Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort (original) (raw)
Abstract
Objective:
Mutations in the FUS gene on chromosome 16 have been recently discovered as a cause of familial amyotrophic lateral sclerosis (FALS). This study determined the frequency and identities of FUS gene mutations in a cohort of Italian patients with FALS.
Methods:
We screened all 15 coding exons of FUS for mutations in 94 Italian patients with FALS.
Results:
We identified 4 distinct missense mutations in 5 patients; 2 were novel. The mutations were not present in 376 healthy Italian controls and thus are likely to be pathogenic.
Conclusions:
Our results demonstrate that FUS mutations cause ∼4% of familial amyotrophic lateral sclerosis cases in the Italian population.
GLOSSARY
ALS
= amyotrophic lateral sclerosis;
FALS
= familial amyotrophic lateral sclerosis;
FTD
= frontotemporal dementia;
gDNA
= genomic DNA;
LMN
= lower motor neuron;
NLS
= nuclear localization signal;
UMN
= upper motor neuron.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease of adult life characterized by a selective loss of upper motor neurons (UMN) and lower motor neurons (LMN). The cause of the disease is largely unknown. Approximately 10% of all ALS cases are familial (FALS), of which ∼20% are caused by mutations in the SOD1 gene.1 Variants in TARDBP, which encodes the DNA/RNA binding protein TDP-43, account for an additional ∼5% of cases.2–4 Pathogenic mutations in 5 other genes (ALS2, SETX, VAPB, DNCT1, and ANG) have also been associated with atypical ALS phenotypes in isolated families.5,6 Recently, mutations in the FUS gene, which also encodes a DNA/RNA binding protein, have been identified in ∼5% of patients with FALS who tested negative for SOD1 and TARDBP mutations.7,8 The similarities between TARDBP and FUS suggest that alterations in RNA processing may play an important role in ALS pathogenesis. The FUS gene was initially isolated analyzing chromosomal translocations in myxoid liposarcoma9 and subsequently found in a variety of cancer-associated fusion genes. The protein plays a role in transcription, splicing,10 and shuttling of RNA from the nucleus to the cytoplasm.11 FUS also acts as a transcriptional regulatory sensor of DNA damage signals and thus is required in maintaining the integrity of the genome.12 FUS is diffusely expressed in the CNS where it regulates the morphology of hippocampal dendritic spines and stabilizes the structure of the synapse.13 To further define the frequency and spectrum of FUS mutations, we have screened a cohort of Italian patients with FALS.
METHODS
Patients and controls.
Peripheral blood samples were collected from 94 unrelated patients with FALS (53 men and 41 women) in the period between 2002 and 2007. All the patients were of Italian descent and were diagnosed with probable or definite ALS according to the El Escorial revised criteria.14 Familial history was considered positive for ALS if the proband had at least 1 affected relative within 3 generations. The disease onset was bulbar in 24% of patients and spinal in 76%. Six patients (4 men and 2 women) developed frontotemporal dementia (FTD). In agreement with previous epidemiologic surveys on motor neuron disease in Italy,15 an initial involvement of bulbar motor neurons was slightly more frequent in women (30%) than men (19%). The average age at onset in our cohort was 46.8 ± 15.2 years. Mutations in SOD1, ANG,16 and TARDBP17 were excluded in all samples in the present study. Control DNA was obtained from 376 unrelated, age-matched Italian subjects with a negative personal and familial history for neurodegenerative diseases.
Standard protocol approvals, registrations, and patient consents.
We received approval for this study from the ethical standards committees on human experimentation of the IRCCS Istituto Auxologico Italiano and of the Fondazione IRCCS Istituto Neurologico “Carlo Besta.” Written informed consent was obtained from all patients and healthy subjects participating in the study (consent for research).
Molecular analysis.
