ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells (original) (raw)
Abstract
Next-generation sequencing has greatly increased the scope and the resolution of transcriptional regulation study. RNA sequencing (RNA-Seq) and ChIP-Seq experiments are now generating comprehensive data on transcript abundance and on regulator–DNA interactions. We propose an approach for an integrated analysis of these data based on feature extraction of ChIP-Seq signals, principal component analysis, and regression-based component selection. Compared with traditional methods, our approach not only offers higher power in predicting gene expression from ChIP-Seq data but also provides a way to capture cooperation among regulators. In mouse embryonic stem cells (ESCs), we find that a remarkably high proportion of variation in gene expression (65%) can be explained by the binding signals of 12 transcription factors (TFs). Two groups of TFs are identified. Whereas the first group (E2f1, Myc, Mycn, and Zfx) act as activators in general, the second group (Oct4, Nanog, Sox2, Smad1, Stat3, Tcfcp2l1, and Esrrb) may serve as either activator or repressor depending on the target. The two groups of TFs cooperate tightly to activate genes that are differentially up-regulated in ESCs. In the absence of binding by the first group, the binding of the second group is associated with genes that are repressed in ESCs and derepressed upon early differentiation.
Keywords: ChIP binding, pluripotency, RNA-Seq, transcription regulation
The recent development of ultra-high-throughput RNA sequencing (RNA-Seq) technology holds the promise to provide more accurate gene expression measurements (1, 2). Compared with microarray, RNA-Seq is able to measure absolute concentration of transcripts (1). Meanwhile, chromatin immunoprecipitation (ChIP) coupled with microarray (ChIP-chip) or sequencing (ChIP-Seq) technologies have been developed to identify whole-genome localization of protein–DNA binding sites (3, 4). These data sets provide the raw materials to study the regulatory functions of transcription factors (TFs) on gene expression.
Predictive modeling (such as regression) is a statistical strategy to predict an outcome from one or more explanatory variables (predictors). In the study of transcriptional regulation, a predictive model can be constructed in which the gene expression profile under a certain condition is regarded as the response variable and various features related to TFs are taken as the predictors. Examples of such features include counts of motifs recognized by the TFs (5), sum of motif occurrences weighted by their distances from the target gene (6), motif scores based on position-specific weight matrices (7), and ChIP-chip log ratios (8). Most models are based on linear regression, but there are also extensions that include network component analysis (9), multivariate adaptive regression splines (10, 11), Bayesian error analysis model (12), boosting and Bayesian additive regression trees (13), partial least squares (14), motif expression decomposition (15), and further generalizations to pairwise interactions between TFs (6, 10, 11). Thus far, the fraction of variation in gene expression (_R_2) explained by TF binding has been very moderate, varying between 9.6% and 36.9% on various datasets from yeast to human (5, 7, 10, 11), even after considering TF–TF interaction. The low _R_2 reported in these studies may be due to insufficient data or suboptimal models or both. In any case, accurate quantitative modeling of expression from binding location data has not been demonstrated.
In this paper, we show that accurate quantitative modeling of gene expression data in a mouse cell type is possible provided that TF-DNA binding locations have been measured by ChIP-Seq experiments for multiple transcriptional regulators. An important first step in this analysis is to extract suitable features from the ChIP-Seq data to serve as explanatory variables in the modeling of gene expression. From each TF, we construct a TF association strength (TFAS) for each gene by computing the weighted sum of the corresponding ChIP-Seq signal strength, where the weights reflect the proximity of the signal to the gene. The use of TFAS as a predictor variable allows us to explain a much higher proportion of gene expression variation than traditional predictors, such as the binary indicators of whether the gene is the closest gene to a ChIP-Seq peak.
Although high predictive power is a desirable property, it is important that a model has enough interpretability so that it offers insight on the regulatory roles of the TFs. One major concern is the modeling of combinatorial regulation in an efficient manner. Traditional linear prediction models are inadequate in this regard in the sense that the best linear model is represented by a single linear combination of the TF-specific predictors. If the coefficient for a TF is positive, then this model implies that the binding of this TF always results in up-regulation of the target gene; i.e., it can only serve as a positive regulator. Conversely, if the coefficient is negative, then its effect is always to repress transcription. However, it is known that a TF may contribute to the activation of some of its target genes while, at the same time, also participate in the repression of other target genes. The divergent regulatory effects of the TF may be due to differences in the binding of cofactors and/or the chromatin context. Such divergent effects are difficult to model, and current methods are limited to very low-order interactions of TFs (16). An additional difficulty of linear regression arises when the predictors are highly correlated. Since two or more TFs may cooperate to regulate many genes, we expect that the corresponding TFASs will vary in a coordinated manner across different genes; i.e., they are correlated as predictor variables. In the presence of such “multicollinearity,” it is easy for the contribution of one TF to be attributed to another TF with a correlated TFAS, and vice versa. In other words, the coefficients of the TFASs in the fitted model are highly unstable and therefore not suitable for interpretation.
