Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles (original) (raw)
. Author manuscript; available in PMC: 2010 Oct 15.
Published in final edited form as: Nature. 2010 Mar 21;464(7291):1067–1070. doi: 10.1038/nature08956
Abstract
Therapeutics that are designed to engage RNA interference (RNAi) pathways have the potential to provide new, major ways of imparting therapy to patients.1,2 Fire et al. first demonstrated that long, double stranded RNAs mediate RNAi in Caenorhabditis elegans,3 and Elbashir et al. opened the pathway to the use of RNAi for human therapy by showing that small interfering RNAs (siRNAs: ca. 21 base pair double stranded RNA) can elicit RNAi in mammalian cells without producing an interferon response.4 We are currently conducting the first-in-human Phase I clinical trial involving the systemic administration of siRNA to patients with solid cancers using a targeted, nanoparticle delivery system. Here we provide evidence of inducing an RNAi mechanism of action in a human from the delivered siRNA. Tumor biopsies from melanoma patients obtained after treatment reveal: (i) the presence of intracellularly-localized nanoparticles in amounts that correlate with dose levels of the nanoparticles administered (this is a first for systemically delivered nanoparticles of any kind), and (ii) reduction in both the specific mRNA (M2 subunit of ribonucleotide reductase (RRM2)) and the protein (RRM2) when compared to pre-dosing tissue. Most importantly, we detect the presence of an mRNA fragment that demonstrates siRNA mediated mRNA cleavage occurs specifically at the site predicted for an RNAi mechanism from a patient who received the highest dose of the nanoparticles. These data when taken in total demonstrate that siRNA administered systemically to a human can produce a specific gene inhibition (reduction in mRNA and protein) by an RNAi mechanism of action.
A major challenge with the use of siRNAs in mammals is their intracellular delivery to specific tissues and organs that express the target gene. The first demonstrations of siRNA-mediated gene silencing in mammals through systemic administration were accomplished using naked siRNA and methods of administration not compatible with clinical application.5–7 Since then, several delivery vehicles have been combined with siRNAs to improve their delivery in animal models.1,2 Soutschek et al. were the first to provide direct evidence for the siRNA mechanism of action by using a modified 5’-RACE (rapid amplification of cDNA ends) PCR technique providing positive identification of the specific mRNA cleavage product.8 Human clinical trials with synthetic siRNAs began in 2004, utilizing direct intraocular siRNA injections for patients with blinding choroidal neovascularization (CNV). Subsequently, other clinical trials have initiated2 and early clinical data are beginning to appear.9,10 While there are animal studies that do support the possibility of an RNAi mechanism of action from administered siRNA,11 other animal data from siRNAs injected into the eyes of mice for the treatment of CNV suggest non-RNAi mechanisms of action for CNV suppression.12 At this time, no direct evidence for an RNAi mechanism of action in humans from siRNA administered either locally or systemically has been reported.
We are currently conducting the first siRNA clinical trial that utilizes a targeted nanoparticle delivery system (clinical trial registration number NCT00689065).13 Patients with solid cancers refractory to standard-of-care therapies are administered doses of targeted, nanoparticles on days 1, 3, 8 and 10 of a 21-day cycle via a 30-minute i.v. infusion. The nanoparticles consist of a synthetic delivery system containing (Fig. 1a): (i) a linear, cyclodextrin-based polymer (CDP), (ii) a human transferrin protein (hTf) targeting ligand displayed on the exterior of the nanoparticle to engage Tf receptors (hTfR) on the surface of the cancer cells, (iii) a hydrophilic polymer (polyethylene glycol (PEG) used to promote nanoparticle stability in biological fluids), and (iv) siRNA designed to reduce the expression of the M2 subunit of ribonucleotide reductase (RRM2: sequence used in the clinic was previously denoted siR2B+5).14 The TfR has long been known to be up-regulated in malignant cells,15 and RRM2 is an established anti-cancer target.16 These nanoparticles (clinical version denoted as CALAA-01) have been shown to be well tolerated in multi-dosing studies in non-human primates.17 While a single patient with chronic myeloid leukemia has been administered siRNA via liposomal delivery,18 our clinical trial is the initial human trial to systemically deliver siRNA with a targeted delivery system and to treat patients with solid cancer.13
Figure 1.
