li et al., 2003; Liao et al., 2004; Ni et al., 2004; Ni et al., 2000). Likewise, mutations in patient samples 727, 993 and 1645 would remove the entire MATH domain. In the ANBL6 cell line, the C-terminal 144 amino acids would be lost due to the mutation, producing a protein in which the MATH domain is disrupted. The mutation in patient sample 83 would similarly truncate the protein within the MATH domain. Of note, all TRAF3 mutations were homozygous, suggesting a strong selective pressure for these mutations in MM.
CYLD deletion in NF-κB-positive MM
Among genes that were silenced in outlier cases, the NF-κB regulator most significantly associated with high NF-κB signature expression was CYLD, a known negative regulator of NF-κB signaling (Brummelkamp et al., 2003; Kovalenko et al., 2003; Regamey et al., 2003; Trompouki et al., 2003). Six MM cases had expression of CYLD mRNA that was, on average, 37-fold below the median (Figures 3B, 3F, S3). CYLD outliers had high expression of the NF-κB signature (Figures, 3B, G).
We quantified CYLD copy number by qPCR in 5 available samples from CYLD outliers and found bi-allelic loss of the locus in all (Figure 5E). Analysis of published aCGH data from some of these cases (Carrasco et al., 2006), confirmed that the sixth CYLD outlier also had a biallelic CYLD deletion (Figure 5D). In summary, homozygous CYLD deletions were present in all CYLD outliers and are thus a recurrent genetic event in multiple MM that is associated with high NF-κB activity.
CD40 overexpression in MM
CD40 signaling activates both the classical and alternative NF-κB pathways (Berberich et al., 1994; Coope et al., 2002; Qing et al., 2005; Ramakrishnan et al., 2004). Six outlier cases expressed CD40 mRNA levels that were 34-fold above the median (Figure 3B, 3F, S3). CD40 outliers had high expression of the NF-κB signature (Figures, 3B, G). Of note, the NF-κB-dependent cell line XG2 expressed CD40 mRNA at a 24-fold higher level than the average in other cell lines, suggesting that it may have originated from a CD40 outlier MM case. Since CD40 is an NF-κB target gene in certain lymphoma types (Lam et al., 2005), we investigated whether the high CD40 expression in the outliers could be due to their high NF-κB signature expression. We chose 60 control cases with an average NF-κB signature expression equivalent to that of the outliers. In these controls, the average CD40 expression was 23-fold lower than that of the outliers (p=5.2 × 10−9). Therefore, the high CD40 expression in the outliers is not primarily due to their NF-κB activity.
qPCR of the CD40 locus in 3 of the outlier cases did not reveal high-level amplification nor did the XG2 cell line have CD40 amplification by aCGH (data not shown). The mechanism of CD40 overexpression in these cases is currently unknown, but could be due to chromosomal translocation or trans-acting regulatory mechanisms.
Other genetic abnormalities affecting the NF-κB pathway in MM
Several cell lines had high NF-κB signature expression but lacked genetic alterations in NIK, TRAF3, CD40 or CYLD (Figure 2F). We therefore examined these cell lines for alterations in other components of the NF-κB pathway. OCI-My5 and KMM1 had high expression of NFKB1, with mRNA levels that were 4.5-fold and 18-fold higher expression than the median, respectively. aCGH revealed a ~25-fold amplification of the NFKB1 gene in of OCI-My5 (Figure S5A).
Two other cell lines, CAG and JK6L, had genetic abnormalities in NFKB2, encoding the p100 and p52 NF-κB subunits. Western blot revealed truncated forms of the NFKB2 protein (Figure S5B). JK6L had a homozygous frameshift mutation in NFKB2, disrupting the C-terminal ankyrin repeat domains, whereas CAG had a genomic deletion involving the NFKB2 3′ exons that truncated the mRNA and protein (Figures S5B,C,D,E). Such alterations were previously described in MM and other hematological malignancies, and lead to nuclear forms of NFKB2 protein that are presumably constitutively active (Migliazza et al., 1994).
Examination of NF-κB target gene expression revealed that KMS18 and KMS28PE had exceedingly low expression of BIRC3, encoding the ubiquitin ligase c-IAP2. These two cell lines also had low expression of BIRC2, a gene that is next to BIRC3 in the genome and encodes c-IAP1, a ubiquitin ligase that inhibits TNFR signaling (Li et al., 2002). qPCR revealed that KMS18 and KMS28PE have homozygous deletions of the BIRC2/BIRC3 chromosomal locus (Figure S5F). Six MM patient samples with low outlier expression of BIRC3 and BIRC2 (see above), and bi-allelic deletions in the BIRC2/BIRC3 locus were detected by aCGH and/or qPCR (Figure S5F,G). All cell lines and patient samples with BIRC2/BIRC3 deletions had NF-κB signature expression above the median (Figures 2F, 3B), suggesting that inactivation cIAP1 and/or cIAP2 may modulate the NF-κB pathway in MM.
NIK protein expression in MM cell lines
To explore the molecular mechanisms by which NIK overexpression activates the NF-κB pathway in MM, we first investigated the expression of NIK protein in cell lines. A high level of NIK protein was detected in 5 cell lines that were sensitive to IKKβ inhibition, including 3 with NIK genomic aberrations (EJM, JJN3, L363) but also 2 with no evidence of NIK aberrations (KMS28PE, KMS18) (Figure 6A). JJN3 cells had a 132kD NIK protein instead of the 104kD wild type protein, which presumably is the EFTUD2-NIK fusion protein. A low level of NIK protein was detected in 4 other NF-κB-dependent cell lines (ANBL6, OCI-MY1, XG2, U266). Other NF-κB-dependent cell lines (OCI-My5, LP1, KMS28BM) lacked detectable NIK protein as did all 3 NF-κB-negative cell lines (H929, JIM3, UTMC2).