To address these difficulties, we consider a scenario where there are several types of coordinated interactions among the TFs and that each type of coordination is relevant for the regulation of many genes. The TFASs for the target genes under a specific type of coordinated regulation should then show a characteristic pattern that may be extracted through unsupervised learning from the set of TFAS vectors. Here each gene has a TFAS vector whose _i_th coordinate is the TFAS between this gene and the _i_th TF. In this paper, we use principal component analysis (PCA) to extract uncorrelated characteristic patterns in the TFAS vectors. The resulting patterns are called TF principal components (TFPCs). By using the TFPCs as covariates in a regression model (PC-regression), we can often select a small number of TFPCs that capture almost all of the predictable variations in the gene expression. This reduces the predictor space to a low dimensional subspace in which more sophisticated nonlinear analysis can be pursued, e.g., classification tree analysis as in our analysis of the ESC data below. One important advantage of this approach is that a TF can have a positive coefficient in one selected TFPC but a negative coefficient in another selected TFPC, which allows it to have different regulatory effects on different genes depending on which TFPC is dominant for a given gene. Furthermore, because the TFPCs are orthogonal vectors, they are stably defined even in the presence of multicollinearity.
We apply the method to the gene regulatory system in mouse ESCs. ESCs can maintain self-renewal and pluripotency, i.e., having the ability to differentiate to any adult cell type. Many regulators have been identified as relevant for pluripotency. Among them, Oct4, Nanog, and Sox2 are the most important ones (17). RNAi screening of TFs identifies new regulators, such as Esrrb (18). Mass spectrometry analysis of Oct4 and Nanog interacting proteins suggests their potential partners, such as Nr0b1, Nac1, and Zfp281 (19). Although experimental studies have revealed the importance of many of these regulators, a quantitative dissection of the functional roles of these regulators is still lacking. The availability of genome-wide gene expression and TF binding data provides an unprecedented opportunity to investigate this problem.
Results
ChIP-Seq Accurately Predicts Absolute Gene Expression.
The number of reads per kilobase of exon region per million mapped reads (RPKM) derived from RNA-Seq data is shown to be approximately proportional to the absolute abundance of mRNAs in cells (1). We calculated the RPKM values of mouse ESCs based on a very deep sequencing data [supporting information (SI) Dataset S1] (2). To predict gene expression, we used the ChIP-Seq data of 12 sequence-specific TFs: Smad1, Stat3, Sox2, Oct4, Nanog, Esrrb, Tcfcp2l1, Klf4, Zfx, E2f1, Myc, and Mycn (20). The binary and continuous TFAS profiles were calculated (Materials and Methods). For the latter, the TFAS was based a weighted summation of TF binding peaks where those with higher reads intensity or location proximity to the transcription start site (TSS) were given higher weights. Fig. 1 displays the binding peaks of E2f1 around the TSSs of three genes. The continuous TFAS values of the three genes are calculated as 324, 19.3, and 0.1, which quantitatively measure the strength of E2f1 binding on these genes. On the contrary, the binary TFAS values fail to distinguish the three genes because they are all equal to 1, which suggests that the continuous TFAS may capture more relevant TF binding information than the binary approach. The normalized continuous TFAS profiles of the 12 TFs are listed in Dataset S2.
Fig. 1.

Illustration of the binding peaks of E2f1 around three genes. The vertical axis represents the amplitude of the ChIP-Seq signals.
To assess the capability of TF binding for the prediction of absolute gene expression, we compare the performance of predicting RNA-Seq gene expression by the PC-regression model (Materials and Methods) using the binary and continuous TFAS, respectively. Briefly, we first decomposed the TFAS profiles into 12 principal components by PCA. Then we performed a log-linear regression on gene expression using the extracted principal components. As shown in Table 1, the model fitting using the continuous TFAS (_R_2 = 0.650) is much better than that of the binary TFAS (_R_2 = 0.425). The prediction power of the continuous TFAS is also much higher than the binary TFAS (_CV-R_2 = 0.639 of the former versus 0.404 of the latter). This demonstrates that the continuous TFAS captures more quantitative information on TF binding than the binary TFAS, as shown in Fig. 1.
