Mutations in the Small GTPase Gene RAB39B Are Responsible for X-linked Mental Retardation Associated with Autism, Epilepsy, and Macrocephaly (original) (raw)
Abstract
Human Mental Retardation (MR) is a common and highly heterogeneous pediatric disorder affecting around 3% of the general population; at least 215 X-linked MR (XLMR) conditions have been described, and mutations have been identified in 83 different genes, encoding proteins with a variety of function, such as chromatin remodeling, synaptic function, and intracellular trafficking. The small GTPases of the RAB family, which play an essential role in intracellular vesicular trafficking, have been shown to be involved in MR. We report here the identification of mutations in the small GTPase RAB39B gene in two male patients. One mutation in family X (D-23) introduced a stop codon seven amino acids after the start codon (c.21C > A; p.Y7X). A second mutation, in the MRX72 family, altered the 5′ splice site (c.215+1G > A) and normal splicing. Neither instance produced a protein. Mutations segregate with the disease in the families, and in some family members intellectual disabilities were associated with autism spectrum disorder, epileptic seizures, and macrocephaly. We show that RAB39B, a novel RAB GTPase of unknown function, is a neuronal-specific protein that is localized to the Golgi compartment. Its downregulation leads to an alteration in the number and morphology of neurite growth cones and a significant reduction in presynaptic buttons, suggesting that RAB39B is required for synapse formation and maintenance. Our results demonstrate developmental and functional neuronal alteration as a consequence of downregulation of RAB39B and emphasize the critical role of vesicular trafficking in the development of neurons and human intellectual abilities.
Introduction
Human Mental Retardation (MR) is a common and highly heterogeneous pediatric disorder with a severe social impact. Accounting for a large, but not well-defined, portion of all MR forms, genetic defects range from 25%–50% of the total. Family studies have demonstrated a relatively large number of X-linked forms (XLMR) that seem to explain why the incidence of MR in males is about 30% higher than in females.1 Because large families are not so common and many autosomal genes are expected, the newly identified genes have served as important tools for understanding the molecular basis of MR and have provided novel breakthroughs on the mechanisms and pathways leading to development of the cognitive functions altered in MR.
As a result, at least 215 XLMR conditions and mutations have been characterized in 83 different genes. Research on the role of proteins encoded by these genes led to the hypothesis that XLMR is a synaptopathy-like disorder that is due, in part, to the fact that most of these proteins localize to pre- and/or post-synaptic neuronal terminals.
The identification of guanine nucleotide dissociation inhibitor gene, GDI1 (MIM 300104) as one of the genes causing human XLMR suggested that vesicular trafficking is an important pathway for the development of cognitive functions.2–4 αGDI, the protein encoded by GDI1, is physiologically involved in regulating RAB GTPase proteins.
More than 60 RAB GTPases are encoded by the human genome and are localized to different subcellular compartments.5,6 Through a GDP-GTP exchange cycle, they act as molecular switches to coordinate vesicular transport. The guanine nucleotide-binding status is modulated by three classes of regulatory proteins, the guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and the guanine nucleotide dissociation inhibitor proteins (GDIs), which retrieved GDP-bound, inactive RAB proteins from the membranes.7 RAB and RAB-associated proteins have been shown to play an important role in a number of diseases.8 Thus, the association of GDI1 mutations with XLMR led us to hypothesize a role for X-linked RAB genes in XLMR. Three out of four X-linked RAB genes are specific to the brain, as shown in the SymAtlas database, and we identified novel loss-of-function mutations in the RAB39B (MIM 300774) gene in two XLMR families. RAB39B is one of the RAB GTPase proteins of unknown function, and understanding how the lack of RAB39B is involved in the pathology of XLMR is therefore particularly relevant for understanding the pathogenesis of the disease.
Material and Methods
Patients and Analysis of the RAB39B Gene
In total, 22 DNA samples from males with XLMR mapped to Xq28 were obtained from the European Mental Retardation Consortium in Nijmegen, The Netherlands, from the Greenwood Genetic Center in North Carolina, USA, and from the MRX72 family in Italy.9 We also analyzed 136 DNA samples from unmapped males with XLMR from the Greenwood Genetic Center and the Women's and Children's Hospital, Adelaide, Australia and 110 DNA samples from unmapped males with XLMR from the European Mental Retardation Consortium in Nijmegen, The Netherlands. From these samples, 92 were from patients with XLMR and autism spectrum disorder, 94 from patients with autism spectrum disorder only, and 94 from Greenwood Genetic Center patients with epileptic seizures only.
Control DNAs were from a previous collection of adult European males. All patients and controls were recruited and studied after appropriate ethical approval was obtained. Sequences of the two exons of RAB39B were obtained by PCR with the primers R39b1exF/R39b1exR and R39b2exF/R39b2exR. The same primers were used for direct sequencing on a 3730 automated sequencing apparatus. Analysis was performed with the SeqScape sequence analysis software (Applied Biosystems). Sequences were numbered according to the RAB39B cDNA NM_171998 and protein NP_741995. Primer sequences are listed in Table S1 available with this article online.
Mutated cDNA Cloning, Transfection, and Immunoblotting
Total RNA from a normal subject, family X (D-23), and MRX72 male patients was extracted from lymphoblastoid cell lines with the RNeasy kit (QIAGEN). RNA was reverse transcribed with M-MLV enzyme (Invitrogen). cDNAs were amplified with primers R39b1exF/R39b2exR, inserted into pGEM-T easy vector (Promega), and amplified with specific primers HindhR39bF/BamhR39bR so that they could be cloned in frame into HindIII and BamHI sites of pCMV2-FLAG plasmid (Sigma-Aldrich). pFLAG-RAB39B constructs were transfected into HeLa cells with Lipofectamine 2000 (Invitrogen), as recommended by the supplier. Two days after transfection, cells were harvested and lysed in 1% SDS. Total protein extracts (30 μg) were loaded onto a 10% polyacrilamide gel and then transferred on a nitrocellulose membrane. Polyclonal anti-FLAG antibody (Sigma-Aldrich) was used for detecting the presence of the protein, and beta-Tubulin was used for normalizing the total amount of protein. Primer sequences are listed in Table S1.
RAB39B Expression Profile in Human and Mouse Tissues by qRT-PCR
Human RNA from various tissues was purchased from Clontech. Mouse tissue RNAs were obtained from different C57BL/6N adult mouse organs (Charles River). Total RNA was isolated with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. For the production of cDNA, 1 μg of total RNA was reverse transcribed with M-MLV enzyme (Invitrogen) according to the manufacturer's instructions.