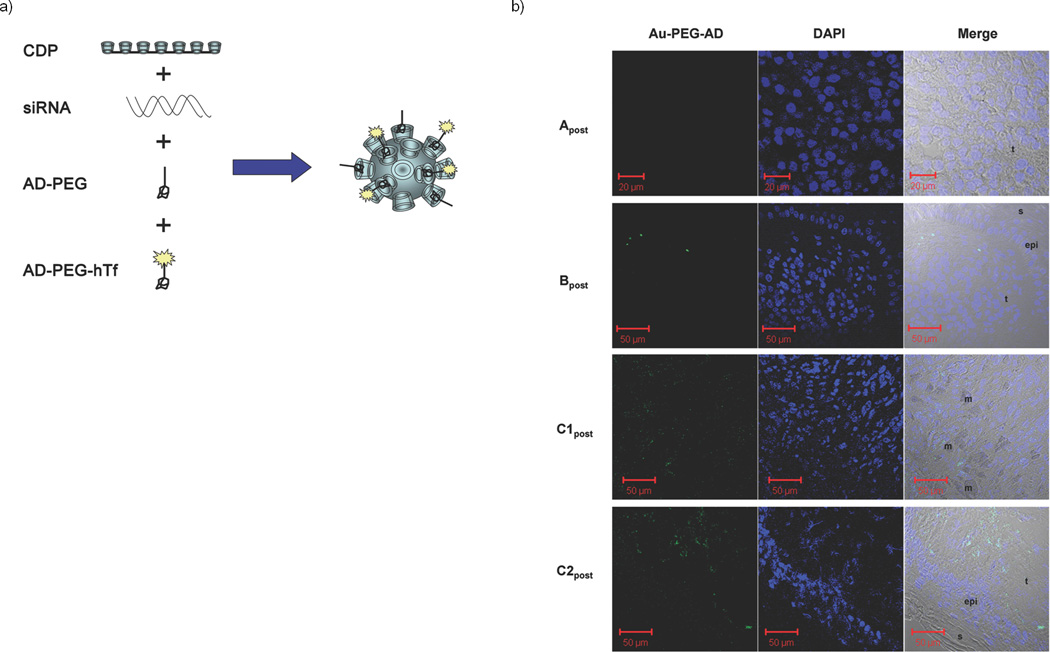
Detection of targeted nanoparticles in human tumors. (a). Schematic representation of the targeted nanoparticles. The polyethyleneglycol (PEG) molecules are terminated with adamantane (AD) that form inclusion complexes with surface cyclodextrins in order to decorate the surface of the nanoparticle with PEG for steric stabilization and PEG-hTf for targeting. (b). Confocal images of post-treatment biopsy sections from patients A, B and C: Au-PEG-AD stain (left), DAPI stain (middle), merged images of the left and right panels with the bright field (right). Image labels: epi = epidermis, t = tumor side, s = skin side, m = melanophage.
In order to ascertain whether the targeted delivery system can provide effective delivery of functional siRNA to human tumors, we investigated biopsies from three patients from three different dosing cohorts; patients A, B and C, all of whom had metastatic melanoma and received doses of CALAA-01 of 18, 24 and 30 mg-siRNA/m2, respectively. Given the highly experimental nature of this protocol, the regulatory process at both the local and federal levels explicitly precluded a provision for mandatory biopsies in all patients. Therefore, biopsies were obtained on a voluntary basis. Biopsies in these three patients were collected after the final dose of cycle 1 (denoted Apost, Bpost and C1post) and compared to archived tissue (denoted Apre, Bpre and C1pre). Patient C continued on therapy beyond one cycle and provided another set of biopsy materials (C2pre that was obtained approximately one month after the final dose of cycle 1 and C2post that was collected on the day of the final dose of cycle 2). Because of limited sample amount, only immunohistochemistry (IHC) and staining for the nanoparticles could be performed on the C1pre and C1post samples, and protein (for Western blot analyses) was only available from the C2pre and C2post samples. Details of this clinical trial will be reported elsewhere when completed.
The targeted nanoparticles (ca. 70 nm diameter) were administered i.v., as they are designed to circulate and then to accumulate and permeate in solid tumors.13 Within the tumor, the hTf targeting ligand assists in directing the nanoparticles into tumor cells overexpressing hTfR.19 To detect the nanoparticles in tumor cells, sections of the tumor tissue were stained for the presence of the nanoparticles using a 5 nm gold particle that is capped with thiolated PEG containing adamantane (AD) at the end distal to the thiol (AD-PEG-Au) to allow for multivalent binding to the cyclodextrins (Supplementary Scheme SI 2). The function of the stain has been previously confirmed using other cyclodextrin-containing particles,20 and is demonstrated here for the targeted nanoparticles carrying siRNA in vitro (Supplementary Fig. 1) and in vivo (Supplementary Figs. 2 and 3). Transmission electron microscopy (TEM) images of the nanoparticles confirm that in mice, the nanoparticles are intracellular (Supplementary Fig. 2). Samples A, B and C1, analyzed in a blinded fashion, demonstrated a heterogeneous distribution of nanoparticles only in post-dosing tumor tissue (Fig. 1 for post-dosing and Supplementary Fig. 4 for pre-dosing). The nanoparticles can localize intracellularly in tumor tissue and are not found in the adjacent epidermis (Fig. 1). In these biopsies TEM images were dominated by melanosomes21 inhibiting the identification of the nanoparticles (data not shown). Samples C1post and C2post reveal the highest number and intensity of stained regions, Bpost exhibits a decreased amount of staining relative to samples C1post and C2post (Fig. 1b), Apost does not reveal the presence of the stain (Fig. 1b), and all the pre-dosing samples are completely negative for the stain (Supplementary Fig. 4). This is the first example of a dose-dependent accumulation of targeted nanoparticles in tumors of humans from systemic injections for nanoparticles of any type.
