TRPM2 Functions as a Lysosomal Ca2+-Release Channel in β Cells (original) (raw)
. Author manuscript; available in PMC: 2010 May 19.
Published in final edited form as: Sci Signal. 2009 May 19;2(71):ra23. doi: 10.1126/scisignal.2000278
Abstract
TRPM2 is a Ca2+-permeable cation channel that is specifically activated by adenosine diphosphoribose (ADPR). Channel activation in the plasma membrane leads to Ca2+ influx and has been linked to apoptotic mechanisms. The primary agonist, ADPR, is produced both extra- and intracellularly and causes increases in intracellular calcium concentration ([Ca2+]i), but the mechanisms involved are not understood. Using short interfering RNA and a knockout mouse, we report that TRPM2, in addition to its role as a plasma membrane channel, also functions as a Ca2+-release channel activated by intracellular ADPR in a lysosomal compartment. We show that both functions of TRPM2 are critically linked to hydrogen peroxide–induced β cell death. Additionally, extracellular ADPR production by the ectoenzyme CD38 from its substrates NAD+ (nicotinamide adenine dinucleotide) or cADPR causes IP3-dependent Ca2+ release via P2Y and adenosine receptors. Thus, ADPR and TRPM2 represent multimodal signaling elements regulating Ca2+ mobilization in β cells through membrane depolarization, Ca2+ influx, and release of Ca2+ from intracellular stores.
Introduction
TRPM2 (transient receptor potential channel, melastatin subfamily type 2) is a nonselective, Ca2+-permeable cation channel with unique gating properties that are conferred by a functional adenosine diphosphoribose (ADPR) hydrolase domain in its C terminus (1–3). TRPM2 is synergistically activated and regulated by multiple signaling pathways through various adenine dinucleotides [ADPR, cyclic ADPR (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP)] and intracellular calcium concentration ([Ca2+]i) (1, 2, 4–9), agonists that vary in structure as well as in source. Although all of these molecules function intracellularly to activate TRPM2, some of them, in particular the primary TRPM2 agonist ADPR, can be produced extracellularly through the multifunctional ectoenzyme CD38 (10). Intracellularly, ADPR can be released from mitochondria (6) or through production of free ADPR during extreme DNA damage through the poly(ADP-ribose) polymerase (PARP)–poly(ADP-ribose) glycohydrolase (PARG) pathway (11). Moreover, apoptotic stimuli that mediate oxidative stress, such as hydrogen peroxide (H2O2) or tumor necrosis factor–α (TNF-α), have emerged as (patho)physiological stimuli for TRPM2-mediated signaling, mediating Ca2+ entry in Jurkat T cells, neutrophils, microglia, and pancreatic β cells (12). When applied extracellularly, the primary cytosolic agonist ADPR can also elicit Ca2+ release in cells that express TRPM2. This mechanism is not well understood, but has been suggested to result from inositol 1,4,5-trisphosphate (IP3) production (13). Here, we examine the signaling mechanisms of ADPR in heterologous expression systems and in natively TRPM2-expressing pancreatic β cells, where we find that TRPM2 serves a dual role as plasma membrane Ca2+-influx channel and as a previously unidentified intracellular Ca2+-release channel, with both functions playing a critical role in H2O2-induced β cell death.
Results
Extracellular ADPR activates P2Y receptors in HEK293 cells
Extracellular nucleosides and different ribosylated nucleotide derivatives have been shown to activate Ca2+ signaling pathways through IP3-producing receptors (13). Thus, before we investigated the possible effects of intracellular ADPR on Ca2+ release from internal stores, we first explored the signaling function of extracellular ADPR. Wild-type human embryonic kidney (HEK) 293 cells do not express native TRPM2 (1). Nevertheless, addition of ADPR to intact HEK293 cells consistently produced a transient Ca2+ signal in both wild-type cells (Fig. 1B) and cells heterologously expressing TRPM2 (TRPM2 HEK293; Fig. 1A). The Ca2+ signal was elicited at a threshold concentration of ∼100 μM ADPR and required neither extracellular Ca2+ (Fig. 1A) nor TRPM2 expression (Fig. 1B), indicating that it arose from release of Ca2+ from intracellular stores through a preexisting signaling pathway that is independent of TRPM2. Consistent with this notion, emptying intracellular stores with thapsigargin before the ADPR challenge resulted in complete loss of ADPR-mediated Ca2+ release (fig. S1).
Fig. 1.

ADPR acts as a purinergic receptor agonist and TRPM2 acts as a Ca2+-release channel when heterologously expressed in HEK293 cells. (A) Average Ca2+ signals measured in intact HEK293 cells heterologously expressingTRPM2 channels (TRPM2 HEK293) in response to application of extracellular ADPR in the presence (1 mM, black trace, n = 8) or absence (blue trace, n = 7) of extracellular Ca2+ in the external solution. The concentration of ADPR was 1 mM in the presence of Ca2+ and 100 μM in the absence of Ca2+. The red trace represents the average Ca2+ signal measured in response to application of 100 μM ADPR in the absence of extracellular Ca2+ with 100 μM suramin (n = 6). Application started as indicated by the arrow and was maintained throughout the experiment. Cells were loaded with 5 μM fura-2-AM at 37°C for 30 min. (B) Average Ca2+ signals in intact wild-type HEK293 cells in response to application of 1 mM ADPR (black trace, n = 7) in the absence of extracellular Ca2+. Application and fura-2-AM loading as described in (A). (C) Average Ca2+ signal measured in intact fura-2-AM–loaded TRPM2 HEK293 cells in response to application of 100 μM ATP (black bar) followed by application of 100 μM ADPR (red bar) in the absence of extracellular Ca2+ (n = 6). (D) The traces depict balanced fura-2 experiments, in which TRPM2 HEK293 cells were preloaded with fura-2-AM and the patch pipette contained 200 μM fura-2 to enable continuous measurements of [Ca2+]i. Whole-cell break-in was just before application of 100 μM ADPR in the absence of extracellular Ca2+ as indicated by the arrow (black trace, n = 4). The internal solution was supplemented with either heparin (100 μg/ml; blue trace, n = 6) or 500 μM GDP-b-S (green trace, n = 5). The red trace represents Ca2+ measurements in intact cells exposed to 10 μM U73122 in the bath (n = 5). (E) Balanced fura-2 experiments with internal perfusion of ADPR. Average Ca2+ signal in whole-cell patch-clamped TRPM2 HEK293 cells preloaded with fura-2-AM. Whole-cell break-in was at the time indicated by the red arrow. Cells were kept in 0 Ca2+ external solution and perfused with internal solution containing 200 μM fura-2 and supplemented with either 1 mM ADPR (black trace, n = 6), 100 μM ADPR (blue trace, n = 7), or 100 μM ADPR and 1 mM AMP (red trace, n = 8). (F) Balanced fura-2 experiments in wild-type HEK293 cells preloaded with fura-2-AM. Whole-cell break-in was achieved at the time indicated by the red arrow. Internal solution contained 200 μM fura-2 supplemented with 1 mM ADPR (n = 6).
In HEK293 cells, adenosine 5′-triphosphate (ATP) causes IP3-dependent Ca2+ release through a phospholipase C (PLC) signaling pathway mediated by P2Y-type purinergic receptors (14–16). When TRPM2-expressing cells were preincubated for 15 min with 100 μM suramin, a nonselective P2Y receptor antagonist, ADPR-induced Ca2+ signals were completely suppressed (Fig. 1A). When HEK293 cells were sequentially exposed to ATP (100 μM) and then to ADPR, the ATP-induced Ca2+ release abrogated a subsequent Ca2+ signal by ADPR (Fig. 1C). Thus, ADPR appears to engage the PLC signaling pathway. We tested this possibility by inhibiting the heterotrimeric guanine nucleotide–binding protein (G protein)–PLC-IP3 signal transduction pathway in patch-clamp experiments (Fig. 1D). Interfering with G-protein coupling by internally perfusing HEK293 cells with 500 μM guanosine 5′-_O_-(2′-thiodiphosphate) (GDP-β-S) blocked ADPR-induced Ca2+ release. Likewise, ADPR-mediated Ca2+ release was eliminated by preincubation of cells with the PLC inhibitor U73122 (10 μM). Lastly, direct block of IP3 receptors with intracellular heparin (100 μg/ml) also prevented ADPR-induced Ca2+ release. These data indicate that ADPR can act as a first messenger through G protein–coupled P2Y receptors that activate the PLC signaling pathway.