Genomic DNA (gDNA) was extracted from peripheral blood leukocytes using standard procedures and was subsequently amplified using the IllustraTM GenomiPhi HY DNA Amplification Kit (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK). For all FALS samples, the 15 exons of FUS were amplified by touchdown PCR using primers located in adjacent intronic or noncoding regions. PCR primers and conditions have been described elsewhere.7 PCR products were subsequently purified by incubation with Exonuclease I and Shrimp Alkaline Phosphatase, sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and then resolved by capillary electrophoresis on an ABI 3730XL DNA Analyzer (Applied Biosystems). Sequence analysis was performed using the PHRED/PHRAP/Consed software suite (www.phrap.org/phredphrapconsed.html),18–21 and variations in the sequences were identified with the Polyphred v6.15 software (http://droog.gs.washington.edu/polyphred).22 All identified mutations were further confirmed by bidirectional sequencing of the original gDNA sample. The screening of the c.701G>T mutation in control samples was performed using a custom TaqMan SNP Genotyping Assay using the File Builder 3.1 software (Applied Biosystems) which consisted of a mix of unlabeled PCR forward and reverse primers (forward: 5′-GCGGCGGTGGTGGTT-3′; reverse: 5′-CACGACCTCTGGGTTCATAGC-3′) and 2 dye-labeled probes (allele X: VIC-CACTGCTGCGGTTGT-NFQ; allele Y: FAM-CACTGCTGAGGTTGT-NFQ). Since the TaqMan assay designed for the c.467G>A mutation failed to generate consistent results, exon 5 of the FUS gene was directly sequenced in all control samples.
Bioinformatic analysis.
The effect on splicing of the noncoding and synonymous variants identified was assessed using the electronic tools ESEfinder 3.0 (http://rulai.cshl.edu/tools/ESE/),23,24 NNSplice (http://www.fruitfly.org/seq_tools/splice.html),25 and SpliceView (http://bioinfo.itb.cnr.it/oriel/splice-view.html).26 In silico prediction of the putative functional effects of the missense mutations was obtained with the SNAP (http://cubic.bioc.columbia.edu/services/SNAP/)27 and PMut (http://mmb2.pcb.ub.es:8080/PMut/)28 software.
RESULTS
To identify novel FUS mutations and further define the frequency of all FUS gene variants in FALS, we sequenced the coding region (exons 1–15) and the intron/exon boundaries in 94 FALS samples of Italian descent. Our sequence analysis identified 2 previously described missense mutations in the FUS gene. The mutation c.1561C>T produces an arginine to cysteine change at amino acid 521 (p.R521C) and was observed in 2 patients. The c.1561C>G mutation was identified in a single patient and alters the same base pair but changes the arginine residue to a glycine (p.R521G). In addition, we have identified 2 novel missense mutations. The mutation c.467G>A was identified in a single patient; it changes a glycine to a glutamic acid at position 156 (p.G156E). The mutation c.701G>T, also identified in a single patient, changes an arginine residue to a leucine at position 234 (p.R234L). The pedigrees of the patients carrying FUS mutations are shown in figure 1. Chromatograms displaying the mutations are shown in figure e-1 on the _Neurology_® Web site at www.neurology.org. Unfortunately, since all the affected relatives of the probands are deceased, we could not evaluate the segregation of the mutations in families. In addition to the missense mutations, we also identified a novel synonymous substitution (c.1566G>A [p.R522R]) as well as 5 novel variants in intronic (c.832 + 36A>G, c.833 − 29C>T, c.1066 + 82C>G) and 3′ UTR (c.*10C>T, c.*41G>A) regions of FUS (table e-1). In silico analysis with the ESEfinder 3.0 software showed that the synonymous variant does not disrupt any exonic splicing enhancer motif, while NNSplice and SpliceView programs predicted that the intronic and 3′ UTR variants do not affect splicing mechanisms. These variants were not further studied.

Figure 1 Pedigrees of familial amyotrophic lateral sclerosis cases harboring FUS mutations
This illustrates the structure of the 5 unrelated Italian pedigrees to which the 5 cases studied here belong. Affected individuals are shown in black; unaffected individuals are shown in white. Arrows indicate the individual harboring the mutation.
To determine whether the 2 novel missense mutations are low allele frequency polymorphisms in the population, we genotyped the p.G156E and p.R234L mutations on a panel of 376 healthy control samples of Italian descent. Neither variant was identified in these normal controls. This finding strongly argues that these missense variants represent pathogenic mutations.
The p.G156E is located within the SYQG domain of the FUS protein, which acts as a transcriptional activation domain. Additionally, the p.R234L mutation is located within the G-rich domain, a region in which 3 mutations have previously been reported (figure 2). To further evaluate the pathogenicity of these novel mutations, we used 2 software applications, PMut and SNAP, which predict whether a missense mutation is pathogenic or benign. As expected, the previously described mutations (p.R521C and p.R521G) were predicted to be deleterious by both PMut and SNAP (table). The novel mutations p.G156E and p.R234L are similarly predicted to be deleterious by both PMut and SNAP (table). In support of this prediction, we note that the residues affected by these mutations (G156 and R234) are highly conserved, being present in mammals, reptiles, and zebra fish (figure 3). This observation also supports the view that the corresponding missense mutations are adverse and likely to be pathogenic.