Real-time PCR was performed with SYBR Green Universal Mix (Sigma-Aldrich) on a Light Cycler 480 (Roche Diagnostics). Samples were run in triplicate, and data quantification was done via the comparative threshold cycle (Ct) method with formula 2-(inputCt-controlCt), assuming that the efficiency was 2. Histone H3 with mHistone3F/mHistone3R primers was used for normalization. Human RAB39B and the mouse Rab39b cDNAs were amplified with the same primer pairs, mR39bF/mR39bR. Mouse Rab39a was amplified with primers R39aF/R39aR. Primer sequences are listed in Table S1.
In Situ Hybridization
In situ hybridization was performed on P90 mouse brains with anti-sense and sense RNA probes containing part of the 3′UTR of Rab39b, as previously described.3
Cell Cultures
Primary cultures of hippocampal neurons at embryonic day E18 were prepared as described by Banker and Cowan.10 Cells were grown in MEM supplemented with 10% FBS, 1% glutamine, and 1% penicillin and streptomycin.
cDNA Constructs and MISSION shRNA Validation
For transfection experiments, different plasmid backbones were generated.
For production of pEGFP-RAB39B, the PCR product obtained from mouse Rab39b cDNA clone (BC052472) was cloned with primers R39bgfpF/R39bgfpR into XhoI and HindIII sites of pEGFP-C3 (Clontech).
For production of pEGFP-RAB39A, the PCR product obtained from C57Bl/6N total brain cDNA was cloned with primers R39AF/R39AR into XhoI and HindIII sites of pEGFP-C3.
For production of pFLAG-RAB39B, the PCR product obtained from the BC052472 clone was cloned with primers R39bflagF/R39bflagR into NotI and KpnI sites of pCMV2-FLAG (Sigma-Aldrich).
For generation of pFLAG-RAB39BRescue, the Rab39b sequence of pFLAG-RAB39B was modified with the QuickChange Site-Directed Mutagenesis Kit (Stratagene) and primer pairs RescueR39bF/RescueR39bR. The sequence GCCTACTACAGGAATTCAGTA was replaced with the sequence GCGTATTACCGGAACTCCGTC, which did not change the amino acid composition of RAB39B but rendered the construct insensitive to the _Rab39b_-specific shRNA.
VSV-pseudotyped third-generation lentiviruses were produced as previously described.11 Rab39b was excised via AgeI and SalI sites from pEGFP-RAB39B and cloned into the AgeI and SalI sites of pCCL.sin.cPPT.PGK.GFP.WPRE plasmid (pCCL-RAB39B) carrying a PGK promoter-GFP cassette.11
For the Rab39b silencing experiments, four MISSION shRNAs recognizing different sequences on the Rab39b cDNA (code TRC000046, 47, 48, and 49) and control scramble shRNA were purchased from Sigma-Aldrich. HeLa cells transfected with pEGFP-RAB39B or pEGFP-RAB39A and transduced with the different shRNAs at a multiplicity of infection (MOI) of 1 or 3 were analyzed by both immunoblot and qRT-PCR so that the capacity of shRNAs to specifically knock down Rab39b expression could be tested. Three out of four shRNAs were able to downregulate Rab39b, but not Rab39a, by about 60%. Control-scramble shRNA (shScramble), TRC000047, and 49 shRNAs (shRab39b) were introduced into pCCL.sin.cPPT.PGK.GFP.WPRE lentiviral vector.
Primer sequences are listed in Table S1.
Cell Transfection and Transduction
For the intracellular localization of RAB39B by immunofluorescence experiments, N2A, HeLa, and Cos7 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. So that overexpression of RAB39B did not occur, different amounts of plasmids encoding pEGFP-RAB39B were tested. The convenient condition was 350 ng for N2A and 100 ng for HeLa and Cos7 cells instead of 4 μg as suggested by the supplier.
So that overexpression of RAB39B in hippocampal neurons did not occur, cells were transduced with pCCL-RAB39B lentivirus at an MOI of 1 immediately after plating.
For the rescue experiment, N2A cells were transduced with shRab39b or shScramble lentiviral particles at an MOI of 3, and after 4 days they were transfected for 24 hr with 130 ng of pFLAG-RAB39BRescue or pFLAG-RAB39B. The Rab39b level was analyzed by qRT-PCR as previously described.
BFA Treatment
Two days after transfection with pEGFP-RAB39B, N2A, HeLa or Cos7 cells were treated with 2.5 mg/ml of Brefeldin A (Sigma-Aldrich) for 30 min at 37°C.
FM4-64 Uptake Assay
For FM4-64-uptake experiments, neurons were incubated with 10 μM FM4-64 (Molecular Probes) diluted in Krebs-Ringer-HEPES solution (KRH) containing 55 mM KCl (depolarizing solution) and 1 mM TTX for 1 min at room temperature (RT), rinsed three times by complete substitution with KRH containing 50 mM CNQX and 1 mM TTX over a course of 10 min, and then live imaged.
Immunofluorescence and Microscopy
Cells were fixed with 120 mM phosphate buffer containing 4% paraformaldehyde and 4% sucrose, washed in PBS 1x, and then incubated overnight at 4°C with primary antibodies diluted in GSDB 1x (16% Goat Serum, 0.3% Triton X-100, 20 mM phosphate buffer, and 450 mM NaCl). Primary antibodies against the following proteins were used for immunofluorescence: GM130 (BD Transduction), GIANTIN (gift from M.A. De Matteis), RAB3A, VAMP4, STX16, STX13 (SySy), RAB11 (Transduction Laboratories), Flag (Sigma), AP2 (Affinity BioReagents, clone AP6), SYN1 (gift from F. Valtorta), and SHANK1 (gift from C. Sala). After cells were washed in a high-salt solution (20 mM phosphate buffer and 500 mM NaCl), appropriate rhodamine (TRITC)- or Cy5-conjugated secondary antibodies (Jackson Laboratories) were added for 1 hr at RT. DAPI was used for visualizing cell nuclei. TRITC-conjugated phalloidin (Sigma-Aldrich) was used for visualizing F-Actin. Slides were mounted on Vectashield (VectaLab).
Images were acquired by UltraVIEW ERS spinning-disk confocal microscope (Perkin Elmer).
Image Analysis
ImageJ Analysis Software was used for measuring the area of neuronal growth cones. Sholl analysis was performed as previously described12 so that the degree of neuronal differentiation could be analyzed.
Statistical Analysis
Factorial or repeated-measures analysis of variance (ANOVA) with an alpha value of 5% was used (SAS Institute, Cary, NC). Normal distribution and homogeneity of variances among data samples was tested with the K-S normality test and Bartlett's test for homogeneity. Main effects were verified with t tests. Data points represent the mean, and bars represent the standard error.
Animals
All experiments with animals were performed in strict accordance with the guidelines of the Institutional Animal Care and Use Committee of the Department of Biological and Technological Research, San Raffaele Scientific Institute.