Intracellular ADPR elicits Ca2+ release in TRPM2-expressing HEK293 cells
We next investigated whether intracellular ADPR can act as second messenger to mediate Ca2+ release from intracellular stores. We eliminated ADPR-mediated Ca2+ release through the P2Y pathway and Ca2+ influx through TRPM2 channels, respectively, by adding suramin to and removing Ca2+ from the extracellular medium. Cells were loaded with fura-2 acetoxymethyl ester (fura-2-AM) and then patch-clamped in the whole-cell configuration to introduce 0.1 to 1 mM ADPR intracellularly. Patch pipettes also contained 200 μM fura-2 to maintain the ability to measure [Ca2+]i during whole-cell recording. TRPM2 HEK293 internally perfused with 100 μM or 1 mM ADPR responded with Ca2+ release signals that were inhibited by intracellular adenosine monophosphate (AMP) (Fig. 1E), an established inhibitor of ADPR-gated TRPM2 channels (5). However, ADPR failed to elicit Ca2+ signals in wild-type cells (Fig. 1F), suggesting that ADPR induced Ca2+ release through TRPM2 channels located in intracellular stores.
TRPM2 is natively expressed in INS-1 β cells
Because TRPM2-like currents have been reported in rat RINm5F and CRI-G1 β cell lines (12), we extended our investigation to the rat β pancreatic cell line INS-1. Internal perfusion of INS-1 cells with ADPR caused rapid activation (Fig. 2A, open circles) of a linear current with biophysical and pharmacological characteristics typical of TRPM2 (Fig. 2B). These ADPR-mediated currents were activated in a concentration-dependent manner with a half-maximal effective concentration (EC50) of ∼100 μM ADPR (Fig. 2C) and were completely suppressed by 1 mM AMP (Figs. 2A, closed circles, and 2B, red trace). These data confirm that TRPM2 is functionally expressed in INS-1 cells and acts as a plasma membrane ion channel.
Fig. 2.

ADPR activates TRPM2, purinergic receptors, and adenosine receptors in INS-1 β cells. (A) Average development of TRPM2 currents assessed by whole-cell patch-clamp measurements in INS-1 cells. Cells were internally perfused with either 100 μM ADPR (black symbols, n = 11) or 100 μM ADPR + 1 mM AMP (red symbols, n = 9). Current amplitudes were assessed at −80 mV, normalized for cell size, averaged and plotted versus time of the experiment. The standard voltage protocol was ramping from −100 mV to +100 mV over 50 ms and at 0.5 Hz. Holding potential was 0 mV. Error bars indicate SEM. (B) Typical current-voltage (I-V) relationship of currents evoked by 1 mM ADPR (black trace), or 100 μM ADPR + 1 mM AMP (red trace) taken from example cells and recorded 100 s into the experiment. (C) Dose-response behavior of TRPM2 currents in INS-1 cells at various internal ADPR concentrations. Current amplitudes were measured at −80 mV, averaged, normalized to cell size, and plotted against the respective ADPR concentration (n = 5 to 11). A dose-response fit to the data yielded an EC50 value of 110 μM with a Hill coefficient of 1. (D) Average Ca2+ signals measured in intact fura-2-AM–loaded INS-1 cells in response to increasing concentrations of extracellular ADPR applied in the absence of extracellular Ca2+ [100 nM (black trace, n = 6), 1 μM (red trace, n = 6), 10 μM (blue trace, n = 6), 30 μM (green trace, n = 6)]. (E) Average Ca2+ signals measured in intact fura-2-AM–loaded INS-1 cells in the absence of extracellular Ca2+ and stimulated by 30 μM extracellular ADPR (black trace, control, n = 11) or 100 μM ADPR plus either 100 μM suramin (green trace, n = 8) or 1 μM CGS-15943 (blue trace, n = 11) or both 100 μM suramin and 1 μM CGS-15943 (red trace, n = 6). (F) Average Ca2+ signals measured in intact fura-2-AM–loaded INS-1 cells in the absence of extracellular Ca2+ and stimulated by 10 μM ADPR plus either 100 μM suramin (black trace, n = 6) or 1 μM CGS-15943 (red trace, n = 6).
Extracellular ADPR activates P2Y and adenosine receptors in INS-1 β cells
Before addressing the possibility that intracellular ADPR induced Ca2+ release in INS-1 cells, we first assessed the Ca2+ signaling mechanisms of extracellular ADPR in these cells. As in HEK293 cells, application of extracellular ADPR in Ca2+-free solution elicited Ca2+ release in INS-1 cells, but compared to HEK293 cells, at a much lower threshold concentration of 1 μM (Fig. 2D). Although the P2Y antagonist suramin reduced the 100 μM ADPR–induced Ca2+ signal, it did not abolish it (Fig. 2E). This indicated that there was another receptor type responsive to ADPR. β cells also express A-1 adenosine receptors (17–19), which might account for the suramin-resistant Ca2+ release with ADPR. We confirmed this possibility by using the broadly acting adenosine receptor antagonist CGS-15943. Like suramin, CGS-15943 reduced the ADPR-mediated Ca2+ signal without abolishing it (Fig. 2E). However, the combination of CGS-15943 and suramin completely suppressed the ADPR-mediated Ca2+ release signal (Fig. 2E). Because both P2Yand adenosine receptors can stimulate the classical G protein–coupled receptor–G protein–PLC-IP3 pathway (20, 21), it is likely that the responses to extracellular ADPR in INS-1 cells are mediated by IP3-induced Ca2+ release.
We next examined whether the enhanced ADPR sensitivity of INS-1 cells compared to HEK293 cells was mediated through adenosine or P2Y receptors by stimulating cells with 10 μM ADPR in the presence of CGS-15943 or suramin. Suramin was considerably more effective than CGS-15943 in suppressing the response to the low concentration of ADPR (Fig. 2F), suggesting that P2Y receptors are primarily responsible for the higher sensitivity of INS-1 cells.
Intracellular ADPR elicits Ca2+ release in INS-1 β cells
We next investigated the possibility that native TRPM2 channels in INS-1 cells mediate intracellular Ca2+ release. Intracellular perfusion of cells with ADPR in the absence of extracellular Ca2+ and with both suramin and CGS-15943 in the bath produced a concentration-dependent increase in [Ca2+]i (Fig. 3A). Because TRPM2 is a downstream target of reactive oxygen species (ROS) (1), we tested whether H2O2 can also mediate Ca2+ release in these cells. Perfusing cells with 100 μM H2O2 (plus heparin to inhibit IP3 receptors) indeed evoked Ca2+ release (Fig. 3B). Furthermore, Ca2+ release induced by direct ADPR perfusion was not prevented by inhibiting IP3 receptors with heparin (100 μg/ml), or ryanodine receptors with 25 μM ryanodine (Fig. 3B). We confirmed that the ADPR-mediated responses involved TRPM2 by molecular knockdown of TRPM2 with short interfering RNA (siRNA). TRPM2-specific siRNA, but not a scrambled control siRNA, caused a significant suppression of ADPR-induced Ca2+ release with P < 0.03, as assessed by peak amplitude changes in Ca2+ release (Fig. 3C and table S1). We confirmed the efficacy of specific siRNA knockdown of TRPM2 by monitoring TRPM2 channel activity in the plasma membrane and observed nearly complete suppression of functional channel activity (Fig. 3D). Together, these data show that TRPM2 proteins in INS-1 β cells function as both Ca2+-permeable cation channels in the plasma membrane and as Ca2+-release channels in intracellular stores.
Fig. 3.