The cell lines KMS18, KMS28PE and JJN3 expressed NIK mRNA at an average level (Figure 6B), suggesting that these cell lines have translational or post-translational mechanisms to elevate NIK protein levels. To investigate the latter possibility, we blocked protein synthesis with cycloheximide and monitored NIK protein abundance over time. The NIK protein in L363 cells was labile, with a half-life of less than 5 minutes, but the NIK proteins in KMS18, KMS28PE, and JJN3 were more stable, with half-lives greater than 30 minutes (Figure 6C). Treatment of L363 cells with the proteasome inhibitor bortezomib increased NIK protein levels dramatically, consistent with rapid protein turnover by ubiquitin-mediated proteolysis (Figure 6D). Proteosomal inhibition in KMS18 cells increased NIK protein levels to a much more modest extent (Figure 6D).
These data suggest that the high NIK protein levels in KMS18, KMS28PE and JJN3 cells are achieved by post-translational stabilization of NIK protein. In JJN3, the EFTUD2-NIK fusion protein lacks amino acids 78-84 of NIK that are required for TRAF3-mediated degradation of NIK, which could account for the stability of this protein. In KMS18 and KMS28PE, no alterations in the TRAF3 binding domain were present (data not shown). Intriguingly, both of these lines lack expression of BIRC2 and BIRC3 due to genomic deletions (Figure S5C), raising the possibility that cIAP1 and/or cIAP2 may play a role in NIK protein stability.
NIK overexpression activates NF-κB signaling and blocks cell death in MM
To examine the functional significance of NIK overexpression in myeloma cell lines, we designed short hairpin RNAs (shRNAs) targeting NIK and expressed them in cell lines in an inducible fashion using retroviruses (Ngo et al., 2006). Two shRNAs against NIK were identified, one targeting the coding region and the other the 3′ untranslated region (UTR), which were capable of knocking down the wild type NIK mRNA and protein expression (Figures 7A,B) as well as the EFTUD-NIK fusion protein in JJN3 (Figure 7F).
Figure 7.
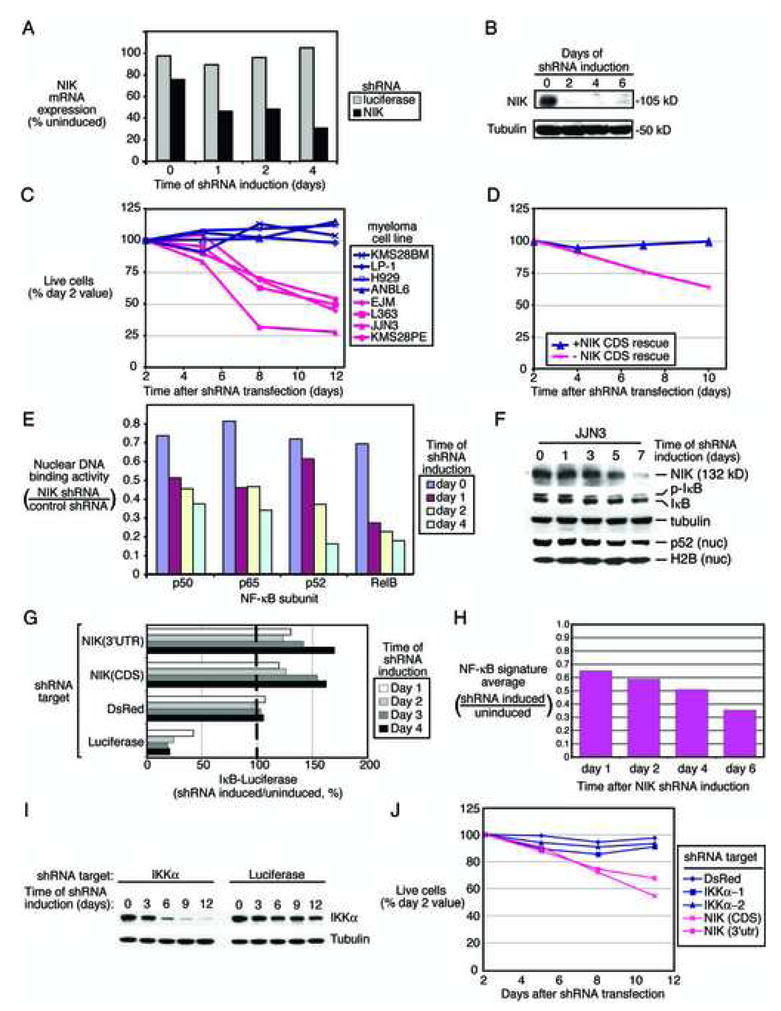
Knockdown of NIK expression by RNA interference inhibits the NF-κB pathway in MM. A: Quantitative RT-PCR measurement of NIK mRNA levels following induction of the NIK shRNA in EJM cells. B: Western blot of NIK protein levels following induction of the NIK shRNA in EJM cells. C: Knockdown of NIK is toxic to NIK-overexpressing MM cell lines. The indicated cell lines were transduced with a retrovirus expressing the NIK shRNA (see Methods). Live cells were enumerated by FACS and normalized to the value at day 2 following retroviral infection. D: Expression of the NIK coding sequence rescues cell lines transduced with an shRNA targeting the NIK 3′UTR. EJM cells expressing a NIK coding region cDNA or control EJM cells were transduced with a retrovirus expressing an shRNA targeting the NIK 3′UTR. Live cells were enumerated as in C. E: Knockdown of NIK inhibits nuclear NF-κB DNA binding. DNA-binding activity by the indicated NF-κB subunits was quantified by ELISA. F: Knock down of the EFTUD2-NIK fusion protein in JJN3 cells affects NF-κB signaling via classical and alternative pathways. G: Inhibition of IKK activity by knock down of NIK in MM cell lines. EJM cells expressing an IkBα-luciferase fusion protein were superinfected with the indicated shRNAs. Induction of shRNAs targeting NIK caused a rise in luciferase activity indicating inhibition of IKK activity. Negative control shRNA targets DsRed; positive control shRNA targets luciferase. H: Inhibition of NF-κB target gene expression following knockdown of NIK in EJM cells. Relative expression of the NF-κB signature in shRNA induced versus uninduced cells is depicted. I: Western blot analysis of IKKα expression after induction of shRNAs targeting IKKα or luciferase. EJM cells were transduced with retroviral vectors expressing the indicated shRNAs and expression of IKKα and tubulin were monitored at the indicated times. J: Knockdown of IKKα is not toxic to a NIK-overexpressing myeloma. EJM cells were transduced with shRNAs targeting IKKα, NIK, or DsRed. Live cells were enumerated as in C.