Results
Detection of Mutations in the RAB39B Gene
We examined 22 XLMR families that mapped to Xq28 to determine whether RAB39B was responsible for XLMR. From the genomic sequence of RAB39B, primers were designed for direct sequencing of the two exons and the flanking splice sites of this gene. A nonsense mutation (NM_171998, c.21C > A) in family X (D-23) introduced a premature stop codon seven amino acids after the start codon (TAA; NP_741995, p.Y7X; Figure 1A). In the MRX72 family, a nucleotide substitution (NM_171998, c.215+1G > A) altered the 5′ splice junction of intron 1 (Figure 1B). Using RT-PCR, we demonstrated that a cryptic 5′ splice donor site (GT at 46–47 bp of cDNA, NM_171998) was activated in the first exon and produced an aberrant mRNA that could lead to synthesis of a putative novel peptide of 52 amino acids in length (data not shown). In both cases, immunoblotting of HeLa cells transfected with FLAG-tagged mutated and control cDNAs did not detect any mutant protein (Figure 1C). The mutations segregated with the disease in both families as shown in the family pedigrees (Figures 1D and 1E). Sequencing of 150 normal chromosomes demonstrated that the mutations were not common polymorphisms.
Figure 1.
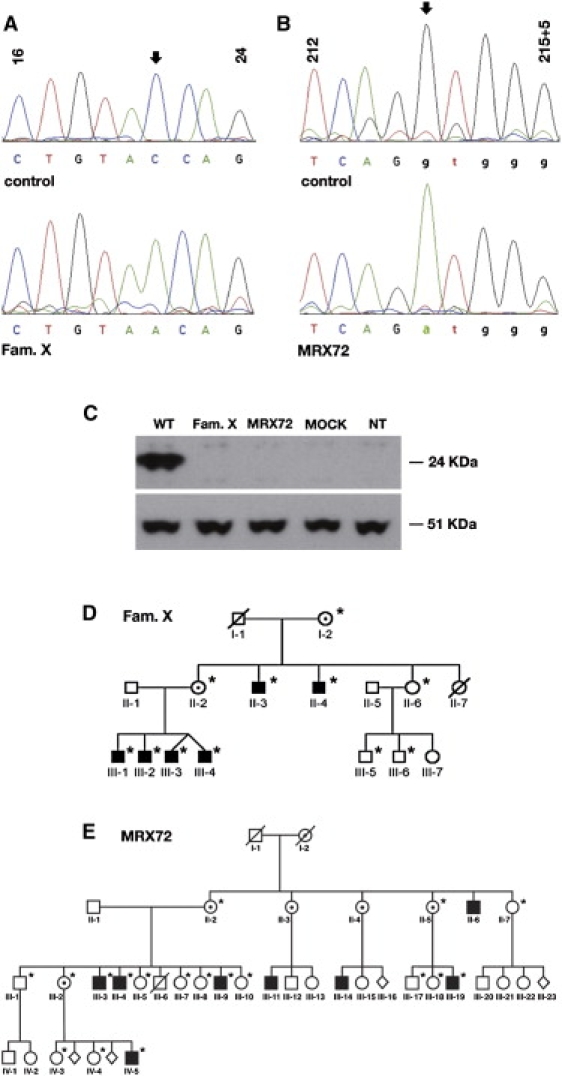
RAB39B Mutations in Family X (D-23) and MRX72
(A and B) The upper panels show the corresponding sequence from a normal individual; the lower panels are from the patients. Arrows indicate the mutations. Sequence numbers refer to the human RAB39B cDNA (NM_171998).
(C) Immunoblot analysis performed on 30 μg of total protein extracted from HeLa cells transfected with pFLAG-RAB39B constructs containing the wild-type RAB39B cDNA (WT), family X (D-23) mutated cDNA, or MRX72 cDNA. The 24-kDa band corresponds to FLAG-RAB39B fusion protein, and the 51-kDa band corresponds to beta-tubulin. MOCK: cells transfected with pCMV2-FLAG. NT: cells not transfected.
(D) Pedigree of family X (D-23).
(E) Pedigree of MRX72. Black squares indicate males with mental retardation, dotted circles indicate carrier females, and open symbols indicate unaffected individuals. Asterisks indicate family members who have been sequenced in this study.
Patients in both families presented with moderate to severe MR, as well as additional features; in the MRX72 family, III-9, III4, and III-5 had seizures, and IV-5 presented with autism spectrum disorder (Figure 1E).9 In family X (D-23), four brothers (III-1, III-2, III-3, and III-4) and two maternal uncles (II-3 and II-4) were mentally retarded, and these six patients presented with macrocephaly, as described in Table 1.
Table 1.
Clinical Evaluation of Family X (D-23)
| Clinical Feature | II-3 | II-4 | III-1 | III-2 | III-3 | III-4 |
|---|---|---|---|---|---|---|
| Age (yr) | 52 | 50 | 19 | 16 | 13 | 13 |
| Height (cm) | 163 (<3rd percentile) | 170 (3rd percentile) | 178.5 (50th percentile) | 177.5 (50th percentile) | 145.5 (3rd percentile) | 145.7 (3rd percentile) |
| OFC | 59 (>97th percentile) | 62.5 (>97th percentile) | 60.5 (>97th percentile) | 56.5 (>97th percentile) | 57 (>97th percentile) | 56.5 (>97th percentile) |
| Degree of MR | mild | severe | moderate | moderate | moderate | severe |
| Additional features | - | - | obesity | - | autism | autism |
Because of the presence of seizures and autism spectrum disorder in family X (D-23) and MRX72, we analyzed 526 DNA samples from unmapped X-linked patients with seizures and autism spectrum disorder. We found the same synonymous variation (c.822A > G, p.T181T) that Tarpey et al.13 described previously in two unrelated autistic males.
RAB39B Is Highly Enriched in the Brain
We performed qRT-PCR on several human and mouse tissues to analyze RAB39B expression. We demonstrated that RAB39B is enriched in the brain (Figures 2A and 2B).
Figure 2.