Table 1.
Statistical assessments of the PC-regression model on gene expression prediction in mouse ESCs using the binary and continuous TFAS
| Expression platform | TFAS | R2 | CV-R2 |
|---|---|---|---|
| RNA-Seq | Continuous | 0.650 | 0.639 |
| Binary | 0.425 | 0.404 | |
| Microarray | Continuous | 0.529 | 0.524 |
| Binary | 0.262 | 0.254 |
We performed the same PC-regression analyses on another expression dataset based on the Affymetrix MOE430 V2 array (21), where the average gene expression profile of three ESC samples was regarded as the response variable. The continuous TFAS model again outperforms the binary TFAS model (Table 1). Note that the _R_2 and _CV-R_2 are 12–15% less than those of the RNA-Seq data. It is known that microarray has an intrinsic limitation to measure absolute mRNA concentration because of probe affinity effects (22). The microarray expression index was plotted against the RPKM of the RNA-Seq data in Fig. S1. It is seen that for lowly expressed genes the microarray values are more condensed and less distinguishable, suggesting that microarrays are less sensitive to detect lowly expressed genes than deep RNA-Seq.
In addition to the _R_2 and _CV- R_2 statistics, it is also informative to plot the predicted versus observed gene expression values. As shown in Fig. 2 A–D, the observed gene expression values and the predicted values of the continuous TFAS model are well matched (Pearson correlation coefficients, r = 0.806 for RNA-Seq and 0.727 for microarray), while those of the binary TFAS do not. Considering that the ChIP-Seq data do not directly measure transcript abundance, this is a surprisingly high correlation which is comparable to those observed between measurements made on the same samples by different types of expression arrays (23, 24). We further noticed that a small number of principal components of the continuous TFASs are able to capture almost all of the predictable variations in the gene expression. These TFPCs are sorted by their capability to explain gene expression as shown in Fig. 2E. The top ones are TFPC1, TFPC2, and TFPC11, which can account for 46.8%, 8.7%, and 6.5% of the gene expression variation, respectively. The other 9 TFPCs, combined together, account for <3% of the gene expression variation.
Fig. 2.

Model assessments. (A) Predicted versus observed ESC gene expression values for the RNA-Seq dataset on the binary TFAS. (B) RNA-Seq dataset on the continuous TFAS. (C) Microarray dataset on the binary TFAS. (D) Microarray dataset on the continuous TFAS. r is the Pearson correlation coefficient. (E) The _R_2 statistics of individual TFPCs for the prediction of RNA-Seq gene expression. (F) The overall _R_2 statistics for the predictions of gene expression under various conditions from the ESC ChIP-Seq data.
To investigate whether this high predictive power of TF binding on gene expression is biologically significant, we used the same ChIP-Seq data to predict other gene expression profiles from a number of early differentiated cell types and terminally differentiated tissue samples of mice (Fig. 2F). Among the datasets, the ESC, embryoid body (EB), and adult tissue profiles are based on RNA-Seq (1, 2); others are microarray-based. For each condition, we used the continuous TFAS profiles and calculated the _R_2. Results showed that the _R_2 in ESCs is the highest among all of the conditions. The _R_2 in EBs is clearly lower than that in ESCs although the difference is small due to the high similarity between the two expression profiles (r = 0.942). The comparison of the _Oct4_-high and _Oct4_-low profiles shows a significantly lower _R_2 in the latter, consistent with the role of Oct4 as the master regulator of ESCs. In the retinoic acid (RA) induction series, the _R_2 consistently decreases (with the switch of day 3 and day 4) as the number of days after induction increases, where the cells become more and more differentiated. In the three terminally differentiated tissue samples, the values of the _R_2 are only slightly above 0.2. This suggests that the ChIP-Seq data reflect ESC-specific TF binding.
TFPCs Discriminate Differentially Expressed Genes.