TRPM2 functions as Ca2+-release channel in INS-1 cells. (A) Balanced fura-2 experiments showing average Ca2+ signals in whole-cell patch-clamped INS-1 cells preloaded with fura-2-AM. Whole-cell break-in was at the time indicated by the red arrow. Cells were kept in 0 Ca2+ external solution supplemented with 1 μM CGS-15943 and 100 μM suramin and perfused with internal solution containing 200 μM fura-2 and supplemented with either 100 μM ADPR (blue trace, n = 9), 30 μM ADPR (green trace, n = 8), or 10 μM ADPR (black trace, n = 6). (B) Balanced fura-2 experiments, showing average Ca2+ signals in whole-cell patch-clamped INS-1 cells preloaded with fura-2-AM. Whole-cell break-in was at the time indicated by the red arrow. Cells were kept in 0 Ca2+ external solution containing 1 μM CGS-15943 and 100 μM suramin. Cells were perfused with internal solution containing 200 μM fura-2 and supplemented with either 100 μM H2O2 plus heparin (100 μg/ml; red trace, n = 10), 100 μM ADPR plus heparin (100 μg/ml; black trace, n = 7), or 100 μM ADPR with 25 μM external ryanodine (blue trace, _n_= 6). (C) Balanced fura-2 experiments showing average Ca2+ signals in response to internal ADPR in whole-cell patch-clamped INS-1 cells preloaded with fura-2-AM. Whole-cell break-in was at the time indicated by the red arrow. Cells were kept in 0 Ca2+ external solution supplemented with 100 μM suramin and 1 μM CGS-15943 and perfused with internal solution containing 200 μM fura-2 and supplemented with 100 μM ADPR. Traces represent Ca2+ signals from cells treated with scrambled control siRNA (black trace, n = 10) or TRPM2-specific siRNA (red trace, n = 10). (D) Average TRPM2 currents assessed by whole-cell patch-clamp measurements in INS-1 cells treated with scrambled control siRNA (black symbols, n = 8) or TRPM2-specific siRNA (red symbols, n = 14). Currents were analyzed as described in Fig. 2A.
TRPM2 is a lysosomal Ca2+-release channel in INS-1 β cells
We next assessed the subcellular localization of TRPM2 in INS-1 cells by immunofluorescence and observed both peripheral and intracellular localization of TRPM2 (Fig. 4A). These data revealed that TRPM2 rarely, if ever, colocalized with the endoplasmic reticulum (ER). Instead, TRPM2 showed a punctate distribution throughout the cytoplasm, indicating localization in a vesicular compartment. Because lysosomal organelles contain Ca2+ and have been implicated in Ca2+ release (22, 23), we investigated the distribution of both TRPM2 and lysosomes in INS-1 cells with specific antibodies against TRPM2 and lysosome-associated membrane protein–1 (LAMP-1), a specific marker for lysosomes (24). Confocal images of INS-1 cells revealed dense regions of punctate labeling for both proteins that exhibited a high degree of overlap (Fig. 4B), although a few vesicular structures were labeled by only TRPM2. We assessed this store functionally by using bafilomycin A, a macrolide antibiotic that selectively inhibits the vacuolar H+-dependent adenosine triphosphatase at nanomolar concentrations and empties lysosomal Ca2+ stores without affecting ER Ca2+ concentrations (22, 25). Indeed, preincubation of cells with 100 nM bafilomycin A for 30 min. completely suppressed Ca2+ release by 300 μM ADPR subsequently introduced through the patch pipette (Fig. 4C). Control experiments showed that IP3-mediated Ca2+ release from the ER of these cells after stimulation of muscarinic receptors with 300 μM carbamylcholine was only slightly reduced by bafilomycin A pretreatment (Fig. 4D). Together, these data indicate that ADPR-dependent TRPM2-mediated Ca2+ release occurs predominantly from a lysosomal store.
Fig. 4.
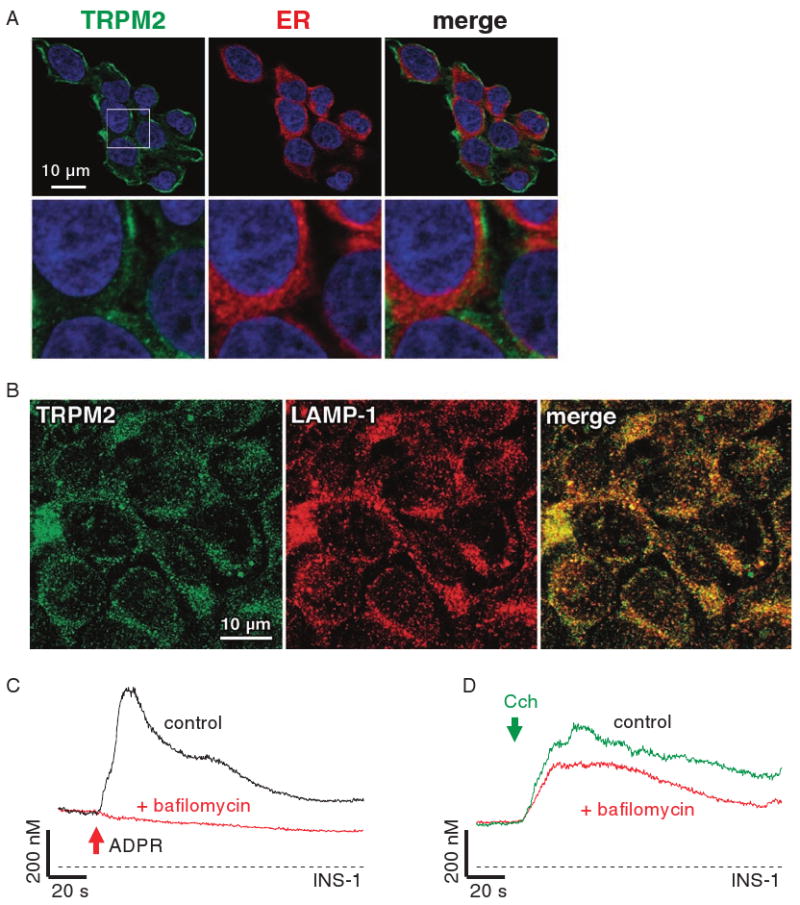
TRPM2 is a lysosomal Ca2+-release channel in INS-1 cells. (A) Detection and cellular localization of TRPM2 by immunofluorescence. Polyclonal antibodies directed against mouse TRPM2 specifically recognizes a protein in INS-1 cells with cytosolic, as well as plasma membrane distribution (left panels, green). Intracellular TRPM2 label is largely excluded from the ER (middle panels, red) network, as evidenced by the merged image (right panels, note absence of yellow spots). DAPI (4′,6-diamidino-2-phenylindole) was used as a nuclear counterstain (blue). Images of cells that are representative of the entire population are shown (63× magnification). The white rectangle indicates the area of expanded view depicted in the respective lower panels. Note the punctuated appearance of intracellularly located TRPM2, indicating vesicular localization. (B) Immunofluorescence of TRPM2 (left panel, green) and LAMP-1 (middle panel, red) with antibodies directed against TRPM2 (3) and LAMP-1. The right panel represents the merged image, suggesting that both proteins have largely overlapping localizations (yellow), with just a few vesicles showing only TRPM2 fluorescence. Cells were visualized with a confocal laser scanning microscope with 63× objective and images are representative of the entire population. (C) Bafilomycin A inhibits intracellular ADPR-mediated Ca2+ release. Balanced fura-2 experiments showing average Ca2+ signals in whole-cell patch-clamped INS-1 cells preloaded with fura-2-AM. Whole-cell break-in was at the time indicated by the red arrow. Cells were kept in 0 Ca2+ external solution containing 100 μM suramin and 1 μM CGS-15943 in the absence (control, black trace, n = 9) or presence of 100 nM bafilomycin A (red trace, n = 16) and perfused with internal solution containing 100 μM ADPR or 300 μM ADPR, respectively. (D) Average Ca2+ signals measured in intact fura-2-AM-loaded INS-1 cells in response to 300 μM carbamylcholine (CCh) in the absence of extracellular Ca2+ and in the presence of 100 μM suramin and 1 μM CGS-15943 (control, black trace, n = 20) in the external solution. The red trace (n = 17) represents cells treated identically, but preincubated with 100 nM bafilomycin A for 30 min.