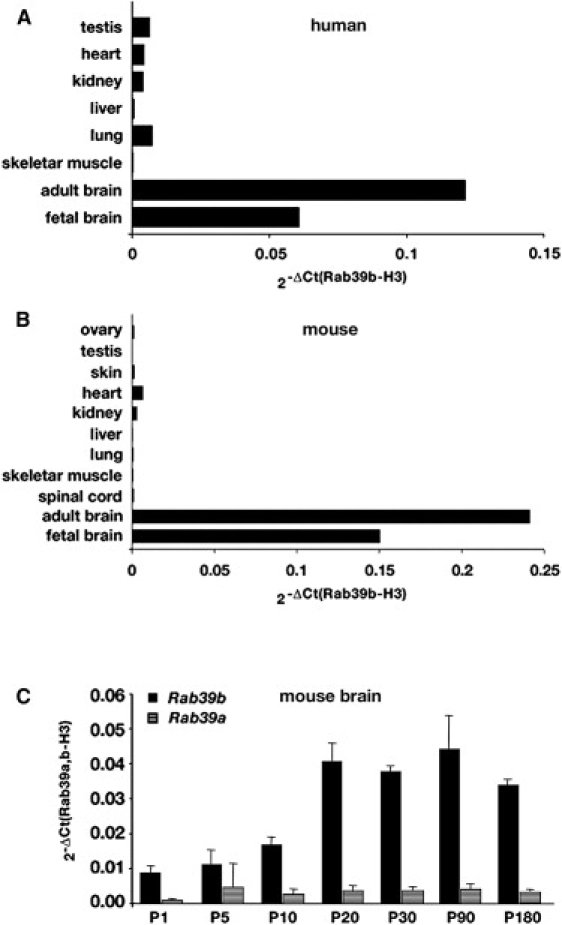
Expression Analysis on Different Human and Mouse Tissues
(A) Expression of RAB39B transcript in different human tissues.
(B) Expression of Rab39b transcript in mouse tissues.
(C) Expression profile of Rab39b and Rab39a in mouse total brain during post-natal (P) development (from P1 to P180; for each age point, n = 8). Data were expressed as Rab39b or Rab39a expression normalized to Histone-H3 (2-ΔCt(_Rab39b_–H3)) (±SE).
During postnatal (P) mouse brain development, Rab39b expression is constant between P1 and P10, as well as between P20 and P180. A significant difference was observed between P10 to P20, where Rab39b was upregulated 2-fold as calculated by the t test (p = 0.003). We also analyzed the expression profile of the Rab39a gene, which has 72% and 76% identity at the cDNA and protein levels, respectively. The Rab39a transcript was expressed at levels ten times less than Rab39b and does not increase at all stages analyzed. These results demonstrated that Rab39b is a brain-specific gene with an important role (Figure 2C). Expression analysis of the Gdi1 null brains revealed no change, suggesting that absence of αGDI did not alter Rab39b RNA levels, as previously shown for other RAB proteins3 (data not shown).
Rab39b Is a Neuron-Specific Gene
Rab39b appeared to be ubiquitously expressed in brain, as determined by in situ hybridization on sagittal sections of adult (P90) mouse brain. The highest expression was observed in the hippocampus, as shown in Figures 3A–3F.
Figure 3.
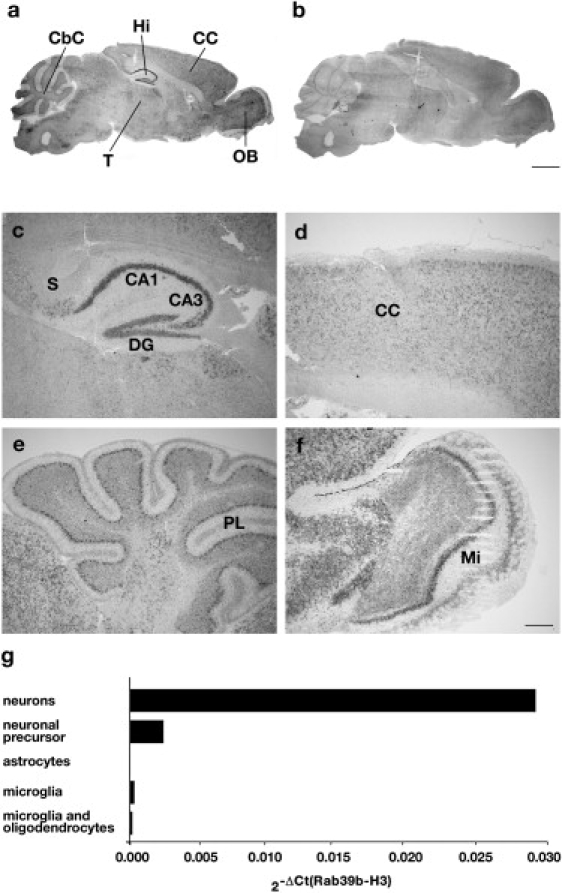
Rab39b Is Specifically Expressed in Neurons
(A) In situ hybridization analysis of Rab39b on P90 mouse brain. Light-microscopic images showing the distribution of Rab39b mRNA in 20 μm mouse brain sagittal sections. Anti-sense (A) and sense (B) Rab39b probe.
(C–F) Enlargements of specific regions: CC, cerebral cortex; CbC, cerebellar cortex; Hi, hippocampus; OB, olfactory bulb; T, thalamus; S, subiculum; CA1 and CA3, fields of the hippocampus; DG, dentate gyrus; PL, Purkinje layer; and Mi, mitral cells. Scale bars represent 2 mm (A and B) and 0.1 mm (C–F).
(G) Rab39b expression normalized to Histone-H3 (2-ΔCt(_Rab39b_–H3)) in neuronal cell types. P2 mouse cerebral cortices were dissected, dissociated in trypsin medium, and plated. Different neuronal cell types were prepared as described.14Rab39b is specifically expressed in neuronal precursors and neurons.
We further analyzed the Rab39b neuronal cellular specificity. We dissected and dissociated P2 mouse cerebral cortices to obtain different neuronal cell types such as microglia, oligodendrocytes, astrocytes, neuronal precursors, and neurons, as described.14 By qRT-PCR, we quantified the Rab39b expression in these cell types and showed that Rab39b was specifically expressed in neuronal precursors and neurons only (Figure 3G).
RAB39B Localizes to the Golgi Compartment
We analyzed the intracellular localization of RAB39B in pEGFP-RAB39B-transfected Cos7 and HeLa cells (data not shown), and in a pCCL-RAB39B-transduced neuroblastoma cell line, N2A (Figure S1a), and primary hippocampal neurons (Figure 4). In all instances, RAB39B appeared to localize to the Golgi compartment, as judged from the colocalization with the Golgi markers, GIANTIN and GM130. We confirmed the Golgi localization by treating N2A cells for 30 min with or without brefeldin A (+ BFA and − BFA), respectively (Figure S1b). GFP-RAB39B and anti-GIANTIN immunostaining, as well as GFP-RAB39B immunostaining after BFA treatment, showed dispersion of the Golgi compartment. We also observed a partial colocalization with markers that cycle from the cell surface to the trans-Golgi network (TGN) via sorting and recycling endosomes such as VAMP4, SYNTAXIN16, and RAB11.15–17 The latter subcellular colocalization suggests a possible role of RAB39B in the recycling pathway connecting the plasma membrane, endosomes, and TGN.
Figure 4.