Tumor RRM2 mRNA levels were measured by quantitative real time polymerase chain reaction (qRT-PCR) and were performed in a blinded fashion.22 Reduction in RRM2 mRNA is observed in the post-treatment samples (Fig. 2). Since samples Apre and Bpre are from tissues collected many months before the initiation of siRNA treatment, the fraction of the overall reduction in mRNA observed in Apost and Bpost attributable to the nanoparticle treatment cannot be directly ascertained. Unfortunately, we were not able to perform PCR on the C1 samples. However, the PCR data from the C2pre vs. C2post samples (collected 10 days apart) provide direct evidence for RRM2 mRNA reduction via the treatment of the patient with the nanoparticles.
Figure 2.
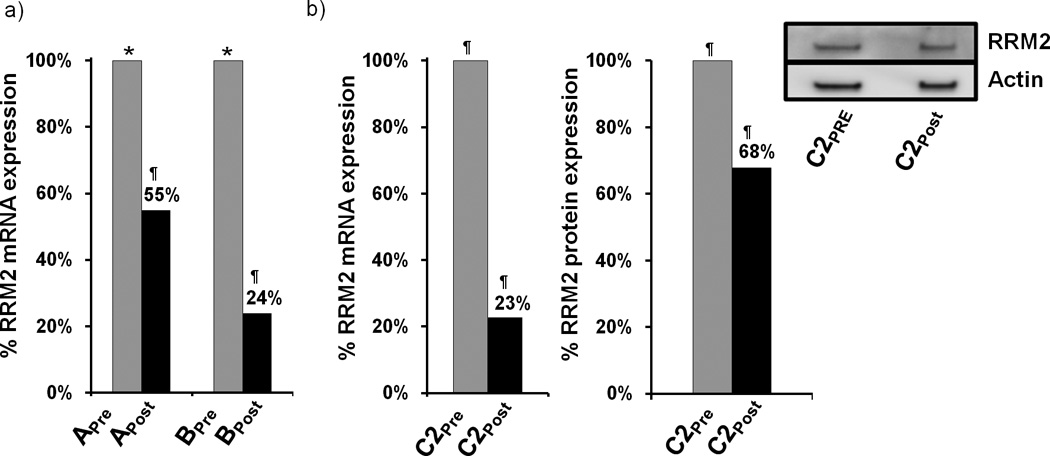
RRM2 mRNA and protein expression in tumor tissue. (a). qRT-PCR analysis of RRM2 mRNA levels in samples from patients A and B before and after dosing. RRM2 mRNA levels are normalized to TBP mRNA levels. Results are presented as percentage of the pre-dosing RRM2/TBP mRNA levels for each patient. (b). qRT-PCR and Western blot analysis of RRM2 protein expression from patient samples C2pre and C2post. Bar graph is average volume of Western blot bands from two independent experiments; one representative blot is pictured. Archived samples are indicated by (*); samples obtained during the trial are indicated by (¶).
To ascertain whether the RRM2 protein level is reduced in the tumor because of the siRNA treatment, IHC and Western blotting were employed as previously described in mice.23 Since RRM2 protein expression is largely restricted to the late G1/early S phase of cell cycle, not all of the tumor cells will be expressing RRM2. Figure 3 shows IHC data for RRM2 and TfR proteins in C1pre and C1post samples (IHC analyses were performed in a blinded fashion and 10 random regions of each sample were analyzed). Significant reduction in RRM2 is observed (mean scoring of RRM2 from the 10 sections was reduced 5-fold) after treatment while TfR levels are somewhat elevated (mean scoring of TfR from the 10 sections was increased 1.2-fold) in the C1pre and C1post samples. The low level of RRM2 that is observed by IHC in the C1post sample is maintained in the C2pre and C2post samples (by IHC). Western blot analyses of the C2pre and C2post samples reveal a reduction in the level of the RRM2 protein that is due to the siRNA treatment (RRM2 mRNA reductions exceeded the reduction levels obtained from protein but this could be due to post-transcriptional mechanisms that have been observed previously24). The decreases in the RRM2 mRNA and protein observed after treatment (Fig. 2b) suggest the siRNA treatment remains effective after several cycles of dosing. The IHC data from patient A do not reveal changes in RRM2 expression after dosing, while results from patient B are indicative of reductions in maximal RRM2 expression (IHC scoring of the regions of maximal expression showed a 1.5-fold decrease) but the overall mean expression levels remained relatively constant (IHC scoring of the 10 sections).