TRPM2 is natively expressed in primary mouse β cells
Although the INS-1 cell line represents a widely used model for pancreatic β cells, cell lines do not fully reflect the properties of primary cells. We therefore extended our analysis to primary pancreatic β cells isolated from C57BL/6 mice. First, we evaluated the potency of ADPR at activating TRPM2-like currents in the plasma membrane. Experiments were performed 24 to 72 hours after isolation of pancreatic β cells. Cells were maintained under the same conditions as the INS-1 cell line and subjected to the same experimental protocols with identical ionic composition of internal and external solutions. We internally perfused cells with various concentrations of ADPR and observed rapid activation of linear currents with biophysical and pharmacological characteristics typical of TRPM2 (Fig. 5B). These currents reached peak amplitudes of about −80 pA/pF at −80 mV (Fig. 5C) within 30 to 50 s and could be suppressed by AMP (Fig. 5A). The ADPR-induced currents were concentration dependent, with an EC50 of ∼360 μM ADPR (Fig. 5C). Thus, TRPM2 is expressed as a functional ion channel in primary mouse β cells.
Fig. 5.

ADPR activates purinergic receptors and elicits Ca2+ influx, as well as Ca2+ release through TRPM2 in mouse pancreatic β cells. (A) Average TRPM2 currents in mouse pancreatic β cells isolated from C57BL/6 or TRPM2 KO mice. Cells were perfused with either 300 μM ADPR (green symbols, _n_= 7), 300 μM ADPR plus 1 mM AMP (blue symbols, n = 9) or 3 mM ADPR (black symbols, n = 6). TRPM2 KO cells were perfused with 1 mM ADPR (red symbols, n = 6). Current amplitudes were assessed as described in Fig. 2A. Error bars indicate SEM. (B) Typical current-voltage (I-V) relationship of currents evoked by 1 mM ADPR (black trace), 300 μM ADPR (green trace), or 300 μM ADPR + 1mM AMP (blue trace) taken from representative cells and recorded 100 s into the experiment. (C) Dose-response behavior of TRPM2 currents in mouse β cells as a function of internal ADPR concentration. Current amplitudes were measured at −80 mV, averaged, normalized to cell size, and plotted versus the respective ADPR concentration (n = 5 to 7). A dose-response fit to the data resulted in an EC50 value of 360 μM with a Hill coefficient of 1. (D) Average Ca2+ signals measured in intact fura-2-AM–loaded mouse β cells in response to increasing concentrations of extracellular ADPR and in the absence of extracellular Ca2+ [1 μM (red trace, n = 4), 10 μM (blue trace, n = 5), 30 μM (green trace, n = 6), 100 μM (black trace, _n_= 6)]. Start of application indicated by black arrow. (E) Average Ca2+ signals measured in intact fura-2-AM–loaded mouse β cells in response to application of 200 μM ADPR in the absence of extracellular Ca2+ and in the presence of either 100 μM suramin (red trace, n = 6) or 1 μM CGS-15943 (black trace, n = 8) in the external solution. (F) Balanced fura-2 experiments showing average Ca2+ signals in whole-cell patch-clamped C57BL/6 mouse pancreatic β cells (black trace, n = 7) or β cells isolated from TRPM2 KO mice (red trace, n = 10) preloaded with fura-2-AM. Whole-cell break-in indicated by red arrow. Cells were kept in 0 Ca2+ external solution and perfused with internal solution containing 300 μM ADPR and 200 μM fura-2. The gray traces show two representative responses measured in individual wild-type (WT) cells.
Extracellular ADPR activates P2Y receptors in primary mouse β cells
Before investigating TRPM2-dependent Ca2+ release, we assessed the extracellular effects of ADPR in primary β cells. With a threshold concentration of ∼10 μM, ADPR produced multiple Ca2+-release transients when applied to intact cells (Fig. 5D). Although the averaged Ca2+ signal obscures the oscillatory pattern of Ca2+ release in individual cells, a more quantitative analysis of Ca2+-release signals at the single-cell level over 100 s, with a fitting routine based on a convolution of a Gaussian peak and exponential decay (Supplementary Materials and Methods and table S1), revealed that application of extracellular ADPR induced two to three Ca2+ oscillations in both INS-1 and primary β cells (fig. S4A). Increasing ADPR concentrations both shortened the delay (fig. S4B) and increased the peak of the first Ca2+ transient (fig. S4C). CGS-15943 had no effect on the response, and suramin alone at 100 μM completely abolished extracellularly mediated ADPR effects (Fig. 5E), suggesting that primary mouse β cells lack adenosine receptors and that Ca2+ signals in these cells are mediated by P2Y receptors.
Ca2+ release by extracellular cADPR and NAD+ requires CD38 in primary mouse β cells
Extracellular production of ADPR is mediated by the ectoenzyme CD38 with NAD+ (the oxidized form of nicotinamide adenine dinucleotide) or cADPR as substrate (10). Nevertheless, in contrast to ADPR, even high millimolar concentrations of these compounds were ineffective at producing Ca2+ signals in HEK293 (Fig. 6A), indicating that neither NAD+ nor cADPR act as P2Y receptor agonists in these cells. In contrast, both NAD+ and cADPR triggered Ca2+ release transients in INS-1 cells, although cADPR did so more potently and effectively than NAD+ (Fig. 6B). The threshold concentration for cADPR was ∼10 μM (fig. S2A) and for NAD+ ∼30 μM (fig. S2C), just 10 to 30 times higher than that of ADPR. Like ADPR, both cADPR and NAD+ effects were mediated through P2Y and adenosine receptors, because the combined suppression of these receptors by suramin and CGS-15943 completely blocked the response (figs. S2B and S2C, respectively). cADPR showed a pharmacological profile similar to that of ADPR, because suramin was more effective than CGS-15943 at suppressing the response to cADPR (fig. S2B). However, cADPR, even at 100 μM, failed to release Ca2+ in the presence of the ADPR antagonist 8-bromo-ADPR [8-Br-ADPR; 100 μM, fig. S3; (26)].
Fig. 6.

The ectoenzyme CD38 is key in cADPR-induced Ca2+ signals. (A) Average Ca2+ signals in intact wild-type HEK293 cells in response to extracellular application of 10 mM cADPR (red trace, n = 8) or 10 mM NAD+ (black trace, n = 6) in the absence of extracellular Ca2+. Application and fura-2-AM loading as described in Fig. 1A. (B) Average Ca2+ signals in intact fura-2-AM–loaded INS-1 cells in response to 30 μM external cADPR (red trace, n = 5) or 100 μM NAD+ (black trace, n = 10) in the absence of extracellular Ca2+. Application start is indicated by the arrow. (C) Average Ca2+ signals measured in intact fura-2-AM–loaded primary mouse β cells in response to external application of either 300 μM cADPR (red trace, n = 6) or 1 mM NAD+ (black trace, n = 8). (D) Average Ca2+ signals measured in intact fura-2-AM–loaded pancreatic β cells isolated from CD38 knockout mice (28) in response to external application of either 100 μM ADPR (black trace, n = 4), 100 μM ATP (blue trace, n = 8), or 300 μM cADPR (red trace, n = 20). (E) Average Ca2+ signals measured in intact fura-2-AM–loaded mouse pancreatic β cells. As indicated by the arrow, 100 μM ADPR was co-applied with either 30 μM NAD+ (black trace, n = 6) or 100 μM NAD+ (red trace, n = 6) in a 0 Ca2+ solution. (F) Average Ca2+ signals measured in wild-type HEK293 cells in response to application of extracellular ADPR (100 μM) in the presence of either 1 mM NAD+ (red trace, n = 9) or 300 μM NAD+ (black trace, n = 7) and in the absence of extracellular Ca2+. Application started as indicated by the arrow and was maintained throughout the experiment.