We next study how differentially expressed genes are regulated by the TFs. By combining the RNA-Seq profiles in ESCs and EBs, and the microarray profiles of the _Oct4_-high and _Oct4_-low samples, we collected four sets of genes with characteristic expression patterns. They are 668 genes highly expressed in both ESCs and differentiated cells (Uniform High), 838 genes lowly expressed in both (Uniform Low), 782 genes up-regulated in ESCs (ES Up), and 831 genes down-regulated in ESCs (ES Down). For detailed selection criteria, see Materials and Methods, Fig. S2, and SI Text. For the full lists of the four gene sets, see Table S1. We performed functional annotation on the four combined gene sets using DAVID (25). The four gene sets are enriched in specific function categories (Table S2). ES Up and ES Down genes are enriched in developmental processes. The ES Up genes include some well-known ESC markers, such as Pou5f1 (Oct4), Sox2, Nanog, Esrrb, Dppa2, Zfp42, Nr0b1, and Nr5a2. The ES Down genes include early developmental regulators, such as Hand2, Mesp1, Foxa2, Sox17, and Gata4/Gata6.
We sought to infer quantitative rules of TF binding governing the regulation of differential gene expression in ESCs. Visualization in the TFPC1–TFPC2 plane shows that the four sets of genes form clear clusters (Fig. S3_A_), suggesting that they are regulated by different combinations of the TFs. The Classification and Regression Tree (CART) algorithm (26) was applied to discriminate the four sets of genes based on the top three TFPCs that most explain the gene expression variation (i.e., TFPC1, TFPC2, and TFPC11) (Fig. 2E). A classification tree with nine splits was learned (Fig. S3 B and C). We computed the misclassification error rate of the learned tree for discriminating the four gene sets. As the baseline, the error rate is 75% in random guessing of the class of a gene. The learned tree is able to reduce the error rate to 37.1% (P value < 5 × 10−200, one-sided Z test; see Fig. S3_D_ for detailed classification results). The regulatory rules learned in this way are combinations of TFPCs. For example, the Uniform Low gene set can be determined by TFPC1 < −0.77 AND TFPC11 < 0.25. The major rule on the ES Down gene set is −0.77 ≤ TFPC1 < 0.06 AND TFPC2 < 1.47. The major rule governing the Uniform High gene set is TFPC1 ≥ 0.06 AND TFPC2 ≥ 0.45. And the major rule on the ES Up gene set is TFPC1 ≥ 0.75 AND TFPC2 < −0.64.
TFPCs Provide Information on the Roles of Regulators.
We now discuss the roles of the 12 TFs in gene expression regulation revealed from the PC-regression model based on the ESC RNA-Seq data. To better illustrate this, we compared the sets of regression coefficients of the model using individual TFASs as predictors with those using TFPCs. In the model using the individual TFs as predictors, it is notable that E2f1 dominates the regression with a very large coefficient, while all of the other TFs have coefficients of small magnitude (Fig. 3A). The coefficients of Nanog, Sox2, Stat3, and Oct4 are all nearly zero. Thus standard regression method failed to reveal the roles of even these regulators that are generally believed to be the master ESC regulators. In contrast, in the model using TFPCs as predictors, the roles of individual TFs are implicated in the top TFPCs with relatively larger regression coefficients (Fig. 3B). We plotted the loadings of the individual TFs in the top TFPCs, weighted by the fractions of variance in TFASs explained by the TFPCs (Fig. 3C), which approximately reflect the relative importance of the roles of the individual TFs. Bootstrap estimation of the 95% confidence intervals indicates that the loadings are robust (SI Text). Importantly, key regulators such as Oct4 and Nanog have significant loadings in the top TFPCs, and hence their importance is not masked by the effect of E2f1. In TFPC1 (accounting for 52% of the variance in TFASs), all of the 12 TFs have positive loadings, indicating that their primary roles are activating gene expression. In TFPC2 (accounting for 10% of the variance in TFASs), the loadings of E2f1, Myc, Mycn, and Zfx (group I TFs) are positive, while those of Oct4, Nanog, Sox2, Smad1, Stat3, Tcfcp2l1, and Esrrb (group II TFs) are negative, suggesting that the latter group may play a role as repressors for genes associated with high TFPC2 values. TFPC11 (accounting for 2% of the variance in TFASs) is dominated by E2f1, suggesting that a small percentage of genes may be activated by E2f1 alone, or by other TFs not studied here. This analysis reveals diverse roles of the TFs. We speculate that the group I TFs activate gene expression in general, while the group II TFs may activate or repress gene expression depending on the targets. This is consistent with the reported experimental result that E2f1 binding is proximate to the TSSs of a large number of highly expressed genes, especially those cell cycle regulated genes needed for self-renewal (27). Myc occupancy is close to TSSs and is associated with large scale modification of chromatin structure (28, 29). Oct4 and Nanog are associated with multiple repression complexes such as the Polycomb, NuRD, Sin3A and Pml complexes (30–32), in addition to their well known activation roles in ESCs.