Induction of NIK shRNAs proved toxic to 4 cell lines with high NIK protein expression but not to 4 cell lines with low or absent NIK expression (Figure 7C). To demonstrate that NIK shRNA toxicity was due to knockdown of NIK protein, we performed a complementation experiment. EJM cells were engineered to express an exogenous NIK mRNA containing the coding region but lacking the 3′ UTR. Expression of the 3′UTR-directed NIK shRNA in these cells was predicted to knock down the endogenous NIK mRNA but spare the exogenous NIK mRNA. Whereas the NIK shRNA was toxic for control EJM cells, cells expressing the exogenous NIK mRNA were rescued from this toxicity (Figure 7D). This experiment demonstrates that the NIK shRNA does not have appreciable off-target toxicity and supports the conclusion that NIK expression is required for the survival of NIK-expressing cell lines.
We next investigated whether knock down of NIK affected constitutive NF-κB signaling in MM cells. When NIK shRNA expression was induced, nuclear extracts had reduced DNA binding activity of p50, p65, p52 and RelB, suggesting that NIK knockdown affected both the classical and alternative NF-κB pathways (Figure 7E). Likewise, knock down of the EFTUD2-NIK fusion protein in JJN3 decreased phosphorylation of IkBα and abundance of nuclear p52, suggesting that this NIK protein also activates the classical and alternative pathways (Figure 7F).
The effect of NIK knock down on IkBα kinase activity was measured in EJM cells engineered to express an IkBα-Photinus luciferase fusion protein, which increases in abundance when IKK activity is inhibited (Lam et al., 2005; Ngo et al., 2006). Induction of NIK shRNA increased luciferase activity, whereas an shRNA targeting DsRed had no effect and an shRNA targeting Photinus luciferase decreased the luciferase signal (Figure 7G). These data were consistent with the NF-κB DNA binding assays and suggest that NIK functions upstream of IKKβ in this cell lines. Accordingly, NF-κB target gene expression decreased in EJM cells during NIK knockdown (Figure 7H).
Since NIK can activate the alternative NF-κB pathway via IKKα, we tested whether IKKα depletion was toxic to the NIK-expressing cell line EJM. Two IKKα shRNAs strongly inhibited the expression of IKKα protein (Figure 7I). In contrast to the NIK shRNAs, these IKKα shRNAs were not toxic to EJM cells (Figure 7J) or other NIK-expressing cells (L363, JJN3, KMS28PE; data not shown), suggesting that the toxicity of NIK shRNAs was not due to blockade of the alternative NF-kB pathway.
TRAF3 inactivation promotes NF-κB signaling and cell survival in MM
To investigate the influence of TRAF3 loss on NF-κB signaling in MM, we engineered two TRAF3-deficient cell lines, OCI-My1 and LP1, to express TRAF3 in a doxycycline-inducible fashion (Figure 8A). In both cell lines, TRAF3 induction was followed by a decrease in IkBα phosphorylation together with a decrease in total IkBα levels (Figure 8A). TRAF3 induction also led to a decrease in NF-κB signature expression (Figure 8B) and an increase in the IkBα-luciferase reporter (Figure 8C), indicative of IKK inhibition. DNA binding activities of p50, p65, p52 and RelB in the nuclei of OCI-My1 cells were all decreased by TRAF3 induction (Figure 8D), suggesting that TRAF3 negatively regulates both the classical and alternative NF-κB pathways in these cells. Finally, induction of TRAF3 was toxic to both OCI-My1 and LP1, but not to H929, KMS28PE and KMS28BM, which do not have TRAF3 abnormalities (Figure 8E). Together, these data suggest that TRAF3 inactivation in MM is likely to promote cell survival via NF-κB pathway activation.
Figure 8.