Figure 3.
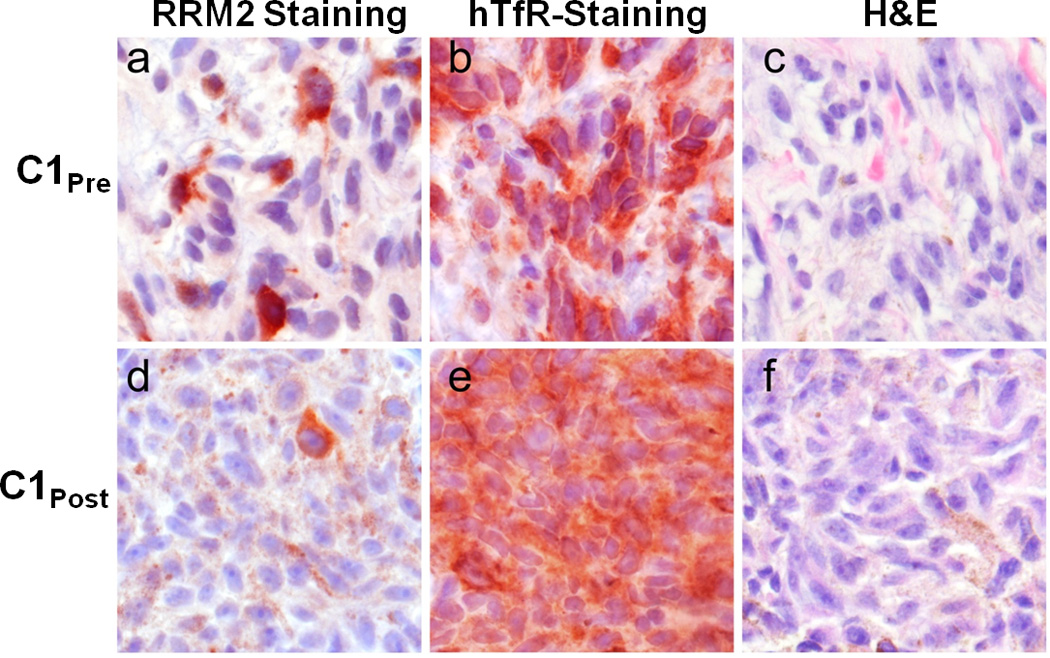
Ribonucleotide reductase (RRM2) and tranferrin receptor (TfR) protein expression in C1pre and C1post samples. Photomicrographs of malignant melanoma belonging to a, b, c, pre-treatment and d, e, f post-treatment samples. Protein expression is represented as brick-red (Nova Red) chromagen staining in immunohistochemically-treated slides (a, d: RRM2; b, e:TfR). c, f, The same tissues are stained with Hematoxylin and Eosin (H&E). d, e, f, Brown, diffuse, finely granular color seen in these images is the endogenous pigment of this lightly melanized tumor. Photomicrographs were captured using a 40× objective.
To demonstrate that the siRNA delivered via the targeted nanoparticles can engage the RNAi machinery, the mRNA cleavage products were characterized using a modified 5’-RNA ligand-mediated rapid amplification of cDNA ends (5’-RLM-RACE) PCR technique (Fig. 4). A RRM2 mRNA fragment, whose 5’ end matches the predicted cleavage site (10 base pairs from the 5’ end of the antisense strand), was detected in the C2pre and C2post samples, but not from Bpost, and Apost or their corresponding pre-treatment samples. RACE does not provide a quantitative measure of the amount of the fragments so the intensities of the bands cannot be correlated with amounts in the tissue samples. The presence of this RRM2 mRNA fragment from patient C indicates siRNA delivered via targeted nanoparticles can engage the RNAi machinery in a solid tumor of a human and induce the desired mRNA cleavage. Furthermore, this result suggests that at least a portion of the RRM2 mRNA and protein reductions observed from the C2 samples are due to a bona fide RNAi mechanism. The presence of the RRM2 mRNA fragment in the C2pre sample suggests that siRNA can provide an RNAi mechanism for several weeks (mRNA cleavage in the C2pre sample must originate from cycle one dosing) as the RRM2 protein levels remained relatively constant when compared to the C1post sample (IHC). We have shown that the length of the RNAi effects of delivered siRNA in both cells and animals (mice) is dependent on the doubling time of the cells being analyzed (longer inhibition times with longer cell doubling times).25 Gene silencing by siRNA can occur on the timescale observed here, ca. one month, provided the cell doubling times are long.25 Patient C had stable disease between these biopsies, and these mostly quiescent tumors have very slow growth kinetics that would be suitable to experience lengthy RNAi effects.25 Additionally, we do not know how long the nanoparticles reside within the cells and release siRNA. Since the nanoparticles are observed in the sample C1post and not the sample C2pre, they must disassemble within one month. Thus, the pharmacodynamics of the RNAi effects could be due to the combination of the nanoparticle disassembly time and the time that the siRNA resides within the RNAi machinery.