Fig. 3.

TFPCs capture the roles of TFs. (A) Regression coefficients of the model using individual TFASs as predictors. (B) Regression coefficients using TFPCs as predictors. (C) Loadings of TFs in the top three selected TFPCs, weighted by the fractions of variance in TFASs explained by the TFPCs. The error bars give the 95% bootstrap confidence intervals.
We next show that the regulatory rules for differential gene expression can be re-written as combinations of the group I and group II TFs. Mathematically, we transform TFPCs by: _g_1 = ½(_TFPC_1 + _TFPC_2), _g_2 = ½(_TFPC_1 − _TFPC_2) and _g_3 = _TFPC_11. Roughly speaking, _g_1 and _g_2 are combinations of the group I TFs and group II TFs, respectively, and _g_3 represents E2f1. Then the regulatory rules can be reexpressed as follows. Uniform Low: _g_1 + _g_2 < −0.77 AND _g_3 < 0.25; ES Down: −0.77 ≤ g1 + g2 < 0.06 AND _g_2 − g1 > −1.47; Uniform High: _g_1 + _g_2 ≥ 0.06 AND _g_1 − _g_2 ≥ 0.45; and ES Up: _g_1 + _g_2 ≥ 0.75 AND _g_2 − _g_1 > 0.64. It can be seen that (_g_1 + _g_2) increases in the order of Uniform Low, ES Down, Uniform High, and ES Up, which means the cooperation between the group I and II TFs becomes more and more extensive. Notably, although both the Uniform High and ES Up gene sets are highly expressed in ESCs, we found that the former favors the group I TFs while the latter favors the group II TFs as reflected by the signs of (_g_1 − _g_2). And although both the Uniform Low and ES Down gene sets are lowly expressed in ESCs, genes in the former may simply not be bound by any of the TFs, while genes in the latter show detectable TF binding events and the group II TF binding is preferred. Recall that the group II TFs include typical ESC-specific regulators such as Oct4, Sox2, Nanog, Stat3, and Esrrb. This is consistent with the hypothesis that the group II TFs are important for the control of differential gene expression in ESCs.
As discussed in the above, group II TFs such as Esrrb can be either an activator (e.g., on ES Up genes) or a repressor (e.g., on ES Down genes). To test this hypothesis, we took the top 10 genes with the strongest Esrrb TFAS values from the correctly predicted ES Up genes, and also the same from the ES Down genes. We asked whether the knockdown of Esrrb expression will down-regulate the expression levels of the first group and up-regulate the expression levels of the second group, as predicted by our model. We obtained the relevant expression data after Esrrb knockdown from ref. 18, which is an independent data set not used in the construction of our model. As shown in Table 2, the expression levels of 9 of the 10 ES-Up-_Esrrb_-bound genes decreased with the knockdown of Esrrb, and 8 of the 10 ES-Down-_Esrrb_-bound genes increased. This provides an independent, experimental validation of the model.
Table 2.
Experimental validation by the Esrrb RNAi knockdown data (18)
| Gene symbol | FC | Gene symbol | FC |
|---|---|---|---|
| ES-Up-_Esrrb_-bound | ES-Down-_Esrrb_-bound | ||
| Ndg2 | 0.62 | Trpc3 | 2.12 |
| Icam1 | 0.34 | Zfpm1 | 2.63 |
| Klk1 | 0.93 | Sema3f | 4.29 |
| Mreg | 0.62 | Gpr124 | 3.54 |
| Ldhb | 0.50 | St6galnac4 | 1.14 |
| Mybl2 | 0.79 | Ryr2 | 0.98 |
| 6430514L14Rik | 1.16 | G0s2 | 2.09 |
| Lrrc2 | 0.22 | Apobec2 | 0.80 |
| Mylpf | 0.71 | Pxmp3 | 1.08 |
| Sept1 | 0.73 | Evx1 | 1.16 |
Combinatorial Gene Regulation of ESC Pluripotency.