In primary β cells, extracellular NAD+ did not elicit any response even at 1 mM (Fig. 6C). However, cADPR at 300 μM produced clear Ca2+-release transients when applied to intact cells (Fig. 6C). Because mouse β cells express CD38 (27) and cADPR acts as a substrate of the ectoenzyme CD38 to produce ADPR (10), we used a CD38 knockout mouse (28) to test whether the efficacy of cADPR relied on the presence of this enzyme. This was indeed the case; CD38-deficient β cells did not respond to cADPR, but retained responsiveness to ADPR (Fig. 6D). CD38-deficient β cells also retained functional P2Y receptors as evidenced by the Ca2+-release transient induced by 100 μM ATP (Fig. 6D), suggesting that responsiveness to extracellular cADPR indeed requires CD38. Together, these data indicate that ADPR, and not cADPR, represents the primary P2Y receptor agonist.
We were puzzled by the lack of effect of NAD+, which is the major substrate for ADPR production by CD38 (10). This lack of effect could be explained if NAD+ acts as a competitive inhibitor to ADPR-induced Ca2+ release in mouse β cells. Indeed, addition of 100 μM NAD+ in combination with 100 μM ADPR (1:1 ratio) to intact mouse β cells (Fig. 6E) completely suppressed the Ca2+ release normally induced by 100 μM ADPR (Figs. 5, D and E, and 6D). However, a robust Ca2+ signal was apparent with a NAD+ concentration of 30 μM (Fig. 6E). Thus, although neither NAD+ nor cADPR act as P2Y agonists, NAD+ seems to act as a competitive inhibitor of ADPR at P2Y receptors in mouse β cells. NAD+ also inhibited ADPR-induced Ca2+ release in HEK293 cells, albeit with lower potency than in primary β cells (Fig. 6F). Although 100 μM ADPR combined with 300 μM NAD+ still elicited Ca2+ release, release was abolished by 1 mM NAD+, a 1:10 ratio of the two compounds.
Intracellular ADPR elicits Ca2+ release in primary mouse β cells
Next, we investigated whether TRPM2 could mediate Ca2+ release from intracellular stores of primary β cells. Figure 5F shows that intracellular perfusion of mouse β cells with 300 μM ADPR evoked Ca2+ release transients. Unlike the IP3-mediated Ca2+ oscillations elicited by extracellular ADPR (Fig. 5D), internally applied ADPR typically gave rise to a single transient both in INS-1 cells and primary β cells (fig. S4D). However, increasing internal ADPR concentrations shortened the delay of this Ca2+ transient and enhanced the overall change in peak amplitude of Ca2+ release in INS-1 cells (fig. S4F). We further confirmed that the responses mediated by intracellular ADPR involved TRPM2 by investigating mouse primary β cells isolated from TRPM2 knockout mice (TRPM2 KO) (29). As illustrated in Fig. 5A, even 1 mM intracellular ADPR failed to evoke currents in these cells, confirming the absence of functional TRPM2 channels in the plasma membrane. Furthermore, Ca2+-release signals were completely absent in TRPM2 KO β cells perfused with 1 mM ADPR in the absence of extracellular Ca2+ (Fig. 5F; P < 0.026 as assessed by change in peak Ca2+ release, table S1).
Lysosomal Ca2+ release through TRPM2 contributes to apoptosis in INS-1 β cells
To examine the possible physiological function of TRPM2-mediated Ca2+ release in pancreatic β cells, we tested INS-1 cells for H2O2-induced susceptibility to cell death assessed by analysis of propidium iodide (PI) staining in flow cytometry. Unfortunately, any molecular knockdown of TRPM2 would affect the expression of this channel in both plasma membrane and lysosomal compartments. We therefore compared the effect of TRPM2 on H2O2-induced cell death in the presence and absence of extracellular Ca2+. The latter abolishes Ca2+ influx through TRPM2 but leaves internal Ca2+ release through this channel intact, representing a functional knockout of plasma membrane TRPM2. These data were compared to results obtained from cells treated with TRPM2-specific siRNA, thereby isolating the severity of cell death linked to TRPM2-mediated Ca2+ release. Exposure to 100 μM H2O2 significantly enhanced cell death in INS-1 cells treated with control siRNA [directed against glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] in the presence of extracellular Ca2+ (P < 0.001). However, cells with suppressed TRPM2 expression were 72% less affected by H2O2-induced cell death (Fig. 7, A and B; P < 0.001). As Fig. 7A further illustrates, H2O2 was able to induce significant cell death in control siRNA (GAPDH)-treated INS-1 cells even in the absence of extracellular Ca2+ (P < 0.001), albeit with reduced severity. However, this was still linked to TRPM2 expression, because H2O2-induced cell death in the absence of extracellular Ca2+ was 68% further reduced (Fig. 7, A and C; P < 0.001) in cells transfected with TRPM2 siRNA. This indicates that not only Ca2+ influx through plasma membrane TRPM2 but also TRPM2-dependent lysosomal Ca2+ release plays a critical role in H2O2-mediated β cell death.
Fig. 7.
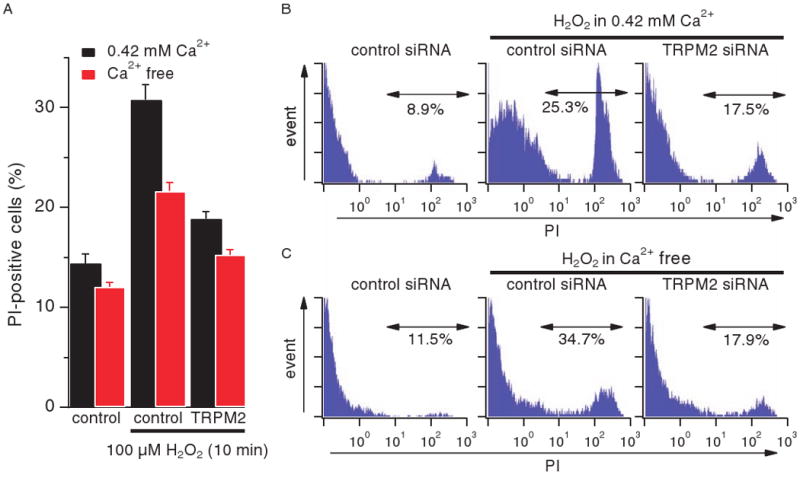
TRPM2-mediated Ca2+ release induces cell death under oxidative stress. (A) Average values for percent of PI-positive cells. INS-1 cells transfected with control (GAPDH) or TRPM2 siRNA were treated with 100 μM H2O2 for 10 min either in RPMI 1640 medium that contained 0.42 mM Ca2+ (black bars, n = 5) or was Ca2+-free (red bars, n = 11). PI-positive cells were analyzed by flow cytometry. Data points are mean ± SEM. Comparing H2O2-treated and -untreated control (GAPDH) cells, H2O2-treated control (GPDH) and TRPM2 siRNA cells, or untreated control (GAPDH) cells with H2O2-treated TRPM2 siRNA cells showed a statistical significance of P < 0.001 in each case, both in the presence and absence of extracellular Ca2+. (B) Representative PI profile of cells tested in the presence of extracellular Ca2+. (C) Representative PI profile of cells tested in the absence of extracellular Ca2+.
Discussion
We here establish that TRPM2, in addition to its known role as a plasma membrane–resident Ca2+-influx channel, can also function as an intracellular Ca2+-release channel in lysosomes of pancreatic β cells. Intracellular ADPR is capable of activating these TRPM2 functions, and both pathways contribute to H2O2-induced β cell death. Independent of its intracellular agonistic action on TRPM2, extracellular ADPR acts as a primary P2Y and adenosine receptor agonist, resulting in IP3 formation and Ca2+ release from the ER. NAD+ and cADPR are ineffective as direct agonists of either P2Yor adenosine receptors, but can indirectly cause IP3-dependent Ca2+ release through metabolic conversion to ADPR by the ectoenzyme CD38. Although NAD+ does not act as a P2Y receptor agonist in the cells investigated here, it functions as a competitive antagonist of ADPR at the P2Y subtypes expressed in HEK293 and primary mouse β cells.