Figure 4.
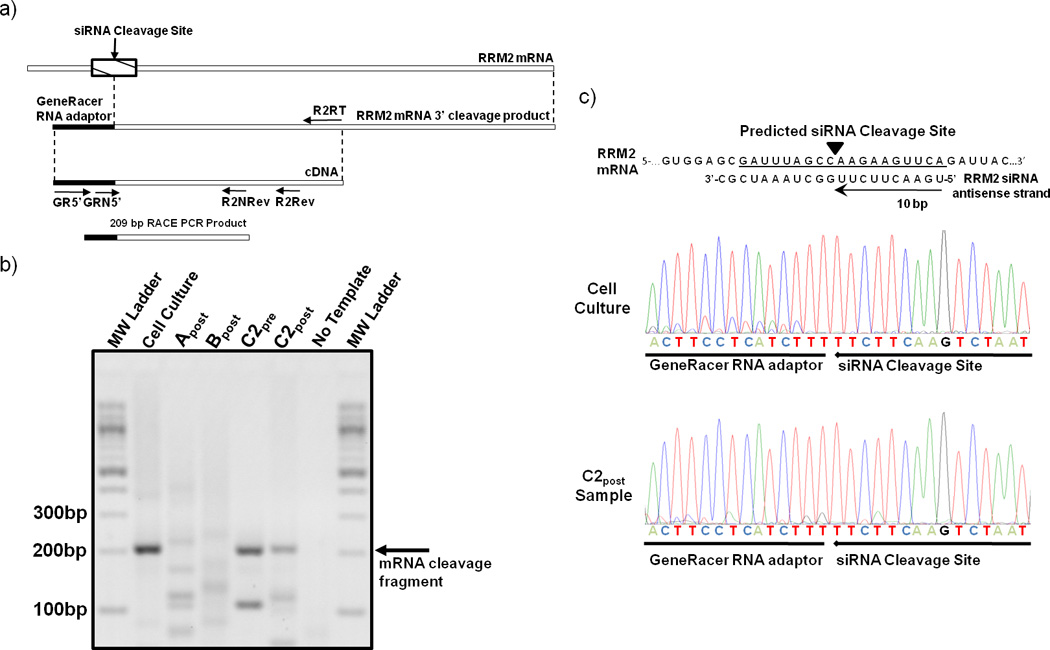
5’-RLM-RACE detection of siRNA induced mRNA cleavage fragment. (a) Schematic depicting the location of the predicted anti-RRM2 siRNA cleavage site and the primers used for PCR amplification of the cleavage fragment. (b) Agarose gel of 5’-RLM-RACE PCR amplification products from post treatment samples (A post, B post, C2post) and in vitro positive control (cell culture). (c) The RRM2 mRNA sequence and siRNA antisense strand are illustrated to show where the cleavage occurs with an RNAi mechanism. The sequence chromatographs obtained from an in vitro cell culture experiment with HT-144 melanoma cells and the patient C2post sample are illustrated.
The data presented here when taken together provide the first mechanistic evidence of RNAi in a human from an administered siRNA. Moreover, these data demonstrate the first example of dose dependent accumulation of targeted nanoparticles in human tumors. The reduction of the RRM2 mRNA and protein by the RRM2-specific siRNA is observed, and the results from 5’-RLM-RACE analyses reveal that the delivered siRNA engages the RNAi machinery. These data demonstrate that RNAi can occur in a human from a systemically delivered siRNA, and that siRNA can be used as a gene-specific therapeutic.
Methods Summary
Detection of targeted nanoparticles in biopsy samples
Snap-frozen patient biopsy samples were embedded in Tissue-Tek O.C.T. compound (Sakura) for the generation of 6 µm-thick cryosections. Upon immersion in PBS at 37 °C for 1 h to remove any surface O.C.T., and subsequent fixation with acetone at −20 °C for 20 minutes to permeabilize the cell membrane, sections received staining of PEGylated, AD-modified gold nanoparticles (Au-PEG-AD; see Supplementary Information for a description of their preparation) in the dark for 2 hours. Brief rinses with PBS were used to remove any nonspecifically bound Au-PEG-AD before mounting with ProLong Gold antifade reagent and staining with DAPI (Invitrogen, Carlsbad, CA). A Zeiss LSM 510 confocal scanning microscope used to collect the images (DAPI-excitation: 370 nm, emission: 440 nm; Au-PEG-AD-excitation: 488 nm, emission: 507 nm).