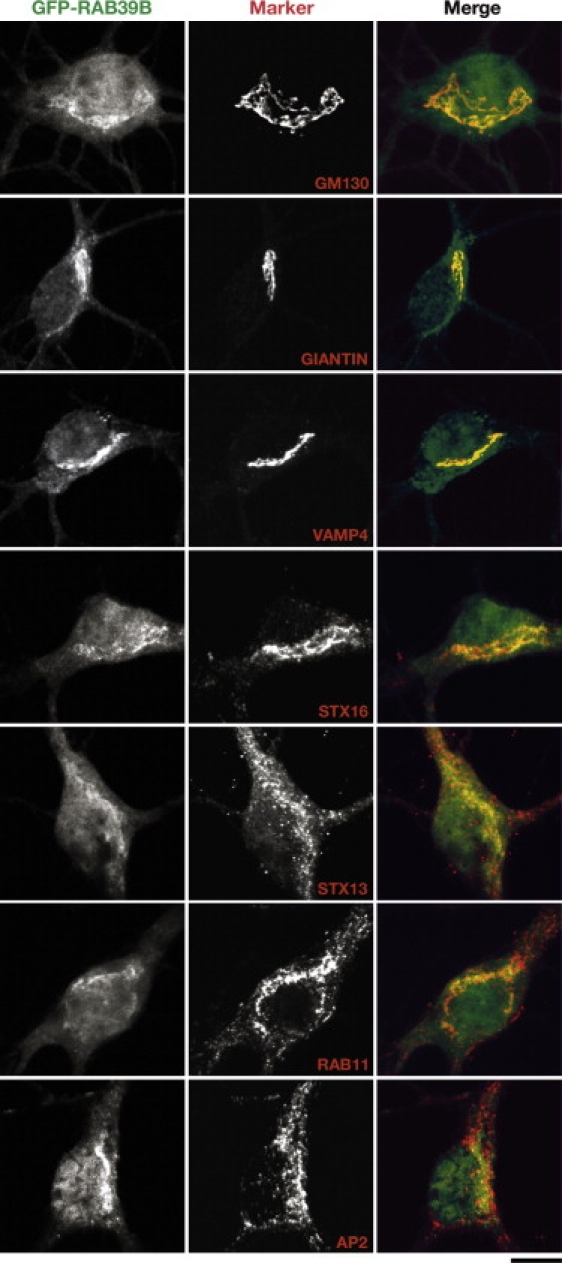
RAB39B Intracellular Localization in Mouse Hippocampal Neurons
Neurons were transduced at 3 DIV with pCCL-RAB39B lentiviral particles (green) at an MOI of 1 and analyzed at 7 DIV. The major colocalization was with GM130 (_cis_-Golgi) and GIANTIN (Golgi complex), VAMP4 and SYNTAXIN16 (STX16)—involved in trafficking from the cell surface to the trans-Golgi network (TGN)—and SYNTAXIN13 (STX13) and RAB11, which are present in recycling endosomes. No colocalization was observed with the adaptor protein 2 (AP2). The scale bar represents 10 μm.
Downregulation of Rab39b by shRNA
Undifferentiated N2A cells were infected with two independent shRNAs against Rab39b at an MOI of 3. As determined by qRT-PCR after 5 days of infection, both shRNAs were able to downregulate the endogenous Rab39b expression by 45%, expressed as n-fold changes (± standard error [SE]) relative to untreated cells (shRab39b: 0.55 ± 0.02). In contrast, shScramble did not have any effect (shScramble: 0,91 ± 0.006) (Figure S2a).
The specificity of shRab39b was determined by a knockdown-rescue experiment. N2A cells were transduced with the shRab39b lentiviral particles for 4 days and then transfected with the full-length wild-type Rab39b (pFLAG-RAB39B) or pFLAG-RAB39BRescue constructs. In cells treated with shRab39b and transfected with pFLAG-RAB39B, the wild-type pFLAG-RAB39B expression was downregulated by 45% in comparison to pFLAG-RAB39B-transfected cells as determined by qRT-PCR (shRab39b + pFLAG-RAB39B: 0.55 ± 0.1; value expressed as n-fold changes ± SE). We also showed that shRab39b was not able to downregulate the rescue Rab39b transcript (Figures S2b and S2c).
After 4 days of treatment with the shRab39b lentivirus, we observed a change in N2A cell morphology. To further investigate the morphological phenotype, we stained N2A shRab39b lentivirus-treated cells with phalloidin as a marker for actin cytoskeleton morphology. We found clumps of actin and reduced and/or altered membrane protrusions that were not present in shScramble-treated N2A cells (Figure S2d). After 4 days, transfection of shRab39b-treated N2A cells with pFLAG-RAB39BRescue for 24 hr was able to restore the normal actin cytoskeleton morphology.
These results suggested that the downregulation of Rab39b might alter intracellular vesicular trafficking steps important for membrane remodeling.
Downregulation of Rab39b Gene Alters Neuronal Differentiation
To mimic the loss-of-function mutations identified in patients, we knocked down Rab39b expression in mouse primary hippocampal neurons. Immediately after plating, hippocampal neurons were transduced with shRab39b and shScramble lentiviral particles at an MOI of 1. ShRAB39b was able to reduce Rab39b expression by 40%, as determined at 3 days in vitro (DIV) by qRT-PCR. Morphological analysis from 80 shScramble and 124 shRab39b randomly sampled images from four independent experiments showed a significant alteration in morphology and the number of growth cones (GCs; Figure 5A). GCs were decreased in number at each neurite terminal. The mean number of GCs per neurite (±SE) was 3.1 (±0.1) for shRAB39b and 3.8 (±0.1) for shScramble neurons. The difference was significant (ANOVA: F [1,202] = 8.2; p = 0.005). The mean area of GCs (±SE) was 38.7 (±2.8) for shRab39b and 30.8 (±2.4) for shScramble neurons. The difference was significant (ANOVA: F [1,202] = 3.8, p = 0.05) (Figures 5B and 5C; see the enlargement in the right panel).
Figure 5.
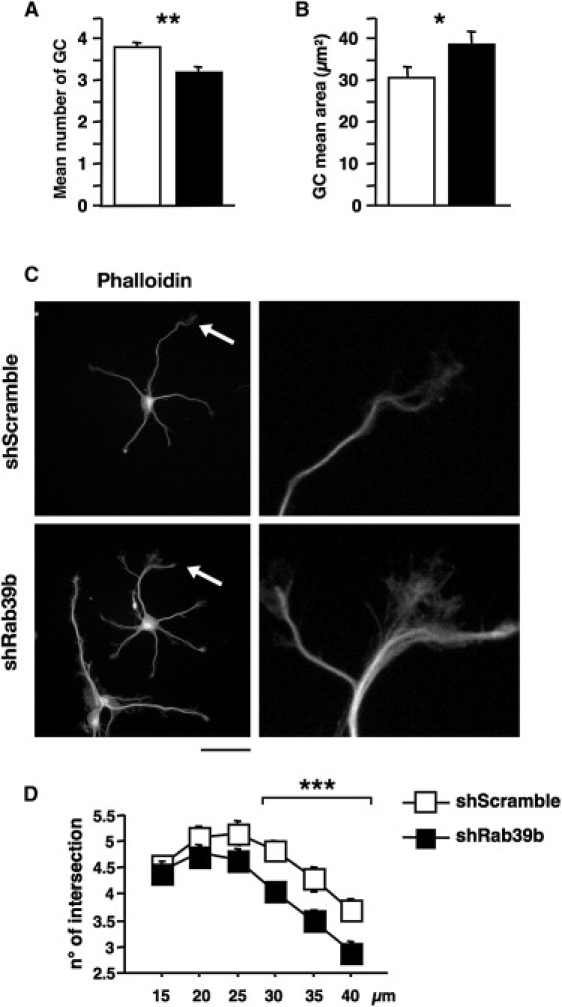
RAB39B Is Involved in Neurite Differentiation
Hippocampal neurons were transduced with lentiviral particles containing shRab39b and shScramble at an MOI of 1. The downregulation of the endogenous Rab39b RNA level was quantified at 3 DIV by qRT-PCR, and 40% reduction was observed in shRab39b-transduced neurons.