Figure 2 Location of FUS mutations in familial amyotrophic lateral sclerosis
(A) Gene structure of FUS indicating coding exons (thick bars) and untranslated regions (medium-height bars). Red asterisks indicate the exons where mutations have been identified. (B) Linear protein structure of FUS indicating the different domains. Amino acid changes are shown above the protein with arrows indicating their position. Mutations found in Italian patients are shown in red.
Table Phenotypic description of patients with FUS mutations
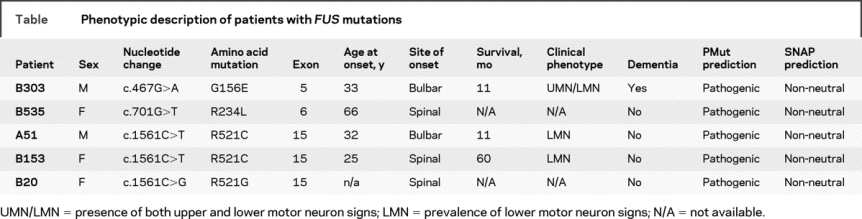
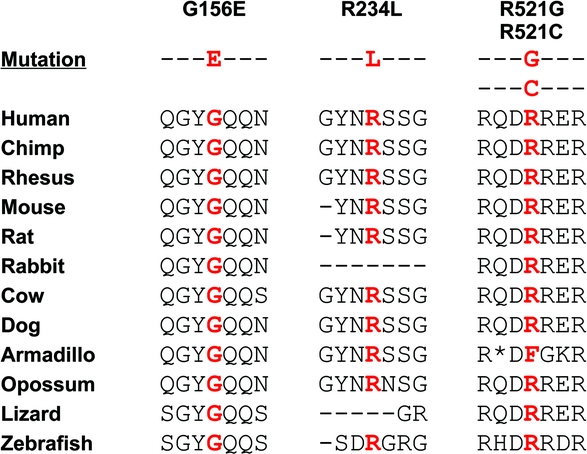
Figure 3 Evolutionary conservation of FUS mutations
The evolutionary conservation of all 4 identified mutations is shown. For each, the mutated amino acid is shown in red.
Phenotypic information for the 5 patients carrying FUS mutations is provided in the table. Detailed clinical records were available for 3 (B303, A51, and B153) of the 5 patients and other affected family members, and are reported in appendix e-1. Patient B303 harbors the p.G156E mutation whereas both A51 and B153 have the p.R521C mutation. Notably, each affected individual in the 3 pedigrees developed early symptoms of symmetric weakness of the scapular or pelvic girdles and of the proximal muscles of the upper or lower limbs. Also prominent was the involvement of the axial muscles of the neck and trunk.
In total, we have identified 5 missense mutations within a panel of 94 Italian patients with FALS which were prescreened and found not to have mutations in the SOD1 and TARDBP genes (which represent ∼25% of all FALS cases). We therefore estimate that ∼4% of FALS cases within the Italian population are caused by mutation in the FUS gene. This frequency is similar to that reported in a recent screening in another FALS cohort.29
DISCUSSION
FUS mutations were initially identified in 20 cases of FALS.7,8 A vast majority of the alterations (16 out of 20) were missense mutations clustered in 5 arginine residues located in exon 15 of the FUS gene. Interestingly, a p.H517Q homozygous mutation within exon 15 was found in a family presenting a recessive inheritance pattern; all other identified families displayed a dominant inheritance pattern. Additional mutations were identified in exon 5 (p.174–175insGG, p.173–174delGG), exon 6 (p.R244C), and exon 14 (p.R514G). The predicted amino acid sequence of the FUS protein is highly evolutionarily conserved and consists of an N-terminal SYQG-rich region that acts as a transcriptional activation domain, two G-rich regions, an RNA binding domain, a Cys2/Cys2-zinc finger motif, and a C-terminal RGG-rich region, which contains the nuclear localization signal (NLS).30,31
In our cohort of 94 unrelated Italian patients with FALS, we identified FUS mutations in 5 patients. The observed mutational frequency in our Italian cohort is similar to that observed in the 2 previous studies of FUS mutations in ALS.7,8 Previous mutational screenings have suggested that the prevalence of SOD1 mutations in Italian ALS families is about 15%.32,33 ANG mutations account for ∼2% of all familial cases,16 while a recent study identified TARDBP mutations in 6 out of 125 Italian patients with FALS (4.8%).17 To date, in the Italian population, mutations in the ALS2 gene have been reported only in an handful of families affected by infantile ascending hereditary spastic paralysis34 or juvenile primary lateral sclerosis,35 while a screening for VAPB variants in patients with ALS was negative.36 To our knowledge, there are no reports of SETX or dynactin mutations in Italian patients with ALS. Our findings thus suggest that FUS mutations represent the third most significant cause of FALS in Italy after SOD1 and TARDBP.