5’ RNA ligand mediated-RACE
5’-RLM-RACE was performed according to the Invitrogen GeneRacer manual with modifications. 2–8 µg of total RNA was ligated directly to 250ng GeneRacer RNA adaptor using T4 RNA ligase. Following phenol extraction and ethanol precipitation the purified ligation products were reverse transcribed using SuperScriptIII (Invitrogen) and a RRM2 gene specific reverse transcription primer (5’-CTCTCTCCTCCGATGGTTTG-3’). 5’RLM RACE PCR was performed using the GeneRacer 5’ and a RRM2 gene specific reverse primer (5’-GGCCAGGCATCAGTCCTCGTTTCTTG-3’). PCR was performed using a Bio-Rad MJ Mini personal thermocycler using PCR conditions of 95°C for 3 minutes (1 cycle), 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 1 minutes (40 cycles), 72°C for 10 minutes (1 cycle). A second round of nested PCR was then performed using the GeneRacer 5’ nested primer and an RRM2 gene-specific nested primer (5’-GGCCCAGTCTGCCTTCTTCTTGAC-3’). PCR products were run on a 2% agarose gel and stained with 1µg/µL ethidium bromide. PCR products were excised from gel and sequenced directly to confirm RACE band identities.
Methods
Detection of targeted nanoparticles in biopsy samples
Snap-frozen patient biopsy samples were embedded in Tissue-Tek O.C.T. compound (Sakura) to generate 6 µm-thick cryosections. Upon immersion in PBS at 37 °C for 1 h to remove any surface O.C.T., and in acetone at −20 °C for 20 min to permeabilize the cell membrane, sections received staining of PEGylated, adamantane-modified gold nanoparticles (Au-PEG-AD; see Supplementary Information for their preparation) in the dark for 2 h. Brief rinses with PBS were used to remove any nonspecifically bound Au-PEG-AD before mounting with ProLong Gold antifade reagent and staining with DAPI (Invitrogen, Carlsbad, CA). A Zeiss LSM 510 confocal scanning microscope (with a Plan-Neofluar 40X/0.75 objective and up to 2× digital zoom) was used to collect the images (DAPIexcitation: 740 nm (two-photon laser), emission filter: 390–490 nm; Au-PEG-AD-excitation: 488 nm (argon laser), emission filter: BP 500–550 nm IR). The measured resolution at which images were acquired is 512 × 512 pixels, and the image bit-depth is 8-bit. The Zeiss LSM Image Browser Software allows the extraction of images.
RNA Extraction
Patient samples preserved in RNALater (Ambion, Austin, TX) were suspended in TRIzol reagent (Invitrogen) and homogenized in a FastPrep-24 Tissue Homogenizer (MP Biomedicals, Solon, OH). Total RNA was purified from the aqueous phase of TRIzol extract using the PureLink RNA Mini Kit (Invitrogen) following manufacturer recommendations. RNA was extracted from archived patient samples using RecoverAll total nucleic acid isolation kit (Ambion) following manufacturer instructions.
5’ RNA ligand mediated-RACE
5-RLM-RACE was performed according to the Invitrogen GeneRacer manual with modifications. 2–8 µg of total RNA was ligated directly to 250ng GeneRacer RNA adaptor (5’-CGACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA-3’) using T4 RNA ligase (5 units) for 1 h at 37°C. Following phenol extraction and ethanol precipitation the purified ligation products were reverse transcribed using SuperScriptIII (Invitrogen) and a RRM2 gene specific reverse transcription primer (5’-CTCTCTCCTCCGATGGTTTG-3’) at 55°C for 45 min followed by inactivation at 70°C. 5’RLM-RACE-PCR was performed using the GeneRacer 5’ primer (5’-CGACTGGAGCACGAGGACACTGA-3’) and a RRM2 gene specific reverse primer (5’-GGCCAGGCATCAGTCCTCGTTTCTTG-3’). PCR was performed using a Bio-Rad MJ Mini personal thermocycler using PCR conditions of 95°C for 3 min (1 cycle), 95°C for 30 s, 60°C for 30 s, 72°C for 1 min (40 cycles), 72°C for 10 min (1 cycle). A second round of nested PCR was then performed using the GeneRacer 5’ nested primer (5-GGACACTGACATGGACTGAAGGAGTA-3’) and a RRM2 gene-specific nested primer (5’-GGCCCAGTCTGCCTTCTTCTTGAC-3’). PCR products were run on a 2% agarose gel and stained with 1µg/µl ethidium bromide. PCR products were excised from gel and sequenced directly to confirm RACE band identities. For the cell culture RACE experiments, 500,000 HT-144 melanoma cells were transfected with 20 nM RRM2 siRNA using Lipofectamine RNAiMax (Invitrogen). RNA was extracted for the RLM-RACE as describe above 48 h after transfection.