(A) There was a significant difference (p = 0.005) in the mean number of growth cones (GCs) at each neurite terminal (±SE) in shRab39b neurons (3.1 ± 0.1) versus shScramble neurons (3.8 ± 0.1) (mean ± SEM).
(B) There was a significant difference (p = 0.05) in the mean area of GCs (±SE) in shRab39b neurons versus shScramble neurons (38.7 ± 2.8 [mean ± SEM] and 30.8 ± 2.4, respectively).
(C) TRITC-Phalloidin immunostaining revealed altered GC morphology (enlargement of GC morphology in right panel). The scale bar represents 10 μm.
(D) For Sholl analysis,12 as a measure of neuronal differentiation, a mask of concentric circles 5 μm apart was created and superimposed on neuronal cells. The first 10 μm from the center of the mask corresponds to the neuronal cell body. Repeated-measures ANOVA revealed significant differences between treatments, where F (1,147) = 6.6, p = 0.01, and between treatment and Sholl category (μm from the cell body), where F (1,5) = 2.3, p = 0.04. Factorial ANOVA for each Sholl category revealed a significant p value for 30 μm [F (1,147) = 9.3, p = 0.003], 35 μm [F (1,147) = 7.6, p = 0.006], and 40 μm [F (1,147) = 7.2; p = 0.007].
∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Data are expressed as means ±SE.
Sholl analysis,12 as a measure of neuronal differentiation, was performed on 62 shScramble and 87 shRab39b randomly sampled images from three independent experiments and also revealed a significant decrease in the number of neuronal branches: repeated-measures ANOVA between treatments had a value of F [1,147] = 6.6, p = 0.01 and showed an interaction between treatment and Sholl category (μm from the cell body): F [1,5] = 2.3, p = 0.04. Factorial ANOVA for each Sholl category revealed significant p values for 30 μm (F [1,147] = 9.3, p = 0.003), 35 μm (F [1,147] = 7.6, p = 0.006), and 40 μm (F [1,147] = 7.2; p = 0.007) (Figure 5D). Taken together, these data suggest a global alteration in neurite extension.
RAB39B Was Localized to GCs
To test whether RAB39B was present in GCs, we transduced primary hippocampal neurons at 3 DIV with pCCL-RAB39B lentiviral particles. We found that GFP-RAB39B signals were spread in the whole GC area, as shown by phalloidin staining (Figure 6A), and some discrete signals colocalized with the Golgi-derived vesicles, as shown in the double labeling with GIANTIN (Figure 6B). All these data suggested that RAB39B could be involved in intracellular trafficking related to membrane recycling of Golgi-derived vesicles; its reduction may impede such trafficking and thus lead to an accumulation in the membrane content and disorganized growth of GCs.
Figure 6.
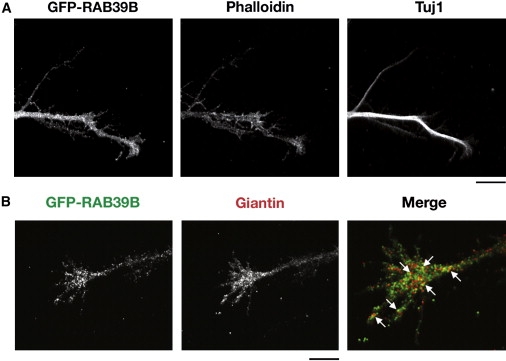
RAB39B Localizes to Golgi-Derived Vesicles
(A) Hippocampal neurons transduced immediately after being plated with pCCL-RAB39B lentiviral particles at an MOI of 1 and immunostained at 3 DIV. TRITC-Phalloidin and anti-Tuj1 immunostaining revealed that RAB39B occupied a region similar to that of the GC actin domain.
(B) A partial colocalization was observed with GIANTIN, as a marker for the Golgi-derived vesicles, as shown by the arrows in the enlargement in the merged right panel.
The scale bar represents 10 μm.
RAB39B Is Involved in Synapse Formation
Next, we looked at the subsequent formation of pre-synaptic terminals of primary neurons transduced immediately after plating with shRab39b and shScramble lentiviral particles at an MOI of 1 until 7 DIV. The downregulation of endogenous Rab39b RNA levels was quantified at 7 DIV by qRT-PCR, and we found that the Rab39b expression was 70% reduced in shRab39b-treated neurons compared with shScramble-treated neurons.
Analysis of SYNAPSIN1-positive puncta, a marker of the presynaptic compartment, revealed 49% reduction in the mean number of synapses. The quantification of the mean number of synapses (±SE) obtained from 56 shRab39b-treated neurons was 115 (±9.2), compared with 223 (±17.7) from 53 shScramble randomly sampled images from three independent experiments. Factorial ANOVA revealed F [1,97] = 32.7 and p < 0.0001 (Figures 7A and 7B). We then measured FM4-64 uptake to analyze the ability of the synaptic vesicles in the remaining synapses to engage in exo- and endocytosis. The quantification of 85 shRab39b and 93 shScramble randomly sampled images from three independent experiments revealed that the recycling of synaptic vesicles appeared to be unaffected because no gross differences in exo- or endocytosis activity were observed. Instead, FM4-64-positive labeling (±SE) showed a 40% reduction in shRab39b neurons (163 ± 8.9) compared to shScramble-treated (272 ± 12) ones, and a significant difference was observed by factorial ANOVA (F [1,151] = 50.7, p < 0.0001) (Figure 7C). These data suggest that the alteration observed in neurite extension at 3 DIV might lead to defective synapse formation or instability.
Figure 7.