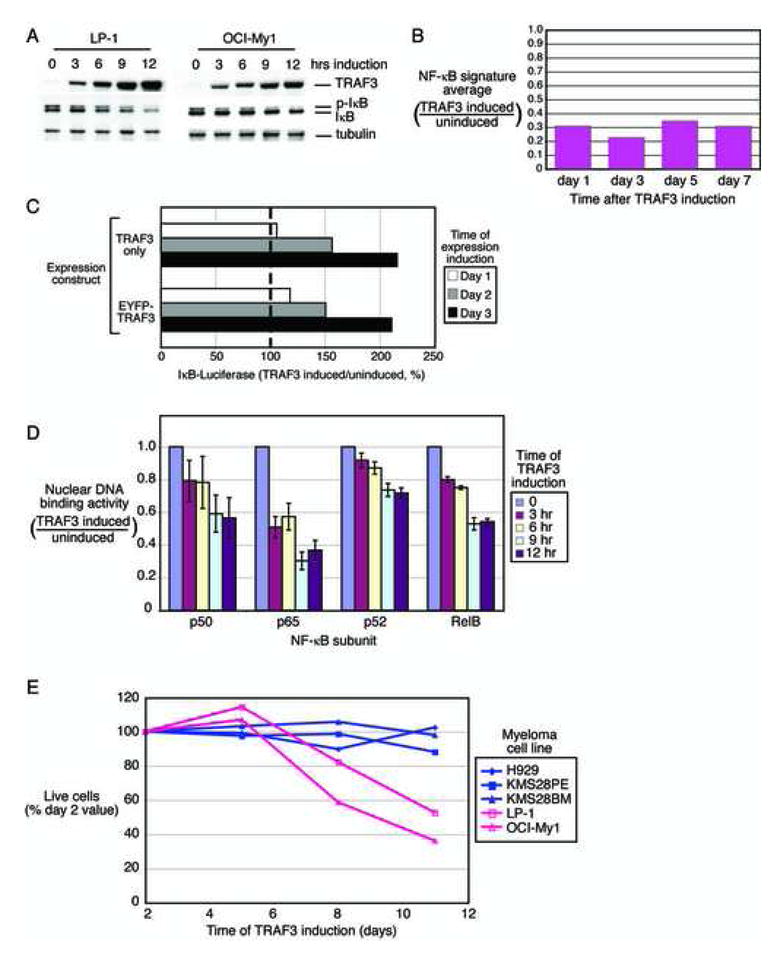
Effect of re-expression of TRAF3 in cell lines with TRAF3 inactivation. A: Inducible expression of TRAF3 in LP-1 and OCI-My1 cells decreases phosphorylated-IkBα (pIkB). B: Inhibition of NF-κB target gene expression following expression of TRAF3 in LP-1 cells. Relative expression of the NF-κB signature in TRAF3 induced versus uninduced cells is depicted. C: Inhibition of IKK activity by expression of TRAF3. OCI-My1 cells expressing an IkBα-luciferase fusion protein were superinfected with retroviruses expressing either wild type TRAF3 or an EGFP-TRAF3 fusion protein. Induction of TRAF3 caused a rise in luciferase activity indicating inhibition of IKK activity. D: Effect of TRAF3 expression in OCI-My1 cells on the nuclear DNA-binding activity of the NF-κB p50, p65, p52, and RelB subunits. Binding to an oligonucleotide containing the NF-κB consensus sequence was measured in nuclear extracts prepared from cell lines induced to express TRAF3 relative to uninduced cells for the indicated times. DNA-binding activity was quantified by colorimetry (mean +/− SD). E. Toxicity of TRAF3 expression in LP-1 and OCI-My1, cell lines with TRAF3 inactivation. The indicated myeloma cell lines were transduced with a retrovirus expressing TRAF3. Live cells were enumerated by FACS and normalized to the value at day 2 following retroviral infection.
Discussion
The present study defined diverse molecular mechanisms activating NF-κB in MM. Most primary MM cases had high expression of the NF-κB signature. This observation suggests frequent engagement of the NF-κB pathway in myeloma for several reasons. First, the NF-κB signature genes were defined based on IKKβ inhibition and coexpression in myeloma cell lines, yet were nevertheless highly correlated in expression across primary patient samples. Second, the presence of the NF-κB p65 subunit in the nucleus of primary MM cells correlated with NF-κB signature expression. Finally, the genetic and epigenetic alterations that we defined were significantly skewed towards high NF-κB signature expression, in both MM patient samples (p=1.01 × 10−9) and cell lines (p=1.08 × 10−17) (Figures 2F, 3B, C, G).
Some NF-κB pathway activation is likely related to signals that PCs receive in the bone marrow microenvironment. Indeed, among B cell subsets, normal PCs expressed the highest level of the NF-κB signature, presumably due to BAFF- and APRIL-mediated signaling (Hideshima et al., 2002; Marsters et al., 2000; Moreaux et al., 2005; O’Connor et al., 2004). Blockade of BAFF and APRIL decreases the number of bone marrow PCs in normal mice (O’Connor et al., 2004), raising the possibility that similar pharmacological inhibition might prove toxic to those MM cells that retain a microenvironmental dependence for NF-κB pathway activation.
However, the most compelling evidence for a critical role of the NF-κB pathway in myeloma pathogenesis was provided by the multiple, recurrent genetic abnormalities that we uncovered in the pathway. In cell lines and primary patient samples, NIK overexpression arose due to amplification or translocation of the NIK locus or due to enhanced NIK protein stability. An additional NF-κB-activating mechanism involved loss of functional TRAF3, either by homozygous deletion, inactivating mutations or epigenetic silencing. Other genetic events associated with high NF-κB activity in MM included CYLD or BIRC2/BIRC3 deletion, CD40 or NFKB1 amplification, and NFKB2 C-terminal truncation. Most of these genetic events increased classical and alternative NF-κB activity in MM cell lines, leading to pathway addiction and sensitivity to IKKβ inhibition. Taken together, these myriad genetic abnormalities strongly support the development of IKKβ inhibitors for the treatment of multiple myeloma.