qRT-PCR
Patient RNA samples were reversed transcribed using SuperScriptIII reverse transcriptase. 0.4–200ng of white blood cell (WBC) cDNA was used as PCR template for standard curves of RRM2 and Tata Binding Protein (TBP). 2 µl of prepared sample cDNA/standard cDNA was used for triplicate Taqman Real time-PCR as described elsewhere.25 Concentrations of RRM2 and TBP in samples were calculated from the WBC cDNA standard curve, and RRM2 levels were normalized to TBP levels within the same sample.
Western blots
Total protein was recovered from the phenol/chloroform phase of TRIzol extraction (see description of the RNA extraction above). Samples were diluted to equivalent protein concentration and denatured via addition of beta-mercaptoethanol-containing Laimmli sample buffer. The primary antibodies were goat polyclonal anti-RRM2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), mouse polyclonal anti-actin antibody (BD Biosciences, San Jose, CA). The secondary antibodies were horseradish peroxidase-conjugated donkey anti-goat antibody and rabbit anti-mouse antibody (Santa Cruz Biotechnology). Development was done using SuperSignal West Dura Extended Duration Substrate (Thermo-Fisher, Waltham, MA). Blot images were captured using a Molecular Imager VersaDoc 3000 system (Bio-Rad, Hercules, CA). Band quantification was performed using Image-Quant TL software (GE/Amersham Biosciences, Piscataway, NJ).
Tissues and immunohistochemical assay
Formalin-fixed, paraffin-embedded (FFPE) human tissue samples from patient–matched pre- and post-treatment cases were obtained under UCLA IRB approval. Immunohistochemical (IHC) assays were performed using a Dako Autostainer Plus (Dako, Carpinteria, CA) with fresh sections of pre- and post-treatment cases stained at the same time. Tissue sections 4 µm thick were deparaffinized in xylene and rehydrated in graded alcohols. The sections were then placed in a pressure cooker (17.5 PSI, 122 °C; Biocare Decloaking Chamber, Biocare Medical LLC., Concord, CA) in 0.01M sodium citrate buffer (pH 6.0) or 0.1M Tris-HCl buffer (pH 9.0) for 10 min for heat antigen retrieval of RRM2 and TfR antigens, respectively. Endogenous peroxidase was quenched with 3% hydrogen peroxide at room temperature. Primary goat anti-human R2 polyclonal antibody, (catalog # sc-10846; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), was applied for 30 min at room temperature at a final concentration of 1.0 µg/ml (1:200). Mouse anti-human TfR monoclonal IgG1 antibody (clone H68.4, catalog number 13–6800; Invitrogen, Camarillo, CA) was applied for 30 min at room temperature at a final concentration of 0.5 µg/ml (1:1000). Antigen detection was accomplished using the Vectastain ABC Elite Goat HRP kit (catalog number PK-6105, Vector Labs, Burlingame, CA, USA) or the Dako Envision goat anti-mouse IgG secondary antibody with attached HRP-labeled dextran polymer (catalog number K4001; Dako, Carpinteria, CA), for RRM2 and TfR, respectively. All tissues were either amelanotic or only lightly melanized, therefore bleaching was not performed and Nova Red (catalog number SK-4800; Vector Labs, Burlingame, CA) was used as the chromagen to easily discern staining from any endogenous pigment. The sections were then counterstained with Meyer’s hematoxylin, followed by dehydration in graded alcohols, xylene, and cover-slipping. Human tonsil and colon cancer served as positive assay controls. Negative controls consisted of duplicate tissue sections stained with either non-immune pooled goat IgG (catalog number I-5000, Vector Labs, Burlingame, CA) or monoclonal mouse IgG1 (catalog number 02–6100; Invitrogen, Camarillo, CA) applied at identical final concentrations as used for RRM2 and TfR primary antibodies, respectively. For each sample, 10 random tumor regions were scored for maximal expression and mean expression.
Supplementary Material
1
Acknowledgements
We thank H. Sazegar, E. Seja, A. Villanueva, and the G-CRC nursing staff at UCLA, L. Kalinoski, J. Peterkin, S. Rele, Y. Liang and J.Y.C. Liu at Calando Pharmaceuticals for their assistance in conducting the clinical trial. We thank Dr. J. S. Economou at UCLA for performing the tumor biopsies, and Drs. B. Chmielowski and Z. Wainberg, also at UCLA, for patient referrals. Histotechnology support at the UCLA Biomarker Innovations Laboratory was provided by C. Savina and J. Reiss. We also thank Dr. J. Rossi at the City of Hope for discussions on RACE analyses. This work was supported in part by the National Cancer Institute Grant CA U54 119347 and the Daljit S. & Elaine Sarkaria Biomarker Laboratory Fund.