The Polycomb Repressive Complex 2 (PRC2) plays an important role in maintaining ESC pluripotency by gene repression (30, 31). The ChIP-Seq data of Suz12, a key subunit of PRC2, is available along with the 12 sequence-specific TFs (20). But because it does not bind DNA sequences directly, we did not include it in the above analysis. Instead, we studied the overlaps between its binding targets and the four gene sets, as well as the predicted gene sets based on the inferred regulatory rules. We identified 1938 Suz12 bound genes with TFAS ≥ 2. Suz12 bound genes are strongly enriched in the ES Down gene sets (Enrichment level R = 3.9, P value = 6 × 10−119), while not enriched in any of the other three sets (Fig. S4 A–D), confirming its association with ESC-repressed genes. They are also enriched in the predicted set of ES Down genes (R = 3.2, P value = 7 × 10−86), suggesting potential interaction between Suz12 and the sequence-specific TFs. This is consistent with the observation that a significant subset of PRC2 target genes are co-occupied by the ESC regulators Oct4, Sox2 and Nanog (31). It is interesting to note that there are 71 predicted ES Down genes which are not in the observed ES Down gene set but are actually bound by Suz12 (Table S3). Some of them may be up-regulated in certain cell differentiation lineages (and thus become ES Down) not covered in this analysis. For example, the Olig1 and Olig2 genes are important for neural progenitor cells to develop into mature neurons, and are up-regulated in neural progenitor cells versus ESCs (30).
The above analyses suggest combinatorial roles of the TFs on regulating ESC pluripotency. E2f1, Myc, Mycn, and Zfx may contribute to rapid proliferation of ESCs. Since they maintain their expression when ESCs differentiate to early differentiation stages (data from the RNA-Seq and the array profiles), they may continue to activate the expression of genes that are essential for proliferation. The group II TFs, on the other hand, maintain ESC pluripotency through activating genes that are otherwise silent in differentiated cells. The maintenance of ESC pluripotency also involves inhibition of genes expressed in differentiated cells. The regulatory rules we found above suggest that the 12 TFs may interact with PRC2 to repress differentiation related genes in the absence of the group I TFs. For example, the ESC up-regulated gene Gdf3 is bound by both the group I and II TFs (Fig. S5_A_), while the ESC down-regulated gene Foxa2 is only bound by the group II TFs plus Suz12 (Fig. S5_B_).
Discussion
By joint modeling of ChIP-Seq and gene expression data (RNA-Seq and microarray) we quantified the contribution of TF binding on gene expression regulation. We demonstrated that binding signals of sequence-specific TFs have remarkably high predictive power on absolute mRNA abundance. We found that gene expression indices measured by RNA-Seq have a noticeable higher correlation with TF binding than those measured by microarray.
We have studied the roles of TF binding on regulating differential gene expression in ESCs. We found that a few simple rules defined by the combinations of TFs are able to summarize the major modes of cooperation among these TFs. The first principal component of the ChIP-Seq signal is essentially uniform across all 12 TFs studied here. This is consistent with the finding that many genomic loci are bound by a large number of different TFs (33, 34). We found that some TFs (group II TFs) have divergent roles in regulating gene expression, i.e., they contribute to activation on some genes but repression on other genes. Understanding why they act as both activators and repressors will help to illustrate the mechanism of pluripotency. Also, the interaction between sequence-specific TFs and PRC2 should be further investigated to clarify how PRC2 recognizes and silences its target genes.
The approach of first extracting characteristic TFAS patterns and then select among them the ones with good predictive power can be generalized. Here we have used principal component analysis for simplicity, but other methods such as _k_-means clustering can also be considered. The basic feature space can be expanded, so that instead of defining one TFAS variable for each TF, one may construct predictor variables designed to capture more fine-scaled co-localization among different TFs. While the simple approach offered here seems already effective in quantitative modeling of gene expression in ESCs based on ChIP-Seq data, further refinement and extension of the basic approach may be needed to fully extract the rich information in these massive data sets.
Materials and Methods
ChIP-Seq and Gene Expression Data.