NIK overexpression in MM cell lines activated both the classical and alternative NF-κB pathways. In normal mouse hematopoietic cells, loss of NIK does not affect activation of the classical pathway by inflammatory stimuli (Yin et al., 2001). However, NIK is required for the activation of the classical NF-κB pathway by certain TNF receptor family members, including CD40 (Ramakrishnan et al., 2004). Further, experimental overexpression of NIK can activate IKKβ and the classical pathway via phosphorylation of IKKβ in its activation loop (Delhase et al., 1999; O’Mahony et al., 2000). Indeed, NIK can form a complex with IKKα and IKKβ (Woronicz et al., 1997), perhaps by virtue of its ability to directly bind IKKα (Regnier et al., 1997). Studies conflict on the exact mechanism of IKKβ activation by NIK (Delhase et al., 1999; O’Mahony et al., 2000), but concur that NIK overexpression can activate IKKβ, supporting our finding that high NIK levels activate the classical NF-κB pathway in some myelomas.
We propose that the major mechanism by which NIK promotes tumor cell survival in MM is by stimulating IKKβ and activating the classical NF-κB pathway for several reasons. First, the toxicity of MLN for NIK-expressing MM cells argues for an essential role of IKKβ signaling: MLN is highly selective for IKKβ, with more than 1000-fold greater inhibitory activity for IKKβ than for 30 other cellular kinases, including IKKα (Nagashima et al., 2006). The IkBα-super-repressor, which also specifically turns off the classical NF-kB pathway, resulted in similar toxicity and gene expression changes in these MM cells. Second, NIK depletion by shRNA inhibited IkBα kinase activity. Since IKKβ is a far more potent IkBα kinase than IKKα, this result suggests that NIK is activating IKKβ in these cells. Third, NIK knock down decreased nuclear DNA binding activity of the classical NF-κB subunits p50 and p65, consistent with an effect on IKKβ activity. Fourth, two shRNAs that effectively decreased IKKα protein expression were not toxic for NIK-expressing cell lines. These data support the argument that NIK acts through IKKβ to stimulate the classical NF-κB pathway, thereby promoting MM survival. Nonetheless, the alternative NF-κB pathway was also affected by NIK knockdown since p52 and RelB DNA binding activity was reduced. It is therefore conceivable that activity of the alternative NF-κB pathway could contribute to the effect of NIK on cell survival.
Multiple mechanisms lead to high NIK protein expression in MM. The wild type NIK protein had a very rapid turnover, and proteasome inhibition caused accumulation of NIK protein, consistent with a ubiquitin-mediated degradative pathway involving TRAF3, as described (Liao et al., 2004). Nevertheless, when wild type NIK mRNA was expressed at extraordinarily high levels due to chromosomal aberrations, as in L363 and EJM cells, wild type NIK protein accumulated. Cells with intermediate NIK mRNA levels engage various mechanisms to stabilize NIK protein. In JJN3 cells, a chromosomal translocation precisely removed the region of NIK that TRAF3 requires to destabilize NIK protein (Liao et al., 2004), highlighting the interrelationship of TRAF3 and NIK in MM pathogenesis. KMS28PE and KMS18 had enhanced stability of NIK protein without evident alterations in the TRAF3 binding domain of NIK or in TRAF3 itself. Both KMS18 and KMS28PE had genomic deletions of BIRC2/BIRC3 genomic locus, which encodes the ubiquitin ligases cIAP1 and cIAP2. cIAP1 attenuates NF-κB signaling from TNFR2 (Li et al., 2002), raising the possibility that this protein or cIAP2 may also inhibit NF-κB signaling downstream of NIK by decreasing NIK protein stability.
Another recurrent mechanism of NF-κB activation in myeloma was silencing, homozygous deletion, or somatic mutation of TRAF3. In primary MM patient samples, 4.4% had a TRAF3 abnormality, and these cases had higher NF-κB signature expression than other myelomas. Homozygous TRAF3 deletions occurred in 2 primary cases and in the OCI-My1 cell line. In addition, 2 NF-κB-dependent cell lines and 4 primary patient samples carried homozygous mutations creating truncated TRAF3 proteins that should be incapable of interacting with NIK or TNFR superfamily members. In cases lacking deletion or mutation of TRAF3, low TRAF3 mRNA expression may be due to epigenetic silencing, a frequent event in cancer.
Experimental re-expression of TRAF3 in cell lines lacking functional TRAF3 decreased IKKβ activity and both classical and alternative NF-κB signaling, resulting in cell death. The precise mechanism by which TRAF3 modulates IKK activity in these cell lines remains to be elucidated. The cell lines with inactive TRAF3 do not have high NIK protein expression. Moreover, one of the TRAF3 mutant cells, LP1, was not affected by the NIK shRNA. In this cell line, therefore, TRAF3 must affect IKKβ activity by a NIK-independent mechanism.
In primary MM, the full consequence of TRAF3 inactivation may only be evident in cells dependent on NF-κB signaling from a TNFR family member, that can be negatively regulated by TRAF3 (Hauer et al., 2005). The most logical candidate cytokines to propagate TRAF3-deficient MM are BAFF and APRIL, which are abundant in the bone marrow and can signal through TACI and BCMA, two TNFRs that are highly expressed in a subset of MM (Moreaux et al., 2005).