Footnotes
Author Contributions M.E.D., J.E.Z., A. R. and J.D.H planned the experiments, J.E.Z., D.S., C.H.J.C., C.A.A., Y.Y., A.T. and A.R. conducted the experiments, M.E.D., J.E.Z., J.D.H, D.S., C.H.J.C. and A.R. analyzed the data, and M.E.D., J.E.Z. and A.R. wrote the paper.
Author Information Preprints and permissions information is available at www.nature.com/reprints.
References
- 1.Bumcrot D, Manoharan M, Koteliansky V, Sah DWY. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006;2:711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 4.Elbashir SM, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 5.McCaffrey AP, et al. RNA interference in adult mice. Nature. 2002;418:38–39. doi: 10.1038/418038a. [DOI] [PubMed] [Google Scholar]
- 6.Lewis DL, Hagstrom JE, Loomis AG, Wolff JA, Herweijer H. Efficient delivery of siRNA for inhibition of gene expression in postnatal mice. Nat. Genet. 2002;32:107–108. doi: 10.1038/ng944. [DOI] [PubMed] [Google Scholar]
- 7.Song E, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 2003;9:347–351. doi: 10.1038/nm828. [DOI] [PubMed] [Google Scholar]
- 8.Soutschek J, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 9.DeVincenzo J, et al. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV) Antivir. Res. 2008;77:225–231. doi: 10.1016/j.antiviral.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Leachman SA, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol. Ther. 2010;18:442–446. doi: 10.1038/mt.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez R, et al. RNAi-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent anti-viral strategy. Antimicrob. Agents Chemother. 2009;53:3952–3962. doi: 10.1128/AAC.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinman ME, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol. Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 14.Heidel JD, et al. Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin. Cancer Res. 2007;13:2207–2215. doi: 10.1158/1078-0432.CCR-06-2218. [DOI] [PubMed] [Google Scholar]
- 15.Gatter KC, Brown G, Strowbridge IS, Wollston RE, Mason DY. Transferrin receptors in human tissues: their distribution and possible clinical relevance. J. Clin. Pathol. 1983;36:539–545. doi: 10.1136/jcp.36.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerqueira NMFSA, Pereira S, Fernandes PA, Ramos MJ. Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumor therapy. Curr. Med. Chem. 2005;12:1283–1294. doi: 10.2174/0929867054020981. [DOI] [PubMed] [Google Scholar]
- 17.Heidel JD, et al. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc. Nat. Acad. Sci. USA. 2007;104:5715–5721. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koldehoff M, Steckel NK, Beelen DW, Elmaagacli AH. Therapeutic application of small interfering RNA directed against bcr-abl transcripts to a patient with imatinib-resistant chronic myeloid leukaemia. Clin. Exp. Med. 2007;7:47–55. doi: 10.1007/s10238-007-0125-z. [DOI] [PubMed] [Google Scholar]
- 19.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc. Nat. Acad. Sci. USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schluep T, et al. Pharmacokinetics and tumor dynamics of the nanoparticle IT-101 from PET imaging and tumor histological measurements. Proc. Nat. Acad. Sci. USA. 2009;106:11394–11399. doi: 10.1073/pnas.0905487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudiger R, Schartl M, Kollinger G. Comparative studies on the ultrastructure of malignant melanoma in fish and human by freeze-etching and transmission electron microscropy. J. Cancer Res. Clin. Oncol. 1984;107:21–31. doi: 10.1007/BF00395486. [DOI] [PubMed] [Google Scholar]
- 22.Juhasz A, Vassilakos A, Chew HK, Gandara D, Yen Y. Analysis of ribonucleotide reductase M2 mRNA levels in patient samples after GTI-2040 antisense drug treatment. Oncol. Rep. 2006;15:1299–1304. [PubMed] [Google Scholar]
- 23.Bartlett DW, Davis ME. Impact of tumor-specific targeting and dosing schedule on tumor growth inhibition after intravenous administration of siRNA-containing nanoparticles. Biotechnol. Bioeng. 2008;99:975–985. doi: 10.1002/bit.21668. [DOI] [PubMed] [Google Scholar]
- 24.McClarty GA, Chan AK, Engstrom Y, Wright JA, Thelander L. Elevated expression of M1 and M2 components and drug-induced posttranscriptional modulation of ribonucleotide reductase in a hydroxyurea-resistant mouse cell line. Biochemistry. 1987;26:8004–8011. doi: 10.1021/bi00398a068. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucl. Acids Res. 2006;34:322–333. doi: 10.1093/nar/gkj439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1