In our cohort, we found 2 previously identified mutations, p.R521G and p.R521C. The substitution of the arginine at codon 521 disrupts the NLS and leads to an aberrant subcellular distribution of FUS, with retention and apparent aggregation of the mutant protein in the cytoplasm.7 The 2 novel mutations observed in our cohort are located in the N-terminal SYQG-rich domain (p.G156E) and in the first G-rich region (p.R234L), where other mutations have been found7 (figure 2B). No data are available on the functional role of these variants, since they are not located in the NLS and thus presumably do not alter the subcellular localization of FUS. Moreover, very little information is available on the activity of both the SYQG-rich and G-rich domain of FUS. We hypothesize, however, that p.G156E and p.R234L are pathogenic for several reasons. The amino acids at these positions are highly evolutionarily conserved (figure 3) and these variants were not detected in 376 Italian healthy controls (752 chromosomes). Finally, both in silico SNAP and PMut software analyses predicted that p.G156E and p.R234L affect protein function (table), although these data should be interpreted with caution.
We note that the clinical presentation of the Italian patients with FUS gene mutations (symmetric, proximal, and axial weakness at onset) was distinct from typical ALS, which often begins in a single limb distally.37 Severe weakness of neck extensor muscles at onset, as seen in one Italian case, is present in only about 1% of patients with ALS,38 and thus is also unusual. We also note that in his fourth decade, patient B303 (p.G156E mutation) developed FTD concurrently with motor neuron disease; patients with ALS in the 2 previous reports on FUS mutations were not described as having concomitant dementia. It will be of interest to determine whether FUS mutations consistently correlate with these distinctive clinical phenotypes in other patients with ALS.
Given the complexity of cellular functions and pathways in which FUS is involved, further studies will be required to delineate the explicit mechanisms whereby these FUS mutations cause motor neuron death. In particular, it will be of interest to assess if the pathogenic pathways involving FUS and TARDBP overlap. The 2 genes encode for DNA/RNA binding proteins involved in gene regulation and RNA processing. Mutations in FUS and TARDBP may alter the binding of their nucleic acid targets, resulting in altered mRNA splicing. Additionally, some mutations have been shown to impair the capability of both proteins to shuttle between the nucleus and the cytoplasm.39 Widespread defects in RNA processing as a result of either, or both, of these mechanisms could lead to motor neuron death. The precise pathogenic mechanism notwithstanding, our study provides strong evidence that mutations in the FUS gene represent a significant cause of familial ALS in the Italian population. It also suggests that some patients carrying FUS mutations may have uncommon clinical phenotypes, a possibility that merits further analysis as more opportunities arise to correlate FUS genotypes with clinical phenotypes.
AUTHOR CONTRIBUTIONS
Writing team: N.T., V.S., A.L., P.K, R.H.B., J.L.; all others received and approved the manuscript. Patient selection, preliminary genetic screening, and record collection: N.T., V.S., A.R., C.G., F.T., P.S. Sequencing and genotyping: N.T., A.L., P.K. Data analysis: N.T., V.S., R.H.B., J.E.L. Scientific planning and direction: N.T., V.S., A.L., T.K., R.H.B., J.E.L.
ACKNOWLEDGMENT
The authors thank the Italian patients with ALS and their caregivers and the Peviani family for support.