It is also notable that extreme overexpression of CD40, a TNFR family member, was associated with NF-κB activation in MM. CD40 and other members of the TNFR superfamily can activate NF-κB when overexpressed, even in the absence of ligand (Lee et al., 1996; Rothe et al., 1995b; Yamamoto et al., 1998). It is therefore plausible that the 34-fold average overexpression of CD40 in the CD40 outlier cases is sufficient to activate NF-κB in a cell-autonomous fashion.
A key negative regulator of NF-κB signaling is CYLD, which was homozygously deleted in primary MM cases with high NF-κB signature expression. CYLD is a deubiquitinating enzyme that removes ubiquitin moieties from activated TRAF2, TRAF6, IKKγ and BCL3, thereby interfering with NF-κB signaling at multiple regulatory levels (Brummelkamp et al., 2003; Kovalenko et al., 2003; Massoumi et al., 2006; Regamey et al., 2003; Trompouki et al., 2003). Of note, CYLD was ineffective in inhibiting NF-κB activity induced by NIK overexpression (Kovalenko et al., 2003).
It therefore appears that diverse upstream signaling pathways activate IKKβ in MM. Further, some myelomas may bypass the requirement for IKK altogether by amplifying and overexpressing NFKB1, encoding the p50 subunit of the classical NF-κB pathway, or by creating truncated and constitutively active NFKB2 proteins. The recurrent yet varied nature of these genetic abnormalities suggests strongly that activity of the NF-κB pathway is the phenotype that is selected rather than any particular genetic lesion.
Overall, we documented genetic abnormalities in the NF-κB pathway in 13 (28%) MM cell lines and 41 (9%) of the primary MM samples. Considering that we resequenced TRAF3 in only 10% of the patient samples, a larger number of TRAF3 mutations probably exist within our sample set. Therefore, the total number of genetic abnormalities in the genes we studied can be predicted to be ~16%. Given the large number and variety of genetic abnormalities discovered in the present report, there may well be genetic aberrations in other components of the NF-κB pathway that we have not studied. As already mentioned, however, many MM cases with high NF-κB signature expression may not have genetic abnormalities in the NF-κB pathway but rather may derive activating signals from the microenvironment. Irrespective of whether the activation of the NF-κB pathway is cell-intrinsic or cell-extrinsic, the MM cell may nonetheless depend upon this pathway for survival.
A clear message from these recurrent yet varied genetic abnormalities is that the NF-κB pathway plays a pervasive role in the pathogenesis of MM, providing a strong impetus to the development of NF-κB pathway inhibitors for the therapy of this malignancy. Currently, IKKβ inhibitors are under development for clinical use and are likely to have manageable on-target side effects (Nagashima et al., 2006). That the majority of MM cell lines tested retained a dependence on NF-κB signaling for survival is encouraging and consistent with previous results (Hideshima et al., 2006). The NF-κB signature provides a basis to develop biomarkers of response to NF-κB pathway inhibitors. A gauge of NF-κB activity, such as quantitative RT-PCR to measure the 11-gene NF-κB signature, could be assessed in bone marrow MM samples both pre-treatment and during administration of the drug. Coupling biomarkers with pathway-targeted therapeutics will ultimately allow treatment of MM patients to be tailored to the genetic abnormalities of their cancers.
Experimental Procedures
Primary MM patient samples
Primary MM cells were purified from bone marrow aspirates obtained under approval of the Institutional Review Board of the University of Arkansas for Medical Sciences, Little Rock, AR. Written informed consent was documented. MM plasma cells were isolated by CD138 immunomagnetic bead selection (Zhan et al., 2006).
Cell culture, retroviral constructs and transduction
MM cell lines were maintained in RPMI 1640 or ACL-4 medium supplemented with 10% fetal calf serum (Hyclone) and penicillin/streptomycin (Invitrogen) with or without 10 ng/ml IL-6 (R&D Systems). The IKKβ inhibitor MLN120b was provided by Millennium Pharmaceuticals (Cambridge, MA). For efficient retroviral transductions, cell lines were engineered to express the murine ecotropic retroviral receptor (Lam et al., 2005). Certain cell lines were also engineered to express the bacterial tetracycline repressor (TETR) (Ngo et al., 2006). All inducible constructs in TETR lines were activated by doxycycline (20 ng/mL).
To assess toxicity of an shRNA, retroviruses that co-expressed GFP were used as described (Ngo et al., 2006). In brief, flow cytometry was performed 2 days after retroviral infection to determine the initial GFP-positive proportion of live cells for each shRNA, then cells were subsequently cultured with doxycycline to induce shRNA and sampled over time. The GFP-positive proportion at each time was normalized to that for the control shRNA (against luciferase), and further normalized to the initial value.
The cell-based assay for IKK activity was performed as described (Ngo et al., 2006) using a TETR cell line engineered to express Renilla luciferase and a fusion protein of IκBα and firefly luciferase (IκBα-Photinus); after infection with shRNA vectors and puromycin selection, shRNA was induced and samples measured for the two reporters using the Dual-Glo luciferase assay system (Promega).
Fluorescence in situ hybridization (FISH)
Three color FISH assays of metaphase chromosomes derived from MM cell lines were performed as described (Shou et al., 2000). In addition to the previously described immunoglobulin probes, a 92 kb PAC (AC003963, version gi:2978476) was used as a probe for NIK. Using the same probes, NIK copy number alterations and NIK-IGH and NIK-IGL gene fusions were evaluated by triple color interphase FISH on mononuclear cells from bone marrow aspirates of newly diagnosed disease (Hanamura et al., 2006).