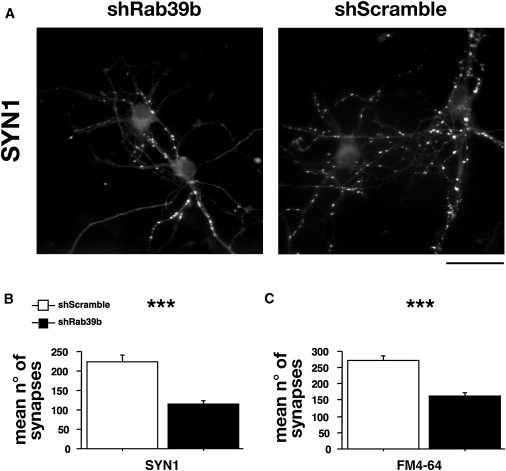
RAB39B Is Involved in Synapse Formation
Hippocampal neurons were transduced immediately after being plating with shRab39b and shScramble lentiviral particles at an MOI of 1. The downregulation of the endogenous Rab39b RNA level was quantified at 7 DIV by qRT-PCR, and 70% reduction was observed in shRab39b-transduced neurons. (A and B) SYNAPSIN1 immunostaining and quantification showed a 49% reduction in the SYN1 pre-synaptic compartment on shRab39b compared to shScramble neurons.
(C) FM4-64-positive labeling showed a 40% reduction for shRab39b neurons compared to shScramble.
The scale bar represents 10 μm. ∗∗∗p < 0.001. Data are expressed as means ±SE.
Discussion
The four X-linked RAB genes were considered as candidate genes for XLMR. Mutation analysis was started on the RAB39B gene, and here we have demonstrated that when mutated, the gene can cause XLMR. We report the identification of two independent point mutations in two unrelated families affected with XLMR. These mutations disrupt the synthesis of RAB39B by introducing a premature stop codon in the open reading frame. The mutations cosegregated with the disease and were not found in 150 controls. Some of the affected males from both families primarily presented with mental retardation associated with autism spectrum disorder, epileptic seizures, and macrocephaly. This adds further evidence that XLMR is a highly heterogeneous condition in which the genetic background and/or environmental factors contribute to determining the clinical phenotype.
More than 60 RAB GTPases are encoded by the human genome and localize to different subcellular compartments.6 Through a GDP-GTP exchange cycle, they act as molecular switches to coordinate vesicular transport. The association of these proteins and their regulators with many diseases reflects the physiological importance of RAB GTPases, which is not surprising in light of their central role in membrane trafficking. To date, mutations in two RAB GTPases have been found in association with hereditary neurological disorders. RAB7 (MIM 602298) mutations are responsible for Charcot-Marie-Tooth type 2B neuropathy (CMT2B [MIM 600882])18 and ulcero-mutilating neuropathy,19 and Rab23 (MIM 606144) mutations are responsible for Carpenter syndrome (ACPSII [MIM 201000]) characterized by cranial-suture defects.20 Among X-linked genes and in addition to GDI1 (MIM 300104), mutation analysis in the three X-linked RAB genes could be helpful to highlight new XLMR genes and further evaluate the prevalence of mutations in this gene family in MR. Along the same lines, we should also mention the finding of mutations in the Rab3 p130 GAP gene (MIM 602536) in Warburg Micro Syndrome (WARBM [MIM 600118])21 and Martsolf Syndrome (MIM 212720),22 severe autosomal-recessive disorders characterized by developmental defects and mental retardation that are possibly also due to an alteration in neuronal intracellular trafficking.
The exact cellular localization, tissue expression profile, and physiological function of many RAB GTPases is still unknown, and this is the case for RAB39B. We showed that Rab39b is a brain-enriched gene that is specifically expressed in neurons. A polyclonal antibody against the C terminus hypervariable amino acid sequence of RAB39B was produced but failed to work in immunofluorescence experiments. We investigated the cellular localization by using a GFP-RAB39B fusion protein, and we tried to minimize the overexpression by using lentiviral infection at an MOI of 1 and at least three different cell types. We observed a diffuse staining in the cytoplasm and in the nucleus in our experiments; this staining was possibly caused by GFP-RAB-fused proteins, as shown by several papers reporting a similar diffuse pattern after transfection of RAB proteins to determine intracellular localization.23,24 We cannot demonstrate that this is the case, but we think this is an unspecific staining possibly due to GFP tagging.
We showed that GFP-RAB39B fusion protein partially colocalized with known proteins of the Golgi compartment and with markers that cycle from the cell surface to the trans-Golgi network (TGN) via sorting and recycling endosomes. The latter subcellular colocalization suggests a possible role for RAB39B in the recycling pathway connecting the plasma membrane, endosomes, and TGN. Searching for interacting RAB39B proteins led to the identification of different proteins, including one Golgi-associated protein (unpublished data). These preliminary data further support the localization of RAB39B to the Golgi compartment.
We investigated the effect of loss-of-function mutations identified in XLMR patients. We used the RNA interference technology (shRNA) to knock down the Rab39b gene in mouse primary hippocampal neurons and investigate a possible effect on neuronal development and connectivity. To test a possible off-target silencing effect25 and downregulation specificity, we used three shRNAs targeting different portions of the Rab39b cDNA in N2A cells and performed a knockdown-rescue experiment. Our data showed that all shRNAs were able to downregulate the expression of Rab39b but not Rab39a, which shares high sequence homology with Rab39b. N2A shRNA-treated cells revealed clumps of actin and altered membrane protrusions that returned to normal in the knockdown-rescue experiment, as a model of membrane remodeling.
In primary hippocampal neurons, Rab39b downregulation resulted in the alteration of growth cone morphology and neuronal branching and a global alteration in neurite arborization. Localization of RAB39B to the growth cones, and the essential role of Golgi-derived vesicles for the successful generation of a functional GC,26 suggest that RAB39B could be involved in intracellular trafficking related to membrane recycling throughout Golgi-derived vesicles. The reduction of RAB39B might impede such trafficking and lead to an accumulation in the membrane and disorganized growth of GCs. The observed alteration in neurite extension might be the cause of defective synapse formation or instability, as shown in adult hippocampal neurons. Rab39b downregulation resulted in a drastic reduction in the total number of synapses without gross differences in synaptic-vesicle recycling in the remaining synapses.
In conclusion, we identified mutations in the neuron-specific RAB39B gene, a new gene for XLMR associated with autism spectrum disorder, epileptic seizures, and macrocephaly.
We showed that RAB39B seems to play a key role in synapse formation, possibly by mediating an intracellular pathway connecting the plasma membrane, endosomes, and TGN and possibly in the organization of neuronal circuits. Further experiments are needed to elucidate the biological function of RAB39B and the alterations in neuronal organization and connectivity caused by its absence.
Supplemental Data
Supplemental Data include two figures and one table and can be found with this article online at http://www.cell.com/AJHG.