DISCLOSURE
Dr. Ticozzi has received research support from the Italian Ministry of Health (Malattie Neurodegenerative, ex Art. 56, n533F/N1). Dr. Silani serves on the editorial advisory boards of Amyotrophic Lateral Sclerosis, European Neurology, and the Journal of Pediatric Neurology, and receives research support from the Italian Ministry of Health (Malattie Neurodegenerative, ex Art. 56, n533F/N1). A.L. Leclerc and P. Keagle report no disclosures. Dr. Gellera has received research support from the Italian Ministry of Health (#RF2007/INN644440). Dr. Ratti has received research support from the Italian Ministry of Health (Malattie Neurodegenerative, ex Art. 56, n533F/N1). Dr. Taroni serves on the scientific advisory board of the Italian Ataxia Association (AISA); serves on the editorial board of the Journal of Neurology; and receives research support from ApoPharma Inc. [EudraCT No. 2007-003331-23 and No. 2009-010865-22 (Unit PI)], the Telethon Foundation (Telethon and Telethon-UILDM Project Coordinator), and the Italian Ministry of Health [AIFA EudraCT No. 2007-003357-85 (PI) and MoH RF2007/INN644440)]. Dr. Kwiatkowski has served as consultant and received funding for international travel from Asia Ventures Management; receives license fee payments as a contributor to US Patents 5,741,645 (issued: 1998) and 5,834,183 (issued: 1998) entitled “Gene sequence for spinocerebellar ataxia type 1 and method for diagnosis” and may accrue revenue from US patent 61/135,689, “Genetic test for FUS ALS” (filed: 2008); has served as consultant and received honoraria from Athena Diagnostics, Inc.; and receives research support from the NIH [R01NS050557 (Co-PI)] and from the ALS Therapy Alliance, the Angel Fund and Project ALS, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, and the ALS Family Charitable Foundation. D.M. McKenna-Yasek receives salary support from the NIH [RO1-NS050641-04]. P.C. Sapp has received research support from the Howard Hughes Medical Institute (grant awarded to Prof. H. Robert Horvitz). Dr. Brown serves on scientific advisory boards for Biogen Idec, Acceleron Pharma, Link Medicine, and AviTx Inc.; has filed/holds US Patent 5,843,641, “Methods for the diagnosis of familial amyotrophic lateral sclerosis” (filed: 1993); receives royalties from publishing Principles of Neurology (McGraw-Hill, 2005); receives research support from the NIH [NINDS R01NS050557 (PI) and NINDS UO1NS052225-02 (PI)], the ALS Therapy Alliance, Project ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, and the ALS Family Charitable Foundation; receives board of directors compensation from AviTx Inc. (co-founder); and receives license fee payments from Athena Diagnostics, Inc. related to diagnostic blood tests. Dr. Landers has received payments from MIT as an inventor of US Patent 6,703,228; has been deposed concerning a lawsuit between Affymetrix, Inc. and E8/MIT regarding this patent; and has received research support from the ALS Therapy Alliance, Project ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, and the ALS Family Charitable Foundation. His spouse is an employee of Bristol-Myers Squibb.
Supplementary Material
[Data Supplement]
Address correspondence and reprint requests to Dr. John E. Landers, LRB604, 364 Plantation St., Worcester, MA 01605 john.landers@umassmed.edu
Editorial, page 1172
See also page 1176
Supplemental data at www.neurology.org
e-Pub ahead of print on September 9, 2009, at www.neurology.org.
Supported by the ALS Therapy Alliance, Project ALS, the Angel Fund, the Pierre L. de Bourgknecht ALS Research Foundation, the Al-Athel ALS Research Foundation, the ALS Family Charitable Foundation, and the National Institute of Neurological Disorders and Stroke (NS050557 and NS050641). N.T., V.S., and A.R. were supported by the Italian Ministry of Health (Malattie Neurodegenerative, ex Art.56, n.533F/N1). F.T. and C.G. were supported by the Italian Ministry of Health grant RF2007/INN644440. P.S. was supported by the Howard Hughes Medical Institute (HHMI) through the auspices of Prof. H. Robert Horvitz, an Investigator in the HHMI. R.H.B. is a cofounder of AviTx, which targets development of therapies.
Disclosure: Author disclosures are provided at the end of the article.
Received March 19, 2009. Accepted in final form July 1, 2009.