The ChIP-Seq binding peak data of the 12 TFs in mouse ESCs were obtained from Chen et al. (20). The peaks in the neighborhood of peaks of a control GFP (20) were filtered out to eliminate false positives due to nonspecific binding. We obtained mapped mRNA sequencing data for mouse ESCs and EBs (2). The gene expression values of the sequencing data were calculated by modifying the code of the ERANGE program, based on the RPKM definition (1). The RNA-Seq expression profiles of mouse brain, liver and muscle were used for comparison (1). The Affymetrix MOE430 V2 array profiles of the Oct4 sorted series were obtained from Zhou et al. (21), in which 8 are _Oct4_-high samples (three ESC profiles and five EB profiles with cells selected by high Oct4 expression) and the rest are _Oct4_-low samples (eight EB profiles with cells selected by low Oct4 expression). The Affymetrix MOE430 AB array profiles of the RA induction and Esrrb RNAi knockdown series are from Ivanova et al. (18). Our analysis focused on the well annotated mouse Refseq genes (N ≈ 19,000). The genomic coordinates of the mm8 mouse Refseq genes were obtained from the UCSC GoldenPath database.
TF Association Strength.
Binary.
Traditionally, a TF binding peak is usually associated with a gene so that the distance between the peak and the gene (usually the TSS) is the nearest out of all Refseq genes (35). Denoting the binary TFAS as aij, aij is equal to 1 if gene i is associated with a peak of TF j; otherwise aij is equal to 0. This approach does not take into account the intensity of the peaks and the relative distance between peaks and genes.
Continuous.
We integrate the peak intensity and the proximity to genes to define the association strength between a TF and a gene. We assume that the association strength of TF j on gene i is a weighted sum of intensities of all of the peaks of TF j:

where gk is the intensity (number of reads aligned to the coordinate) of the _k_th binding peak of the TF j, dk is the distance (number of nucleotides) between the TSS of gene i and the _k_th binding peak in the reference genome, and _d_0 is a constant. In theory, the summation is over all binding peaks of a given TF. But the effect of a peak decays exponentially when dk increases where the speed depends on _d_0. When dk/_d_0 is very large the contribution of the peak will be effectively zero. We set _d_0 = 500 bps for E2f1 and 5,000 bps for other TFs because E2f1 tends to be closer to TSSs. To save computation time, we only consider peaks within a sufficiently large distance (say, 1 Mbps) of a gene. The TFAS values are then log-transformed and quantile-normalized. For N genes and M TFs the TFAS profiles are denoted by an N × M matrix A.
PC Regression.
We use PCA to extract characteristic patterns (TFPCs) from the TFAS profiles of multiple TFs. After having been centered and standardized, the TFAS matrix A is decomposed by the singular value decomposition (SVD) A = U_Σ_VT, where both U (an N × M matrix) and V (an M × M matrix) are orthogonal matrices, and Σ (an M × M matrix) is diagonal. The loading matrix V consists of the weights of individual TFs in the TFPCs. We denote X = _U_Σ, where the TFPC scores are specified. PCA has been used to capture characteristic modes of gene expression profiles, where the principal components are called eigengenes (36, 37), and to study the clustering property of TFs based on their genomic distributions (38). Here we aim to use TFPCs to predict gene expression.
Given a single condition, the gene expression is expressed by the log-linear regression model
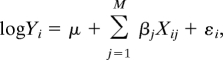
where Yi is the absolute expression of gene i, μ is the basal expression, Xij is the score of the j_th TFPC on gene i, β_j is the regression coefficient of the j_th TFPC, and ε_i is a gene-specific error term. To avoid taking the logarithm of zero, a small positive constant is added to Yi. In this model, each TF contributes to the prediction through the TFPCs. Thus the same TF can have positive effect on gene expression through one TFPC and negative effect through a different TFPC. This allows interpreting a TF as both activator and repressor, depending on the TF combinations. Another advantage of using TFPCs as the predictors is that the regression coefficients do not affect each other and each TFPC can explain a unique fraction of the variance of gene expression since the TFPCs are uncorrelated. Thus these TFPCs can be ordered in a decreasing manner by their importance for gene expression. We use the _R_2 to measure how much variation in gene expression can be explained by the TF binding data. To evaluate the predictive power of the model on new datasets, we employ ten-fold cross-validation to calculate the average _R_2 in the ten independent test sets which is denoted by _CV-R_2.
Gene Selection Criteria.
We apply the SVD to analyze gene expression patterns upon ESC differentiation. The top two gene expression principal components are denoted by GPC1 and GPC2. The Uniform High or Uniform Low genes are those with higher or lower 2.5% values in GPC1. The ES Up or ES Down genes are those with higher or lower 2.5% values in GPC2. The respective unions of the four sets of genes from the RNA-Seq dataset and those from the array dataset are taken as the final gene sets (SI Text).
Classification Tree.