Supplemental Data
Document S1. Two Figures and One Table
Web Resources
The URLs for data presented herein are as follows:
- X-linked mental retardation, http://www.ggc.org/xlmr.htm and http://xlmr.interfree.it/home.htm
- Gene expression, http://symatlas.gnf.org/SymAtlas/symform
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.org/Omim
- Statistic analysis, http://www.statview.com
- European Mental Retardation Consortium, http://www.euromrx.com
Acknowledgments
We thank the subjects who participated in this study and their parents. We also thank L. Wrabetz and B. Oostra for their helpful discussion, comments, and critical reading of the manuscript; R. Cassinari for graphic support; ALEMBIC for the microscopy technical support; L. Naldini, L. Sergi Sergi for the help in lentiviral production; and A. De Matteis for providing us with a surplus of antibodies. This work was supported by funding from Telethon, Italy (TCP04015 to P.D.). M.G. is supported by a fellowship from the Italian Ministry of Research and Health (PRIN 2007). V.B. is supported by a Ph.D. fellowship from Vita Salute San Raffaele University, Milan, Italy. This study was also supported, in part, by grants from the National Institute of Child Health and Human Development (HD26202 to C.E.S.) and the South Carolina Department of Disabilities and Special Needs.
References
- 1.Herbst D.S. Nonspecific X-linked mental retardation I: A review with information from 24 new families. Am. J. Med. Genet. 1980;7:443–460. doi: 10.1002/ajmg.1320070406. [DOI] [PubMed] [Google Scholar]
- 2.D'Adamo P., Menegon A., Lo Nigro C., Grasso M., Gulisano M., Tamanini F., Bienvenu T., Gedeon A.K., Oostra B., Wu S.K. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat. Genet. 1998;19:134–139. doi: 10.1038/487. [DOI] [PubMed] [Google Scholar]
- 3.D'Adamo P., Welzl H., Papadimitriou S., Raffaele di Barletta M., Tiveron C., Tatangelo L., Pozzi L., Chapman P.F., Knevett S.G., Ramsay M.F. Deletion of the mental retardation gene Gdi1 impairs associative memory and alters social behavior in mice. Hum. Mol. Genet. 2002;11:2567–2580. doi: 10.1093/hmg/11.21.2567. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi V., Farisello P., Baldelli P., Meskenaite V., Milanese M., Vecellio M., Muhlemann S., Lipp H.P., Bonanno G., Benfenati F. Cognitive impairment in Gdi1-deficient mice is associated with altered synaptic vesicle pools and short-term synaptic plasticity, and can be corrected by appropriate learning training. Hum. Mol. Genet. 2009;18:105–117. doi: 10.1093/hmg/ddn321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pereira-Leal J.B., Seabra M.C. The mammalian Rab family of small GTPases: Definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J. Mol. Biol. 2000;301:1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- 6.Pereira-Leal J.B., Seabra M.C. Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 7.Pfeffer S., Aivazian D. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 2004;5:886–896. doi: 10.1038/nrm1500. [DOI] [PubMed] [Google Scholar]
- 8.Corbeel L., Freson K. Rab proteins and Rab-associated proteins: Major actors in the mechanism of protein-trafficking disorders. Eur. J. Pediatr. 2008;167:723–729. doi: 10.1007/s00431-008-0740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo S., Cogliati F., Cavalleri F., Cassitto M.G., Giglioli R., Toniolo D., Casari G., Larizza L. Mapping to distal Xq28 of nonspecific X-linked mental retardation MRX72: Linkage analysis and clinical findings in a three-generation Sardinian family. Am. J. Med. Genet. 2000;94:376–382. doi: 10.1002/1096-8628(20001023)94:5<376::aid-ajmg6>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 10.Banker G.A., Cowan W.M. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 11.De Palma M., Naldini L. Transduction of a gene expression cassette using advanced generation lentiviral vectors. Methods Enzymol. 2002;346:514–529. doi: 10.1016/s0076-6879(02)46074-0. [DOI] [PubMed] [Google Scholar]
- 12.Sholl D.A. A discussion on the measurement of growth and form; The theory of differential growth analysis. Proc. R. Soc. Lond. B Biol. Sci. 1950;137:470–474. doi: 10.1098/rspb.1950.0056. [DOI] [PubMed] [Google Scholar]
- 13.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butovsky O., Ziv Y., Schwartz A., Landa G., Talpalar A.E., Pluchino S., Martino G., Schwartz M. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Khvotchev M.V., Ren M., Takamori S., Jahn R., Sudhof T.C. Divergent functions of neuronal Rab11b in Ca2+-regulated versus constitutive exocytosis. J. Neurosci. 2003;23:10531–10539. doi: 10.1523/JNEUROSCI.23-33-10531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amessou M., Fradagrada A., Falguieres T., Lord J.M., Smith D.C., Roberts L.M., Lamaze C., Johannes L. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. J. Cell Sci. 2007;120:1457–1468. doi: 10.1242/jcs.03436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran T.H., Zeng Q., Hong W. VAMP4 cycles from the cell surface to the trans-Golgi network via sorting and recycling endosomes. J. Cell Sci. 2007;120:1028–1041. doi: 10.1242/jcs.03387. [DOI] [PubMed] [Google Scholar]
- 18.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houlden H., King R.H., Muddle J.R., Warner T.T., Reilly M.M., Orrell R.W., Ginsberg L. A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004;56:586–590. doi: 10.1002/ana.20281. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins D., Seelow D., Jehee F.S., Perlyn C.A., Alonso L.G., Bueno D.F., Donnai D., Josifova D., Mathijssen I.M., Morton J.E. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aligianis I.A., Johnson C.A., Gissen P., Chen D., Hampshire D., Hoffmann K., Maina E.N., Morgan N.V., Tee L., Morton J. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005;37:221–223. doi: 10.1038/ng1517. [DOI] [PubMed] [Google Scholar]
- 22.Aligianis I.A., Morgan N.V., Mione M., Johnson C.A., Rosser E., Hennekam R.C., Adams G., Trembath R.C., Pilz D.T., Stoodley N. Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am. J. Hum. Genet. 2006;78:702–707. doi: 10.1086/502681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barbero P., Bittova L., Pfeffer S.R. Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J. Cell Biol. 2002;156:511–518. doi: 10.1083/jcb.200109030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh T., Fujita N., Kanno E., Yamamoto A., Yoshimori T., Fukuda M. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell. 2008;19:2916–2925. doi: 10.1091/mbc.E07-12-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao D.D., Vorhies J.S., Senzer N., Nemunaitis J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009;61:746–759. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Erez H., Malkinson G., Prager-Khoutorsky M., De Zeeuw C.I., Hoogenraad C.C., Spira M.E. Formation of microtubule-based traps controls the sorting and concentration of vesicles to restricted sites of regenerating neurons after axotomy. J. Cell Biol. 2007;176:497–507. doi: 10.1083/jcb.200607098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Two Figures and One Table