Polymorphism to distinguish NIK alleles
A CT polymorphism at base pair 3020 (reference mRNA NM_003954) in the 3′ untranslated region of NIK RNA was identified from the human EST database. Sequence analysis of PCR products from genomic DNA was determined for 16 MM cell lines and 14 primary MM tumors. Nine samples each had only C or T alleles and 12 samples had both C and T. When both alleles were present, the assay was repeated using cDNA to distinguish mono- vs. bi-allelic expression.
Cloning of translocation breakpoints
Southern blotting was used to identify rearranged HpaI genomic fragments within or near the NIK gene for the L363 and JJN3 cell lines. Genomic libraries were made from HpaI fragments modified with BamHI adaptors that were cloned in the λDash II vector (Stratagene), and then screened with the probes used for the Southern blots. The sequence of inserts was obtained directly (L363) or from a PCR product (JJN3).
Western blot and ELISA
Protein was harvested from MM cell lines and fractionated using a Nuclear/cytosol fractionation kit (BioVision). Protein was quantified using the BCA method (Pierce) and separated by SDS-PAGE on a 4–12% acrylamide gradient. Nuclear protein was analyzed for NF-κB activity using the TransAm NF-κB family kit (Active Motif). The following antibodies were used: NIK, phospho-IkBα (Cell signaling), IkBα, p65 (SantaCruz Biotechnologies), p52/p100 (Upstate), B-tubulin (Sigma), histone H2B (Imgenex).
qPCR
Copy number of TRAF3, BIRC2, BIRC3 and CYLD was measured in DNA extracted from CD138+ MM samples or mononuclear cells purified from peripheral blood of normal donors. qPCR was performed and normalized to the PRKCQ locus as described (Rosenwald et al., 2003). Primer sequences are listed in the Methods supplement.
aCGH
DNA from MM cell lines was labeled fluorescently with Cy5 and normal control genomic DNA was labeled with Cy3. These probes were co-hybridized to 400,000-element Nimblegen whole genome arrays. Intensity of hybridized probes was determined using an Axon scanner and data were analyzed with Nimblegen Scanarray software. Previous aCGH data on 47 MM samples was downloaded for comparison with gene expression (Carrasco et al., 2006).
Gene expression profiling and analysis
Lymphochip DNA microarrays were utilized as described (Alizadeh et al., 2000). Normal human B cell subsets were purified as described (Alizadeh et al., 2000; Nilsson et al., 2000) and profiled following linear mRNA amplification (see Supplemental Methods for details). Microarray data from primary MM samples was obtained from http://www.ncbi.nlm.nih.gov/geo/ (accession GSE2658) (Shaughnessy et al., 2006; Zhan et al., 2006).
Genes comprising the NF-κB signature in MM were those that were decreased in expression by >40% in at least 6 of 8 time points following treatment of L363 cells with MLN for 8–24 hours in 3 separate experiments (accession GSE8487). Genes were chosen if they correlated in expression across the MM cell lines (r > 0.5). See Supplemental data for Affymetrix U133plus2.0 probsets used for analysis.
Analysis of outlier gene expression was performed as described (Tomlins et al., 2005). Briefly, data from each Affymetrix probe set were median centered across all samples. The absolute difference from the median was calculated for each sample, and the data were scaled such that the median of these values was 1 (log2). Outlier gene expression was defined to be any scaled values greater than 5 (log2). In other words, compared to the median deviation of samples from the median, outlier cases had >32-fold larger deviation. For a set of cases with outlier expression of a particular gene, the association of this outlier profile with the NF-κB signature was calculated by comparing expression of the NF-κB signature in outliers versus other cases using a t-test. To adjust for the influence of gene expression differences among known MM subgroups (Zhan et al., 2006), we fitted an analysis of variance model for log-signal of the NF-κB signature, including MM subgroup as a categorical variable and outlier status as a two-categorical variable.
The p-value reported the association of NF-κB pathway abnormalities with high NF-κB signature expression is based on a Wilcoxon signed rank test.
Supplementary Material
01
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (LMS, WMK), NIH grant CA55819 (JDS) and the Lebow Fund for the Cure (JDS). LD is an employee of Millennium Pharmaceuticals, Inc., Cambridge, MA. The authors would like to acknowledge the technical contributions of Leslie Brents. WMK thanks Leif Bergsagel for suggesting the possibility of a NIK translocation in JJN3 cells.