We applied the CART algorithm (26) to learn a classification tree to distinguish the four gene sets using the TFPCs as predictors. We use 10-fold cross-validation to choose the most parsimonious tree with a CV error within one standard error of the minimum (26). For detailed description of the CART algorithm, see SI Text.
Supplementary Material
Supporting Information
Acknowledgments.
We thank Michael Zhang for a careful reading of the manuscript and Zhen-Su She for comments. This work was supported by National Institutes of Health Grants R01HG004634 and R01HG003903, National Science Foundation Grant DMS-0805491, and California Institute for Regenerative Medicine (CIRM) Grant RC1-00133. Z.O. thanks the CIRM for a predoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. T.S. is a guest editor invited by the Editorial Board.
References
- 1.Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 2.Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 3.Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 5.Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 6.Keles S, van der Laan M, Eisen MB. Identification of regulatory elements using a feature selection method. Bioinformatics. 2002;18:1167–1175. doi: 10.1093/bioinformatics/18.9.1167. [DOI] [PubMed] [Google Scholar]
- 7.Conlon EM, Liu XS, Lieb JD, Liu JS. Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci USA. 2003;100:3339–3344. doi: 10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao F, Foat BC, Bussemaker HJ. Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data. BMC Bioinformatics. 2004;5:31. doi: 10.1186/1471-2105-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao JC, et al. Network component analysis: Reconstruction of regulatory signals in biological systems. Proc Natl Acad Sci USA. 2003;100:15522–15527. doi: 10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc Natl Acad Sci USA. 2004;101:16234–16239. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das D, Nahl Z, Zhang MQ. Adaptively inferring human transcriptional subnetworks. Mol Syst Biol. 2006;2:msb4100067. doi: 10.1038/msb4100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun N, Carroll RJ, Zhao H. Bayesian error analysis model for reconstructing transcriptional regulatory networks. Proc Natl Acad Sci USA. 2006;103:7988–7993. doi: 10.1073/pnas.0600164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Q, Liu JS. Extracting sequence features to predict protein-DNA interactions: a comparative study. Nucleic Acids Res. 2008;36:4137–4148. doi: 10.1093/nar/gkn361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulesteix AL, Strimmer K. Predicting transcription factor activities from combined analysis of microarray and ChIP data: A partial least squares approach. Theor Biol Med Model. 2005;2:23. doi: 10.1186/1742-4682-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen DH, D'Haeseleer P. Deciphering principles of transcription regulation in eukaryotic genomes. Mol Syst Biol. 2006;2:msb4100054. doi: 10.1038/msb4100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madar A, Bonneau R. Learning global models of transcriptional regulatory networks from data. Methods Mol Biol. 2009;541:1–30. doi: 10.1007/978-1-59745-243-4_9. [DOI] [PubMed] [Google Scholar]
- 17.Chambers I, Smith A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene. 2004;23:7150–7160. doi: 10.1038/sj.onc.1207930. [DOI] [PubMed] [Google Scholar]
- 18.Ivanova N, et al. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Q, Chipperfield H, Melton D, Wong WH. A gene regulatory network in mouse embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:16438–16443. doi: 10.1073/pnas.0701014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irizarry RA, et al. Multiple-laboratory comparison of microarray platforms. Nat Methods. 2005;2:345–350. doi: 10.1038/nmeth756. [DOI] [PubMed] [Google Scholar]
- 23.Shi L, et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo WP, et al. A sequence-oriented comparison of gene expression measurements across different hybridization-based technologies. Nat Biotechnol. 2006;24:832–840. doi: 10.1038/nbt1217. [DOI] [PubMed] [Google Scholar]
- 25.Dennis G, Jr, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 26.Hastie T, Tibshirani R, Friedman JH. The Elements of Statistical Learning. New York: Springer; 2001. [Google Scholar]
- 27.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guccione E, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 31.Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang J, et al. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat Cell Biol. 2008;10:731–739. doi: 10.1038/ncb1736. [DOI] [PubMed] [Google Scholar]
- 33.Moorman C, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci USA. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li XY, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji H, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holter NS, et al. Fundamental patterns underlying gene expression profiles: simplicity from complexity. Proc Natl Acad Sci USA. 2000;97:8409–8414. doi: 10.1073/pnas.150242097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang ZD, et al. Statistical analysis of the genomic distribution and correlation of regulatory elements in the ENCODE regions. Genome Res. 2007;17:787–797. doi: 10.1101/gr.5573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information