REFERENCES
- 1.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362: 59–62. [DOI] [PubMed] [Google Scholar]
- 2.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008;40:572–574. [DOI] [PubMed] [Google Scholar]
- 4.Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 2008;7:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006;7:710–723. [DOI] [PubMed] [Google Scholar]
- 6.Beleza-Meireles A, Al-Chalabi A. Genetic studies of amyotrophic lateral sclerosis: controversies and perspectives. Amyotroph Lateral Scler 2009;10:1–14. [DOI] [PubMed] [Google Scholar]
- 7.Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 8.Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993;363:640–644. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 1998;273:27761–27764. [DOI] [PubMed] [Google Scholar]
- 11.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 1997;110(Pt 15):1741–1750. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Arai S, Song X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008;454:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujii R, Okabe S, Urushido T, et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 2005;15:587–593. [DOI] [PubMed] [Google Scholar]
- 14.Miller RG, Munsat TL, Swash M, Brooks BR. Consensus guidelines for the design and implementation of clinical trials in ALS: World Federation of Neurology committee on Research. J Neurol Sci 1999;169:2–12. [DOI] [PubMed] [Google Scholar]
- 15.Beghi E, Millul A, Micheli A, Vitelli E, Logroscino G. Incidence of ALS in Lombardy, Italy. Neurology 2007;68:141–145. [DOI] [PubMed] [Google Scholar]
- 16.Gellera C, Colombrita C, Ticozzi N, et al. Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics 2008;9:33–40. [DOI] [PubMed] [Google Scholar]
- 17.Corrado L, Ratti A, Gellera C, et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat 2009;30:688–694. [DOI] [PubMed] [Google Scholar]
- 18.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred: I: accuracy assessment. Genome Res 1998;8:175–185. [DOI] [PubMed] [Google Scholar]
- 19.Ewing B, Green P. Base-calling of automated sequencer traces using phred II: error probabilities. Genome Res 1998;8:186–194. [PubMed] [Google Scholar]
- 20.Gordon D, Desmarais C, Green P. Automated finishing with autofinish. Genome Res 2001;11:614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res 1998;8:195–202. [DOI] [PubMed] [Google Scholar]
- 22.Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res 1997;25:2745–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 2006;15:2490–2508. [DOI] [PubMed] [Google Scholar]
- 24.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003;31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol 1997;4:311–323. [DOI] [PubMed] [Google Scholar]
- 26.Rogozin IB, Milanesi L. Analysis of donor splice sites in different eukaryotic organisms. J Mol Evol 1997;45:50–59. [DOI] [PubMed] [Google Scholar]
- 27.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 2007;35:3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 2005;21:3176–3178. [DOI] [PubMed] [Google Scholar]
- 29.Chiò A, Restagno G, Brunetti M, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging 2009;30:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morohoshi F, Ootsuka Y, Arai K, et al. Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene 1998;221:191–198. [DOI] [PubMed] [Google Scholar]
- 31.Zakaryan RP, Gehring H. Identification and characterization of the nuclear localization/retention signal in the EWS proto-oncoprotein. J Mol Biol 2006;363:27–38. [DOI] [PubMed] [Google Scholar]
- 32.Gellera C, Castellotti B, Riggio MC, et al. Superoxide dismutase gene mutations in Italian patients with familial and sporadic amyotrophic lateral sclerosis: identification of three novel missense mutations. Neuromuscul Disord 2001;11:404–410. [DOI] [PubMed] [Google Scholar]
- 33.Battistini S, Giannini F, Greco G, et al. SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study J Neurol 2005;252:782–788. [DOI] [PubMed] [Google Scholar]
- 34.Lesca G, Eymard-Pierre E, Santorelli FM, et al. Infantile ascending hereditary spastic paralysis (IAHSP): clinical features in 11 families. Neurology 2003;60:674–682. [DOI] [PubMed] [Google Scholar]
- 35.Panzeri C, De Palma C, Martinuzzi A, et al. The first ALS2 missense mutation associated with JPLS reveals new aspects of alsin biological function. Brain 2006;129:1710–1719. [DOI] [PubMed] [Google Scholar]
- 36.Conforti FL, Sprovieri T, Mazzei R, et al. Sporadic ALS is not associated with VAPB gene mutations in Southern Italy. J Negat Results Biomed 2006;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007;68:1571–1575. [DOI] [PubMed] [Google Scholar]
- 38.Gourie-Devi M, Nalini A, Sandhya S. Early or late appearance of “dropped head syndrome” in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2003;74:683–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell 2009;136:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Data Supplement]