TRPM2 has been extensively characterized as a plasma membrane ion channel that is specifically activated by ADPR (1–3). TRPM2 sensitivity to ADPR is regulated by facilitatory cofactors such as cytosolic Ca2+ or production of cADPR and NAADP (5, 7). These molecules act synergistically and sensitize TRPM2 to ADPR so that, for example, the apparent EC50 of ADPR of ∼100 μM in HEK293 cells is reduced to EC50 concentrations as low as 90 nM in the presence of cADPR (5). In addition, different cellular systems exhibit different ADPR sensitivity of TRPM2 for unknown reasons (1, 5, 7). Although high micromolar concentrations of agonist may occur during pathophysiological events, it is possible or even likely that under physiological conditions, synergistic events may facilitate TRPM2 activation.
The present study now adds previously unrecognized facets to the function of TRPM2 and its natural ligands by establishing TRPM2 as an intracellular Ca2+-release channel and showing that ADPR and its precursors NAD+ and cADPR also exhibit extracellular activity as receptor agonists and antagonists. Although ADPR has been shown to activate Ca2+ signaling through IP3-producing receptors, the specific receptor species involved has not yet been identified (13). Our results show that ADPR can activate P2Y receptors in the three cell types investigated in this study (HEK293, INS-1, and primary mouse β cells), as well as adenosine receptors in INS-1 cells. However, ADPR was about two orders of magnitude more potent in β cells than in HEK293 cells. Possible reasons for this greater sensitivity include species differences in P2Y sensitivity, different complements of P2Y receptor subtypes, or both. HEK293 cells mainly express P2Y subtypes 1, 2, and 4 (16), although a slightly differing P2Y receptor complement has also been reported for these cells (30). INS-1 cells express subtypes 1, 2, 4, 6, and 12 in similar amounts (16, 31). Thus, a specific P2Y receptor subtype complement, possibly involving subtypes 6 or 12 or both, might be responsible the high-affinity response to ADPR in INS-1 cells.
ADPR can be produced extracellularly from its precursors NAD+ or cADPR through the action of the ectoenzyme CD38 (10). Two recent studies report that NAD+ itself may be an agonist for P2Y11 receptors in granulocytes (32) and P2Y1 receptors in visceral smooth muscle (33). However, even at high millimolar concentrations, neither NAD+ nor cADPR produced Ca2+ signals in HEK293 (Fig. 6A). This suggests that these molecules are not effective agonists for the P2Y receptor subtypes endogenously expressed in HEK293 cells or that these cells may not express enough CD38 to produce substantial amounts of ADPR from these precursors.
In INS-1 cells, however, NAD+ and cADPR caused Ca2+ release with an ∼10 times increased efficiency compared to that of HEK293 cells. This is either due to a genuine agonistic action of these compounds on cell surface receptors or caused by exogenous metabolic conversion to ADPR through CD38, which is abundant in β cells (34). Although CD38 accepts both NAD+ and cADPR as substrates and converts them to the common product ADPR, it appears to process NAD+ more efficiently than cADPR (35–37). cADPR induced Ca2+ release in both INS-1 and primary β cells, albeit 10 times less potently than ADPR. Its ability to do so can be linked to metabolic conversion of cADPR to ADPR through CD38; cADPR was ineffective in eliciting Ca2+ release in primary β cells of transgenic mice deficient in CD38, despite an intact P2Y pathway (Fig. 6D). NAD+ was ineffective in triggering Ca2+ release in primary β cells, likely because it acted as a competitive antagonist to ADPR, suppressing specific P2Y receptor–mediated Ca2+ signals in primary β cells (Fig. 6E) and HEK293 cells (Fig. 6F). Although NAD+ may well be converted to ADPR by CD38, the action of ADPR is prevented by the concomitant inhibition of P2Y receptors by NAD+ itself. Nevertheless, extracellular NAD+ does cause Ca2+ release in the INS-1 cell line. Although this effect could be due to the specific P2Y subtype expression pattern in these cells or, alternatively, caused by direct adenosine receptor activation, our data are most consistent with a metabolic conversion of NAD+ to ADPR, which activates adenosine receptors present in INS-1 cells; not only is there a substantial delay in the Ca2+ release response to NAD+ application (Fig. 6B), the signal itself is smaller than that elicited by ADPR (Fig. 2D). Thus, the precise P2Y receptor subtype composition of a cell, as well as the expression of adenosine receptors and CD38, would determine the strength of the resulting Ca2+ signal elicited in the presence of ADPR and NAD+. Given that the extracellular effects of ADPR are entirely mediated through membrane receptors, it appears that ADPR is not transported across the plasma membrane to an extent necessary to activate TRPM2.
The present study identifies TRPM2 as an intracellular Ca2+-release channel localized in lysosomal compartments. Our data reveal that intracellular ADPR causes Ca2+ release only in TRPM2-expressing cells, but not in wild-type HEK293 cells, and that this Ca2+ release is sensitive to the TRPM2-specific antagonist AMP. Furthermore, endogenously expressed TRPM2 also functions as a Ca2+-release channel in rat INS-1 and primary mouse β cells, because intracellular ADPR-induced Ca2+ release is reduced in siRNA experiments suppressing TRPM2 in INS-1 cells and absent in β cells isolated from TRPM2 knockout mice. Both immunofluorescence and functional data confirm the presence of TRPM2 predominantly in a lysosomal compartment rather than the ER. Accordingly, neither heparin nor ryanodine interfered with the Ca2+ release activity induced by intracellular ADPR, although the response to ADPR was somewhat blunted by ryanodine (Fig. 3B). This could mean that ryanodine at the relatively high concentration used here has nonspecific effects on TRPM2 channels or that ryanodine receptors may partially colocalize with TRPM2 in a store subcompartment, possibly indicating some cross talk between lysosomal and ER Ca2+ stores due to a small component of Ca2+-induced Ca2+ release.
Oxidative stress due to production of ROS is thought to play a central role in β cell death and development of diabetes types 1 and type 2 (38, 39). β cells have only modest capacity for self-protection against ROS, including H2O2, because of their low expression of antioxidant enzymes, in particular glutathione peroxidase and catalase, which decompose H2O2 (40). Exposure of MIN6 β cells to H2O2 has been reported to induce Ca2+-dependent cell death involving both extracellular Ca2+ and release from Ca2+ stores (41). Although some reports have linked TRPM2 to H2O2-induced Ca2+ influx and cell death (42, 43), we here identify this channel as a novel mechanism in this process through its function as a lysosomal Ca2+-release channel. Because release of Ca2+ from lysosomes is critical for the redistribution of phosphatidylserine (PS) from the inner plasma membrane leaflet to the cell surface (44), TRPM2-mediated Ca2+ release may not only contribute to apoptosis itself, but additionally represent a crucial element for the externalization of PS, a key recognition ligand for the ultimate elimination of apoptotic cells.
In summary, TRPM2 and its agonist ADPR are multifunctional elements of the β cell Ca2+ signaling machinery. External ADPR functions as a first messenger for receptor-mediated IP3 signaling through P2Yand adenosine receptors and activation of TRPM2 by internal ADPR contributes both to Ca2+ entry across the plasma membrane and Ca2+ release from lysosomes, affecting Ca2+-dependent apoptosis.
Materials and Methods
Cells
Full-length TRPM2 complementary DNAwas cloned into a modified version of the pCDNA4/TO vector (Invitrogen) with an N-terminal Flag epitope tag and electroporated into HEK293 cells previously transfected with the pCDNA6/TR construct for Tet repressor expression as described (1). Pancreatic β cells were isolated from C57BL/6 wild-type or CD38 knockout mice as described (45) and as approved by the Institutional Animal Care and Use Committee, University of Hawaii, and the Animal Care Committee, The Queen's Medical Center.