Footnotes
See Supplemental material for additional methods.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, Walker R, Crowley J. Treatment of multiple myeloma. Blood. 2004;103:20–32. doi: 10.1182/blood-2003-04-1045. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Berberich I, Shu GL, Clark EA. Cross-linking CD40 on B cells rapidly activates nuclear factor-kappa B. J Immunol. 1994;153:4357–4366. [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- Carrasco DR, Tonon G, Huang Y, Zhang Y, Sinha R, Feng B, Stewart JP, Zhan F, Khatry D, Protopopova M, et al. High-resolution genomic profiles define distinct clinico-pathogenetic subgroups of multiple myeloma patients. Cancer Cell. 2006;9:313–325. doi: 10.1016/j.ccr.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GG, Johnston LH, Ley SC. CD40 regulates the processing of NF-kappaB2 p100 to p52. Embo J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive Nuclear Factor kappaB Activity Is Required for Survival of Activated B Cell-like Diffuse Large B Cell Lymphoma Cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, Hollmig K, Zangarri M, Pineda-Roman M, van Rhee F, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–1732. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer J, Puschner S, Ramakrishnan P, Simon U, Bongers M, Federle C, Engelmann H. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Neri P, Tassone P, Yasui H, Ishitsuka K, Raje N, Chauhan D, Podar K, Mitsiades C, Dang L, et al. MLN120B, a novel IkappaB kinase beta inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin Cancer Res. 2006;12:5887–5894. doi: 10.1158/1078-0432.CCR-05-2501. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- Lam LT, Davis RE, Wright G, Rosenwald A, Hurt EMXY, Pierce J, Hepperle M, Xu Y, Adams J, et al. Small Molecule Inhibitors of IkB-Kinase are Selectively Toxic for Subgroups of Diffuse Large B Cell Lymphoma Defined by Gene Expression Profiling. Clin Cancer Res. 2005;11:28–40. [PubMed] [Google Scholar]
- Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. CD30/TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci U S A. 1996;93:9699–9703. doi: 10.1073/pnas.93.18.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Norris PS, Ni CZ, Havert ML, Chiong EM, Tran BR, Cabezas E, Reed JC, Satterthwait AC, Ware CF, Ely KR. Structurally distinct recognition motifs in lymphotoxin-beta receptor and CD40 for tumor necrosis factor receptor-associated factor (TRAF)-mediated signaling. J Biol Chem. 2003;278:50523–50529. doi: 10.1074/jbc.M309381200. [DOI] [PubMed] [Google Scholar]
- Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–347. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol. 2000;10:785–788. doi: 10.1016/s0960-9822(00)00566-2. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Migliazza A, Lombardi L, Rocchi M, Trecca D, Chang CC, Antonacci R, Fracchiolla NS, Ciana P, Maiolo AT, Neri A. Heterogeneous chromosomal aberrations generate 3′ truncations of the NFKB2/lyt-10 gene in lymphoid malignancies. Blood. 1994;84:3850–3860. [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Munshi NC, Anderson KC. Focus on multiple myeloma. Cancer Cell. 2004;6:439–444. doi: 10.1016/j.ccr.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Moreaux J, Cremer FW, Reme T, Raab M, Mahtouk K, Kaukel P, Pantesco V, De Vos J, Jourdan E, Jauch A, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood. 2005;106:1021–1030. doi: 10.1182/blood-2004-11-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima K, Sasseville VG, Wen D, Bielecki A, Yang H, Simpson C, Grant E, Hepperle M, Harriman G, Jaffee B, et al. Rapid TNFR1-dependent lymphocyte depletion in vivo with a selective chemical inhibitor of IKKbeta. Blood. 2006;107:4266–4273. doi: 10.1182/blood-2005-09-3852. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, Staudt LM. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- Ni CZ, Oganesyan G, Welsh K, Zhu X, Reed JC, Satterthwait AC, Cheng G, Ely KR. Key molecular contacts promote recognition of the BAFF receptor by TNF receptor-associated factor 3: implications for intracellular signaling regulation. J Immunol. 2004;173:7394–7400. doi: 10.4049/jimmunol.173.12.7394. [DOI] [PubMed] [Google Scholar]
- Ni CZ, Welsh K, Leo E, Chiou CK, Wu H, Reed JC, Ely KR. Molecular basis for CD40 signaling mediated by TRAF3. Proc Natl Acad Sci U S A. 2000;97:10395–10399. doi: 10.1073/pnas.97.19.10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson N, Ingvarsson S, Borrebaeck CA. Immature B cells in bone marrow express Fas/FasL. Scand J Immunol. 2000;51:279–284. doi: 10.1046/j.1365-3083.2000.00701.x. [DOI] [PubMed] [Google Scholar]
- O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Mahony A, Lin X, Geleziunas R, Greene WC. Activation of the heterodimeric IkappaB kinase alpha (IKKalpha)-IKKbeta complex is directional: IKKalpha regulates IKKbeta under both basal and stimulated conditions. Mol Cell Biol. 2000;20:1170–1178. doi: 10.1128/mcb.20.4.1170-1178.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Qu Z, Xiao G. Stabilization of basally translated NF-kappaB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-kappaB2 p100. J Biol Chem. 2005;280:40578–40582. doi: 10.1074/jbc.M508776200. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan P, Wang W, Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Regamey A, Hohl D, Liu JW, Roger T, Kogerman P, Toftgard R, Huber M. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J Exp Med. 2003;198:1959–1964. doi: 10.1084/jem.20031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995a;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995b;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2006 doi: 10.1182/blood-2006-07-038430. in press. [DOI] [PubMed] [Google Scholar]
- Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IkappaB kinase-beta: NF-kappaB activation and complex formation with IkappaB kinase-alpha and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Xiao G, Sun SC. Negative regulation of the nuclear factor kappa B-inducing kinase by a cis-acting domain. J Biol Chem. 2000;275:21081–21085. doi: 10.1074/jbc.M002552200. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kishimoto T, Minamoto S. NF-kappaB activation in CD27 signaling: involvement of TNF receptor-associated factors in its signaling and identification of functional region of CD27. J Immunol. 1998;161:4753–4759. [PubMed] [Google Scholar]
- Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, Pineda-Roman M, Tricot G, van Rhee F, Zangari M, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007;109:1692–1700. doi: 10.1182/blood-2006-07-037077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99:1745–1757. doi: 10.1182/blood.v99.5.1745. [DOI] [PubMed] [Google Scholar]
- Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01