Electrophysiology
External solution contained (in mM) 140 NaCl, 2.8 KCl, 1 CaCl2, 2 MgCl2, 10 glucose, 10 HEPES-NaOH (pH 7.2 adjusted with NaOH). Internal solution contained (in mM) 140 cesium glutamate for INS-1 and primary β cells, 140 potassium glutamate for HEK293, 8 NaCl, 1 MgCl2, 10 Hepes·Cs/KOH. ADPR was added as appropriate. Reagents were from Sigma-Aldrich except Suramin (Fluka). 8-Br-ADPR was synthesized as described (26). Relative purity of 8-Br-ADPR was assessed by HPLC analyses and found greater than 95%. No traceable 8-Br-NAD+ or 8-Br-cADPR contaminants were detected (46). Patch-clamp experiments were performed as described (1). Error bars indicate SEM with n determinations.
Fluorescence measurements
Fluorescence signals were sampled at a rate of 5 Hz with a photomultiplier-based system using a monochromatic light source (TILL Photonics, Gräfelfing, Germany). Emission was detected with a photomultiplier whose analog signals were sampled by a digital-analog interface (ITC-16, Instrutech, New York) and processed by X-Chart software (HEKA, Lambrecht, Germany). Fluorescence ratios were converted into free intracellular Ca2+ concentration based on calibration parameters derived from patch-clamp experiments with calibrated Ca2+ concentrations. Three different kinds of fluorescence experiments were performed. In experiments combining patch-clamp and fluorescence experiments, cells were perfused with standard intracellular pipette solution containing 200 μM fura-2. Balanced fura-2 experiments were performed by preloading cells with fura-2-AM at 5 μM for 30 minutes. In the subsequent whole-cell patch clamp experiments 200 μM fura-2 had been added to the standard internal solution to assure continuous fura-2 signals. For intact-cell Ca2+ measurements, cells were loaded with 5 μM fura-2-AM for 30 minutes.
For detailed descriptions, see Supplementary Materials and Methods.
Supplementary Material
www.sciencesignaling.org/cgi/content/full/2/71/ra23/DC1
Materials and Methods
Fig. S1. Thapsigargin prevents Ca2+ release induced by extracellular ADPR. Average Ca2+ signals measured in INS-1 cells in response to application of 100 μM ADPR in the absence of extracellular Ca2+ and presence of 1 μM CGS-15943, 100 μM suramin, and 500 nM thapsigargin (n = 11). Cells were loaded with 5 μM Fura-2-AM at 37° C for 30 min.
Fig. S2. Extracellular cADPR and NAD+ evoke Ca2+ signals in INS-1 β-cells. (A) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to increasing concentrations of extracellular cADPR [1 μM (black trace, n = 5), 10 μM (blue trace, n = 8), 30 μM (red trace, n = 5)] and in the absence of extracellular Ca2+. Application start as indicated by the arrow. (B) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to application of 100 μM cADPR in the absence of extracellular Ca2+ and in the presence of either 100 μM suramin (blue trace, n = 6) or 1 μM CGS-15943 (black trace, n = 5) or both suramin and CGS-15943 (red trace, n = 6) in the external solution. (C) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to application of 10 μM (black trace, n = 5), 30 μM (blue trace, n = 7), or 100 μM (green trace, n = 10) NAD+ in the absence of extracellular Ca2+. The red trace indicates application of 100 μM NAD+ in the presence of 100 μM suramin and 1 μM CGS-15943 (n = 7) in the external solution.
Fig. S3. 8-Br-ADPR application inhibits Ca2+ release by cADPR. Average Ca2+ signals measured in INS-1 cells in response to application of extracellular 8-Bromo-ADPR (100 μM) and, shortly thereafter, 8-Bromo-ADPR and 100 μM cADPR without extracellular Ca2+ (n = 12) in the standard external solution. Application started as indicated by arrows and was maintained throughout the experiment. Cells were loaded with 5 μM Fura-2 AM at 37° C for 30 min.
Fig. S4. Statistical analysis of Ca2+ signals induced by external or internal ADPR in β-cells. Ca2+ signals were analyzed for number of oscillations using a Gaussian fitting routine (panels A,D). The first oscillation (if any) was assessed for time-to-peak (panels B,E) and Ca2+-peak amplitude (Ca2+ peak; panels C,F). Black circles are data for INS-1 cells, red circles are data for primary mouse β-cells. Error bar indicates S.E.M. Data plotted from Table S1.
Table S1. Statistical analysis of agonist-induced Ca2+ signals in TRPM2 HEK and β-cells. Where feasible, Ca2+ signals were analyzed for number of oscillations (no. of peaks). The first oscillation was analyzed for time-to-peak (TTP) and Ca2+-peak amplitude (Ca2+ peak) ± S.E.M. with n determinations. The first column indicates figures, panels and conditions. The last column details the cell type used.
Acknowledgments
We are grateful to F. E. Lund for generous provision of CD38−/− mice. We thank D. L. Ko'omoa for establishing the primary β cell isolation and M. Bellinger, A. Love, and H. Bhagat for technical support. This study was supported by I. v.F. McKee Fund, Hawaii Community Foundation (I.L.), NIH GM063954 (R.P.), and NIH GM070634 (A.F.). The findings and conclusions of this study do not necessarily represent the views of The Queen's Medical Center.
Footnotes
The authors declare no conflict of interest.
References and Notes
- 1.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 2.Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 3.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 4.McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- 5.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 6.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 7.Beck A, Kolisek M, Bagley LA, Fleig A, Penner R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006;20:962–964. doi: 10.1096/fj.05-5538fje. [DOI] [PubMed] [Google Scholar]
- 8.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADPR-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–1815. doi: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starkus J, Beck A, Fleig A, Penner R. Regulation of TRPM2 by extra- and intracellular calcium. J Gen Physiol. 2007;130:427–440. doi: 10.1085/jgp.200709836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Partidá-Sanchez S, Rivero-Nava L, Shi G, Lund FE. CD38: An ecto-enzyme at the crossroads of innate and adaptive immune responses. Adv Exp Med Biol. 2007;590:171–183. doi: 10.1007/978-0-387-34814-8_12. [DOI] [PubMed] [Google Scholar]
- 11.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 12.Eisfeld J, Lückhoff A. Handbook of Experimental Pharmacology. Vol. 179. Springer; Berlin, Germany: 2007. TRPM2; pp. 237–252. [DOI] [PubMed] [Google Scholar]
- 13.Ishii M, Shimizu S, Hagiwara T, Wajima T, Miyazaki A, Mori Y, Kiuchi Y. Extracellular-added ADP-ribose increases intracellular free Ca2+ concentration through Ca2+ release from stores, but not through TRPM2-mediated Ca2+ entry, in rat β-cell line RIN-5F. J Pharmacol Sci. 2006;101:174–178. doi: 10.1254/jphs.scj06001x. [DOI] [PubMed] [Google Scholar]
- 14.Schachter JB, Sromek SM, Nicholas RA, Harden TK. HEK293 human embryonic kidney cells endogenously express the P2Y1 and P2Y2 receptors. Neuropharmacology. 1997;36:1181–1187. doi: 10.1016/s0028-3908(97)00138-x. [DOI] [PubMed] [Google Scholar]
- 15.Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 16.Fischer W, Franke H, Gröger-Arndt H, Illes P. Evidence for the existence of P2Y1,2,4 receptor subtypes in HEK-293 cells: Reactivation of P2Y1 receptors after repetitive agonist application. Naunyn Schmiedeberg's Arch Pharmacol. 2005;371:466–472. doi: 10.1007/s00210-005-1070-6. [DOI] [PubMed] [Google Scholar]
- 17.Hillaire-Buys D, Bertrand G, Gross R, Loubatières-Mariani MM. Evidence for an inhibitory A1 subtype adenosine receptor on pancreatic insulin-secreting cells. Eur J Pharmacol. 1987;136:109–112. doi: 10.1016/0014-2999(87)90786-2. [DOI] [PubMed] [Google Scholar]
- 18.Hillaire-Buys D, Chapal J, Bertrand G, Petit P, Loubatières-Mariani MM. Purinergic receptors on insulin-secreting cells. Fundam Clin Pharmacol. 1994;8:117–127. doi: 10.1111/j.1472-8206.1994.tb00788.x. [DOI] [PubMed] [Google Scholar]
- 19.Verspohl EJ, Johannwille B, Waheed A, Neye H. Effect of purinergic agonists and antagonists on insulin secretion from INS-1 cells (insulinoma cell line) and rat pancreatic islets. Can J Physiol Pharmacol. 2002;80:562–568. doi: 10.1139/y02-079. [DOI] [PubMed] [Google Scholar]
- 20.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 21.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- 23.Gerasimenko JV, Sherwood M, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J Cell Sci. 2006;119:226–238. doi: 10.1242/jcs.02721. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda M, Viitala J, Matteson J, Carlsson SR. Cloning of cDNAs encoding human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Comparison of their deduced amino acid sequences. J Biol Chem. 1988;263:18920–18928. [PubMed] [Google Scholar]
- 25.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Partida-Sanchez S, Gasser A, Fliegert R, Siebrands CC, Dammermann W, Shi G, Mousseau BJ, Sumoza-Toledo A, Bhagat H, Walseth TF, Guse AH, Lund FE. Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by ADP-ribose, the major product generated by the CD38 enzyme reaction. J Immunol. 2007;179:7827–7839. doi: 10.4049/jimmunol.179.11.7827. [DOI] [PubMed] [Google Scholar]
- 27.Okamoto H, Takasawa S, Tohgo A. New aspects of the physiological significance of NAD, poly ADP-ribose and cyclic ADP-ribose. Biochimie. 1995;77:356–363. doi: 10.1016/0300-9084(96)88146-6. [DOI] [PubMed] [Google Scholar]
- 28.Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, Murray R, Schuber F, Howard MC. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood. 1998;92:1324–1333. [PubMed] [Google Scholar]
- 29.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirkner K, Schweigel J, Gerevich Z, Franke H, Allgaier C, Barsoumian EL, Draheim H, Illes P. Adenine nucleotides inhibit recombinant N-type calcium channels via G protein-coupled mechanisms in HEK 293 cells; involvement of the P2Y13 receptor-type. Br J Pharmacol. 2004;141:141–151. doi: 10.1038/sj.bjp.0705588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lugo-Garcia L, Filhol R, Lajoix AD, Gross R, Petit P, Vignon J. Expression of purinergic P2Y receptor subtypes by INS-1 insulinoma β-cells: A molecular and binding characterization. Eur J Pharmacol. 2007;568:54–60. doi: 10.1016/j.ejphar.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, Usai C, Meis S, Kassack MU, Zocchi E, De Flora A. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281:31419–31429. doi: 10.1074/jbc.M606625200. [DOI] [PubMed] [Google Scholar]
- 33.Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM. β-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104:16359–16364. doi: 10.1073/pnas.0705510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato I, Takasawa S, Akabane A, Tanaka O, Abe H, Takamura T, Suzuki Y, Nata K, Yonekura H, Yoshimoto T, Okamoto H. Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic β cells. Enhanced insulin secretion in CD38-expressing transgenic mice. J Biol Chem. 1995;270:30045–30050. doi: 10.1074/jbc.270.50.30045. [DOI] [PubMed] [Google Scholar]
- 35.Berthelier V, Tixier JM, Muller-Steffner H, Schuber F, Deterre P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem J. 1998;330(Pt 3):1383–1390. doi: 10.1042/bj3301383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Structural basis for the mechanistic understanding of human CD38-controlled multiple catalysis. J Biol Chem. 2006;281:32861–32869. doi: 10.1074/jbc.M606365200. [DOI] [PubMed] [Google Scholar]
- 38.Mandrup-Poulsen T. Apoptotic signal transduction pathways in diabetes. Biochem Pharmacol. 2003;66:1433–1440. doi: 10.1016/s0006-2952(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 39.Rhodes CJ. Type 2 diabetes—A matter of β-cell life and death? Science. 2005;307:380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 40.Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- 41.Choi SE, Min SH, Shin HC, Kim HE, Jung MW, Kang Y. Involvement of calcium-mediated apoptotic signals in H2O2-induced MIN6N8a cell death. Eur J Pharmacol. 2006;547:1–9. doi: 10.1016/j.ejphar.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 42.Ishii M, Shimizu S, Hara Y, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat β-cell line RIN-5F. Cell Calcium. 2006;39:487–494. doi: 10.1016/j.ceca.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci. 2006;101:66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 44.Mirnikjoo B, Balasubramanian K, Schroit AJ. Mobilization of lysosomal calcium regulates the externalization of phosphatidylserine during apoptosis. J Biol Chem. 2009;284:6918–6923. doi: 10.1074/jbc.M805288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 46.Massullo P, Sumoza-Toledo A, Bhagat H, Partida-Sánchez S. TRPM channels, calcium and redox sensors during innate immune responses. Semin Cell Dev Biol. 2006;17:654–666. doi: 10.1016/j.semcdb.2006.11.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
www.sciencesignaling.org/cgi/content/full/2/71/ra23/DC1
Materials and Methods
Fig. S1. Thapsigargin prevents Ca2+ release induced by extracellular ADPR. Average Ca2+ signals measured in INS-1 cells in response to application of 100 μM ADPR in the absence of extracellular Ca2+ and presence of 1 μM CGS-15943, 100 μM suramin, and 500 nM thapsigargin (n = 11). Cells were loaded with 5 μM Fura-2-AM at 37° C for 30 min.
Fig. S2. Extracellular cADPR and NAD+ evoke Ca2+ signals in INS-1 β-cells. (A) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to increasing concentrations of extracellular cADPR [1 μM (black trace, n = 5), 10 μM (blue trace, n = 8), 30 μM (red trace, n = 5)] and in the absence of extracellular Ca2+. Application start as indicated by the arrow. (B) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to application of 100 μM cADPR in the absence of extracellular Ca2+ and in the presence of either 100 μM suramin (blue trace, n = 6) or 1 μM CGS-15943 (black trace, n = 5) or both suramin and CGS-15943 (red trace, n = 6) in the external solution. (C) Average Ca2+ signal measured in intact Fura-2 AM-loaded INS-1 cells in response to application of 10 μM (black trace, n = 5), 30 μM (blue trace, n = 7), or 100 μM (green trace, n = 10) NAD+ in the absence of extracellular Ca2+. The red trace indicates application of 100 μM NAD+ in the presence of 100 μM suramin and 1 μM CGS-15943 (n = 7) in the external solution.
Fig. S3. 8-Br-ADPR application inhibits Ca2+ release by cADPR. Average Ca2+ signals measured in INS-1 cells in response to application of extracellular 8-Bromo-ADPR (100 μM) and, shortly thereafter, 8-Bromo-ADPR and 100 μM cADPR without extracellular Ca2+ (n = 12) in the standard external solution. Application started as indicated by arrows and was maintained throughout the experiment. Cells were loaded with 5 μM Fura-2 AM at 37° C for 30 min.
Fig. S4. Statistical analysis of Ca2+ signals induced by external or internal ADPR in β-cells. Ca2+ signals were analyzed for number of oscillations using a Gaussian fitting routine (panels A,D). The first oscillation (if any) was assessed for time-to-peak (panels B,E) and Ca2+-peak amplitude (Ca2+ peak; panels C,F). Black circles are data for INS-1 cells, red circles are data for primary mouse β-cells. Error bar indicates S.E.M. Data plotted from Table S1.
Table S1. Statistical analysis of agonist-induced Ca2+ signals in TRPM2 HEK and β-cells. Where feasible, Ca2+ signals were analyzed for number of oscillations (no. of peaks). The first oscillation was analyzed for time-to-peak (TTP) and Ca2+-peak amplitude (Ca2+ peak) ± S.E.M. with n determinations. The first column indicates figures, panels and conditions. The last column details the cell type used.